

Abstract
Gene families differ in composition, expression, and chromosomal organization between conifers and angiosperms, but little is known regarding nucleotide polymorphism. Using various sequencing strategies, an atlas of 212k high-confidence single nucleotide polymorphisms (SNPs) with a validation rate of more than 92% was developed for the conifer white spruce (Picea glauca). Nonsynonymous and synonymous SNPs were annotated over the corresponding 13,498 white spruce genes representative of 2,457 known gene families. Patterns of nucleotide polymorphisms were analyzed by estimating the ratio of nonsynonymous to synonymous numbers of substitutions per site (A/S). A general excess of synonymous SNPs was expected and observed. However, the analysis from several perspectives enabled to identify groups of genes harboring an excess of nonsynonymous SNPs, thus potentially under positive selection. Four known gene families harbored such an excess: dehydrins, ankyrin-repeats, AP2/DREB, and leucine-rich repeat. Conifer-specific sequences were also generally associated with the highest A/S ratios. A/S values were also distributed asymmetrically across genes specifically expressed in megagametophytes, roots, or in both, harboring on average an excess of nonsynonymous SNPs. These patterns confirm that the breadth of gene expression is a contributing factor to the evolution of nucleotide polymorphism. The A/S ratios of Medicago truncatula genes were also analyzed: several gene families shared between P. glauca and M. truncatula data sets had similar excess of synonymous or nonsynonymous SNPs. However, a number of families with high A/S ratios were found specific to P. glauca, suggesting cases of divergent evolution at the functional level.
Keywords: Picea, Medicago, nucleotide polymorphism, synonymous and nonsynonymous substitutions, expression profiles, selection
Introduction
Assessing the genetic bases of adaptation and speciation is an important challenge to understand the evolution of functional diversity. Numerous interdependent factors have been shown to affect protein evolution in simple model organisms such as yeasts or microbial systems (reviewed by Pál et al. 2006). In plants, the development of next-generation sequencing has recently enabled the analysis of nucleotide polymorphism patterns at a genome-wide scale to address fundamental questions related to genome evolution (Cao et al. 2011; Horton et al. 2012; Weigel 2012), physiological specialization (Branca et al. 2011), domestication, and selection (Lam et al. 2010; Xu et al. 2011; Zheng et al. 2011). These studies have focused on a few angiosperm species with a fully sequenced genome, given the difficulty in distinguishing orthologous from paralogous single nucleotide polymorphisms (SNPs) when a reference sequence is not available. The understanding of these processes may be broadened by comparing gymnosperms and angiosperms, which diverged nearly 300 million years ago (Savard et al. 1994). Wide-scale studies of the patterns of nucleotide polymorphism in gymnosperm genes are still lacking and would represent a useful starting point to identify divergent and shared evolutionary trends between gene families and between angiosperm and gymnosperm lineages.
Conifers, a major order of the gymnosperms, carry genomes characterized by many distinctive and unique features, including their very large size (Murray 1998), the scarcity of recent whole-genome duplication (Jiao et al. 2011; Nystedt et al. 2013) and relative paralysis of their genome macrostructure (Pavy et al. 2012a), a generally low recombination rate at the chromosome level (Jaramillo-Correa et al. 2010), the abundance of retroelements (Morgante and De Paoli 2011; Nystedt et al. 2013), and the size of some gene families overpopulated in comparison to angiosperms (Rigault et al. 2011). In conifers, genetic diversity has only been analyzed for a few to about 100 genes (e.g., Palmé et al. 2008; Wachowiak et al. 2009; Namroud et al. 2010; Pavy et al. 2012b). Studies have found a lower mutation rate compared with annuals and angiosperm model species (e.g., Palmé et al. 2008). Rates of molecular evolution may be linked to the life history where perennial species such as conifers and trees have different evolutionary rates than modern herbaceous plants (Bousquet et al. 1992; Gaut et al. 1992; Smith and Donoghue 2008). Part of this variation may be associated with more neutral demographic factors, but part of it may also be linked to specific functional mechanisms related to adaptation. For instance, it has been shown that gene expression may be a driver of nucleotide polymorphism in plants (Yang and Gaut 2011).
This study aimed at determining the landscape of nucleotide polymorphism across a large part of the transcriptome of a conifer to provide a broad view of its distribution across the spectrum of gene families involved in various physiological functions. We focused on white spruce (Picea glauca [Moench] Voss), which is a largely distributed transcontinental boreal conifer species in North America with important ecological and economic roles. Most of its transcriptome were identified and coding sequences were assembled into unique gene representatives (Rigault et al. 2011). We used these sequence data to build a high-confidence SNP atlas using a new procedure and extensive validation through genotyping. We classified 13,500 P. glauca expressed genes carrying high-confidence SNPs according to their molecular functions, gene families, and expression patterns and analyzed the differential distribution of their coding SNPs across these classes. We also compared the P. glauca landscape of nucleotide polymorphism with that of the angiosperm Medicago truncatula to delineate contrasting patterns. This study represents an investigation of unprecedented scale for a nonflowering plant.
Material and Methods
Plant Material, Reference Data Set, and Sequences
We sampled 212 white spruce individuals (Picea glauca [Moench] Voss) from natural populations and germplasm collections (supplementary table S1, Supplementary Material online). Sequences were obtained from 48 different cDNA libraries representing a wide variety of tissues and treatments, with the Sanger technology (Pavy et al. 2005; Ralph et al. 2008; Rigault et al. 2011) and next-generation sequencing technologies (Rigault et al. 2011) (supplementary table S1, Supplementary Material online). Each library was assembled from as many as 40 unrelated individuals. We processed 64.5 million reads to obtain 33.5 million quality reads representing 2.9 billion bp of sequence that were used to search for SNPs (supplementary table S2, Supplementary Material online). All of the sequence data from expressed sequence tag and cDNA clusters were previously described and released (supplementary table S2, Supplementary Material online) (Pavy et al. 2005; Rigault et al. 2011).
We performed a reference-guided alignment against a catalog of 27,720 P. glauca cDNA clusters (Rigault et al. 2011). This reference set was obtained from Sanger sequences and included 23,589 full-length insert cDNAs (FLICs); it is considered as a robust reference set (Rigault et al. 2011) that strengthens SNP discovery. They comprised 99.5% of next-generation sequences (454 GS and Illumina GAII) distinct from those used to develop the reference data set (supplementary table S2, Supplementary Material online). The 454 GS libraries (3.2% of the sequences) included 80 unrelated individuals from natural populations and germplasm collections from Quebec; the Illumina GAII sequenced libraries (96.3% of the sequences) were from a population of 30 individuals collected in germplasm collections from Quebec and representative of trees from natural populations (supplementary table S1, Supplementary Material online). Procedures for sequence processing, quality filtering, and alignments are described in supplemental materials (supplementary methods S1 Supplementary Material online).
SNP Prediction
Variant calling was done with the VarScan software (version 2.2) (Koboldt et al. 2009) with the following parameter settings: min-coverage = 2; min-reads2 = 1; min-avg-qual = 10; min-var-freq = 0.0; P = 2.0. Given the number of individuals represented in the sampling, singleton SNPs and SNPs with a minor allele frequency (MAF) <0.01 were presumed to be sequencing errors and were discarded. For each SNP, VarScan computed a P value representing the significance of variant read count versus expected baseline error of 0.001; it is based on Fisher’s exact test on the read counts supporting reference and specified variant alleles. VarScan also computed the frequency of the variant allele, defined as the fraction of the read counts of the specified variant within the sum of the read counts of the supporting reference; the read counts of the other variants, if present, are dismissed in the calculation.
Genotyping relying on the Infinium iSelect platform (Illumina, San Diego, CA) was used to assess the validity of a subset of 5,938 predicted SNPs (Pavy et al. 2013), which is an unusually large validation sample. The true positive (TP) rate was defined as the rate of the predicted SNPs which were polymorphic with at least two genotypic classes represented in the genotyping data obtained.
Coding SNP Analysis
Coding sequences (cds) were determined in FLICs by using the getorf program from EMBOSS applications, version 6.4.0.0 (Rice et al. 2000), and the longest cds was retained. SNPs positioned in cds were classified as nonsynonymous or synonymous by comparing the translated amino acids from the reference codon and the codon containing the SNP. Multi-allelic SNPs were excluded from analyses. A python script was developed to calculate numbers of nonsynonymous and synonymous sites in each cds for sites with a depth of 10 reads or more, with the method of Hartl and Clark (2007) (as Novaes et al. 2008). In a coding region, the number of synonymous sites was defined as the number of 4-fold degenerate sites plus one-third of the number of 2-fold degenerate sites. Similarly, the number of nonsynonymous sites was defined as the number of nondegenerate sites plus two-thirds of the number of 2-fold degenerate sites. We compared the number of substitutions per nonsynonymous site (A) and the number of substitutions per synonymous site (S) and calculated the A/S ratio for each gene. The A/S ratio is largely correlated with the Ka/Ks ratio usually derived from alignments made between species or divergent lineages (Liu et al. 2008). The adjusted ratio (A + 1)/(S + 1) was used to include the genes with no synonymous substitutions in the analysis.
Sequence similarities were searched at the protein level with the BlastX program against several databases. Genes with no match (e-value <e−10) against Arabidopsis (TAIR10), rice (MSU6), Amborella (http://www.amborella.org/, last accessed October 11, 2013) but with a match either against pines (unigenes were downloaded from the Plant Genome Database and translated with the getorf program; http://www.plantgdb.org/, last accessed October 11, 2013) or Douglas-fir (assembly of Howe et al. [2013] translated with the getorf program) were declared as conifer-specific genes. Picea glauca gene families were assigned by using similarities with PFAM families (Rigault et al. 2011; e-value <e−10). A BlastX search was performed with Blast2GO using the default parameters (but e-value <e−10) against the nonredundant protein sequence database; the gene ontology (GO) mapping step was run with the plant GO-Slim terms (Conesa et al. 2005). Gene set enrichment analyses (Subramanian et al. 2005) were conducted to identify functional classes with a significant asymmetric distribution toward the extremes of a list of FLICs ranked according to the adjusted A/S ratio using Fatiscan (Al-Shahrour et al. 2007; Medina et al. 2010). Briefly, Fatiscan splits the ordered list of A/S values into 30 partitions of the same size, a number which has been optimized during the development of the software (Al-Shahrour et al. 2007). Then, it sequentially applies two-tailed Fisher’s exact tests over the contingency tables formed with the two sides of different partitions to test for a possible enrichment in gene categories on one side or the other of the list of A/S values (Al-Shahrour et al. 2007). Therefore, P values need to be corrected to account for multiple testing and the correction by the false discovery rate (FDR) was applied (Benjamini and Hochberg 1995). The distributions of the P values obtained from the Fatiscan analyses were skewed toward small values (data not shown). The FDR correction on such data is conservative; therefore, we reported the trends observed from the enrichment gene set analyses both with and without correction.
Picea glauca expression data were retrieved from the PiceaGenExpress database in which genes are classified according to their expression levels across eight tissues or organs (Raherison et al. 2012). Gene expression classes were used for the 13,498 genes considered in the analysis of nucleotide polymorphism to build an expression file for further clustering analysis with the Mev tool (Saeed et al. 2003; http://www.tm4.org/mev.html, last accessed October 11, 2013) by using the Pearson correlation analysis with the K-means method (30 clusters).
Analysis of M. truncatula SNPs
A comparative analysis was carried out with the SNP data set and annotations for 30,769 genes from M. truncatula (Branca et al. 2011), which was chosen as a representative of the angiosperm phylum. The M. truncatula gene SNPs were identified in genomic sequences of 26 accessions from natural or mapping populations, which were self-fertilized (Branca et al. 2011). Medicago truncatula SNPs were appropriate for comparison because 1) they are available and reliable; 2) they were representative of a broad genetic diversity of M. truncatula; 3) the classifications between synonymous and nonsynonymous SNPs and sites were available; and 4) they were well distributed throughout the gene space (Branca et al. 2011). The M. truncatula gene annotations were mined according to A/S ratios; however, to maximize the number of genes considered, the Fatiscan analysis was performed on the adjusted A/S values (discussed earlier).
Results
Using Genotyped SNPs Data to Identify High-Confidence SNPs
A P. glauca SNP atlas was constructed starting from 33.5 million quality reads which were ascribed to 27,645 distinct coding sequences from the P. glauca gene catalog (Rigault et al. 2011) (fig. 1). Then, the identification of high-confidence SNPs considered three main criteria: the sequencing depth, the VarScan P value, and the MAF (table 1). From the sequence alignments, 373,686 nonsingleton SNPs (i.e., a SNP polymorphism that is present in at least two reads) were identified with a MAF ≥0.01. Genotyping data were available for 5,938 of the SNPs (Infinium array PgAS1 in Pavy et al. 2013). They were used to determine the TP rates and assess variations as a function of sequencing depth, the MAF, and the P value obtained with the variant calling software VarScan (table 1). The findings are summarized in supplementary methods S2, Supplementary Material online, and were used to define three main criteria to develop an atlas of high-confidence P. glauca SNPs: 1) selection of nonsingleton SNPs, 2) a sequence depth ≥10, and 3) a VarScan P < 0.10. With these criteria, the overall TP rate was maximized with a value of 92.1%. This validation rate is a conservative estimate, given that the Infinium iSelect (Illumina) genotyping array used, PgAS1, had a maximum success rate of 92.3% for SNPs previously confirmed using genotyping arrays based on the Illumina Golden Gate assay (Pavy et al. 2013). Hence, most of the failures (7.9%) are not due to miscalling SNPs but to the inherent limits of the hyperplex genotyping assay. However, a high false-negative rate (27.8%) was one drawback of the application of stringent criteria aiming to maximize the TP rate, given that 1,662 of the 5,986 in silico SNPs successfully genotyped with the array were not present in our final set of high-confidence SNPs. When we did not apply the alignment depth and the VarScan P value criteria, the TP rate of non-validated SNPs decreased to 87.7% among SNPs tested with the genotyping array.
Fig. 1.—

Tools and data used to delineate the Picea glauca atlas of 212,765 high-confidence SNPs. 1The P. glauca reference gene catalog was described by Rigault et al. (2011). It encompassed 27,720 sequences representative of distinct transcribed genes. 2http://bioinformatics.bc.edu/marthlab/wiki/index.php/Software (last accessed October 11, 2013). 3Koboldt et al. (2009). 4A nonsingleton SNP is a nucleotide polymorphism that is present on at least two reads. 5Pavy et al. (2013) described the genotyping array and released the genotyping data. 6A full-length insert clone (FLIC) sequence represents a sequence encompassing the entire length of a cloned cDNA insert. 7 & 8 RNA transcript sequence completion was determined by Rigault et al. (2011). Complete cds were similar to a reference protein (Arabidopsis, rice, poplar, grape, Swissprot BlastX e-value <e−10). The sequence was declared as confirmed complete cds if it was similar over the entire protein7. It was declared as predicted complete cds if the cds was similar over part of the protein but the transcript extended long enough on either side to cover the entire protein length. 9The other cds were either partial or complete but with no match with a reference protein.
Table 1.
Effect of the Depth of Sequence Alignment, the VarScan P Value, and the MAF upon the Number of Predicted SNPs and the True Positive Rate in P. glauca
| Number of Nonsingleton SNPs with a MAF ≥1% | Number of Nonsingleton SNPs Tested by Infinium iSelect Genotypinga | Number of Nonsingleton SNPs Successfully Genotyped | Minimum TP rate (%)b | |
|---|---|---|---|---|
| Depth of sequence alignment | ||||
| 0–10 | 24,115 | 155 | 117 | 75.5 |
| 10–20 | 40,340 | 332 | 283 | 85.2 |
| 20–30 | 33,934 | 307 | 262 | 85.3 |
| 30–40 | 28,496 | 288 | 250 | 86.8 |
| 40–50 | 24,563 | 292 | 246 | 84.2 |
| 50–100 | 83,399 | 1,154 | 1,018 | 88.2 |
| 100–150 | 50,356 | 862 | 746 | 86.5 |
| 150–200 | 35,992 | 602 | 503 | 83.5 |
| ≥200 | 52,491 | 1,946 | 1,784 | 91.7 |
| Total | 373,686 | 5,938 | 5,209 | 87.7 |
| SNP P value computed by VarScanc | ||||
| 0.00–0.05 | 193,437 | 4,789 | 4,420 | 92.3 |
| 0.05–0.10 | 21,616 | 174 | 150 | 86.2 |
| 0.10–0.15 | 39,025 | 277 | 212 | 76.5 |
| 0.15–0.20 | 0 | 0 | 0 | — |
| 0.20–0.25 | 95,493 | 543 | 310 | 57.1 |
| ≥0.25 | 0 | 0 | 0 | — |
| Total | 349,571 | 5,783 | 5,092 | 88.1 |
| MAFd | ||||
| 0.01–0.05 | 33,176 | 404 | 356 | 88.1 |
| 0.05–0.10 | 37,859 | 586 | 532 | 90.8 |
| 0.10–0.15 | 29,481 | 596 | 548 | 91.9 |
| 0.15–0.20 | 22,667 | 477 | 442 | 92.7 |
| 0.20–0.25 | 19,281 | 544 | 508 | 93.4 |
| 0.25–0.30 | 17,615 | 489 | 453 | 92.6 |
| 0.30–0.35 | 15,052 | 481 | 433 | 90.0 |
| 0.35–0.40 | 13,270 | 472 | 435 | 92.2 |
| 0.40–0.45 | 13,665 | 470 | 439 | 93.4 |
| 0.45–0.50 | 12,987 | 444 | 424 | 95.5 |
| Total | 215,053 | 4,963 | 4,570 | 92.1 |
Note.—The true positive rate was determined from 5,938 genotyped SNPs.
aSNPs predicted by VarScan and genotyped with the PgAS1 Infinium SNP array (Pavy et al. 2013).
bThis is a minimum rate, given that the success rate of the Infinium iSelect high-throughput genotyping array PgAS1 reached 92.3% for P. glauca SNPs previously confirmed by other means (see Pavy et al. 2013).
cSubset of SNPs predicted with a depth of alignment ≥10.
dSubset of SNPs predicted with a depth of alignment ≥10 and a VarScan P < 0.10.
The 215,053 high-confidence SNPs from the atlas populated 17,568 distinct expressed genes and represented 212,765 different polymorphic sites; a slightly lower number of sites was obtained because of those encompassing more than two nucleotides (table 1). The atlas is publicly available and includes the confidence parameters, the SNP annotation, and the gene annotations (supplementary table S3, Supplementary Material online). SNPs have been deposited in dbSNP (http://www.ncbi.nlm.nih.gov/SNP/snp_viewBatch.cgi?sbid=1058878, last accessed October 11, 2013). Because the SNP atlas is anchored onto a gene catalog, it enables the investigation of nucleotide polymorphism in conjunction with predicted gene functions and with gene expression data. The patterns of nucleotide polymorphism were analyzed by targeting SNPs contained in coding sequences of FLICs. It has been shown that SNPs are not randomly distributed along the coding sequence (cds) (Lu et al. 2010) and FLICs are expected to adequately represent the true nucleotide polymorphism in the cds, whereas incomplete cDNAs from 3′ or 5′ sequences may introduce biases. We identified 15,382 FLICs that contained at least 100 sites with a depth ≥10 reads and at least one SNP; half of FLICs (7,786) encompassed a complete coding sequence (fig. 1). We analyzed 13,498 coding sequences (cds) for which the nature of the sites (partially synonymous or nonsynonymous) could be determined over 50% of the codons, which gave a total of 10,712,082 sites (positions with a depth ≥10) and a number of sites that was highly representative of the FLIC length (supplementary fig. S1, Supplementary Material online). The snped FLICs (i.e., FLICs with one SNP or more) had a mean length of 1,256 bp (SD = 479) and an average of 1,079 sites (SD = 523) (redundant positions with depth ≥10) (supplementary fig. S1, Supplementary Material online). They encompassed 55.5% of the known P. glauca genes and represented about half of the gene space estimated at 32.7k genes (Rigault et al. 2011). These genes were representative of 2,457 gene families corresponding to 37.1% of the known conifer gene families. The number of high-confidence SNPs per snped FLIC was 12.7 but was highly variable (SD = 12.7) (supplementary fig. S1, Supplementary Material online). The SNP abundance was 1.24 SNPs per 100 sites on average and also ranged widely (SD = 1.0) (supplementary fig. S1, Supplementary Material online). The SNP abundances were significantly lower (t = 1.0, P < 2.2e−16) in cds (1.15 SNP/100 sites or 1 SNP per 87 sites) compared with untranslated regions (1.29 SNP/100 sites or 1 SNP per 77 sites).
Distribution of SNPs Varies among Functional Classes and Protein Families
Functional classes were investigated to search for groups of genes with A/S ratios that were statistically above (highest) or below (lowest) the overall distribution of A/S ratios. Functional classes were determined following two approaches, that is, based on GO terms and protein families sharing PFAM domains. The A/S ratio was determined as the number of substitutions per nonsynonymous sites (A) over the number of substitutions per synonymous sites (S) (fig. 2). The GO term analysis identified 19 molecular functions, 32 biological processes, and 18 cellular components terms associated with low A/S ratios (P < 0.05), a majority of which were significant after correcting for multiple testing (FDR-adjusted P < 0.05) (see Materials and Methods; supplementary fig. S2, Supplementary Material online). Genes with the lowest A/S ratios belonged to several functional classes including transcription and translation machineries, enzymes, signal transduction, transport, structural molecule activities, several metabolism levels (catabolism, secondary metabolism, and photosynthesis), response to several stimuli and cell death, and a few terms related to development (growth, reproduction, and differentiation). The analysis based on the biological processes showed a differential distribution of the A/S ratios in genes involved in metabolism (carbohydrate metabolism, lipid metabolism, and secondary metabolism) and development (growth, reproduction, and differentiation) (supplementary fig. S2b, Supplementary Material online). Also, the cellular component terms involving membranes and cell walls, plastids, nucleus, and cytoplasm were associated with the lowest A/S ratios (supplementary fig. S2c, Supplementary Material online).
Fig. 2.—

Number of substitutions per nonsynonymous site (A) and number of substitutions per synonymous site (S) for P. glauca (13,498 genes; a) and Medicago truncatula (30,768 genes; b) genes encompassing SNPs. The yellow triangles represent the averages. The dashed line represents the null expectation if substitutions were randomly distributed (A = S). The solid line is the slope averaged over all data (average A for all coding sequences divided by average S for all coding sequences). Data points for genes from two families involved in response to biotic stress (LRR family) and abiotic stress (dehydrins) are colored.
Next, the patterns of nucleotide polymorphism were analyzed across gene families based on 7,622 genes belonging to 2,457 PFAM families. In total, 102 families were significantly differentially distributed (P < 0.05) toward the highest or lowest A/S ratios (supplementary table S4, Supplementary Material online). We focused our analyses on 48 families represented by 10 gene members or more (supplementary table S4, Supplementary Material online, and figs. 3 and 4). The majority of these families could be grouped into only five types of proteins: enzymes, proteins associated with membranes or cytoskeleton, proteins involved in the degradation or protection of proteins, in protein–protein interactions, and in the regulation of gene expression or in DNA folding (fig. 4). As with the GO term analysis, a majority of the families (96.1%) were characterized by the lowest A/S ratios; however, four families were associated with the highest A/S ratios, that is, the AP2 family, the ankyrin repeat family, the leucine-rich repeat (LRR) family, and the dehydrins (figs. 2 and 3a). The LRRs and dehydrins were among genes with the highest A/S ratios with averages of 0.66 and 1.19, respectively (figs. 2 and 3a). For the AP2 and ankyrin repeat families, the A/S ratios were moderate (0.27 and 0.31, respectively, on average).
Fig. 3.—

Relationship between protein families and their imbalance in A/S ratios in Picea glauca (a) and Medicago truncatula (b). For each label (y axis), the bars represent the proportion of genes (%) with the high (blue) or low (green) adjusted A/S ratios (see Materials and Methods). Labels shown are those with statistically significant over- or underrepresentation among genes with high or low A/S ratios (gene set enrichment analysis, Fisher’s exact test, two-tailed, P < 0.05; gene families indicated in red exhibited a more stringent significance at FDR-adjusted P < 0.05). Numbers indicate the average A/S for each data set with high or low adjusted A/S ratios. For example, in P. glauca, 0.06% of the genes in the family of dehydrins were in the lowest A/S partition (with average A/S = 0.36) and 0.31% were in the highest one (with average A/S = 1.19). Boxes highlight families with mostly high A/S ratios (in pink), families shared in the analysis of both P. glauca and M. truncatula (in yellow), and families encoding enzymes (in orange).
Fig. 4.—

Functions of the protein families encompassing 10 members or more in Picea glauca (a) and Medicago truncatula (b) and associated with the lowest (a1; b1) or highest (a2; b2) A/S ratios (gene set enrichment analysis, Fisher’s exact test, two-tailed, P < 0.05).
For all other families, our threshold-free statistical method gave A/S ratios mostly below the 0.15 cutoff often used in similar studies (fig. 3a). The nine families that were enriched among the genes with the lowest A/S ratios (FDR-adjusted P < 0.05) included core histones, multicopper oxidases, kinases, cytochromes P450, and several families related to the degradation of proteins (ubiquitination pathways, proteasome subunits) or polysaccharides (pectate lyases) (fig. 3a).
Next, we focused on genes related to responses to environmental stresses including apoptosis and/or the cell wall degradation to determine whether their A/S ratios could pinpoint particular patterns. We report here on the results by restricting the analysis to the 14 such families with 10 members or more (fig. 3a), but the results of the overall analysis are provided in supplementary table S4, Supplementary Material online. Among families the most populated with genes harboring the highest A/S ratios, the dehydrins, AP2/DREB, and LRR families are known to be involved in response to stresses (fig. 3a). Among families the most populated with genes harboring the lowest A/S ratios, several have been linked to response to biotic stress including programed cell death (NB-ARC, terpene synthases, Miro-like proteins, chitinases I, subtilases, pectate lyases, and inhibitor of pectin methylesterases) or high temperature stress (Hsp70 and Hsp20) or freezing (AWPM-19-like family) (fig. 3a). Cell wall degrading enzymes, some of which are involved in altering the wall of the pathogenic fungi, were also characterized by the lowest A/S ratios (fig. 3a).
Gene Families with Extreme A/S Ratios Partially Overlap between P. glauca and M. truncatula
A similar analysis of A/S ratios among gene families was conducted for an angiosperm model organism by using publicly available M. truncatula data (fig. 3b). M. truncatula was chosen for the availability and high quality of the SNP data set derived from 26 accessions (Branca et al. 2011). An asymetrical distribution of A/S ratios (gene set enrichment analysis; Fisher’s exact test, two-tailed, P < 0.05) was found in 89 M. truncatula gene families; they included 64 families encompassing 10 members or more (supplementary table S5, Supplementary Material online). Fig 3b illustrates a simplified list of 46 nonoverlapping families.
The analyses showed that rates of substitution per site and A/S ratios were different, on average, between P. glauca and M. truncatula (figs. 2 and 3). Over the 13,498 P. glauca genes sampled, the average A was 0.0080 and the average S was 0.0251 for a global ratio A/S of 0.32. Over the 28,019 M. truncatula genes, the average A and S were 0.0228 and 0.0442, respectively, for a global ratio A/S of 0.51. Given this, it is not appropriate to compare directly A/S ratios between P. glauca and M. truncatula for a given gene family, because it is the relative ranking of the A/S ratio of a gene family within each species that is the most informative. For example, the LRR family was characterized by the highest A/S ratios in P. glauca and the lowest ones in M. truncatula, although the highest ratios in P. glauca and the lowest ones in M. truncatula had both averages around 0.66. The subset of LRR genes identified by the gene set enrichment analysis with the highest A/S ratios in M. truncatula had a mean A/S of 2.23 (fig. 3a). There were only two other families characterized by the lowest A/S ratios in M. truncatula but with values more than 0.5 (NB-ARCs, F-box family; fig. 3b). Interestingly, LRR and NB-ARC families belong to a small group of four families harboring diversity levels at synonymous and nonsynonymous sites higher than the other gene families in M. truncatula (Branca et al. 2011). For all the other M. truncatula families, the A/S ratios averaged over the genes with the lowest A/S values was well below 0.5 (fig. 3b).
The comparison between P. glauca and M. truncatula identified 12 families with the lowest A/S ratios in both species: oxygenases, heat shock proteins, ribosomal proteins, proteins involved in ubiquitination, kinases, UDP-glycosyltransferases, cytochromes P450, the NB-ARC family, and the EF-hand calcium-binding proteins (fig. 3b). Several families of enzymes, transporters, receptors, and transcription factors were characterized with the lowest A/S ratios in M. truncatula but not in P. glauca (figs. 3 and 4). In M. truncatula, four families were overrepresented among genes with the highest A/S ratios (the ribonuclease of transposable elements, the WRKY and the homeodomain transcription factors, the armadillo repeat family) (fig. 3b). Three of these families were found in P. glauca but did not show a high average A/S ratio, and P. glauca lacked sequences annotated as transposable element ribonuclease H. However, we observed that several families with the highest A/S ratios in P. glauca and in M. truncatula are involved in protein–protein interactions: the LRR and ankyrin family in P. glauca and the armadillo family in M. truncatula (fig. 4).
Conifer-Specific Genes Are Associated with the Highest A/S Ratios
The PFAM-based approach relied on sequence conservation with known proteins and thus, it excluded the most divergent sequences. The P. glauca data set encompassed 1,911 conifer-specific sequences (see Materials and Methods) and 11,186 sequences that were conserved with angiosperms (BlastX e-value <e−10). According to the gene set enrichment analysis, the most populated list of genes was the one with the lowest A/S ratios (12,001 genes; 86.3%), whereas a minority of genes was characterized with the highest A/S ratios (1,096 genes; 7.9%) (fig. 5a). Interestingly, the conifer-specific genes were more abundant among genes with the highest A/S (45.3%) than among those with the lowest A/S values (11.8%) (fig. 5a). On the opposite, P. glauca genes conserved with an angiosperm homolog (BlastX e-value <e−10) were significantly more populated among the genes with the lowest A/S (gene set enrichment analysis; Fisher’s exact test, two-tailed, adjusted P < 0.01) (fig. 5b). Our approach based on Blast search (e-value <e−10) may have retained spruce sequences sharing short motifs with angiosperms; such motifs could be found in fast-evolving genes. To filter out such cases, we also ran the analysis with a much more stringent e-value cutoff of 0.5 (data not shown). Only 486 putative conifer-specific genes remained; but for this reduced subset, the trend was the same (gene set enrichment analysis; Fisher’s exact test, two-tailed, adjusted P < 0.01) as mentioned above (fig. 5).
Fig. 5.—
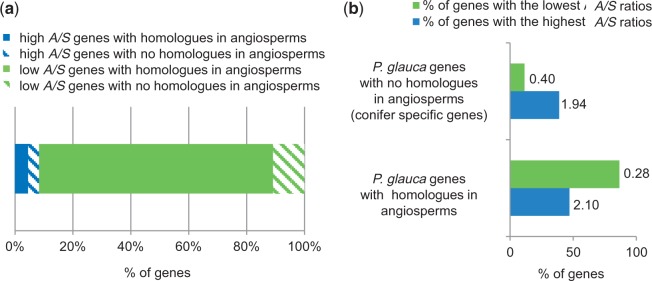
Relationship between sequence conservation with angiosperm genes and A/S ratios in Picea glauca. (a) Distribution of the 13,097 annotated P. glauca genes according to their conservation with angiosperm genes (BlastX e-value <e−10) and their A/S values. Hatched boxes: genes with a homolog in pines or Douglas-fir or both, but not in angiosperms (nor Amborella, nor Arabidopsis, nor rice) (14.6% of the data set) Solid boxes: genes with a homolog in angiosperms (at least with Amborella, or Arabidopsis, or rice) (85.4% of the data set). A total of 401 orphan P. glauca genes with no match (BlastX e-value >e−10) were excluded from this analysis. (b) Histogram illustrating the representation of P. glauca genes in the sets of genes with low (in green) or high (in blue) A/S values, according to their conservation with angiosperm sequences (gene set enrichment analysis, Fisher’s exact test, two-tailed, adjusted P < 0.01). Numbers indicate the average A/S for each data set with high or low A/S ratios.
Relationships between P. glauca A/S Ratios and Gene Expression Level and Specificity
The patterns of nucleotide polymorphism were also analyzed in relation to the differential gene expression profiles across several spruce tissues or organs available in the PiceaGenExpress database (Raherison et al. 2012). Expression profiles could be retrieved for 13,498 genes of the SNP atlas. We compared the distribution of the A/S ratios of 364 genes that were ubiquitously and highly expressed and 554 genes that were strongly preferential or specific to one of the tissues. Genes with ubiquitous and high expression levels were more abundant among the genes with the lowest A/S values (average A/S of 0.12) (gene set enrichment analysis; Fisher’s exact test, two-tailed, adjusted P < 0.01), whereas genes that were strongly preferential or unique to one tissue were more abundant among genes with the highest A/S values (average A/S of 0.63) (gene set enrichment analysis; Fisher’s exact test, two-tailed, adjusted P < 0.01).
We identified 12 different co-expression clusters that had a significant abundance of genes with the highest or lowest A/S ratios (gene set enrichment analysis; Fisher’s exact test, two-tailed, adjusted Ps < 0.01) (fig. 6b). Those co-expression clusters (8 out 12) with the lowest A/S values were overpopulated by genes with high expression levels in several organs (fig. 6a). For example, the 727 genes in cluster 26 were highly expressed in seven tissues but weakly expressed in embryogenic cells; this cluster had 5.5% of its genes with the lowest A/S values (average 0.31) and 1.8% of its genes with the highest A/S values (average 2.12) (fig. 6a). Also, the 737 genes in cluster 23 were highly expressed in buds, roots, megagametophytes, embryogenic cells but weakly to moderately expressed in needles, xylem, and phelloderm; they included more than 12% of genes with the lowest A/S ratios with an average A/S of 0.08 (fig. 6a). On the other hand, the four other clusters had, on average, genes with higher A/S ratios, and they were overpopulated by genes expressed in a single organ (mostly root, needle, or megagametophyte) or in two organs (root and megagametophyte) (fig. 6c). For example, genes in cluster 15 (fig. 6c) were mainly expressed in roots and included 73 genes classified among those with the highest A/S ratios with an average of 1.98, which would be indicative of positive selection (fig. 6b). Sequence similarity searches conducted on this subset of 73 genes resulted in putative functions for only 15 sequences, a fraction (20.5%) which is well below the expected proportion (63.8%) (χ2 = 55.4, adjusted P value <0.01). We identified a defensin sequence with a close homolog expressed in Scots pine roots that was shown to have antimicrobial properties (Kovaleva et al. 2009) and two other sequences related to defense mechanisms (an LRR and a MAP kinase kinase), whereas all the other sequences did not show obvious functional annotations, which suggests that some of these are likely to be conifer-specific genes.
Fig. 6.—

Relationship between co-expressed genes and their imbalance in A/S ratios in Picea glauca. (a) Expression profiles of genes more represented among sequences with the lowest A/S values. The pictures represent the expression patterns in buds (B), needles (N), mature (Mx) or juvenile (Jx) xylem, phloem (P), roots (R), megagametophytes (M), and embryogenic cells (E). Due to space limitation, only four examples are showed out of the eight expression clusters encompassing genes with low A/S values. (b) The histogram illustrates the 12 co-expression clusters with a significant overrepresentation among genes with the highest or lowest A/S values (gene set enrichment analysis, Fisher’s exact test, two-tailed, adjusted Ps < 0.01). (c) Expression profiles of the genes from the four co-expression clusters of genes with the highest A/S values.
Discussion
Nucleotide polymorphism in coding DNA was investigated in natural populations of P. glauca encompassing a total of 13,498 genes and 2,457 gene families, making it the largest analysis of its kind in a nonangiosperm plant. The A/S ratio was determined for each gene and compared among gene families and functional classes and analyzed according to expression profiles. Most of the P. glauca genes had low A/S ratios, especially when compared with the angiosperm M. truncatula. Genes with the highest A/S values were overrepresented in families related to stress response, among conifer-specific sequences and among genes with strong tissue preferential expression profiles, suggesting that gene expression may truly be a contributing factor to the evolution of nucleotide polymorphism.
Construction of a High-Confidence SNP Atlas
To our knowledge, the P. glauca SNP atlas produced herein is the largest one available with such a high quality for a conifer species, and it provides a desirable framework for analyzing the landscape of nucleotide polymorphism across the transcriptome for a nonmodel species lacking a finished genome sequence. High confidence was obtained following several lines of evidence. First, reads were mapped against a reliable P. glauca reference database in which each sequence is a gene representative (Rigault et al. 2011). In this reference database, 85% of the genes are represented by fully sequenced cDNA clones (FLICs) (Rigault et al. 2011), which reduces the biases that could result from undetected paralogs in de novo sequence assemblies. Second, reads corresponding to two genes were excluded. Third, we defined SNP calling criteria that were highly robust as shown with a set of many thousands genotyped SNPs (Pavy et al. 2013). At the expense of loosing some true positives, the selection of conservative prediction parameters increased markedly the overall quality of SNPs derived mostly from next-generation sequencing, compared with that previously obtained in P. glauca by using Sanger reads alone (Pavy et al. 2006).
The average abundance of 1 SNP per 81 sites observed in P. glauca FLICs from sequencing 212 P. glauca individuals was four times that obtained with cDNA clusters derived from 18 P. glauca individuals (Pavy et al. 2006). The overall A/S ratio of 0.32 estimated over 13,498 cds was also higher compared to the value of 0.17 estimated based on 3,590 cDNA clusters (Pavy et al. 2006) but was similar to the A/S ratio of 0.30 obtained in eucalypt from next-generation sequences in a sample of 21 trees and for 2,001 cDNA clusters (Novaes et al. 2008). Another exhaustive conifer SNP resource has been developed for Pseudostuga menziesii (Howe et al. 2013). It contains 279k SNPs with a validation rate of 72% in genotyping and was designed for genomic selection, which did not require anchoring of the SNPs onto a gene catalog. In this study, a total of 373k SNPs could be called, but the severe quality criteria corresponding to a validation rate of 92.1% in genotyping restricted the atlas to 212k high-confidence SNPs.
Several recently initiated projects aim to sequence and characterize conifer genomes both in pines and spruces (Birol et al. 2013; Nystedt et al. 2013). These initiatives present significant challenges for assembling genomes in excess of 20,000 Mbp from heterozygous individuals without physical maps. The P. glauca gene SNP atlas represents a base resource for building high-density genetic maps (e.g., Pavy et al. 2012a) to which genome sequence scaffolds could be robustly anchored through the annotated gene loci in order to create pseudo-chromosome sequences (Ritland et al. 2011). It will also be useful to develop high-throughput genotyping assays (Pavy et al. 2013) for a variety of investigations including ecological/population genomics analyses of adaptive mechanisms and the discovery of transcriptome-wide epistatic effects and gene networks, which is an emerging research issue in relation to monitoring and mitigating the effects of climatic change on biological diversity. It will also serve to develop genomics-assisted breeding methods for these largely undomesticated woody species that are intensively used in reforestation, adding flexibility and reducing reaction time in the context of deploying adapted varieties in a rapidly changing environment.
Patterns of Nucleotide Polymorphism and Variation between Plant Phyla
We analyzed nucleotide polymorphism in functional gene classes by using more than 50% of the known P. glauca transcriptome and complete coding sequences (Rigault et al. 2011). As reported by Buschiazzo et al. (2012), we used a cutoff-free statistical method based on the overall distribution of the A/S ratios to identify gene classes harboring highest A/S ratios (Al-Shahrour et al. 2007), whereas other reports have used arbitrary cutoffs. In plants, a few studies investigated polymorphism patterns at a wide scale but did not report on patterns across gene families or were limited to smaller gene sets (Novaes et al. 2008; Warren et al. 2010; Slotte et al. 2011; Buschiazzo et al. 2012).
The analysis of the distribution of the A/S ratios highlighted a number of spruce genes that may be under positive selection (figs. 3, 5, and 6). Liu et al. (2008) demonstrated a positive relationship between the intraspecific A/S ratio and the interspecific Ka/Ks (dN/dS). However, dN/dS deals with fixed substitutions between species or divergent lineages, whereas A/S deals with segregating substitutions at the intraspecific level. dN/dS is a simple and monotonic function of the selection coefficient, whereas A/S reaches a maximum value when the selection coefficient increases (Kryazhimskiy and Plotkin 2008). Therefore, A/S levels should be interpreted cautiously. To avoid this caveat, our analysis has not relied on any arbitrary A/S cutoff, as mostly done for dN/dS analysis in the literature. Instead, we have used a cutoff-free statistical analysis enabling to identify groups of genes with an asymmetrical A/S distribution along an ordered list of values. At some point, using a A/S cutoff would have been invalid, given that for a selection coefficient ranging from 3 to 10, A/S may not increase and can even slightly decrease depending on the mutation rate (Kryazhimskiy and Plotkin 2008). Nevertheless, it is safe to assume that the P. glauca genes harboring the highest A/S ratios are candidates being under selection, although we would not draw any interpretation about the selection strength. Thus, we believe that these genes deserve further analyses to test for deviations from neutrality. However, let us remind that boreal conifers such as P. glauca have undergone population expansion since the last glacial maximum, making it a challenge to disentangle the effects of selection from demographic effects (e.g., Namroud et al. 2010; Pavy et al. 2012b). Thus, a sizeable set of analyses is further required to test for selection in the candidate genes, including the survey of all SNPs and the use of haplotype tests, which necessitate the resequencing of genes in single individuals or haploid megagametophytes (e.g., Palmé et al. 2008; Pavy et al. 2012b).
When comparing our results with those previously reported with smaller gene sets investigated at the intraspecific (Novaes et al. 2008) or interspecific (Buschiazzo et al. 2012) level, only a partial overlap in gene content was observed (supplementary fig. S2, Supplementary Material online). Thus, we analyzed and compared nucleotide polymorphism in a fully sequenced angiosperm (M. truncatula) by using the same approach applied to P. glauca (fig. 3). Low A/S ratios were observed for most gene families in both P. glauca and M. truncatula genes (figs. 2 and 4), which follows expectations that most of the genes are under purifying selection (e.g., Novaes et al. 2008; Warren et al. 2010). In general, the spectrum of A and S values as well as that of A/S ratios were more restricted in P. glauca than in M. truncatula genes, although a large number of white spruce individuals were considered. This trend might be related to the application of stringent rules defining the high-confidence P. glauca SNPs retained in the estimation of substitution, though such a procedure should apply quite evenly between synonymous and nonsynonymous SNPs. Also, lower substitution rates were previously observed in trees and perennials compared with annual plants (e.g., Bousquet et al. 1992; Gaut et al. 1992; Smith and Donoghue 2008; Gaut et al. 2011) or in conifers compared with angiosperms (Dvornyk et al. 2002; Buschiazzo et al. 2012), which may affect differentially synonymous and nonsynonymous sites. For instance, larger differences in nonsynonymous rates than in synonymous rates were observed between perennial and annual taxa (Bousquet et al. 1992), leading to lower Ka/Ks ratios for perennial and tree taxa, a pattern that is reminiscent of that observed here between P. glauca and M. truncatula. Given that a larger difference was detected in rates of nonsynonymous substitutions than in rates of synonymous substitutions between P. glauca and M. truncatula, such a trend might reflect generally more purifying selection at the genome-wide level in P. glauca or less constrained evolution in M. truncatula. The study of additional taxa including outliers to seed plants would help settle the issue. These differences seen between P. glauca and M. truncatula should not bear major consequences in the present comparative analysis as it did not rely on absolute values of A/S ratios but their relative ranking within each species.
The analysis of variation in P. glauca indicated that many of the GO classes were associated with low A/S ratios (supplementary fig. S2, Supplementary Material online). Seven molecular functions, two processes, and eight cellular components harbored low dN/dS between pine and spruce (Buschiazzo et al. 2012), as well as low A/S ratios for P. glauca. Large classes such as hydrolases, kinases, and nucleotide binding shared the same excess of synonymous SNPs, which may be indicative of strong purifying selection. Our analysis of gene families in P. glauca provided a higher degree of resolution, clearly showing which families were responsible for the patterns observed at the GO level, given that GO classes are large and heterogeneous in nature.
Picea glauca Stress-Response Genes Showing an Excess of Nonsynonymous SNPs
The P. glauca genes with the highest A/S ratios belonged to three families that include biotic stress response genes (LRR, AP2/DREB, and ankyrin repeat families) and one family of cold and drought resistance responsive genes (dehydrins) (fig. 3a). The AP2/DREB family is involved in response to ethylene and organ identity and includes disease resistance regulators (Lu et al. 2013). The ankyrin repeat family is involved in protein–protein interactions that mediate a wide diversity of biological functions and includes BDA1, a critical signaling component of plant immunity (Yang et al. 2012).
Both the ankyrin and the LRR families have a role in protein–protein interactions, which are mediated by a specific amino acid domain shown to be under diversifying selection in some angiosperm LRR genes (e.g., Ellis et al. 2000; Mauricio et al. 2003; Bakker et al. 2006). In general, LRR genes underwent purifying selection, but their different protein domains harbor different evolutionary rates, as was shown in Solanum sequences (Caicedo and Schaal 2004). The pattern of nucleotide polymorphism of several Arabidopsis LRR genes also varied from one domain to the other and was reported to result from balancing selection in several family members (Mauricio et al. 2003; Bakker et al. 2006; Guo et al. 2011). In general, genes involved in biotic and abiotic stress have undergone positive selection in Arabidopsis (Slotte et al. 2011).
The P. glauca dehydrin genes were mostly distributed among genes with the highest A/S values but could not be characterized as such in M. truncatula, although one sequence had an A/S value of 0.94. Comparisons with other angiosperms are not straightforward given that a single or a few dehydrin genes at a time have been considered in the literature. The pattern of nucleotide polymorphism for a dehydrin gene has been reported to result from positive selection in tomato (Xia et al. 2010) and in wild barley (Yang et al. 2009). In Pinus sylvestris, the nucleotide diversity in 10 dehydrin genes was consistent with expectations for positive selection in two genes and balancing selection in a third gene (Wachowiak et al. 2009).
The dehydrin family harbored other characteristics, making it unique in spruce: the family is larger in spruce compared to angiosperms (Rigault et al. 2011), and the expression patterns are highly differentiated among tissues during normal development, with strong expression in roots and in megagametophytes during germination (Raherison et al. 2012). The expression of dehydrin genes is responsive to cold and drought stress in species such as Pinus pinaster (Velasco-Conde et al. 2012) and correlates with bud burst during Spring warming in Picea abies (Yakovlev et al. 2008). Dehydrins are believed to prevent protein denaturation or restore function in denatured proteins (Hara 2010). We found that other families involved in protecting proteins against denaturation, such as the heat shock proteins, mostly had genes with low A/S values both in P. glauca and M. truncatula (fig. 3). Furthermore, gene families involved in defense mechanisms against fungi or insects harbored mostly low A/S in P. glauca, such as the pectin methyl esterase inhibitors (A/S = 0.22), chitinases class I (A/S = 0.20), NB-ARCs (A/S = 0.29), pectinases (A/S = 0.24), and terpene synthases (A/S = 0.19) and a few genes with high A/S ratios, illustrating the diversity of evolutionary rates that can be found within gene families (fig. 3).
Links between Gene Function, Gene Expression, and Nucleotide Polymorphism
In addition to stress-response genes, the present analyses of nucleotide polymorphism highlighted two other groups of conifer genes with excesses of nonsynonymous SNPs. The first group was made of genes lacking similarity with angiosperms, which is of upmost interest to understand divergent evolutionary patterns related to function between conifers and angiosperms. The overrepresentation of nonsynonymous SNPs in a nonnegligible proportion (26.0%) of these genes is all the more intriguing, given that such genes are more likely involved in adaptive features specific to conifers. These genes should deserve the highest scrutiny in the ongoing analyses of conifer genome sequences (Birol et al. 2013; Nystedt et al. 2013).
The second group of genes was characterized by atypical expression patterns. A/S ratios were high in genes with more tissue-specific expression and low among genes with generic expression in a variety of tissues. Such observation correlates well with other results obtained with mammalian (Duret and Mouchiroud 2000) and Arabidopsis (Slotte et al. 2011; Yang and Gaut 2011) genes where Ka/Ks ratios showed strong negative correlations with expression level and breadth. This pattern would arise because the selective pressure on nonsynonymous sites depends on the variety of tissues where the genes are expressed (Duret and Mouchiroud 2000). In yeast, where the notion of expression breadth does not apply, the gene expression level and protein evolution rate are strongly and negatively correlated (Drummond et al. 2005; Wall et al. 2005). Although, the expression level and expression breadth were identified as the factors that most strongly explain the nonsynonymous rate of substitution in Arabidopsis (Yang and Gaut 2011), these studies did not isolate the expression patterns that are most likely associated with the differential mutation rate or selection effects. Our approach based on the screening of the A/S ratios across co-expression clusters enabled to pinpoint which genes are involved in this process. Interestingly, several co-expressed genes with the highest A/S ratios were involved in defense mechanisms and were not identified by the gene family-based approach. Moreover, many co-expressed genes with the highest A/S ratios lacked sequence conservation with known genes, suggesting that they may be specific or highly diverged in spruce. The link between expression specificity and high levels of nonsynonymous SNPs reported in the present study of P. glauca sets the stage for targeted investigations of genes that may contribute significantly to genetic adaptation.
Genes expressed in a large number of tissues may encode essential cellular functions and thus would evolve more slowly. Yet, although this hypothesis makes intuitive sense, Drummond et al. (2005) proposed another model to explain why highly expressed genes might evolve more slowly. The cost of misfolded proteins appears to depend on expression level, but protein misfolding can be counteracted by “translational robustness,” which increases the number of proteins that folds properly despite mistranslation; such a process would slow down the rate of nonsynonymous substitutions (Drummond et al. 2005). Several factors affect protein evolution rate and these factors are interdependent, making difficult the disentangling of evolutionary forces at the molecular level (Pál et al. 2006).
In conclusion, our findings based on the increasing availability of large SNP databases illustrate the potential to investigate patterns of nucleotide polymorphism in phylogenetically remote lineages of seed plants to uncover diverged evolutionary paths and relating these patterns to expression profiles and other molecular attributes when they are available. The ongoing whole-genome resequencing projects in Arabidopsis (Cao et al. 2011; Weigel 2012) and in several other angiosperms as well as genome sequencing and transcriptome resequencing projects in conifers (Birol et al. 2013; Nystedt et al. 2013) should provide more reference taxa for such studies in the near future.
Supplementary Material
Supplementary methods S1 and S2, figures S1 and S2, and tables S1–S5 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank J. Laroche (IBIS, Univ. Laval) and P. Belleau (CRCHUL, Univ. Laval) for fruitful discussions. They also thank two anonymous reviewers for their constructive comments on a previous version of this manuscript. This work was supported by grants from Genome Québec and Genome Canada for the Arborea II and SMarTForests and a grant from Genome Québec for a pilot genome sequencing project to J.M. and J.Bo., by NSERC discovery grants to J.Bo. and J.M., and the Genomics R&D Initiative to N.I. and J.Be.
Literature Cited
- Al-Shahrour F, et al. From genes to functional classes in the study of biological systems. BMC Bioinformatics. 2007;8:114. doi: 10.1186/1471-2105-8-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker EG, Toomajian C, Kreitman M, Bergelson J. A genome-wide survey of R gene polymorphisms in Arabidopsis. Plant Cell. 2006;18:1803–1818. doi: 10.1105/tpc.106.042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995;57:289–300. [Google Scholar]
- Birol I, et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics. 2013;29:1492–1497. doi: 10.1093/bioinformatics/btt178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet J, Strauss SH, Doerksen AH, Price A. Extensive variation in evolutionary rate of rbcL gene sequences among seed plants. Proc Natl Acad Sci U S A. 1992;89:7844–7848. doi: 10.1073/pnas.89.16.7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branca A, et al. Whole-genome nucleotide diversity, recombination, and linkage disequilibrium in the model legume Medicago truncatula. Proc Natl Acad Sci U S A. 2011;108:E864–E870. doi: 10.1073/pnas.1104032108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschiazzo E, Ritland C, Bohlmann J, Ritland K. Slow but not low: genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol Biol. 2012;12:8. doi: 10.1186/1471-2148-12-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo AL, Schaal B. Heterogeneous evolutionary processes affect R gene diversity in natural populations of Solanum pimpinellifolium. Proc Natl Acad Sci U S A. 2004;101:17444–17449. doi: 10.1073/pnas.0407899101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43:956–963. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Drummond DA, et al. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 2005;102:14338–14343. doi: 10.1073/pnas.0504070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D. Determinants of substitution rates in Mammalian genes: expression pattern affects selection intensity but not mutation rate. Mol Biol Evol. 2000;17:68–74. doi: 10.1093/oxfordjournals.molbev.a026239. [DOI] [PubMed] [Google Scholar]
- Dvornyk V, Sirviö A, Mikkonen M, Savolainen O. Low nucleotide diversity at the pal1 locus in the widely distributed Pinus sylvestris. Mol Biol Evol. 2002;19:179–188. doi: 10.1093/oxfordjournals.molbev.a004070. [DOI] [PubMed] [Google Scholar]
- Ellis J, Dodds P, Pryor T. The generation of plant disease resistance gene specificities. Trends Plant Sci. 2000;5:373–379. doi: 10.1016/s1360-1385(00)01694-0. [DOI] [PubMed] [Google Scholar]
- Gaut BS, Muse SV, Clark WD, Clegg MT. Relative rates of nucleotide substitution at the rbcL locus of monocotyledonous plants. J Mol Evol. 1992;35:292–303. doi: 10.1007/BF00161167. [DOI] [PubMed] [Google Scholar]
- Gaut B, Yang L, Takuno S, Eguiarte LE. The patterns and causes of variation in plant nucleotide substitution rates. Annu Rev Ecol Evol Syst. 2011;42:245–266. [Google Scholar]
- Guo Y, et al. Genome-wide comparison of nucleotide-binding site-leucine-rich repeat-encoding genes. Plant Physiol. 2011;157:757–769. doi: 10.1104/pp.111.181990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M. The multifunctionality of dehydrins: an overview. Plant Signal Behav. 2010;5:503–508. doi: 10.4161/psb.11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL, Clark AG. Principles of population genetics. 4th ed. Sunderland (MA): Sinauer Associates; 2007. p. 652. [Google Scholar]
- Horton MW, et al. Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nature. 2012;44:212–216. doi: 10.1038/ng.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe GT, et al. A SNP resource for Douglas-fir: de novo transcriptome assembly and SNP detection and validation. BMC Genomics. 2013;14:137. doi: 10.1186/1471-2164-14-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo-Correa JP, Verdú M, González-Martínez SC. The contribution of recombination to heterozygosity differs among plant evolutionary lineages and life-forms. BMC Evol Biol. 2010;10:22. doi: 10.1186/1471-2148-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, et al. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473:97–100. doi: 10.1038/nature09916. [DOI] [PubMed] [Google Scholar]
- Koboldt DC, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaleva V, et al. Purification and molecular cloning of antimicrobial peptides from Scots pine seedlings. Peptides. 2009;30:2136–2643. doi: 10.1016/j.peptides.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Kryazhimskiy S, Plotkin JB. The population genetics of dN/dS. PLoS Genet. 2008;4:e1000304. doi: 10.1371/journal.pgen.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam HM, et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet. 2010;42:1053–1059. doi: 10.1038/ng.715. [DOI] [PubMed] [Google Scholar]
- Liu J, Zhang Y, Lei X, Zhang Z. Natural selection of protein structural and functional properties: a single nucleotide polymorphism perspective. Genome Biol. 2008;9:R69. doi: 10.1186/gb-2008-9-4-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, et al. Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res. 2010;20:1238–1249. doi: 10.1101/gr.106120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, et al. AaORA, a trichome-specific AP2/ERF transcription factor of Artemisia annua, is a positive regulator in the artemisinin biosynthetic pathway and in disease resistance to Botrytis cinerea. New Phytol. 2013;198:1191–1202. doi: 10.1111/nph.12207. [DOI] [PubMed] [Google Scholar]
- Mauricio R, et al. Natural selection for polymorphism in the disease resistance gene Rps2 of Arabidopsis thaliana. Genetics. 2003;163:735–746. doi: 10.1093/genetics/163.2.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina I, et al. Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res. 2010;38:W210–W213. doi: 10.1093/nar/gkq388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgante M, De Paoli E. Toward the conifer genome sequence. In: Plomion C, Bousquet J, Kole K, editors. Genetics, genomics and breeding of conifers. New York: CRC Press and Edenbridge Science Publishers; 2011. pp. 389–403. [Google Scholar]
- Murray BG. Nuclear DNA amounts in gymnosperms. Ann Bot. 1998;82:3–15. [Google Scholar]
- Namroud M-C, Guillet-Claude C, Mackay J, Isabel N, Bousquet J. Molecular evolution of regulatory genes in spruces from different species and continents: heterogeneous patterns of linkage disequilibrium and selection but correlated recent demographic changes. J Mol Evol. 2010;70:371–386. doi: 10.1007/s00239-010-9335-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novaes E, et al. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics. 2008;9:312. doi: 10.1186/1471-2164-9-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystedt B, et al. The Norway spruce genome sequence and conifer genome evolution. Nature. 2013;497:579–584. doi: 10.1038/nature12211. [DOI] [PubMed] [Google Scholar]
- Pál C, Papp B, Lercher MJ. An integrated view of protein evolution. Nat Rev Genet. 2006;7:337–348. doi: 10.1038/nrg1838. [DOI] [PubMed] [Google Scholar]
- Palmé AE, Wright M, Savolainen O. Patterns of divergence among conifer ESTs and polymorphism in Pinus sylvestris identify putative selective sweeps. Mol Biol Evol. 2008;25:2567–2577. doi: 10.1093/molbev/msn194. [DOI] [PubMed] [Google Scholar]
- Pavy N, Namroud M-C, Gagnon F, Isabel N, Bousquet J. The heterogeneous levels of linkage disequilibrium in white spruce genes and comparative analysis with other conifers. Heredity. 2012b;108:273–284. doi: 10.1038/hdy.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavy N, Parsons LS, Paule C, MacKay J, Bousquet J. Automated SNP detection from a large collection of white spruce expressed sequences: contributing factors and approaches for the categorization of SNPs. BMC Genomics. 2006;7:174. doi: 10.1186/1471-2164-7-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavy N, et al. Generation, annotation, analysis and database integration of 16,500 white spruce EST clusters. BMC Genomics. 2005;6:144. doi: 10.1186/1471-2164-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavy N, et al. A spruce gene map infers ancient plant genome reshuffling and subsequent slow evolution in the gymnosperm lineage leading to extant conifers. BMC Biol. 2012a;10:84. doi: 10.1186/1741-7007-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavy N, et al. Development of high-density SNP genotyping arrays for white spruce (Picea glauca) and transferability to subtropical and nordic congeners. Mol Ecol Res. 2013;13:324–336. doi: 10.1111/1755-0998.12062. [DOI] [PubMed] [Google Scholar]
- Raherison ESM, et al. Transcriptome profiling in conifers and the PiceaGenExpress database show patterns of diversification within gene families and interspecific conservation in vascular gene expression. BMC Genomics. 2012;13:434. doi: 10.1186/1471-2164-13-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph SG, et al. A conifer genomics resource of 200,000 spruce (Picea spp.) ESTs and 6,464 high-quality, sequence-finished full-length cDNAs for Sitka spruce (Picea sitchensis) BMC Genomics. 2008;9:484. doi: 10.1186/1471-2164-9-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rigault P, et al. A white spruce gene catalog for conifer genome analyses. Plant Physiol. 2011;157:14–28. doi: 10.1104/pp.111.179663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritland K, et al. Genetic mapping in conifers. In: Plomion C, Bousquet J, Kole K, editors. Genetics, genomics and breeding of conifers. New York: CRC Press and Edenbridge Science Publishers; 2011. pp. 196–238. [Google Scholar]
- Saeed AI, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- Savard L, et al. Chloroplast and nuclear gene sequences indicate Late Pennsylvanian time for the last common ancestor of extant seed plants. Proc Natl Acad Sci U S A. 1994;91:5163–5167. doi: 10.1073/pnas.91.11.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotte T, et al. Genomic determinants of protein evolution and polymorphism in Arabidopsis. Genome Biol Evol. 2011;3:1210–1219. doi: 10.1093/gbe/evr094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S, Donoghue MJ. Rates of molecular evolution are linked to life history in flowering plants. Science. 2008;322:86–89. doi: 10.1126/science.1163197. [DOI] [PubMed] [Google Scholar]
- Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-Conde T, Yakovlev I, Majada JP, Aranda I, Johnsen Ø. Dehydrins in maritime pine (Pinus pinaster) and their expression related to drought stress response. Tree Genet Genomes. 2012;8:957–973. [Google Scholar]
- Wachowiak W, Balk PA, Savolainen O. Search for nucleotide diversity patterns of local adaptation in dehydrins and other cold-related candidate genes in Scots pine (Pinus sylvestris L.) Tree Genet Genomes. 2009;5:117–132. [Google Scholar]
- Wall DP, et al. Functional genomic analysis of the rates of protein evolution. Proc Natl Acad Sci U S A. 2005;102:5483–5488. doi: 10.1073/pnas.0501761102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren AS, Anandakrishnan R, Zhang L. Functional bias in molecular evolution rate of Arabidopsis thaliana. BMC Evol Biol. 2010;10:125. doi: 10.1186/1471-2148-10-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel D. Natural variation in Arabidopsis: from molecular genetics to ecological genomics. Plant Physiol. 2012;158:2–22. doi: 10.1104/pp.111.189845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Camus-Kulandaivelu L, Stephan W, Tellier A, Zhang Z. Nucleotide diversity patterns of local adaptation at drought-related candidate genes in wild tomatoes. Mol Ecol. 2010;19:4144–4154. doi: 10.1111/j.1365-294X.2010.04762.x. [DOI] [PubMed] [Google Scholar]
- Xu X, et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotechnol. 2011;30:105–111. doi: 10.1038/nbt.2050. [DOI] [PubMed] [Google Scholar]
- Yakovlev I, et al. Dehydrins expression related to timing of bud burst in Norway spruce. Planta. 2008;228:459–472. doi: 10.1007/s00425-008-0750-0. [DOI] [PubMed] [Google Scholar]
- Yang L, Gaut BS. Factors that contribute to variation in evolutionary rate among Arabidopsis genes. Mol Biol Evol. 2011;28:2359–2369. doi: 10.1093/molbev/msr058. [DOI] [PubMed] [Google Scholar]
- Yang Z, Zhang T, Bolshoy A, Beharav A, Nevo E. Adaptive microclimatic structural and expressional dehydrin1 evolution in wild barley, Hordeum spontaneum, at “Evolution Canyon”, Mount Carmel, Israel. Mol Ecol. 2009;18:2063–2075. doi: 10.1111/j.1365-294X.2009.04140.x. [DOI] [PubMed] [Google Scholar]
- Yang Y, et al. The ankyrin-repeat transmembrane protein BDA1 functions downstream of the receptor-like protein SNC2 to regulate plant immunity. Plant Physiol. 2012;159:1857–1865. doi: 10.1104/pp.112.197152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng LY, et al. Genome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor) Genome Biol. 2011;12:R114. doi: 10.1186/gb-2011-12-11-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.