

Abstract
Increasing evidence from sequence data from various environments, including the human gut, suggests the existence of a previously unknown putative seventh order of methanogens. The first genomic data from members of this lineage, Methanomassiliicoccus luminyensis and “Candidatus Methanomethylophilus alvus,” provide insights into its evolutionary history and metabolic features. Phylogenetic analysis of ribosomal proteins robustly indicates a monophyletic group independent of any previously known methanogenic order, which shares ancestry with the Marine Benthic Group D, the Marine Group II, the DHVE2 group, and the Thermoplasmatales. This phylogenetic position, along with the analysis of enzymes involved in core methanogenesis, strengthens a single ancient origin of methanogenesis in the Euryarchaeota and indicates further multiple independent losses of this metabolism in nonmethanogenic lineages than previously suggested. Genomic analysis revealed an unprecedented loss of the genes coding for the first six steps of methanogenesis from H2/CO2 and the oxidative part of methylotrophic methanogenesis, consistent with the fact that M. luminyensis and “Ca. M. alvus” are obligate H2-dependent methylotrophic methanogens. Genomic data also suggest that these methanogens may use a large panel of methylated compounds. Phylogenetic analysis including homologs retrieved from environmental samples indicates that methylotrophic methanogenesis (regardless of dependency on H2) is not restricted to gut representatives but may be an ancestral characteristic of the whole order, and possibly also of ancient origin in the Euryarchaeota. 16S rRNA and McrA trees show that this new order of methanogens is very diverse and occupies environments highly relevant for methane production, therefore representing a key lineage to fully understand the diversity and evolution of methanogenesis.
Keywords: Archaea, evolution, genomics, methanogenesis, Methanoplasmatales, Methanomassiliicoccales
The importance of biogenic methane has drawn scientific attention to several archaeal lineages not only as powerful new energy producers but also as major contributors to the global warming process (Weiland 2010; Montzka et al. 2011). Methanogenesis is an ancient metabolism specific of the Archaea that originated in the Euryarchaeota (Brochier et al. 2004; Bapteste et al. 2005; Gribaldo and Brochier-Armanet 2006). All methanogens belong to six euryarchaeal orders (i.e., Methanococcales, Methanopyrales, Methanobacteriales, Methanosarcinales, Methanomicrobiales, and Methanocellales) unified by their unique ability to gain energy from methane production. Three main methanogenic pathways are classically defined: methanogenesis from H2/CO2, acetoclastic methanogenesis, and methylotrophic methanogenesis, involving a conserved core of enzymes (FWD/FMD, FTR, MCH, MTD/HMD, MER, MTR, and MCR) common to all six orders of methanogens (fig. 1) (for a comprehensive review, see Hedderich and Whitman 2006). The last step of all these pathways consists of the conversion of methyl-coenzyme M (methyl-S-CoM) into CH4 and is performed by the same enzymatic complex in all methanogens, the methyl-coenzyme M reductase (MCR) (fig. 1, red box). The electrons used for this reduction reaction come either from H2 or a reduced cofactor F420 (F420H2) depending on the pathway.
Fig. 1.—
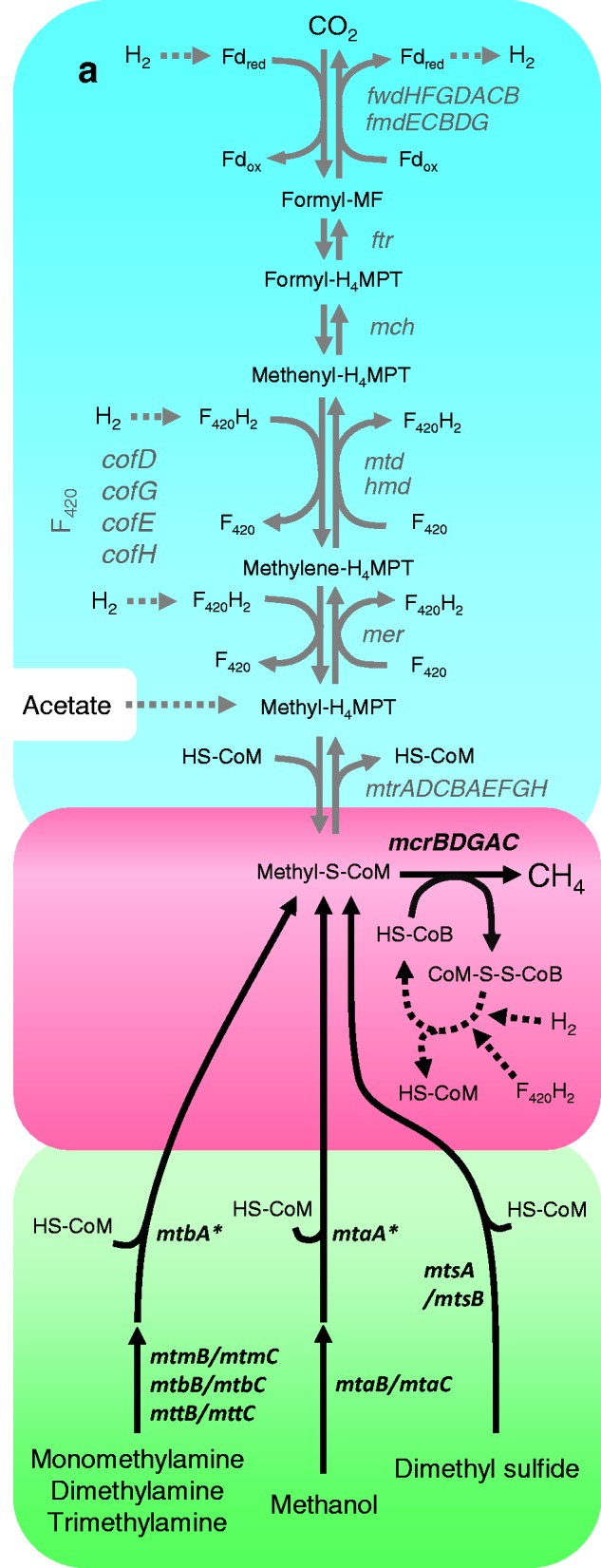
Main methanogenic pathways and associated genes. The blue box highlights the first six steps of methanogenesis from H2/CO2 (downward arrows) and the production of reducing equivalents during methylotrophic methanogenesis without external H2 source (upward arrows), while the dotted line in the blue box indicates the entry of the methyl group in the acetoclastic pathway. The green box highlights the first steps of methylotrophic methanogenesis and H2-dependent methylotrophic methanogenesis. The red box highlights the final step of methanogenesis in all three pathways. The genes absent in the Mx lineage are indicated in gray. The mtaA and mtaB genes are marked with an asterisk to signify that the homologs present in Mx cannot be presently assigned to one or the other enzyme category (see text for details). Dotted arrows designate the presence of steps not detailed on the figure. The oxidation of the carbonyl group of the acetate in the acetoclastic methanogenesis is not apparent on the figure. MFR, methanofuran; H4MPT, tetrahydromethanopterin; HS-CoM, coenzyme M; HS-CoB, coenzyme B, CoM-S-S-CoB, heterodisulfide of HS-CoM and HS-CoB; F420H2, reduced coenzyme F420; Fdred, reduced ferredoxin; Fdox, oxidized ferredoxin. For simplicity, tetrahydrosarcinapterin (H4SPT, an analog of H4MPT) is not displayed.
In methanogenesis from H2/CO2, methyl-S-CoM is produced from CO2 along six steps with formyl-, methenyl-, methylene-, and methyl-coenzymes as intermediates (fig. 1, blue box, downward arrows), and the electrons required to reduce methyl-S-CoM to methane derive from an external H2 source (fig. 1, red box). In acetoclastic methanogenesis, acetate is cleaved and its methyl group is transferred to H4MPT (fig. 1, blue box, dotted arrow) and then to HS-CoM. The electrons required to reduce methyl-S-CoM to methane are derived from the oxidation of the carboxyl group of the acetate. In methylotrophic methanogenesis, methyl-S-CoM is produced via transfer of a methyl group from methanol, methylamines (mono-, di-, and trimethylamine), or dimethyl sulfide to HS-CoM by enzymes specific for each substrate (van der Meijden et al. 1983; Wassenaar et al. 1996; Ferguson et al. 2000; Bose et al. 2008): MtaABC for methanol, MtmBC/MtbA for monomethylamine, MtbBC/MtbA for dimethylamine, MttBC/MtbA for trimethylamine, and MtsA/MtsB for dimethyl sulfide (fig. 1, green box). One methyl group associated with the coenzyme M is oxidized to CO2 along a reverse H2/CO2 methanogenesis pathway (fig. 1, blue box, upward arrows) to produce the reducing equivalents (F420H2 and Fdred) needed to reduce three other methyl-S-CoM to methane. A particular form of this metabolism is H2-dependent methylotrophic methanogenesis, which may be referred to as a fourth pathway (Welander and Metcalf 2005), where all the methyl-S-CoM is reduced into methane, the electrons being obtained from H2 coming from an external source (fig. 1, red box).
Most of the cultured Methanococcales, Methanopyrales, Methanobacteriales, Methanomicrobiales, and Methanocellales perform methanogenesis from H2/CO2 (Liu and Whitman 2008). The order Methanosarcinales comprises the most versatile species, capable of performing all four described methanogenic pathways (some Methanosarcina spp.), although some are uniquely dependent on acetoclastic methanogenesis (Methanosaeta spp.) or H2-dependent methylotrophic methanogenesis by using methanol, mono-, di-, tri-methylamine, and dimethyl sulfide (Methanomicrococcus blatticola [Sprenger et al. 2000]). The ability to use methanol via H2-dependent methylotrophic methanogenesis is also present in a few species belonging to the Methanobacteriales. Among them, Methanosphaera stadtmanae displays obligate H2-dependent methylotrophy from methanol, and it has lost the capability to reduce CO2 to methane or oxidize methanol to CO2, although it has kept most of the corresponding enzymes that might be used in other metabolic pathways (Fricke et al. 2006). As both Methanosphaera stadtmanae and Methanomicrococcus blatticola were isolated from the gut, specialization for H2-dependent methylotrophic methanogenesis could appear as an adaptation to these environments.
Based on the phylogenetic placement of methanogens in the archaeal tree and the analysis of the core enzymes, previous analyses have suggested that methanogenesis has a unique and early origin in the Euryarchaeota, likely after the divergence of Thermococcales, and would have been followed by subsequent multiple independent losses in nonmethanogenic euryarchaeal lineages (Brochier et al. 2004; Bapteste et al. 2005; Gribaldo and Brochier-Armanet 2006). Indeed, remnants of a methanogenic past are found in the genomes of Archaeoglobales, which still harbor the enzymes for the first five steps of methanogenesis from H2/CO2, now involved in the oxidation of lactate to CO2 (Möller-Zinkhan et al. 1989; Klenk et al. 1997). The composite and almost complete genome of an anaerobic methanotroph affiliated to ANME-1 displays the genes coding for the same conserved core of enzymes than the six orders of methanogens, except mer (Meyerdierks et al. 2010). The exact role of all these genes and the possible bypassing of Mer for the methanotrophic pathway were thoroughly discussed but remain to be elucidated (Meyerdierks et al. 2010).
A putative new lineage of methanogens unrelated to any of the six previously known orders was proposed by us a few years ago on the basis of sequence data (16S rRNA genes and a molecular marker of methanogenesis, mcrA) from human stools (Mihajlovski et al. 2008). Closely related phylotypes were reported by several other studies from animal gut, including humans, and various other environments such as soil, paddy fields, lakes, rivers, marine sediments, and subsurfaces (Kemnitz et al. 2005; Janssen and Kirs 2008; Scanlan et al. 2008; Evans et al. 2009; Mihajlovski et al. 2010; Biderre-Petit et al. 2011; Horz et al. 2012). One strain has now been isolated and one enriched from human feces, Methanomassiliicoccus luminyensis (Dridi et al. 2012) and “Candidatus Methanomethylophilus alvus” (Borrel et al. 2012), respectively. Moreover, two other strains have been enriched from termite gut, MpT1 and MpM2 (Paul et al. 2012), and very recently, an additional one from waste treatment sludge, “Candidatus Methanogranum caenicola” (Iino et al. 2013). Methanomassiliicoccus luminyensis has been shown to perform exclusively H2-dependent methanogenesis from methanol (Dridi et al. 2012), similar to Methanosphaera stadtmanae and Methanomicrococcus blatticola. Analyses on the other not-yet isolated strains strongly suggest that they have a similar specialization (Borrel et al. 2012; Paul et al. 2012; Iino et al. 2013). Two recent studies indicated that members of this lineage might also use methylamines based on the presence of the genes for the corresponding enzymes in the “Ca. M. alvus” genome (Borrel et al. 2012) and analysis of their transcripts induced by the addition of trimethylamine in rumen (Poulsen et al. 2013). Because of 16S rRNA phylogenetic proximity with the wall-less thermoacidophilic Thermoplasmatales, the name “Methanoplasmatales” was recently proposed for this order (Paul et al. 2012). However, as highlighted by Iino et al. (2013), the name “Methanomassiliicoccales” should instead be used, in accordance with the rules 9 and 15 of the International Code of Nomenclature of Bacteria (Lapage et al. 1992). Waiting for an agreement on its name, we will refer to this lineage as Mx in the current communication.
The first two genomes of Mx representatives, M. luminyensis and “Ca. M. alvus,” whose 16S rRNA gene sequences are only 87% identical, have recently become available (Borrel et al. 2012; Gorlas et al. 2012). Moreover, more than 40 genomes from all orders of methanogens are now available. This provides an unprecedented opportunity to clarify their evolutionary relationships with the other euryarchaeal lineages by markers alternative to 16S rRNA and to gain genomic insights into their metabolism, which we describe in the following sections.
Phylogenetic Placement of the Mx Order Supports Multiple Losses of Methanogenesis in the Euryarchaeota
Analysis of a concatenation of ribosomal proteins from 84 euryarchaeal genomes and comprising 7,472 amino acid positions robustly shows that M. luminyensis and “Ca. M. alvus” represent a monophyletic lineage that is not phylogenetically associated with any of the previously known orders of methanogens or the anaerobic methanotrophic ANME1 lineage (fig. 2a). Interestingly, the Mx order, albeit evolutionarily close to Thermoplasmatales as previously noted, robustly clusters with two lineages without cultured representatives: the planktonic Marine Group II (MG-II) and the sediment dwelling Marine Benthic Group D (MBG-D) for which the first genomic sequences were recently obtained (Iverson et al. 2012; Lloyd et al. 2013).
Fig. 2.—

Phylogenetic position of the Mx order and inferred losses of methanogenesis. (a) Bayesian phylogeny of Euryarchaeota based on a concatenation of 57 ribosomal proteins comprising 7,472 amino acid positions. Values at nodes represent Bayesian posterior probabilities and bootstrap values based on maximum likelihood analysis and 100 resamplings of the original data set. For clarity, only the values corresponding to the monophyly of orders and their evolutionary relationships are shown. “../” symbols indicate that the corresponding node was not recovered in the maximum likelihood phylogeny. The scale bar represents the average number of substitutions per site. For details on analyses, see Materials and Methods. Red crosses indicate complete loss of methanogenesis. Colored spots indicate presence of the genes of the respective methanogenic pathways/reactions as indicated by the colored boxes in figure 1. Crosses within spots indicate that the enzymes are present but not used to perform methanogenesis from H2/CO2 and/or from methylated compounds. Genomes with a green spot harbor the genes needed to use at least one type of methyl compound. (b) Maximum likelihood phylogeny of methanogens based on a concatenation of McrA-B-C-D-G protein sequences comprising 1,159 amino acid positions. Values at nodes represent bootstrap proportions calculated based on 100 resamplings of the original alignment and Bayesian posterior probabilities. For clarity, only the values corresponding to the monophyly of orders and their evolutionary relationships are shown. The scale bar represents the average number of substitutions per site. For details on analyses, see Materials and Methods.
The phylogenetic position of M. luminyensis and “Ca. M. alvus” in the tree of Euryarchaeota and their clustering with nonmethanogenic lineages pose the question of the origin of their metabolic capabilities. A tree based on a concatenation of the five markers of methanogenesis (McrA-B-C-D-G) that are shared by all methanogens and the ANME1 member (fig. 2b) is largely consistent with the ribosomal protein-based phylogeny, notably by recovering the monophyly of all orders. This strongly indicates that the current distribution of these components is not due to horizontal gene transfer but to vertical inheritance, and strengthens the scenario whereby methanogenesis emerged once, likely after the emergence of Thermococcales, and was subsequently lost multiple times independently during the evolutionary history of Euryarchaeota (red crosses in fig. 2a). Significantly, it also implies that the MG-II and MBG-D lineages descend from a methanogenic ancestor shared with M. luminyensis and “Ca. M. alvus.” Although predominantly found in anoxic sediments, it is unlikely that the MBG-D members are methanogens. The mcrA gene could not be amplified using genomic DNA extracted from the samples dominated by MBG-D in a study on hypersaline lake sediments, and isotopic measurements have indicated that methane production and consumption is negligible in that environment (Jiang et al. 2008). This lineage probably generates energy from metabolism of protein degradation (Lloyd et al. 2013). Accordingly, no genes coding for core methanogenesis enzymes were found in the MBG-D genomes, and the few homologs of the genes involved in energy conservation in methanogenesis from H2/CO2 (mtrAH, hdrABC, and mvhAGD) that are present in these genomes are also shared by nonmethanogenic microorganisms (Lloyd et al. 2013). According to their close relationship with the Mx lineage, the presence of these homologs in MBG-D members may be the vestige of a methanogenic past. In contrast, MG-II representatives live in fully oxygenated waters and do not harbor any remnant of enzymes involved in methanogenesis, which would therefore have been completely lost during adaptation from a strict anaerobic methanogenic lifestyle to this very different new environment.
Comparison of 16S rRNA gene-based and mcrA protein-based trees of sequences obtained from the same environmental samples allows delineating more precisely the current phylogenetic coverage of the Mx lineage (fig. 3). In both trees, “Ca. M. alvus” and M. luminyensis fall into two distinct, well-supported clades, consistent with a recent but less complete analysis (Paul et al. 2012). “Ca. M. alvus” is phylogenetically related to taxa obtained mainly from the gut of different animals including the Rumen Cluster C (RCC) (Janssen and Kirs 2008), whereas the M. luminyensis clade comprises clones reported from various nondigestive environments including representatives of the Rice Cluster III lineage (Kemnitz et al. 2005). It is presently unclear whether these differences in environmental distribution are real or due to a sampling bias. Other clades can also be observed that include phylotypes from diverse environments (fig. 3). Notably, a third cluster distantly related to those of “Ca. M alvus” and M. luminyensis is apparent, represented by multiple 16S and mcrA sequences from the water column of the meromictic Lake Pavin in France (Biderre-Petit et al. 2011). It was previously proposed that some of the McrA sequences closely related to the Mx order might represent the MBG-D lineage (Paul et al. 2012). However, our analysis and the fact that the genomes of MBG-D members do not harbor mcrA support the assignment of current McrA sequences exclusively to the Mx lineage.
Fig. 3.—

Diversity of the Mx order and its evolutionary relationships with other uncultured lineages. (a) Maximum likelihood McrA phylogeny of the candidate Mx order. (b) Maximum likelihood 16S rRNA phylogeny showing the uncultured lineages branching between the Mx lineage and the Thermoplasmatales/DVHE2, MG-II, and MBG-D lineages. Both trees were rooted by using six representatives of Methanobacteriales and Methanococcales that are not shown for clarity. Trees were calculated from a data set of 138 unambiguously aligned amino acid positions (McrA) and 1,113 unambiguously aligned nucleic acid positions (16S rRNA). Values at nodes represent bootstrap support calculated on 100 resamplings of the original data set and Bayesian posterior probabilities calculated by Bayesian analysis. For clarity, only the values corresponding to the monophyly of lineages and their evolutionary relationships are shown. “../” symbols indicate that the corresponding node was not recovered in the Bayesian phylogeny. The scale bar represents the average number of substitutions per site. For details on data set construction and tree calculation, see Materials and Methods. The dotted box delineates the candidate Mx order. Sequences from the complete genomes of “Candidatus Methanomethylophilus alvus” and Methanomassiliicoccus luminyensis are indicated in red; other colored sequences indicate McrA and 16S rRNA sequences retrieved from the same sample in previous studies. Sequences corresponding to the Lake Pavin sample from which we retrieved Mt coding gene sequences are underlined. Other than the already discussed losses of methanogenesis in the Thermoplasmatales/DVHE2, MG-II lineages, and MBG-D, additional putative losses can be inferred (red crosses with a question mark) in two uncultured lineages based on the environment from which the corresponding sequences were retrieved: oxygenated water column for MG-III; oxygenated and extremely acidic environments (pH <3) for TAC. Lineages 20c4, VC-21Arc6, CCA-47 (divided in two subgroup -1 and -2), ANT06-05, ASC21, F2apm1A36, and MKCST-A3 were derived from the July 2012 version (v.111) of the Silva Database (http://www.arb-silva.de/) and are named according to the clone definition. TMEG, Terrestrial Miscellaneous Euryarchaeotal Group; MBG-D, Marine Benthic Group D; MG-III, Marine Group III; MG-II, Marine Group II; TAC, Thermoplasmatales-related Acidic Cluster.
16S rRNA analysis also shows that a large number of presently uncultured lineages from diverse environments branch between the Mx/MGII/MBG-D clade and the Thermoplasmatales/DVHE2 clade (fig. 3b). Although some of these lineages are commonly found in anoxic environments, such as sediments, that are compatible with methanogenesis, others are mostly from aerobic and/or extremely acidic environments that are unsuitable for this metabolism, for example, the Marine Group-III (MG-III) and the Thermoplasmatales-related Acidic Cluster (TAC) (fig. 3b). The identification of the Mx lineage and its specific placement in the archaeal phylogeny therefore suggest that multiple independent losses of methanogenesis have occurred during euryarchaeal evolution even more times than previously proposed.
A Unprecedented Case of Reduction of the Methanogenic Pathway and Specialization on Methylated Compounds: Recent Adaptation or Ancestral Feature?
The wide distribution of Mx members in various environments and the fact they are not evolutionarily affiliated to any other previously known methanogenic order may suggest that they harbor specific characteristics. In particular, information on the metabolic capacities of “Ca. M. alvus” and M. luminyensis is important for further investigations of the role of these organisms in the human gut. Surprisingly, “Ca. M. alvus” and M. luminyensis are the first methanogens that appear to have totally lost all the genes involved in both the first six steps of methanogenesis from H2/CO2 and the oxidative part of methylotrophic methanogenesis (fig. 1, blue box, in gray font). These genes are otherwise present in all genomes from methanogens sequenced to date, including those that do not use the CO2 reduction/methyl group oxidation pathway for methanogenesis, such as Methanosphaera stadtmanae (Fricke et al. 2006) and Methanosaeta spp. (Zhu et al. 2012) (fig. 2a). Moreover, genes encoding the synthesis of the F420 coenzyme, an important electron carrier in methanogens, are also absent. This is in agreement with Paul et al. (2012), who observed a lack of F420 autofluorescence in their enrichments of MpT1 and MpM2. The absence of all these enzymes explains the fact that M. luminyensis is unable to grow on H2/CO2, acetate, or on methanol without an H2 external source (Dridi et al. 2012). Such metabolic profile was also observed for the strains belonging to the Mx lineage enriched from a microbial consortium (Paul et al. 2012; Iino et al. 2013), including “Ca. M. alvus” (Borrel et al. 2012), even if this has to be confirmed in pure cultures. Consistently, “Ca. M. alvus” and M. luminyensis harbor homologs of the enzymes that are involved in H2-dependent methylotrophic methanogenesis from methanol in Methanosarcinales and Methanobacteriales (MtaABC, fig. 1). Homologs of enzymes that may be potentially used to reduce other methylated compounds such as monomethylamine (MtmBC), dimethylamine (MtbBC), and trimethylamine (MttBC) (fig. 1) are also present in the two genomes (Borrel et al. 2012; Poulsen et al. 2013). Additionally, we identified genes for enzymes involved in methanogenesis from dimethyl sulfide (MtsAB) in both the “Ca. M. alvus” and M. luminyensis genomes (fig. 1). Such a potentially large range of substrates is unusual in methanogens and was so far restricted to the Methanosarcinales (Thauer et al. 2008). “Ca. M. alvus” has one copy each of mtaB and mtaC that are located close to each other in the genome as observed in other methanol-using methanogens, whereas M. luminyensis has three mtaBC clusters (fig. 4). The close association of mtaB and mtaC in these genomes suggests that they form a transcription unit as in other methanogens (Sauer et al. 1997; Fricke et al. 2006). Similar to mtaB and mtaC, the genes coding for the enzymes involved in the use of methylamines (mtmBC, mtbBC, and mttBC) are in close association with both genomes (data not shown). “Ca. M. alvus” and M. luminyensis have two and four copies of the mtmBC cluster, respectively, and both genomes have one copy each of the mtbBC and mttBC clusters (data not shown). “Ca. M. alvus” has three homologs of mtaA/mtbA and M. luminyensis has two, and all five genes are located apart in the respective genomes (fig. 4).
Fig. 4.—

Physical maps of mtaA/mtbA, mtaB, and mtaC. Physical maps of mtaA/mtbA, mtaB, and mtaC genes in “Candidatus Methanomethylophilus alvus” Mx1201, Methanomassiliicoccus luminyensis B10, in comparison with Methanosarcina acetivorans CA2 as a representative of Methanosarcinales, and with Methanosphaera stadtmanae DSM 3091 as a representative of Methanobacteriales. The contigs of M. luminyensis (the assembled genome is not yet available) are separated by slashes. MAP, Methyltransferase Activation Protein.
It has been proposed that methanol utilization by Methanosphaera stadtmanae occurred through acquisition of its mtaABC gene cluster via horizontal gene transfer from Methanosarcinales followed by specific gene duplications that multiplied the number of mtaABC copies in this species (Fricke et al. 2006). The large evolutionary distance between “Ca. M. alvus” and M. luminyensis poses the question of whether these two strains developed the capacity to use methyl compounds independently, as a specific adaptation to the gut environment and possibly via horizontal gene transfer from another methylotrophic methanogen, or if it is a more ancient feature of the whole Mx lineage. We retrieved partial homologs of genes involved in the use of methanol, methylamines, and dimethyl sulfide, related to M. luminyensis, from the metagenome of a wood degrading bioreactor, hosting a microbial community naturally associated with the wood at the beginning of the experiment (van der Lelie et al. 2012). Moreover, we successfully amplified and sequenced a genomic fragment containing a nearly complete mtaB gene and a partial mtaC gene affiliated with the Mx lineage from a Lake Pavin sample containing Mx members (underlined in fig. 3a) (see supplementary methods and table S1, Supplementary Material online, for details on these analyses). This indicates that the ability to use methylated compound may be widespread in the Mx lineage and not only restricted to gut representatives. Moreover, phylogenetic analysis of MtaB, MtaC, and MtaA/MtbA shows that the sequences of the Mx lineage are not intermixed with members of Methanosarcinales or Methanobacteriales but form robustly supported monophyletic groups (fig. 5). The fact that “Ca. M. alvus” and M. luminyensis belong to two distantly related clusters (fig. 3) and the branching of nongut environmental sequences in the MtaA/MtbA and MtaB trees (fig. 5) argues against an acquisition of methylotrophy (regardless of dependency on H2) via recent horizontal gene transfer to “Ca. M. alvus” and M. luminyensis would rather suggest their presence in the ancestor of the whole Mx order. Interestingly, the same may be said for Methanobacteriales, weakening the previous hypothesis of a specific horizontal gene transfer from Methanosarcinales to Methanosphaera stadtmanae.
Fig. 5.—

Phylogeny of MtaA/MtbA, MtaB, and MtaC. Unrooted maximum likelihood phylogenies of (a) MtaA/MtbA, (b) MtaB, and (c) MtaC. The Methanosarcinales are indicated in green, the Methanobacteriales in blue, and the Mx lineage in red. Sequences in bold belong to uncultivated microorganisms. Of the environmental sequences from Lake Pavin obtained in this study, only MtaB could be included in phylogenetic analysis because the others were too partial. Also, the MtaC homolog from the bioreactor metagenome was too partial to be included in the tree. However, these are clearly affiliated with the Mx lineage. Values at nodes represent bootstrap support calculated based on 100 resamplings of the original data set. For clarity, only the values corresponding to the monophyly of lineages and their evolutionary relationships are shown. The scale bar represents the average number of substitutions per site. For details on data set construction and tree calculation, see Materials and Methods.
The tree of MtaA/MtbA does not allow assignment of the Mx homologs to one or the other enzyme category (fig. 5a), which will have to wait for a full functional characterization of these proteins. The low sequence similarity between these predicted enzymes in “Ca. M. alvus” (max. 48%) and in M. luminyensis (max. 44%) might reflect a substrate specialization of each copy on either methylamines or methanol. Unfortunately, none of the multiple copies of homologs of mtaA/mtbA in “Ca. M. alvus” and M. luminyensis are adjacent to the mtaBC or their homologs involved in methylamines utilization (not shown), as found in the genomes of Methanobacteriales and Methanosarcinales (fig. 4). This prevents speculation on the putative specialization of any of these enzymes on either methanol or methylamines. Alternatively, each of these enzymes might be implied in both methanol and methylamine utilization, similar to the MtaA of Methanosarcina barkeri, which is predominantly active on methanol but also in the transfer of the methyl group originating from trimethylamine, albeit less efficiently (Ferguson et al. 1996).
Finally, the phylogenies of the enzymes involved in methanogenesis from methylamines and dimethyl sulfide display a pattern consistent with the Mta trees (supplementary fig. S1, Supplementary Material online). The presence of partial homologs affiliated with the Mx lineage in the metagenome of the wood degrading bioreactor (van der Lelie et al. 2012) suggests that versatility to use a large panel of methylated compounds may be a characteristic of the whole order and possibly already present in the Mx ancestor, although the limited taxonomic distribution of homologs prevents a definitive answer.
It appears therefore that the use of methylated compounds, or at least methanol, is not a specific adaptation to the gut environment but is rather an ancestral feature in the Mx lineage. All representatives studied so far, despite their evolutionary distance, seem to display H2-dependent methylotrophic methanogenesis as their sole methanogenic pathway, including “Candidatus Methanogranum caenicola” recently enriched from wastewater sludge (Iino et al. 2013). Obligatory H2-dependent methylotrophic methanogenesis is also reminiscent of that of two gut methanogens (Methanosphaera stadtmanae and Methanomicrococcus blatticola), suggesting that the gut environment might have selected organisms with this metabolism or more versatile methylotrophic methanogens that then evolved to become dependent on an external H2 source as an adaptation to gut conditions. If such adaptation occurred for the two sequenced Mx members, it would have occurred recently in the case of M. luminyensis because this species is closely related to sequences retrieved in a number of environments other than the digestive tracts of animals (fig. 3). However, because the complete loss of the genes involved in the first six steps of methanogenesis from H2/CO2 and the oxidative part of methylotrophic methanogenesis observed in the two Mx genomes likely have required a number of metabolic adjustments, it is possible that it occurred once and not recently. It is therefore tempting to speculate that this loss occurred in the ancestor of the Mx lineage and that all of its members are now obligate H2-dependent methylotrophic methanogens. Genomic data and characterization of additional Mx members, in particular from nongut environments such as those from the “Lake Pavin cluster,” will be important to know if the dependency on H2 for methylotrophic methanogenesis is a recent evolutionary event (i.e., an adaptation to gut environment) or an ancestral feature of the whole lineage.
Materials and Methods
Exhaustive homology searches of the complete set of archaeal ribosomal proteins were performed by BlastP, TBlastN, and different seeds on a local database of 85 complete euryarchaeal genomes obtained from the NCBI, including “Ca. M. alvus” and Methanomassiliicoccus luminyensis (accession numbers CP004049.1 and CAJE01000000.1, respectively). Each protein data set was aligned by using Muscle (Edgar 2004) with default parameters, and unambiguously aligned positions were automatically selected by using the BMGE software for multiple-alignment trimming (Criscuolo and Gribaldo 2010) with a BLOSUM30 substitution matrix. Trimmed alignments were then concatenated by allowing a maximum of 12 missing proteins per data set, giving a final data set of 57 ribosomal proteins and 7,472 amino acid positions for phylogenetic analysis. A Bayesian tree was calculated by PhyloBayes (Lartillot et al. 2009), with the CAT model and four categories of evolutionary rates. Two MCMC chains were run in parallel until convergence, and the consensus tree was calculated by removing the first 25% of trees as burnin. A maximum likelihood tree was also calculated by PhyML (Guindon et al. 2010) and the LG amino acid substitution model (Le and Gascuel 2008) with four rate categories as suggested by the Akaike information criterion (AIC) implemented in Treefinder (Jobb et al. 2004).
Search for homologs of the McrA-B-C-D-G proteins and assembly of a concatenated data set followed the same strategy detailed above, giving a final data set of 1,159 amino acid positions. Maximum likelihood trees were calculated by PhyML and Treefinder and the LG amino acid substitution model with four rate categories, as suggested by the AIC implemented in Treefinder. A Bayesian tree was calculated by PhyloBayes as described above.
Both r-protein and MCR trees were rooted in the branch leading to Thermococcales, as indicated by previously published analyses of large concatenated protein data sets (Brochier-Armanet et al. 2011). This allowed keeping more amino acid positions for analysis and avoiding potential artifacts caused by the introduction of a distant outgroup.
16S rRNA gene and McrA protein sequences closely related to the Mx lineage were retrieved from NCBI and included in a data set of 138 unambiguously aligned amino acid positions (McrA) and 1,113 unambiguously aligned nucleic acid positions (16S rRNA) selected by BMGE. Bayesian trees were calculated by MrBayes (Ronquist and Huelsenbeck 2003) with the mixed model and four rate categories (McrA) and the 4by4 model and four rate categories (16S rRNA), as suggested by the AIC implemented in Treefinder. For Bayesian analysis, two MCMC chains were run in parallel until convergence, and the consensus tree was calculated by removing the first 25% of trees as burnin. Maximum likelihood 16S rRNA and McrA trees were calculated by PhyML with the GTR model and LG model, respectively, and four rate categories, as suggested by the AIC implemented in Treefinder.
Homologs of the enzymes involved in methylotrophic methanogenesis were exhaustively searched against our local database, and additional homologs from related protein families were gathered by BlastP and TBlastN at the NCBI. Preliminary trees including all available homologs allowed extracting monophyletic Mt clusters from methylotrophic archaea that were used to build refined unrooted phylogenies with a restricted taxonomic sampling and more unambiguously aligned positions to the main aim of showing that members of these three orders are not intermixed. Final maximum likelihood trees were calculated by PhyML with the LG model with four rate categories, and 100 bootstrap resamplings of the original data set. Additional mtaA, mtaB, and mtaC sequences were also recovered from the WGS database at the NCBI and in the metagenomes deposited in the public database of MG-RAST (Meyer et al. 2008). Primers targeting the mtaBC gene cluster designed on the sequences of “Ca. M. alvus” and M. luminyensis were used to amplify environmental sequences of mtaBC from genomic DNA previously extracted from the Lake Pavin water column (Denonfoux et al. 2013) (supplementary methods and table S1, Supplementary Material online). The sequences of mtaB and mtaBC were deposited in GenBank under accession numbers KF302411–KF302419 and KF302420–KF302421, respectively.
Conclusions
The growing availability of genomic data from the Archaea is appreciably increasing our knowledge of its diversity and evolutionary history (Brochier-Armanet et al. 2011). The number of lineages that currently lack any cultured or isolated members indicates that much remains to be elucidated, even from within our own gut microbiota. Our phylogenomic analysis justifies the proposal that the Mx lineage represents a seventh order of methanogens because it robustly separates from all previously known orders based on multiple markers analysis and likely harbors specific metabolic abilities (H2-dependent methylotrophy by use of a large variety of methylated compounds) combined with a unique genomic signature (lack of the first six steps of methanogenesis from H2/CO2 and the oxidative part of methylotrophic methanogenesis). Mx representatives were recovered from environments identified as main contributors to atmospheric methane such as digestive systems of ruminants and termites, rice fields, peat bog, and freshwater and marine sediments, highlighting the importance of taking into account this whole new lineage in future culture-independent studies. The ancestral ability for methylotrophy, at least from methanol, in Euryarchaeota seems to parallel that for methanogenesis from H2/CO2. Our analysis therefore expands the range of metabolic capacities of the methanogen that gave rise to the majority of present-day euryarchaeal lineages.
Supplementary Material
Supplementary table S1 and figure S1 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
G.B. and J.-F.B. are grateful to William Tottey for reading a draft version of this manuscript and for useful discussion. P.W.O’T.s lab is supported by Dept. Agriculture, Fisheries and Marine, and Science Foundation Ireland. S.G. is supported by the French Agence Nationale de la Recherche (ANR) through Grant ANR-10-BINF-01-01 “Ancestrome.”
Literature Cited
- Bapteste E, Brochier C, Boucher Y. Higher-level classification of the Archaea: evolution of methanogenesis and methanogens. Archaea. 2005;1:353–363. doi: 10.1155/2005/859728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biderre-Petit C, et al. Identification of microbial communities involved in the methane cycle of a freshwater meromictic lake. FEMS Microbiol Ecol. 2011;77:533–545. doi: 10.1111/j.1574-6941.2011.01134.x. [DOI] [PubMed] [Google Scholar]
- Borrel G, et al. Genome sequence of “Candidatus Methanomethylophilus alvus” Mx1201, a methanogenic archaea from the human gut belonging to a seventh order of methanogens. J Bacteriol. 2012;194:6944–6945. doi: 10.1128/JB.01867-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose A, Pritchett MA, Metcalf WW. Genetic analysis of the methanol-and methylamine-specific methyltransferase 2 genes of Methanosarcina acetivorans C2A. J Bacteriol. 2008;190:4017–4026. doi: 10.1128/JB.00117-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochier-Armanet C, Forterre P, Gribaldo S. Phylogeny and evolution of the Archaea: one hundred genomes later. Curr Opin Microbiol. 2011;14:274–281. doi: 10.1016/j.mib.2011.04.015. [DOI] [PubMed] [Google Scholar]
- Brochier C, Forterre P, Gribaldo S. Archaeal phylogeny based on proteins of the transcription and translation machineries: tackling the Methanopyrus kandleri paradox. Genome Biol. 2004;5:R17–R17. doi: 10.1186/gb-2004-5-3-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo A, Gribaldo S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10:210. doi: 10.1186/1471-2148-10-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denonfoux J, et al. Gene capture coupled to high-throughput sequencing as a strategy for targeted metagenome exploration. DNA Res. 2013;20:185–196. doi: 10.1093/dnares/dst001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dridi B, Fardeau ML, Ollivier B, Raoult D, Drancourt M. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int J Syst Evol Microbiol. 2012;62:1902–1907. doi: 10.1099/ijs.0.033712-0. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PN, et al. Community composition and density of methanogens in the foregut of the Tammar wallaby (Macropus eugenii) Appl Environ Microbiol. 2009;75:2598–2602. doi: 10.1128/AEM.02436-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson DJ, Gorlatova N, Grahame DA, Krzycki JA. Reconstitution of dimethylamine: coenzyme M methyl transfer with a discrete corrinoid protein and two methyltransferases purified from Methanosarcina barkeri. J Biol Chem. 2000;275:29053–29060. doi: 10.1074/jbc.M910218199. [DOI] [PubMed] [Google Scholar]
- Ferguson DJ, Krzycki JA, Grahame DA. Specific roles of methylcobamide: coenzyme M methyltransferase isozymes in metabolism of methanol and methylamines in Methanosarcina barkeri. J Biol Chem. 1996;271:5189–5194. doi: 10.1074/jbc.271.9.5189. [DOI] [PubMed] [Google Scholar]
- Fricke WF, et al. The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J Bacteriol. 2006;188:642–658. doi: 10.1128/JB.188.2.642-658.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlas A, Robert C, Gimenez G, Drancourt M, Raoult D. Complete genome sequence of Methanomassiliicoccus luminyensis, the largest genome of a human-associated Archaea species. J Bacteriol. 2012;194:4745–4745. doi: 10.1128/JB.00956-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribaldo S, Brochier-Armanet C. The origin and evolution of Archaea: a state of the art. Philos Trans R Soc Lond Ser B Biol Sci. 2006;361:1007–1022. doi: 10.1098/rstb.2006.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hedderich R, Whitman W. Physiology and biochemistry of the methane-producing archaea. In: Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, Dworkin M, editors. Prokaryotes: a Handbook on the Biology of Bacteria. New York: Springer; 2006. pp. 1050–1079. [Google Scholar]
- Horz HP, Seyfarth I, Conrads G. McrA and 16S rRNA gene analysis suggests a novel lineage of archaea phylogenetically affiliated with Thermoplasmatales in human subgingival plaque. Anaerobe. 2012;18:373–377. doi: 10.1016/j.anaerobe.2012.04.006. [DOI] [PubMed] [Google Scholar]
- Iino T, et al. Candidatus Methanogranum caenicola: a novel methanogen from the anaerobic digested sludge, and proposal of Methanomassiliicoccaceae fam. nov. and Methanomassiliicoccales ord. nov., for a methanogenic lineage of the class Thermoplasmata. Microb Environ. 2013;28:244–250. doi: 10.1264/jsme2.ME12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson V, et al. Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science. 2012;335:587–590. doi: 10.1126/science.1212665. [DOI] [PubMed] [Google Scholar]
- Janssen PH, Kirs M. Structure of the archaeal community of the rumen. Appl Environ Microbiol. 2008;74:3619–3625. doi: 10.1128/AEM.02812-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HC, et al. Dominance of putative marine benthic Archaea in Qinghai Lake, north-western China. Environ Microbiol. 2008;10:2355–2367. doi: 10.1111/j.1462-2920.2008.01661.x. [DOI] [PubMed] [Google Scholar]
- Jobb G, von Haeseler A, Strimmer K. TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol Biol. 2004;4:18. doi: 10.1186/1471-2148-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kemnitz D, Kolb S, Conrad R. Phenotypic characterization of Rice Cluster III archaea without prior isolation by applying quantitative polymerase chain reaction to an enrichment culture. Environ Microbiol. 2005;7:553–565. doi: 10.1111/j.1462-2920.2005.00723.x. [DOI] [PubMed] [Google Scholar]
- Klenk H-P, et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- Lapage SP, et al. International code of nomenclature of bacteria: bacteriological code, 1990 revision. Washington (DC): ASM Press; 1992. [PubMed] [Google Scholar]
- Lartillot N, Lepage T, Blanquart S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25:2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- Liu Y, Whitman WB. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann N Y Acad Sci. 2008;1125:171–189. doi: 10.1196/annals.1419.019. [DOI] [PubMed] [Google Scholar]
- Lloyd KG, et al. Predominant archaea in marine sediments degrade detrital proteins. Nature. 2013;496:215–218. doi: 10.1038/nature12033. [DOI] [PubMed] [Google Scholar]
- Meyer F, et al. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerdierks A, et al. Metagenome and mRNA expression analyses of anaerobic methanotrophic archaea of the ANME1 group. Environ Microbiol. 2010;12:422–439. doi: 10.1111/j.1462-2920.2009.02083.x. [DOI] [PubMed] [Google Scholar]
- Mihajlovski A, Alric M, Brugere JF. A putative new order of methanogenic Archaea inhabiting the human gut, as revealed by molecular analyses of the mcrA gene. Res Microbiol. 2008;159:516–521. doi: 10.1016/j.resmic.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Mihajlovski A, Dore J, Levenez F, Alric M, Brugere JF. Molecular evaluation of the human gut methanogenic archaeal microbiota reveals an age-associated increase of the diversity. Environ Microbiol Rep. 2010;2:272–280. doi: 10.1111/j.1758-2229.2009.00116.x. [DOI] [PubMed] [Google Scholar]
- Möller-Zinkhan D, Börner G, Thauer R. Function of methanofuran, tetrahydromethanopterin, and coenzyme F420 in Archaeoglobus fulgidus. Arch Microbiol. 1989;152:362–368. [Google Scholar]
- Montzka S, Dlugokencky E, Butler J. Non-CO2 greenhouse gases and climate change. Nature. 2011;476:43–50. doi: 10.1038/nature10322. [DOI] [PubMed] [Google Scholar]
- Paul K, Nonoh JO, Mikulski L, Brune A. “Methanoplasmatales,” Thermoplasmatales-related archaea in termite guts and other environments, are the seventh order of methanogens. Appl Environ Microbiol. 2012;78:8245–8253. doi: 10.1128/AEM.02193-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen M, et al. Methylotrophic methanogenic Thermoplasmata implicated in reduced methane emissions from bovine rumen. Nat Commun. 2013;4:1428. doi: 10.1038/ncomms2432. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Sauer K, Harms U, Thauer RK. Methanol: coenzyme M methyltransferase from Methanosarcina barkeri. Eur J Biochem. 1997;243:670–677. doi: 10.1111/j.1432-1033.1997.t01-1-00670.x. [DOI] [PubMed] [Google Scholar]
- Scanlan P, Shanahan F, Marchesi J. Human methanogen diversity and incidence in healthy and diseased colonic groups using mcrA gene analysis. BMC Microbiol. 2008;8:79. doi: 10.1186/1471-2180-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger WW, van Belzen MC, Rosenberg J, Hackstein J, Keltjens J. Methanomicrococcus blatticola gen. nov., sp. nov., a methanol-and methylamine-reducing methanogen from the hindgut of the cockroach Periplaneta americana. Int J Syst Evol Microbiol. 2000;50:1989–1999. doi: 10.1099/00207713-50-6-1989. [DOI] [PubMed] [Google Scholar]
- Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol. 2008;6:579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- van der Lelie D, et al. The metagenome of an anaerobic microbial community decomposing poplar wood chips. PLoS One. 2012;7:e36740. doi: 10.1371/journal.pone.0036740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meijden P, et al. Methyltransferases involved in methanol conversion by Methanosarcina barkeri. Arch Microbiol. 1983;134:238–242. doi: 10.1007/BF00407765. [DOI] [PubMed] [Google Scholar]
- Wassenaar RW, Daas P, Geerts WJ, Keltjens JT, Van der Drift C. Involvement of methyltransferase-activating protein and methyltransferase 2 isoenzyme II in methylamine: coenzyme M methyltransferase reactions in Methanosarcina barkeri Fusaro. J Bacteriol. 1996;178:6937–6944. doi: 10.1128/jb.178.23.6937-6944.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland P. Biogas production: current state and perspectives. Appl Microbiol Biotechnol. 2010;85:849–860. doi: 10.1007/s00253-009-2246-7. [DOI] [PubMed] [Google Scholar]
- Welander PV, Metcalf WW. Loss of the mtr operon in Methanosarcina blocks growth on methanol, but not methanogenesis, and reveals an unknown methanogenic pathway. Proc Natl Acad Sci U S A. 2005;102:10664–10669. doi: 10.1073/pnas.0502623102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, et al. The genome characteristics and predicted function of methyl-group oxidation pathway in the obligate aceticlastic methanogens, Methanosaeta spp. PLoS One. 2012;7:e36756. doi: 10.1371/journal.pone.0036756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.