

Abstract
Divergence of serially homologous elements of organisms is a common evolutionary pattern contributing to increased phenotypic complexity. Here, we study the genomic intervals affecting the variational independence of fore- and hind limb traits within an experimental mouse population. We use an advanced intercross of inbred mouse strains to map the loci associated with the degree of autonomy between fore- and hind limb long bone lengths (loci affecting the relationship between traits, relationship quantitative trait loci [rQTL]). These loci have been proposed to interact locally with the products of pleiotropic genes, thereby freeing the local trait from the variational constraint due to pleiotropic mutations. Using the known polymorphisms (single nucleotide polymorphisms [SNPs]) between the parental strains, we characterized and compared the genomic regions in which the rQTL, as well as their interaction partners (intQTL), reside. We find that these two classes of QTL intervals harbor different kinds of molecular variation. SNPs in rQTL intervals more frequently reside in limb-specific cis-regulatory regions than SNPs in intQTL intervals. The intQTL loci modified by the rQTL, in contrast, show the signature of protein-coding variation. This result is consistent with the widely accepted view that protein-coding mutations have broader pleiotropic effects than cis-regulatory polymorphisms. For both types of QTL intervals, the underlying candidate genes are enriched for genes involved in protein binding. This finding suggests that rQTL effects are caused by local interactions among the products of the causal genes harbored in rQTL and intQTL intervals. This is the first study to systematically document the population-level molecular variation underlying the evolution of character individuation.
Keywords: autonomy, pleiotropy, constraint, genetic interaction, character
Introduction
The individuation and divergence of parts is fundamental in the evolution of complex organisms, whether at the level of cell types, body segments (e.g., in arthropods), body sides (asymmetry), repeated body parts (fore- and hind limbs), or sexes (sexual dimorphism) (Pavlicev and Wagner 2012). The underlying principle is intuitive: diversification into genetically individualized parts requires that the developmental effects of the genes shared between similar parts become modified differently in the diverging parts. At the population level, the pleiotropic mutations in shared genes cause parts to covary (Lande 1980). Accordingly, the evolutionary divergence of body parts is associated with decreasing genetic covariance of these parts (Berg 1960; Hansen et al. 2003; Young et al. 2010). The evolution of character covariance is thus a key process in evolution of complex organisms. We know from comparative studies that evolution of covariance is common and has adaptive value by refocusing the direction in which variation is produced (Porto et al. 2009; Young et al. 2010; Grabowski et al. 2011). Specifically, integration of phenotypic characters is advantageous when the characters are under correlated selective pressure (Pavlicev et al. 2011). In contrast, a low correlation of mutational effects on the characters is beneficial when independent selection responses are required (Cheverud 1996; Wagner 1996). An example of divergence is limb evolution in birds and bats where the forelimbs are selected for flight, whereas the hind limbs are selected for perching or walking (Young and Hallgrimsson 2005). But what mechanism enables such divergence?
Phenotypic effects of genes are mediated by physical interactions among gene products in development (regulation, protein interaction, signaling, etc.). Thus, it is plausible to suggest that specific genetic interactions, rather than specific genes per se, constitute the unique genetic basis of individual characters. A well-established example is the role of Hox genes in diversification of arthropod body plan. Hox genes determine the identity of body regions along anterior–posterior body axis. Yet, their numerous effects on morphology of segments, such as whether and which extremities are produced, are not inherent to Hox genes alone but are mediated by the interactions with local cofactors (Hughes and Kaufman 2002; Mann et al. 2009; Ohde et al. 2013). These local interactions of local cofactors with Hox genes specify activation or suppression of downstream genes and the segment-specific morphology. Similar local modification is assumed for vertebrate limbs (Ruvinsky and Gibson-Brown 2000; Minguillon et al. 2009; Duboc and Logan 2011a; reviewed in Duboc and Logan 2011b). Moreover, evolution of local interaction may be involved in the initial individuation of a character, for example, the sex comb in Drosophila (Kopp 2011). A pattern of local interactions is furthermore manifest in protein interaction networks, where gene products are often involved in multiple characters, yet in very specific interactions in each character (Han et al. 2004; Luscombe et al. 2004; Bossi and Lehner 2009).
It is, however, unknown how such topology of developmental interactions evolves and how it relates to the variational patterns at the population level. Generally, developmental regulation is thought to naturally result in pleiotropy of, and epistasis between, the effects of genetic polymorphisms on the phenotypic traits (Gibson 1996; Gjuvsland et al. 2007). Here, we build on a recent model of evolution of pleiotropy by epistatic interactions (Pavlicev and Wagner 2012). This model suggests that covariance evolves in two steps: a selection response in a focal trait and epistatic compensatory modification of correlated side effects in another trait (fig. 1B); hence the name selection–pleiotropy–compensation or SPC model. Due to epistasis, the contribution of pleiotropic mutation to trait covariance changes from pre- to post-compensatory genetic background. In this model, genetic individuation of characters occurs by coadaptation between the pleiotropic and the local compensatory gene. When alleles at the pleiotropic and the compensatory locus segregate, such topology is detected as genetic variation in pleiotropic effects.
Fig. 1.—

The topology and evolution of pleiotropy. (A) The phenotypic domain of pleiotropic locus (intQTL; blue) is modified by the local action of rQTL (red). (B) Evolutionary scenario: selection on trait 1 (black arrow) results in a change in pleiotropic locus, which propagates to trait 2 (U-shaped arrow) and is followed in the next step by a local compensation of side effects in non-focal traits.
The underlying genetic loci can be mapped using relationship quantitative trait locus (rQTL) mapping, combined with the mapping of the loci interacting with the rQTL, the so-called intQTL (Cheverud et al. 2004; Pavlicev et al. 2008; Leamy et al. 2009). The capacity of this model to respond to selection and indeed change covariance has been modeled in a population genetic context previously (Pavlicev et al. 2011).
Due to the nature of mapping, the resulting two categories of mapped loci differ in the way they affect the phenotypic variation, either conditional on the second trait (rQTL) or conditional on the genotype at the second locus (intQTL). SPC model provides the hypothesis how this specific variation may be produced, namely as a result of trait-specific modification of more global effect. Here, we show that the loci involved indeed have genomic signatures supporting this model. The loci modified by the rQTL, the intQTL, include genomic regions with high density of nonsynonymous coding mutations, and rQTL, are more likely to have cis-regulatory variation. Furthermore, rQTL intervals are enriched for single-nucleotide polymorphism (SNPs) in limb-specific H3K27ac marks, representing putative limb-specific enhancers. This is, to our knowledge, the first study to systematically connect the variational study of trait divergence to its genomic basis.
Materials and Methods
Population and Phenotyping
Mapping was conducted in the 34th generation of intercross consisting of 1,134 individuals (Wustl: LG, SM-G34). The intercross was initiated by crossing inbred house mouse strains LG/J and SM/J that were previously selected for high and low body weight at 60 days of age. The intercross and the specific generation are described in detail in Norgard et al. (2008, 2011). The limb bones on the right side were removed at necropsy, and the lengths of the femur, tibia, humerus, and ulna were measured immediately to the nearest 0.01 mm. As the mice were part of dietary study, the effect of diet as well as of sex was removed by multiple regression of these variables on the phenotypic traits and addition of the residuals to the trait means.
Heritability
To test the presence of genetic basis in the variational relationships between fore- and hind limb traits, we estimated the family effect on trait covariance using analysis of covariance (ANCOVA). The broad-sense heritabilities of trait relationships are expressed as the percentage of variance in the relationship that is accounted for by the variation between families (= trait × family interaction variance/[interaction variance + residual variance]). This estimates the variance associated with interaction term between family and a covariate (e.g., femur) as a fraction of variance in variable 1 (e.g., humerus) that is not explained by the additive effects of family and covariate. Heritabilities for single relationships range between 50% and 74% (table 1), being somewhat lower than the trait heritabilities (Norgard et al. 2011).
Table 1.
Mean Values of Traits and Heritabilities of Traits and Trait Relationships
| Trait | Mean (mm) | SE (mm) | Heritability | Relationship | Heritability |
|---|---|---|---|---|---|
| Humerus | 12.514 | 0.015 | 0.83 | hum/fem | 0.74 |
| Ulna | 14.176 | 0.017 | 0.68 | fem/hum | 0.74 |
| Femur | 16.092 | 0.018 | 0.81 | tib/uln | 0.50 |
| Tibia | 17.866 | 0.018 | 0.85 | uln/tib | 0.57 |
Genotyping and QTL Mapping
The population was genotyped at 2,842 SNPs, distributed across all 19 autosomes. SNPs were selected from the Oxford/CTC set (http://www.well.ox.ac.uk/mouse/INBREDS/, last accessed October 7, 2013) and scored by the John Hopkins Center of Inherited Disease Research (CIDR; Baltimore, MD), using the Illumina Golden Gate Bead Array (Illumina, San Diego, CA). The R/QTL package (Broman et al. 2009) was used to map SNP positions along autosomes. The density of SNPs in this generation is approximately 1 SNP/8.5 cM (Pavlicev et al. 2008, 2011).
Mapping of Genomic Loci Associated with Variation in Forelimb–Hind Limb Trait Covariance: rQTL
To study genetic variation in the covariance between two corresponding elements in fore- and hind limbs, we used ANCOVA, mapping loci for one phenotypic element while the second entered the model as a covariate. Specifically, in this model, the trait of interest is regressed onto the additive and dominance scores at each locus (subscripts i, j), the score of the second character (subscript k), and the interactions between the second character and additive and dominance scores at the locus. Below, the individual phenotypic value is expressed in terms of population mean and the deviations from the mean due to additive and dominance effect, the covariate, and the interaction terms between the additive, dominance, and covariate effects:
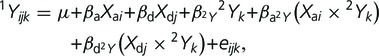 |
where 1Y and 2Y are the phenotypic scores of the two characters (e.g., femur and humerus lengths), µ is the population mean value of character 1Y, Xa and Xd are additive and dominance genotype scores, β are the corresponding regression coefficients, and e is an error term. An rQTL location is detected as the location at which the interaction between the genotype effect and the covariate is significant (the fourth and/or the fifth term above); that is, when the locus differs in its genetic effect on one character at different values of the second character. Statistical significance of the interaction effect was established by comparing the log-likelihoods of two models: one with interaction terms included and the second with only direct genetic and phenotypic effects. This also provides that the effects on the mean character value are not reported in this study if they do not also affect the relationship between the characters. We focused on genetic variation in the following relationships (1Y vs. 2Y): femur versus humerus, humerus versus femur, tibia versus ulna, and ulna versus tibia. When the genetic variation in relationship between characters is due to character-specific genetic effects, these will be detected when that character is included in a model as a response variable, but not when it is a covariate. Therefore, each relationship is addressed twice, exchanging the response variable and the covariate.
Epistasis Mapping
We established in previous studies that the genetic variation in character relationship results from genetic interactions or epistasis between the rQTL and other genomic locations (Cheverud et al. 2004; Pavlicev et al. 2008). In this step, we therefore perform epistasis mapping to reveal these locations. For each rQTL and each character of the relationship (1Y, 2Y) separately, we performed the scan for genetic interactions with rQTL:
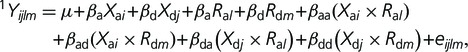 |
where Ra and Rd are the additive and dominance scores at the rQTL (fixed for a particular mapping), Xa and Xd are additive and dominance genotype scores at other genomic loci; i, j, l, m stand for the alleles at two loci; and 1Y (2Y) are characters involved in varying relationship. The locus at which either of the interaction effects (terms 5–8 above) were significant is detected as epistatically interacting with the particular rQTL. As above, the statistical significance of the interaction effects was established by comparing the log-likelihoods of the model with interaction terms included, with the second model with only direct genetic effects of either locus (i.e., only including first four terms above). The chromosome of rQTL was excluded from the scan to avoid associations due to linkage.
Single locus significance thresholds for the association between SNP variation and phenotypes were adjusted for multiple tests to avoid false positives. The adjustment of the threshold significance value was calculated using the correlation between genotypes along the chromosomes, following Li and Ji (2005), resulting in chromosome-wide thresholds of 2.71–3.13 LPR (LPR = −log10[probability of observing the particular F-ratio under null hypothesis of no effect on phenotype]). These chromosome-wide thresholds were used in rQTL mapping as well as in epistasis mapping (discussed later). Note that when an epistasis scan involves a fixed, previously determined, position, the threshold equivalent of single locus scan can be applied. In all subsequent analyses, we refer to the QTL interval meaning the interval around the peak position and including the confidence region spanning one unit LPR drop from the peak.
We used a mixed-effects linear regression model (R; version 2.9.0; package lme4) to estimate the above models. As the individuals in advanced intercross generations are variably related, we furthermore included family as a random variable.
Genomic Analyses of QTL Regions
We identified the genome coordinates of SNPs flanking the QTL intervals and downloaded the sequence (coding regions, introns, 5′-UTR, 3′-UTR, and 10 kb upstream and downstream of the genes) for these genomic intervals from the University of California Santa Cruz Genome Bioinformatics website (http://genome.ucsc.edu/ [last accessed October 7, 2013]; assembly mm9). To derive the polymorphism densities in different genic regions, we intersected QTL intervals and associated genomic annotations with SNPs at which the parental lines LG/J and SM/J differ. These SNPs were previously identified by comparing the full genomic sequences of LG/J and SM/J with C57BL6/J (Lawson et al. 2011). This list of SNPs is available at dbSNP (Sherry et al. 2001) under the handle “cheverud.” We used transcription factor binding site (TFBS) track (Yale/Stanford ENCODE [Euskirchen et al. 2007]) in UCSC genome browser to retrieve predicted TFBSs. Intersections were performed using UCSC Table browser, custom R scripts (ver. 2.15.1), and Galaxy (Giardine et al. 2005; Blankenberg et al. 2010).
To relate the coding SNPs with the numbers of synonymous and nonsynonymous sites in the coding regions (dN/dS), we extracted the coding exons within the QTL intervals from the UCSC and used the maximum likelihood approach in program codonml (package PAML [Yang 1997]) to assess the synonymous and nonsynonymous sites.
All analyses of gene ontology enrichment within sets of genes were conducted with Gostat (http://gostat.wehi.edu.au/, last accessed October 7, 2013 [Beissbarth and Speed 2004]), using Mouse Genome Informatics (MGI) gene collection and the false discovery rate correction for multiple testing.
To inspect the cis-regulatory regions we used 1) MEME and TOMTOM for the analysis of enriched binding motifs and their comparison with the motif databases (for both: http://meme.nbcr.net/meme/, last accessed October 7, 2013) and 2) GREAT to examine the genes potentially regulated by the identified polymorphic cis-regulatory regions (http://bejerano.stanford.edu/great/, last accessed October 7, 2013).
The significance of enrichment of various elements was tested using hypergeometric test. For example, the enrichment of enhancers in rQTL was calculated by estimating the probability of detecting an overlap of k enhancers in n = loci (or total sequence length), given a total number of enhancers K in a total genome.
Results and Discussion
Genetic Basis of Trait Relationship: rQTL and intQTL
We measured femur and tibia lengths to represent the hind limb and humerus and ulna lengths for the forelimb. We refer to these measurements as traits representing the characters, the fore- and hind limbs. The relationships refer to the variational relationships between the corresponding traits in the fore- and hind limbs.
We confirmed the genetic nature of differences in variational relationships between fore- and hind limb traits by estimating the family effect on trait covariance (table 1). To study the genetic basis of covariance in more detail, we map the rQT loci affecting the variational relationships between the corresponding bone elements in the fore- and hind limb (humerus/femur, ulna/tibia). In rQTL mapping, the genotypic value is the regression coefficient of one trait on the other in a given genotype, rather than the trait mean as in standard mapping. Subsequent epistasis mapping reveals loci (intQTL) interacting with rQTL (Pavlicev et al. 2008, 2011). Mappings were carried out in the 34th generation of the intercross between small (SM/J) and large (LG/J) mouse inbred lines.
rQTL
Overall, 26 distinct rQTL exceed the chromosome-wide detection thresholds. Of these, 16 affect the relationship between the stylopodial traits (femur, humerus) and 10 the relationship between zeugopodial traits (ulna, tibia). The loci positions, confidence intervals, and trait relationships affected are detailed in supplementary table S1, Supplementary Material online. We find no overlap between stylopodial and zeugopodial rQTL, suggesting that a different set of loci modify pleiotropic effects in distal and proximal limb parts. Only three rQTL overlap with main-effect loci, which were reported for this population in Norgard et al. (2011): r.4 for humerus/femur, r.23 for tibia/ulna, and r.28 for femur/humerus overlap with main-effect loci Lbn4.3 for femur and humerus, Lbn1.3 for tibia, and Lbn14.1 for femur and humerus. This suggests that rQTL tend to have, at most, small direct effects on the traits, a pattern described previously (Pavlicev et al. 2008).
The genomic intervals within 1 LOD (logarithm of the odds) drop of the peak signal range in length from 0.11 to 11.49 Mbp (median 2.52), encompassing 4–158 annotated genes (median 18). Of a total of 983 genes in all rQTL, 569 are polymorphic between the LG/J and SM/J lines (median 11/locus). This list includes genes with SNPs in exons, introns, UTR regions, and in the 10 kb regions up- and downstream of the gene. 73–83% of the polymorphic genes within rQTL carry SNPs in intronic regions, and those within 10 kb up- and downstream of the gene, SNPs in UTR regions are found in 34% (3′UTR) and 16% (5′UTR) and nonsynonymous exonic SNPs are found in 29% of the polymorphic genes. SNPs in coding regions represent 0.2% of all polymorphisms in the rQTL intervals. According to the MGI database, at least 129 (23%) of the 569 polymorphic genes in rQTL intervals have known expression in limbs in development (table 3).
Table 3.
Polymorphic Limb-Expressed Genes within the rQTL Regions
| rQTL ID | Underlying Polymorphic Genes (Limb Expression Documented) |
|---|---|
| r.1 | Ptprd, Tle1, Megf9 |
| r.2 | Ift74, Tek |
| r.3 | Tns3 |
| r.4 | Dach1, Dis3, Mzt1, Pcdh9 |
| r.8 | Pold1, Lrrc4b, Mybpc2, Nr1h2 |
| r.9 | Gas2, Nav2 |
| r.10 | Git1, Aldoc, Bhlha9, Cpd, Phf12, Spag5, Traf4, Rpl23a, Trp53i13 |
| r.11 | E2f6, Rock2, Trib2 |
| r.12 | Dnajb9, Stxbp6, Lrrn3 |
| r.13 | Plcxd2, Pvrl3 |
| r.15 | Eprs, Bpnt1, Mark1, Mosc2, Rab3gap2 |
| r.16 | Igsf10, Mbnl1, Serp1, Wwtr1 |
| r.17 | Gars, 2410066E13Rik, Aqp1, Fam188b, Ggct, Pde1c, Inmt |
| r.18 | Cotl1, Plcg2, 2310061C15Rik, Cdh13, Cenpn, Gan, Gcsh, Mbtps1, Mphosph6, Sdr42e1, Wfdc1, Bcmo1, Maf |
| r.21 | Boc, Zbtb20, BC002163, Ccdc52 |
| r.23 | Tpr, 1200016B10Rik, BC003331, Ivns1abp, Pdc, Prg4, Pla2g4a, Hmcn1 |
| r.25 | Capn2, Dusp10, Trp53bp2 |
| r.26 | Ltbr |
| r.27 | Apex1, Mrpl52, Prmt5, Psmc6, Supt16h, Ang2, Bmp4, Cnih, Dad1, Ear7 (Thra), Gch1, Gnpnat1, Haus4, Lgals3, Lrp10, Mmp14, Ndrg2, Otx2, Peli2, Pnp, Ptger2, Rem2, Rnase1, Slc7a8, Tep1, Zfp219, Ttc5, Txndc16 |
| r.29 | Apc, Ik, Matr3, Ndufa2, Wdr55, Egr1, Brd8, Cd14, Ctnna1, Dnd1, Fgf1, Hars, Hdac3, Pcdhb10, Pcdhb5, Pcdhga12, Pcdhga9, Pfdn1, Pura, Spry4, Wnt8a, 0610009O20Rik, Cdc23, Pcdhb11, Pcdhb19, Pcdhb3, Slc35a4, Taf7 |
| r.30 | Smc3, Pdcd4, Adra2a, Mxi1, Add3 |
Note.—Genes differentially expressed between forelimb and hind limb at E10.5 are in bold.
For feasibility, we include a subset of 12 nonoverlapping rQTL in epistasis scans for interactions with rQTL. These loci were chosen due to the strongest association with traits. The description of the intervals (supplementary table S1, Supplementary Material online) and the intersection with gene expression data (discussed later) nevertheless refer to the complete set of 26 rQTL.
intQTL
We mapped epistatic interactions with each of the 12 rQTL to uncover genes modified by the rQTL. For example, for each rQTL detected for the relationship humerus/femur, we scanned for interactions between the rQTL and other loci, separately for the effects on humerus and femur. This scan reveals loci, at which the effect on the trait changes with the genotype at rQTL. We detected 48 intQTL, including 11 loci whose interactions with rQTL affect ulna, 8 for tibia, 14 for humerus, and 15 for femur (on average 4 loci/rQTL). All but four interactions detectably affect only one trait of the relationship (92%). Exceptions are two interactions affecting humerus and femur and two interactions affecting ulna and tibia (supplementary table S2, Supplementary Material online); however, the effect size or the pattern of shared interactions differ between traits (i.e., whether additive × additive, additive × dominance, etc., interaction is significant [Routman and Cheverud 1997]).
The genomic intervals within 1-LOD drop of the peak signal range for intQTL from 0.4 to 12.7 Mbp (median 1.79 Mbp). Of the total of 1,371 genes within intQTL, 721 (median 12/locus) are polymorphic between the parental lines, in intronic, exonic, 3′-UTR, 5′-UTR region, or in the 10 kbp intervals up- or downstream from the gene (supplementary table S2, Supplementary Material online). At least 253 of the polymorphic genes (supplementary table S3, Supplementary Material online) are expressed in limbs during development (MGI). Furthermore, 22 of 48 intQTL (46%) overlap with known QTL for bone defects or density, long bone length, body size, or growth. This is noteworthy as these studies involved strains with distinct genetic variation. Also, the known loss-of-function mutations in the intQTL include bone (42 genes), growth (84 genes), and limb-specific defects (30 genes). Only three intQTL (int.13, int.107, and int. 42) overlap with the main effect loci in the same population (Lbn2.1c, Lbn3.2a, and Lbn18.1a [Norgard et al. 2011]). All three have been detected as pleiotropic for the trait pair of concern.
Genomic Characterization of the rQTL and intQTL Intervals
In the genomic analyses, we take advantage of the available information on SNPs between the parental strains (table 2). We compare the distribution of SNPs in both sets of genomic intervals, with the rationale that the pattern of SNPs shared among and distinct between the rQTL and intQTL reveals the role of the two sets of loci and potentially the molecular mechanisms of trait individuation.
Table 2.
Overview of Genomic Analyses and Results
| Feature | rQTL | intQTL |
|---|---|---|
| Median length (range) | 2.52 (0.11–11.5) Mbp | 1.79 (0.42–12.7) Mbp |
| GC content (range) | 0.41 (0.37–0.52) | 0.42 (0.37–0.48) |
| Interval composition | ||
| Intergenic | 67.7% | 60.1% |
| UTR | 1.4% | 1.9% |
| Coding exon | 1.1% | 1.2% |
| Intron | 29.8% | 37.1% |
| SNP density (median) | ||
| nsyn SNP/N | 5.53 E−4 (0–0.01) | 5.96 E−4 (0–0.02) |
| syn SNP/S | 4.54 E−3 (0–0.05) | 4.42 E−3 (0–0.04) |
| SNP/intron | 2.94 E−1 | 1.14 E−3 |
| SNP/3′-UTR | 6.50 E−3 | 6.74 E−4 |
| SNP/5′-UTR | 2.95 E−3 | 6.84 E−2 |
| SNP/TFBS | 1.46 E−3 | 1.20 E−3 |
| Correlation of sequence-type SNP densities | High correlation of SNP densities between all parts of the sequence, except with introns | High correlation of SNP densities between all parts of the sequence, except with exons |
| Overlap with genes differentially expressed in limbs (Cotney et al. 2012) | 22 genes (P = 6.2 E−5) | 30 genes (P = 2.1 E−7) |
| Overlap with limb-specific enhancers (Cotney et al. 2012) | Total enhancers: 535; polymorphic: 224 (high overlap); polymorphic enhancers present in 70% rQTL | Total enhancers: 1,100; polymorphic: 140; polymorphic enhancers present in 12% intQTL |
| Overlap with limb-type -specific enhancers (Cotney et al. 2012) | Total enhancers: 39; polymorphic: 7; polymorphic enhancers present in 15% rQTL | Total enhancers: 69; polymorphic: 8; polymorphic enhancers present in 8% intQTL |
The rQTL intervals are on average longer than intQTL (supplementary tables S1 and S2, Supplementary Material online), mostly due to longer intergenic regions in rQTL. The relative lengths of exonic, intronic, and UTR regions are similar in both types (fig. 2). The loci are also similar in GC content (rQTL: median 0.41, range 0.37–0.52; intQTL: median 0.42, range 0.37–0.48) and in distribution along the chromosomes (supplementary fig. S1, Supplementary material online). The GC content is further uncorrelated with the level of polymorphism in either set of loci.
Fig. 2.—

Composition of QTL intervals in relationship and interaction loci. The mean proportions of sequence types within the rQTL and intQTL. Note that intQTL have greater portion of introns and rQTL greater portion of intergenic regions (“rest”).
SNP Densities in Genic Subregions
To understand whether SNP densities vary consistently across all subregions of the intervals, we tested correlations of SNP densities between the subregions (e.g., exons, introns, and UTRs) within and between the two sets of loci (fig. 3B). If a subregion is related to the function of a QTL type, for example, rQTL, we expect that its SNP density will not follow the overall variation of SNP densities across the other subregions. For rQTL, the SNP densities are highly positively correlated across most subregions, i.e., loci either have high or low SNP densities across all subregions. The exceptions are intronic regions, with lower or no correlation with other regions. This is different in intQTL, where the correlations between the SNP densities in most regions are also uniformly high, the clear exception being the exonic nonsynonymous positions. These are uncorrelated with SNP densities in all other subregions (fig. 3B).
Fig. 3.—

SNP densities in rQTL and intQTL. (A) The density of SNPs in subregions, in rQTL and intQTL. Note the low density of SNPs in rQTL nonsynonymous exonic sites, despite a general trend toward higher SNP densities in rQTL. (B) The Pearson correlation between SNP densities within and across interaction partners. Within-QTL type correlations are in the upper left for intQTL and lower right for rQTL and are higher than the correlations between the corresponding interacting loci. All within-intQTL correlations with nonsynonymous SNP density and all within-intQTL correlations with intronic SNP density are low. This implies a divergence of SNP pattern in rQTL introns and intQTL exons (r.: rQTL, int.: intQTL, s.exon: synonymous sites, ns.exon: nonsynonymous sites, sign: significant at the 95% confidence level).
The correlations of SNP densities between the pairs of interacting rQTL–intQTL reveal that rQTL with high SNPs density in 3′UTR tend to interact with intQTL with high density of SNPs in transcription factor binding sites. The implications of this observation are unclear and will be examined in future studies (fig. 3B).
Figure 4 shows single-gene nonsynonymous versus synonymous substitution rates (dN/dS ratio) within rQTL and intQTL. Although the bulk of genes in rQTL and intQTL have comparable distribution of dN/dS, single genes in intQTL consistently lie above the cloud, indicating that intQTL harbor genes with higher relative density of nonsynonymous SNPs. We also observe a trend to higher density of SNPs in rQTL for most regions, except for the nonsynonymous exonic SNPs. This effect is weak, yet consistent across the regions (fig. 3A; six genic regions analyzed; P = 0.55 = 0.031). Together, these results demonstrate a distinct character of exonic nonsynonymous variation in intQTL and intronic variation in rQTL. Note that the mutations in coding regions are thought to be associated with higher pleiotropy on average than the mutations in noncoding regions (Prud'homme et al. 2007). The detected pattern of exonic SNPs in intQTL and intronic SNPs in rQTL thus is consistent with the prediction of the SPC model (Pavlicev and Wagner 2012) that intQTL cause more pleiotropic and rQTL more character-specific variation.
Fig. 4.—

Exonic SNP densities in rQTL and intQTL. Distribution of dN/dS in coding genes in both intervals. Genes from rQTL are shown as red triangles and genes from intQTL intervals as black dots. Although many genes from both intervals overlap, the genes high in nonsynonymous substitution rate are genes from intQTL intervals.
Consistent differences between QTL types provide evidence that heritable variation in this cross is due to many, rather than single, SNPs in each QTL interval. If, in contrast, the variation were due to single mutations, the signal would be overwhelmed by other variation and we would not observe consistent patterns of SNP density and distribution. Although we cannot exclude single large-effect mutations, these alone cannot explain the observed pattern. The two intercrossed lines thus differ in the multiple alleles involved in trait divergence. Our interpretation of this result is that opposite selection on body size in the parental lines caused changes in size-related pleiotropic genes, and the differences among limbs were maintained within each line by line-specific rQTL changes. These changes nevertheless affected the similar pathways in both lines.
It is perhaps not surprising that the limb divergence is a complex evolutionary step involving many genetic factors. This may complicate the search for the actual genes involved; and it is the value of the present approach to nevertheless characterize the nature of these changes.
Gene Expression in rQTL and intQTL
We suggested that the genes underlying rQTL are involved in character-specific modification of pleiotropic genes (Pavlicev et al. 2011; Pavlicev and Wagner 2012). Character-specific effects can arise from variation in coding regions of locally expressed genes or from variation in cis-regulatory elements driving local gene expression. To test whether QTL intervals encompass genes with limb-type-specific gene expression, we intersected them with the transcriptome (Taher et al. 2011; Cotney et al. 2012) and chromatin mark (Cotney et al. 2012) data for murine fore- and hind limbs.
rQTL and intQTL Encompass Differentially Expressed Genes
We intersected the QTL intervals with the set of genes differentially expressed between fore- and hind limbs (Taher et al. 2011; Cotney et al. 2012). Differential expression was established by the comparisons of early developmental transcriptomes (stages E9.5–E13.5) of fore- and hind limb buds (Taher et al. 2011: microarray; Cotney et al. 2012: RNA Seq). We find 20 of the candidate genes to be significantly differentially expressed between fore- and hind limbs at E10.5 (raw P < 0.05; (supplementary table S2, Supplementary Material online) in Cotney et al. [2012]) and two additional genes in the later stages (Taher et al. 2011). These genes are located in 11 rQTL, of which 6 rQTL are associated with the relationship between proximal and 5 between the distal limb parts (table 3, boldface). Twenty is a significantly higher portion (11%) of differentially expressed genes than expected by sampling random intervals of the same size (hypergeometric test, P < 6.2 × 10−5). In intQTL, 30 genes are differentially expressed (hypergeometric test, P < 2.1 × 10−7). Thus, both rQTL and intQTL are enriched in genes differentially expressed in early development of mice limbs.
rQTL Are Enriched for Polymorphic Limb Enhancers
To address whether the QTL harbor limb-specific polymorphic enhancers, we intersected the intervals with the chromatin mark data reported by Cotney et al. (2012). The authors used chromatin state marks H3K27ac and H3K27me3 to map active and repressed enhancers in limbs during developmental stages E10.5, E11.5. Putative limb-specific enhancers were detected as more strongly marked in limb tissues than in embryonic stem cells or in neuronal progenitor cells.
Intersecting the limb-specific enhancers with rQTL revealed that a total of 535 limb enhancers (160 at E10.5 and 375 at E11.5) map into 23 rQTL. Of these, 224 (42%) are polymorphic in our population, located within 18 rQTL. This is significant enrichment of SNPs in limb-specific enhancers, relative to the rest of the genome (hypergeometric test, P = 1.3 × 10−10). In contrast, we find no enrichment of polymorphic limb enhancers in intQTL: although 1,100 limb enhancers map into intQTL (297 at E10.5 and 803 at E11.5), of these only 140 (38 at E10.5 and 102 at E11.5) are polymorphic, found in six intQTL. This finding is remarkable because intQTL are enriched in limb enhancers (hypergeometric test, P = 1.6 × 10−5), indicating the presence of limb-expressed genes; however, these enhancers tend not to be polymorphic. In summary, we find that 70% of rQTL harbor polymorphic limb-specific enhancers, whereas this is the case in only 12.5% of intQTL (fig. 5). These results suggest that cis-regulatory limb-specific variation underlies rQTL but not intQTL.
Fig. 5.—
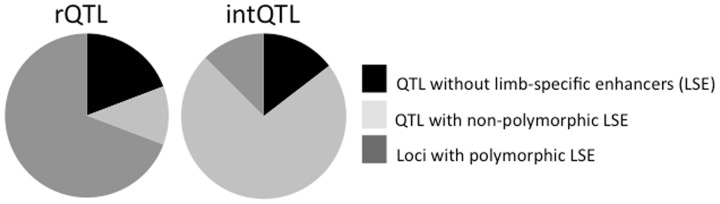
Proportion of loci with SNPs in limb-specific enhancers. The proportion of loci harboring the limb-specifically marked enhancers in the subset of rQTL (N = 26) and intQTL (N = 48). Although comparable proportions of intQTL and rQTL regions harbor limb-specific enhancers, much greater proportion of these is variable in rQTL.
We used GREAT (McLean et al. 2010) to characterize genes associated with the polymorphic limb-specific enhancers in intQTL and rQTL. In intQTL, these genes are enriched 117-fold for genes involved in developmental cell growth, as expected for H3K27ac marking the enhancers near developmental genes. In rQTL, the low number of genes (114) associated with limb-specific enhancers precluded enrichment analysis.
Note that the expression and chromatin mark data used capture early developmental processes, whereas the QTL are mapped on the adult variation. The degree to which these data sets can overlap depends on the developmental stages in which variation and divergence occurs. Although general opinion on the topic is ambiguous (Hall and Miyake 2000; Mariani and Martin 2003; Rolian 2008; Sanger et al. 2011; Cooper et al. 2013), our data suggest that some of it occurs in early developmental stages.
Overall, the data show that cis-regulatory variation in rQTL is likely causing the fore-/hind limb-specific effects. As we find no indication that genes underlying intQTL directly bind to these regions as transcriptional regulators, we speculate that the interaction is downstream of rQTL function, potentially by protein–protein interactions between the products of rQTL and intQTL genes. This interpretation is supported by the enrichment of protein binding functions among the candidate genes in both QTL intervals (GO:0005509; intQTL: P < 5 × 10−3; rQTL: P < 2 × 10−7).
Conclusion
In this study, our goal was to discover the molecular nature of the mutations involved in character individuation, specifically the individuation of fore- and hind limbs in mice. We hypothesized that character individuation results from the character-specific modification of the effects of pleiotropic genes (fig. 6). In our QTL mapping approach, the modifying genes are identified as rQTL and the pleiotropic genes as genes interacting with rQTL, called intQTL. For genes in intQTL intervals, we find sequences with a high density of nonsynonymous SNPs but no enrichment of SNPs in putative cis-regulatory regions. In contrast, rQTL intervals have a genomic signature that suggests cis-regulatory variation. We thus conclude that, in our system, pleiotropic mutations tend to affect protein coding sequences, whereas mutations causing rQTL effects are more likely cis-regulatory. The specific molecular nature of the modification of pleiotropic effects by rQTL genes cannot be inferred from the current data, except that it likely occurs downstream of both types of genes.
Fig. 6.—

The suggested genetic architecture of divergent traits affects trait covariance. (A) Undifferentiated limbs with fully shared genetic basis (left) and coordinated change in the mean values of both traits (right) as the genetic basis changes (I→I′). (B) Local interaction with shared genetic basis (left) generates the potential in forelimb (FL) to individualize the variation as interacting locus varies (R→R′→R″).
Supplementary Material
Supplementary tables S1–S3 and figure S1 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Gloria Fawcett, Elizabeth Norgard, and the Cheverud Lab for data collection. SNPs were scored by the John Hopkins Center of Inherited Disease Research (Baltimore, MD). This work was funded by the Postdoctoral Fellowship at the Konrad Lorenz Institute for Evolution and Cognition Research (to M.P.), the program in Foundational Questions in Evolutionary Biology of the National Philanthropic Trust (grant number RFP12-23 to G.P.W.), the National Science Foundation (grant number GM094780 to J.P.N. and grant number BCS-0725068 to J.M.C.), and the Discovery grant of the Natural Sciences and Engineering Research Council of Canada (grant number 238992 to B.H.).
Literature Cited
- Beissbarth T, Speed TP. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004;20:1464–1465. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
- Berg R. The ecological significance of correlation pleiades. Evolution. 1960;17:171–180. [Google Scholar]
- Blankenberg D, et al. Manipulation of FASTQ data with Galaxy. Bioinformatics. 2010;26:1783–1785. doi: 10.1093/bioinformatics/btq281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi A, Lehner B. Tissue specificity and the human protein interaction network. Mol Syst Biol. 2009;5:260. doi: 10.1038/msb.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Sen S, SpringerLink (Online service) A guide to QTL mapping with R/qtl. New York, London: Springer; 2009. [Google Scholar]
- Cheverud JM. Developmental integration and the evolution of pleiotropy. Am Zool. 1996;36:44–50. [Google Scholar]
- Cheverud JM, et al. Pleiotropic effects on mandibular morphology II: differential epistasis and genetic variation in morphological integration. J Exp Zool B Mol Dev Evol. 2004;302:424–435. doi: 10.1002/jez.b.21008. [DOI] [PubMed] [Google Scholar]
- Cooper KL, et al. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature. 2013;495:375–378. doi: 10.1038/nature11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney J, et al. Chromatin state signatures associated with tissue-specific gene expression and enhancer activity in the embryonic limb. Genome Res. 2012;22:1069–1080. doi: 10.1101/gr.129817.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboc V, Logan MP. Pitx1 is necessary for normal initiation of hindlimb outgrowth through regulation of Tbx4 expression and shapes hindlimb morphologies via targeted growth control. Development. 2011a;138:5301–5309. doi: 10.1242/dev.074153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboc V, Logan MP. Regulation of limb bud initiation and limb-type morphology. Dev Dyn. 2011b;240:1017–1027. doi: 10.1002/dvdy.22582. [DOI] [PubMed] [Google Scholar]
- Euskirchen GM, et al. Mapping of transcription factor binding regions in mammalian cells by ChIP: comparison of array- and sequencing-based technologies. Genome Res. 2007;17:898–909. doi: 10.1101/gr.5583007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G. Epistasis and pleiotropy as natural properties of transcriptional regulation. Theor Popul Biol. 1996;49:58–89. doi: 10.1006/tpbi.1996.0003. [DOI] [PubMed] [Google Scholar]
- Gjuvsland AB, Hayes BJ, Omholt SW, Carlborg O. Statistical epistasis is a generic feature of gene regulatory networks. Genetics. 2007;175:411–420. doi: 10.1534/genetics.106.058859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski MW, Polk JD, Roseman CC. Divergent patterns of integration and reduced constraint in the human hip and the origins of bipedalism. Evolution. 2011;65:1336–1356. doi: 10.1111/j.1558-5646.2011.01226.x. [DOI] [PubMed] [Google Scholar]
- Hall BK, Miyake T. All for one and one for all: condensations and the initiation of skeletal development. Bioessays. 2000;22:138–147. doi: 10.1002/(SICI)1521-1878(200002)22:2<138::AID-BIES5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Han JD, et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature. 2004;430:88–93. doi: 10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- Hansen TF, Pelabon C, Armbruster WS, Carlson ML. Evolvability and genetic constraint in Dalechampia blossoms: components of variance and measures of evolvability. J Evol Biol. 2003;16:754–766. doi: 10.1046/j.1420-9101.2003.00556.x. [DOI] [PubMed] [Google Scholar]
- Hughes CL, Kaufman TC. Hox genes and the evolution of the arthropod body plan. Evol Dev. 2002;4:459–499. doi: 10.1046/j.1525-142x.2002.02034.x. [DOI] [PubMed] [Google Scholar]
- Kenney-Hunt JP, et al. Quantitative trait loci for body size components in mice. Mamm Genome. 2006;17:526–537. doi: 10.1007/s00335-005-0160-6. [DOI] [PubMed] [Google Scholar]
- Kopp A. Drosophila sex combs as a model of evolutionary innovations. Evol Dev. 2011;13:504–522. doi: 10.1111/j.1525-142X.2011.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande R. The genetic covariance between characters maintained by pleiotropic mutations. Genetics. 1980;94:203–215. doi: 10.1093/genetics/94.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson HA, et al. Genetic effects at pleiotropic loci are context-dependent with consequences for the maintenance of genetic variation in populations. PLoS Genet. 2011;7:e1002256. doi: 10.1371/journal.pgen.1002256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leamy LJ, Pomp D, Lightfoot JT. Genetic variation for body weight change in mice in response to physical exercise. BMC Genet. 2009;10:58. doi: 10.1186/1471-2156-10-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ji L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity. 2005;95:221–227. doi: 10.1038/sj.hdy.6800717. [DOI] [PubMed] [Google Scholar]
- Luscombe NM, et al. Genomic analysis of regulatory network dynamics reveals large topological changes. Nature. 2004;431:308–312. doi: 10.1038/nature02782. [DOI] [PubMed] [Google Scholar]
- Mann RS, Lelli KM, Joshi R. Hox specificity unique roles for cofactors and collaborators. Curr Top Dev Biol. 2009;88:63–101. doi: 10.1016/S0070-2153(09)88003-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani FV, Martin GR. Deciphering skeletal patterning: clues from the limb. Nature. 2003;423:319–325. doi: 10.1038/nature01655. [DOI] [PubMed] [Google Scholar]
- McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minguillon C, Gibson-Brown JJ, Logan MP. Tbx4/5 gene duplication and the origin of vertebrate paired appendages. Proc Natl Acad Sci U S A. 2009;106:21726–21730. doi: 10.1073/pnas.0910153106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgard EA, et al. Genetic factors and diet affect long-bone length in the F34 LG,SM advanced intercross. Mamm Genome. 2011;22:178–196. doi: 10.1007/s00335-010-9311-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgard EA, et al. Identification of quantitative trait loci affecting murine long bone length in a two-generation intercross of LG/J and SM/J Mice. J Bone Miner Res. 2008;23:887–895. doi: 10.1359/JBMR.080210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohde T, Yaginuma T, Niimi T. Insect morphological diversification through the modification of wing serial homologs. Science. 2013;340:495–498. doi: 10.1126/science.1234219. [DOI] [PubMed] [Google Scholar]
- Pavlicev M, Cheverud JM, Wagner GP. Evolution of adaptive phenotypic variation patterns by direct selection for evolvability. Proc Biol Sci. 2011;278:1903–1912. doi: 10.1098/rspb.2010.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlicev M, et al. Genetic variation in pleiotropy: differential epistasis as a source of variation in the allometric relationship between long bone lengths and body weight. Evolution. 2008;62:199–213. doi: 10.1111/j.1558-5646.2007.00255.x. [DOI] [PubMed] [Google Scholar]
- Pavlicev M, Norgard EA, Fawcett GL, Cheverud JM. Evolution of pleiotropy: epistatic interaction pattern supports a mechanistic model underlying variation in genotype-phenotype map. J Exp Zool B Mol Dev Evol. 2011;316:371–385. doi: 10.1002/jez.b.21410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlicev M, Wagner GP. A model of developmental evolution: selection, pleiotropy and compensation. Trends Ecol Evol. 2012;27:316–322. doi: 10.1016/j.tree.2012.01.016. [DOI] [PubMed] [Google Scholar]
- Porto A, et al. The evolution of modularity in the mammalian skull I: morphological integraton patterns and magnitudes. Evol Biol. 2009;36:118–135. [Google Scholar]
- Prud'homme B, Gompel N, Carroll SB. Emerging principles of regulatory evolution. Proc Natl Acad Sci U S A. 2007;104(Suppl 1):8605–8612. doi: 10.1073/pnas.0700488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolian C. Developmental basis of limb length in rodents: evidence for multiple divisions of labor in mechanisms of endochondral bone growth. Evol Dev. 2008;10:15–28. doi: 10.1111/j.1525-142X.2008.00211.x. [DOI] [PubMed] [Google Scholar]
- Routman EJ, Cheverud JM. Gene effects on a quantitative trait: two-locus epistatic effects measured at microsatellite markers and at estimated QTL. Evolution. 1997;51:1654–1662. doi: 10.1111/j.1558-5646.1997.tb01488.x. [DOI] [PubMed] [Google Scholar]
- Ruvinsky I, Gibson-Brown JJ. Genetic and developmental bases of serial homology in vertebrate limb evolution. Development. 2000;127:5233–5244. doi: 10.1242/dev.127.24.5233. [DOI] [PubMed] [Google Scholar]
- Sanger TJ, et al. Developmental and genetic origins of murine long bone length variation. J Exp Zool B Mol Dev Evol. 2011;316B:146–161. doi: 10.1002/jez.b.21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taher L, et al. Global gene expression analysis of murine limb development. PLoS One. 2011;6:e28358. doi: 10.1371/journal.pone.0028358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GP. Homologues, natural kinds and the evolution of modularity. Am Zool. 1996;36:36–43. [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Young NM, Hallgrimsson B. Serial homology and the evolution of mammalian limb covariation structure. Evolution. 2005;59:2691–2704. [PubMed] [Google Scholar]
- Young NM, Wagner GP, Hallgrimsson B. Development and the evolvability of human limbs. Proc Natl Acad Sci U S A. 2010;107:3400–3405. doi: 10.1073/pnas.0911856107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.