

Abstract
Obesity is a long-term source of cellular stress that predisposes to chronic kidney disease (CKD). Autophagy is a homeostatic mechanism for cellular quality control through the disposal and recycling of cellular components. During times of cellular stress, autophagy affords mechanisms to manage stress by selectively ridding the cell of the accumulation of potentially toxic proteins, lipids and organelles. The adaptive processes employed may vary between cell types and selectively adjust to the insult by inducing components of the basic autophagy machinery utilized by the cells while not under duress. In this review, we will discuss the autophagic responses of organs to cellular stressors, such as high-fat diet, obesity and diabetes, and how these mechanisms may prevent or promote the progression of disease. The identification of early cellular mechanisms in the advent of obesity- and diabetes-related renal complications could afford avenues for future therapeutic interventions.
Keywords: AMPK, diabetes, high fat diet, insulin resistance, mTOR
INTRODUCTION
Obesity continues to be a widespread public health issue. In the USA, the prevalence of obesity doubled between 1980 and 2002, affecting approximately one in three adults [1]. Adipose tissue is not merely for energy storage but is rather a complex endocrine gland that interacts with other organs. It is largely through these interactions that obesity increases the likelihood of various diseases and associates with diabetes, hypertension, proteinuria and arthrosclerosis, risk factors of chronic kidney disease (CKD). Several studies support this association between obesity and kidney disease [2–4], including a recent meta-analysis, which demonstrated the contribution of obesity to the progressive loss of kidney function in those patients with CKD [5]. In this review, we will briefly describe the links between adiposity and regulation of autophagy, and the role that autophagy plays in kidney diseases associated with obesity and diabetes.
OBESITY
Genetically obese Zucker rats demonstrate early changes in kidney hemodynamics including increases in glomerular filtration rate (GFR) and albuminuria along with changes in renal morphology including glomerular enlargement, mesangial expansion and podocyte injury. These hemodynamic changes are reversible with caloric restriction [6], as are the structural changes [7]. In patients, an increased body mass index (BMI) is not only correlated with albumin excretion, but caloric restriction or bariatric surgery as a means of weight reduction reduces proteinuria [8]. The impact on the kidney beyond hemodynamic, structural and pathologic changes is that obesity amplifies the risk of CKD. Other comorbidities include metabolic syndrome, high blood pressure, diabetes mellitus and cardiovascular disease [9].
One link between caloric excess and CKD involves the nutrient sensor, 5′ AMP-activated protein kinase (AMPK). AMPK is activated when the AMP(ADP):ATP ratio increases, such as in times of caloric restriction or exercise. AMPK is also responsive to the adipokine, adiponectin, which is shown to decrease in obese individuals with insulin resistance. The adiponectin-deficient mouse demonstrates low AMPK activity, albuminuria and podocyte foot process effacement. Agonist induction of AMPK activity attenuates these pathologic conditions [10]. One central, cellular homeostatic mechanism responsive to AMPK is autophagy, and there is a growing body of literature linking autophagy to chronic organ dysfunction.
GENERAL AUTOPHAGY
Autophagy is a conserved catabolic mechanism required for cellular homeostatic quality control and regeneration as well as a cellular stress response mechanism. There are three types of autophagy that differ in their mechanism of delivering their cargo to the lysosome: macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy. Macroautophagy requires the formation of a double-membrane vesicle, the autophagosome, which upon formation sequesters cytosolic components for delivery to the lysosome (Figure 1). CMA is a form of selective autophagy utilizing heat shock chaperone of 70 kDa, hsc70, which recognizes the pentapeptide KFERQ motif upon misfolding of protein complexes. This hsc70-protein complex binds to a lysosomal receptor complex, is unfolded and translocated into the lysosome [11–13]. In microautophagy, cytosolic materials are directly engulfed by invagination of the lysosomal membrane [14].
FIGURE 1:
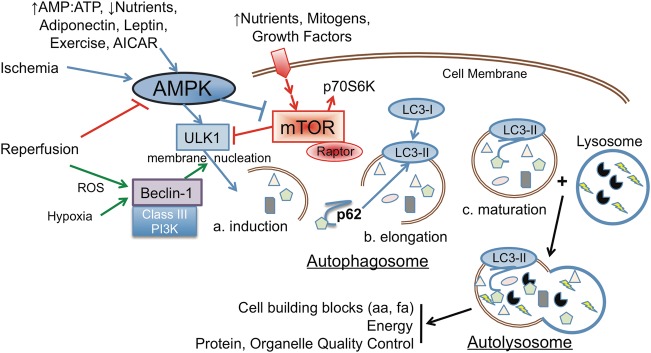
Autophagy: mechanism. Illustrated is a simplified progression of autophagy from initiation, nucleation and membrane elongation and maturation with final fusion with a lysosome. ATG conjugation pathways are not shown for space considerations. Nutrients can affect the AMPK/mTOR balance to modulate autophagy, as can cellular stress in an AMPK independent manner. Probably, the best demonstration of these two divergent initiation pathways in a single model is ischemia-reperfusion, as shown. Arrows are progressive and bars are inhibitory. Small triangles, rectangles and ovals represent cargo for autophagic degradation. Lightning bolts and Pac-Man represent lysosomal enzymes.
Macroautophagy will be simply referred to as autophagy for the remainder of this review. In this form of autophagy, the double membrane autophagosome fuses with a lysosomal vesicle to form an autolysosome where lysosomal proteases can then access and degrade the sequestered cargo of the autophagosome (Figure 1). This process is fundamental in ridding the cell of aged and aggregated proteins and is the only known mechanism for disposing of damaged organelles under normal conditions as well as in cells under stress [15–19]. The capacity to remove aged and damaged cellular components, such as mitochondria and endoplasmic reticulum (ER), can avoid cellular stress, apoptosis and promote cell and organism longevity. In states of normal physiology, there is an active autophagic process working in all cell types of the kidney and is especially prominent in podocytes to protect cellular integrity against the stresses these cells encounter at the filtration barrier [20].
The process of initiation, nucleation and elongation/maturation of the macroautophagy vesicle requires several different AuTophaGy (ATG) proteins. Initiation is controlled by a ULK1-ATG13-FIP200 complex, nucleation requires a Beclin-1-Class III PI3K and associated protein complex, and two conjugation systems are involved in elongation and maturation or closure (Figure 1). One of these conjugation systems entails the cleavage of microtubule-associated proteins light chain 3 (LC3) to cytosolic LC3-I, and the further modification for lipidation to phosphatidylethanolamine in the autophagosome membrane, forming LC3-II. Thus, cytosolic LC3-I and autophagosome-bound LC3-II levels are established indicators of canonical autophagy [21].
Whereas the chaperone hsc70 of CMA binds a pentapeptide motif, KFERQ, in proteins targeted for translocation into the lysosome, macroautophagy was considered a non-selective process engulfing segments of the cytosol. Proteins have since been identified that selectively enhance the autophagosomal cargo with proteins targeted for degradation. The best studied is linker protein p62/SQSTM1 (p62). Binding domains on p62 include an ubiquitin-associated binding domain and an LC3 interacting region. In this way, p62 scavenges ubiquitinated proteins for degradation and binds LC3 to anchor these proteins into the forming autophagosome. An accumulation of p62 into aggregates occurs when autophagy is low or deficient [22]. Aggregates of p62 can stabilize Nrf-2 (nuclear factor (erythroid-derived 2)-like 2), a central transcriptional regulator of enzymes expressed in response to the oxidative cell stress that would occur in low autophagic states [23]. Accumulated p62 also suppresses proteasomal activity [24], the other cellular means of protein degradation. A chronic inability to dispose of toxic cellular aggregates further increases cell stress and death.
REGULATION OF AUTOPHAGY
A fine balance: the reciprocal harmony of mTOR and AMPK
Nutrient input activates the protein kinase, mammalian or mechanistic target of rapamycin (mTOR). There are two distinct mTOR complexes, Complex 1 where mTOR associates with Raptor, PRAS40 and mLST8, or Complex 2 where it associates with Rictor, mSIN-1, Protor-1 and mLST8. The small GTPases Rag, via amino acids or Rheb, via growth factors, activates mTORC1 and degrades Deptor. Downstream targets induce protein and ribosomal biosynthesis and cell growth. mTORC1 suppresses autophagy through phosphorylation of ULK1 and ATG13, as such we will be concerned with mTORC1 in this review.
AMPK is a ubiquitously expressed kinase that plays a key role in energy homeostasis. AMPK is activated in response to low nutrition or energy expenditure that results in an increase in the AMP (ADP)/ATP ratio and the nutritional sensor LKB1. It can also be activated by adipohormones, such as adiponectin. Stimulation of AMPK activates autophagy directly through phosphorylation of the ULK1 autophagy initiation complex (ULK1, ATG13, FIP200), at a different site than mTORC1, and also suppresses mTORC1 activity through phosphorylation of TSC2, which maintains Rheb in an inactive state. Inhibiting mTORC1 also promotes autophagy. AMPK in this way suppresses energy expenditure processes, such as protein synthesis, and also affords mitochondria protection and biogenesis [25], the expression of cellular antioxidant defense proteins [26], suppression of extracellular matrix proteins [27], and induction of eNOS [28] yet suppression of iNOS [29]. For most cells, there is a balance between mTOR, which induces anabolic, energy-utilizing processes and AMPK/autophagy, which promote catabolic, energy-conserving processes.
At times when food is plentiful, AMPK activity will decrease allowing downstream mTORC1 to increase. The cells and organisms will synthesize new proteins and fatty acids and increase metabolic activity. When food intake is limited, activation of AMPK acts to conserve energy expenditure, utilize intracellular energy sources, and degrade aged and damaged proteins and organelles by autophagy to generate building blocks for future mTOR induction of anabolic processes [30]. The biorhythm of these opposing biological cycles is required for longevity. In ischemia-reperfusion (IR) in the heart [31] and kidney (unpublished data), induction of AMPK occurs during the nutrient-deficient ischemic phase, and is down regulated upon reperfusion, where mTOR is induced. Please refer to Figure 1 for a diagrammatic representation of interaction between AMPK and mTOR in the induction of autophagy. Sirt1 plays a role in activating autophagy by deacetylating autophagic factors including ATG5, ATG7 and LC3 [32]. Activation of Sirt1 by an increase in the NAD+:NADH ratio in response to exercise or fasting may be attributed to AMPK.
There is an integral interrelationship between the anabolic, mTOR, and catabolic, autophagy, pathways. Induction of AMPK/autophagy results in the catabolic degradation of cellular components with the resultant generation of amino acids. The amino acids then comprise part of a cycle activating the anabolic mTORC1 pathway via Rag (Figure 2). To accommodate the secretory phenotype in senescent cells, this feedback can be breached whereby both the pathways are activated via a reorganization of the endomembrane system termed the TOR-autophagy spatial coupling compartment (TASCC) [33]. In the TASCC, mature autophagosomes fuse with lysosomes bound with mTOR. The amino acids generated by autophagy are utilized to activate mTOR and recruit it onto the lysosomal membrane and thus promote synthesis of secreted proteins characteristic of senescent cells. In this process, mTOR remains spatially distinct from ULK1 and early autophagosome formation allowing both autophagy and protein synthesis to progress. TASCC was observed in podocytes and may permit protein synthesis in these cells that maintain high constitutive autophagy. The linker protein p62 is another interconnecting component of the catabolic/anabolic cycle. It also binds Raptor and is required for mTORC1 stimulation in response to amino acids [34]. The function of p62 thus serves not only as a selective cargo linker for autophagy, where it gets degraded by autophagy, but also controls autophagy by promoting mTORC1 activity in the presence of amino acids, thus suppressing its own degradation. Induction of mTORC1 is required for lysosomal biogenesis [35], and thus supports autophagy. Deficiency of p62 increases the activity of adipogenic regulator, PPARγ, and also imparts the characteristics of the metabolic syndrome with glucose intolerance, insulin resistance, inflammation, reduced energy expenditure and obesity in mature mice [36].
FIGURE 2:

Autophagy: regulation. A line diagram with arrows as progressive pathways and bars inhibitory is shown. Blue lines are primarily catabolic, whereas black are anabolic. Excessive mTORC1 activity can lead to increases in proteins, growth, adipogenesis and lipogenesis. Combined with an mTORC1 inhibition of autophagy for quality control and removal of dysfunctional proteins and organelles, progression can lead to cell stress. This diagram also illustrates the checks and balances between the catabolic and anabolic pathways, the result if one pathway, mTORC1 as per diabetes, skews that balance, and the reliance of one pathway on the other (i.e. amino acids generated by autophagy for mTORC1 activation, and mTORC1 generation of lysosomes support autophagy).
The proper balance of these pathways affords a nutritional balance for proper cell homeostasis and longevity. Current research shows that a balance in the interregulation of these pathways is required for the reduction of stress and preservation of cell health. This cycle of autophagic degradation, amino acid generation, mTORC1 induction and lysosomal regeneration establishes a pendulum-like interrelationship between anabolic and catabolic cycles, and their dependence upon one another. That chronic inhibition or activation of either the catabolic, autophagy or anabolic, mTORC1, arms of the cycle will negatively affect the entire cycle (Figure 2). This can be appreciated by the disruption of this biorhythm in disease or utilizing pharmacologic or genetic means to disrupt this balance and mimic disease, as will be described later. To further support this biorhythm, the autophagy protein ATG14 is regulated by circadian rhythm [37]. Therefore, to restore the balance pharmaceuticals should be administered with this cycle in mind.
Mitophagy in response to cellular stress
There are different forms of autophagy in response to diverse signaling pathways, and we are only now beginning to appreciate their differences and consequences [38]. As shown in Figure 1, although ischemia would reduce nutrients and activate AMPK, reperfusion increases cellular stress that induces autophagy independent of nutrient status. Cellular stress can damage mitochondria and ER resulting in the generation of ROS, and oxidized and misfolded proteins. An autophagic response can selectively target, acquire and degrade these organelles and affected proteins. The mechanisms involved in selective mitochondrial macroautophagy, or mitophagy, are of particular interest.
Mitochondria are widely considered the cellular power plants with aerobic production of ATP, with other functions including calcium signaling and apoptosis. Cell demand and physiologic conditions play a role in the number and size of these organelles. The high-energy requiring transport responsibilities of the kidney proximal tubule, for example, require abundant mitochondria. Selective degradation occurs by autophagy coupled with fission/fusion events. In disease states, this mechanism might become further taxed to avoid cell death. Damaged or aged mitochondria exhibit a decreased mitochondrial membrane potential that activates the PTEN-induced putative kinase 1 (PINK1) and the E3-ubiquitin ligase, Parkin. PINK1 is constitutively produced and binds the mitochondrial membrane but has a very short half-life. Damage to the mitochondria stabilizes PINK1 proteins, which accumulate on the outer mitochondrial membrane and recruit Parkin. The PINK1/Parkin complex has the dual function of ubiquitinating both mitofusins to prevent mitochondrial fusion, and the voltage-dependent anion channel 1. These polyubiquitinated mitochondria bind the p62 linker protein and then LC3 on the developing autophagosome resulting in sequestration and removal of the mitochondria by autophagy. For a current review of mitophagy, see [39].
A programed process for removing mitochondria during development, as in red blood cells, demonstrates the involvement of the BH3-only proteins Nix (also known as Bnip3L) and Bnip3. These mitochondrial-binding proteins are able to bind to the autophagosome LC3 directly without requiring the ubiquitin-p62 linker bridge. This mechanism was previously shown to respond to stress stimulation and dictate apoptosis or necrosis. It is now thought that this mechanism is utilized to maintain baseline mitochondrial homeostasis by autophagy, but can regulate mitochondrial autophagy, apoptosis or necrosis depending upon the magnitude and duration of the cellular stress [40]. Hypoxia can also induce Bnip3 that can displace Beclin1 from Bcl-2 and promote autophagy [41].
The ER unfolded protein response (UPR) is a cellular stress response pathway engaged to restore ER homeostasis after stress or aging challenges to ER function. The ER UPR employs proteasomal and autophagic mechanisms to dispose of misfolded proteins and damaged ER. ER stress to kidney proximal tubule cells increases the autophagy response [42]. Both PERK [43] and IRE1 [44] UPR pathways induce autophagy. Like mitophagy, ER autophagy, or reticulophagy, appears to be a mechanism for cell protection. ROS may be a common factor in cellular stress induction of autophagic response mechanisms [45].
Lipophagy
Autophagy not only serves as a homeostatic and cellular stress quality control mechanism, but also generates energy for the cell at a time when nutrients are low. Energy derived from amino acids is relatively inefficient, but autophagy was recently shown to be instrumental in the more energy efficient catabolism of intracellular lipid droplets (LDs) [46], or lipophagy. Why there are two systems capable of mobilizing LDs, autophagy and cytosolic lipases is not known and requires further study.
Starvation increases basal lipophagy for energy generation, whereas lipid challenge also increases basal lipophagy and leads to selective sequestration of LD. However, chronic lipid challenge leads to an increase of LD in the cell [46]. This expansion of the LD compartment observed with a 4 month high-fat diet (HFD) is due to the progressive inability of the autophagosome to fuse with the lysosome (Figure 3) [47]. A higher composition of cholesterol in the autophagosome appears to be the culprit, and depletion of cholesterol in fibroblast cells increases autophagy [48]. Thus, a change in membrane lipid composition appears to be the reason for the failure of autophagosome–lysosome fusion that consequently turns an increase in autophagy in response to HFD to suppression of autophagy in the long term [47].
FIGURE 3:
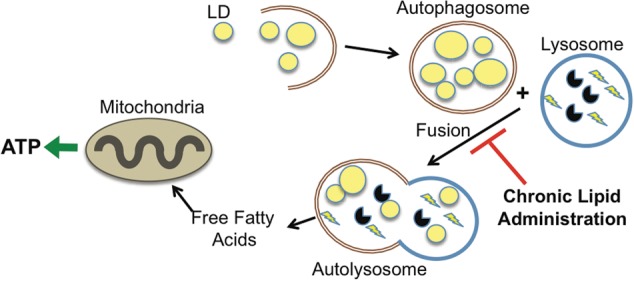
Lipophagy. In either excess lipid load or lack of nutrients autophagy is utilized to mobilize LDs. Lipophagy in the former is to maintain LD capacity/homeostasis, and the latter for energy. Chronic lipid administration can eventually inhibit autophagosome–lysosome fusion causing an accumulation of autophagosome vesicles.
HFD AND OBESITY AFFECT AUTOPHAGY: LINK TO INSULIN RESISTANCE
Studies investigating the impact of HFD on autophagy, and the effects of autophagy on the organ, have been largely in the hypothalamus, liver, heart, pancreatic β-cells and adipocytes. Obesity and HFD negatively affect the balance and response of autophagy and related pathways.
Adipose cells engage in the mobilization of LD not only to supply energy in times of low nutrient supply, but also to prevent enlargement of stores from compromising cellular integrity in response to a large lipid load. In line with this rationale, HFD or obesity leads to an increase in autophagy in adipocytes, and in a study of obese patients autophagy was particularly elevated in omental adipose tissue [49]. Adipocytes from obese type 2 diabetic (T2D) patients displayed a decrease in mTORC1 with a concurrent increase in autophagy, impaired mitochondria, ER stress and cytosolic LD [50]. Adiponectin is an adipokine that decreases with obesity. It was shown to have a negative correlation with albuminuria in obese patients and adiponectin-deficient mice display increased albuminuria and fused podocyte foot processes [10]. Elevated levels of autophagy in adipose tissue in response to obesity-induced ER stress increase degradation and thus reduce levels of adiponectin [51] as well as insulin receptors [52]. Thus, increased autophagic degradation was purported to play a role in insulin resistance. Studies with autophagy gene ATG7 knock-out mice supported this concept. These autophagy-compromised mice displayed smaller adipocytes with multiocular LDs and increased numbers of mitochondria and consequent rates of β-oxidation. That is, these adipocytes resembled brown adipose tissue [53]. Furthermore, the mice were slimmer, more energetic and resistant to HFD-induced obesity and insulin resistance [53, 54]. In obese T2D patients, the inability of insulin to activate mTORC1 in adipocytes contributes to overactive autophagy [50].
Unlike adipocytes, the liver displays a decrease in autophagy and an increase in ER stress in obese (ob/ob) mice [55]. Knocking down ATG7 to suppress autophagy even in lean mice is sufficient to induce ER stress and insulin resistance. Further, restoring autophagy by adenoviral expression of ATG7 in ob/ob mice increased insulin sensitivity and decreased ER stress in these animals relative to the ob/ob mice with control vector. Chronic HFD feeding disturbs the capacity of autophagosomes to fuse with lysosomes in the liver [47]. The relevance of autophagy in obesity was further exemplified in pancreatic β-cells where selectively suppressing autophagy by breeding β-cell-specific ATG7-deficient mice with ob/ob mice resulted in increased ER stress, β-cell death and severe diabetes [56]. Pancreatic β cells also display an accumulation of autophagosomes and decrease in lysosome gene expression in response to HFD or diabetes, which denotes a low autophagic flux [57]. HFD in the hypothalamus also resulted in an autophagy defect that correlated with increased inflammation, ER stress and obesity [58]. HFD induction of autophagy in adipose tissue is detrimental, but that does not appear to extend to these other organs where autophagy proved beneficial. However, a progression to defective autophagy in these organs may present complications offsetting the protective benefits.
AUTOPHAGY IN OBESITY-RELATED CKD
Although little work has been done with HFD and autophagy in the kidney, there has been extensive work in kidney diabetic models and current reviews on the topic of kidney autophagy and diabetes, along with other kidney diseases [59–61].
Aging can contribute to the progression of many diseases, including CKD. Both AMPK activity and autophagy decrease with aging, whereas aging brings increases in cholesterol, free fatty acids, ROS and in response to increased cellular stress a transition to a senescent phenotype. Aging mice with a podocyte-specific deficiency in ATG5 exhibited increased ER stress, accumulation of oxidized and ubiquitinated proteins and organelles, proteinuria and glomerular damage [20]. Podocytes play a pivotal role in maintaining the filtration barrier, which appears to be a vulnerable component of the kidney as podocyte injury can lead to proteinuria and glomerulosclerosis, principal features of diabetic nephropathy. Complicating the issue further, these cells are post-mitotic with limited proliferative capacity and must manage cellular stresses in a highly variable environment. For these reasons, high constitutive autophagic activity may be required in these cells to suppress cellular stress and maintain podocyte integrity.
This high autophagy, low mTORC1 status typical of podocytes is transformed to a low autophagy, high mTORC1 status in many disease states, including diabetic nephropathy. This can lead to an accumulation of damaged and ubiquitinated proteins and organelles that increase cellular stress and podocyte hypertrophy, podocyte injury and loss, and eventual glomerular disease with progressive proteinuria and glomerulosclerosis [62]. To further illustrate this balance, Inoki et al. [63] hyper-activated podocyte selective mTORC1 in mice and observed podocyte damage and glomerular lesions similar to the diabetic phenotype, including proteinuria and mesangial expansion. These data are largely consistent with the findings of Hartleben et al. [20] of aging mice with podocyte-deficient ATG5 mentioned above, except the hyper-active podocyte mTORC1 mice display podocyte overgrowth as well. Conversely, inhibition of mTORC1 with rapamycin largely suppresses these complications and thus the diabetic phenotype [64, 65]. This may, in part, be due to re-activation of homeostatic autophagy. Issues did arise when administering rapamycin to patients, most notably increases in proteinuria that was attributed to the podocytes [66]. It was thought that long-term rapamycin may affect mTORC2 and actin cytoskeletal components required for podocyte slit diaphragm structure, and indeed slit diaphragm and associated proteins were down regulated with rapamycin [67]. Interestingly, deletion of Raptor (mTORC1 specific) alone also resulted in glomerular injury [68], suggesting a more complicated paradigm. Recently, mTOR inhibition in podocytes was shown to suppress new lysosome formation, leading to an accumulation of autophagosomes [69]. This again illustrates an interdependence of the catabolic autophagy pathways with the anabolic mTOR pathways (Figure 2).
SUMMARY
Autophagy carefully balances with anabolic nutrition cycles and is required for cellular homeostasis and orgasm longevity. Because of the natural circadian rhythm of this balancing act, selective agonists or antagonists should also be administered accordingly. Autophagy can also respond to cellular and environmental stressors in the short term, but in the long term a persistent demand of such selective and protective activities may detract from the normal homeostatic processes of breaking down old and damaged proteins and organelles to detoxify the cell and generate new cellular building blocks. HFD and obesity negatively affect the balance and response of these pathways. What starts out as a protective mechanism of breaking down LD in response to a lipid load, in animals maintained on a HFD chronic lipophagy becomes deleterious in adipocytes. A similar issue may arise in diabetes where podocytes shift from a high autophagy, low mTOR status to low autophagy, high mTOR status. Blunt inhibition of mTOR through either pharmacologic or genetic means may prove beneficial in animal models, but because of the interdependence of these pathways such intervention may eventually negatively affect autophagy and increase cellular stress in the long term. Patients may also be more acutely sensitive to these therapeutic strategies if they already have underlying health issues. In all, understanding the interrelationships of these pathways brings to light the beneficial effects of diet and exercise in obese and diabetic patients.
Other forms of autophagy, including CMA [13], peroxisome selective autophagy, pexophagy [70], ER autophagy, reticulophagy, as well as several other proteins and pathways, such as Sirt1 [71], can impact the catabolic anabolic balance but have not been discussed due to length restrictions. I apologize to colleagues whose work has not been cited due to these same limitations.
CONFLICT OF INTEREST STATEMENT
None declared
ACKNOWLEDGEMENTS
Our projects are supported by grants from a VA MERIT Award (KS) and NIDDK (U01 DK060995, DP3 DK094352-01 and DK083142 awards) to K.S., National Institutes of Health Grants DK56248, DK28602, UAB-UCSD O'Brien Center for Acute Kidney Injury Research Grant P30DK079337 (JS), and Department of Veterans Affairs, VA Merit Awards 5101 and 0011, Veterans Health Administration, Office of Research and Development.
REFERENCES
- 1.Ogden CL, Carroll MD, Curtin LR, et al. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–1555. doi: 10.1001/jama.295.13.1549. doi:10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 2.Ejerblad E, Fored CM, Lindblad P, et al. Obesity and risk for chronic renal failure. J Am Soc Nephrol. 2006;17:1695–1702. doi: 10.1681/ASN.2005060638. doi:10.1681/ASN.2005060638. [DOI] [PubMed] [Google Scholar]
- 3.Hsu CY, Iribarren C, McCulloch CE, et al. Risk factors for end-stage renal disease: 25-year follow-up. Arch Intern Med. 2009;169:342–350. doi: 10.1001/archinternmed.2008.605. doi:10.1001/archinternmed.2008.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ogden CL, Yanovski SZ, Carroll MD, et al. The epidemiology of obesity. Gastroenterology. 2007;132:2087–2102. doi: 10.1053/j.gastro.2007.03.052. doi:10.1053/j.gastro.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Chen X, Song Y, et al. Association between obesity and kidney disease: a systematic review and meta-analysis. Kidney Int. 2008;73:19–33. doi: 10.1038/sj.ki.5002586. doi:10.1038/sj.ki.5002586. [DOI] [PubMed] [Google Scholar]
- 6.Maddox DA, Alavi FK, Santella RN, et al. Prevention of obesity-linked renal disease: age-dependent effects of dietary food restriction. Kidney Int. 2002;62:208–219. doi: 10.1046/j.1523-1755.2002.00412.x. doi:10.1046/j.1523-1755.2002.00412.x. [DOI] [PubMed] [Google Scholar]
- 7.Henegar JR, Bigler SA, Henegar LK, et al. Functional and structural changes in the kidney in the early stages of obesity. J Am Soc Nephrol. 2001;12:1211–1217. doi: 10.1681/ASN.V1261211. [DOI] [PubMed] [Google Scholar]
- 8.Afshinnia F, Wilt TJ, Duval S, et al. Weight loss and proteinuria: systematic review of clinical trials and comparative cohorts. Nephrol Dial Transplant. 2010;25:1173–1183. doi: 10.1093/ndt/gfp640. doi:10.1093/ndt/gfp640. [DOI] [PubMed] [Google Scholar]
- 9.Eknoyan G. Obesity and chronic kidney disease. Nefrologia. 2011;31:397–403. doi: 10.3265/Nefrologia.pre2011.May.10963. [DOI] [PubMed] [Google Scholar]
- 10.Sharma K, Ramachandrarao S, Qiu G, et al. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest. 2008;118:1645–1656. doi: 10.1172/JCI32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011;23:184–189. doi: 10.1016/j.ceb.2010.10.009. doi:10.1016/j.ceb.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franch HA. Kidney growth during catabolic illness: what it does not destroy makes it grow stronger. J Ren Nutr. 2007;17:167–172. doi: 10.1053/j.jrn.2007.01.018. doi:10.1053/j.jrn.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 13.Sooparb S, Price SR, Shaoguang J, et al. Suppression of chaperone-mediated autophagy in the renal cortex during acute diabetes mellitus. Kidney Int. 2004;65:2135–2144. doi: 10.1111/j.1523-1755.2004.00639.x. doi:10.1111/j.1523-1755.2004.00639.x. [DOI] [PubMed] [Google Scholar]
- 14.Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69:1125–1136. doi: 10.1007/s00018-011-0865-5. doi:10.1007/s00018-011-0865-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol. 2008;44:654–661. doi: 10.1016/j.yjmcc.2008.01.010. doi:10.1016/j.yjmcc.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–455. doi: 10.1146/annurev.pathmechdis.2.010506.091842. doi:10.1146/annurev.pathmechdis.2.010506.091842. [DOI] [PubMed] [Google Scholar]
- 17.Levine B, Kroemer G. Autophagy in aging, disease and death: the true identity of a cell death impostor. Cell Death Differ. 2009;16:1–2. doi: 10.1038/cdd.2008.139. doi:10.1038/cdd.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. doi:10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pallet N, Anglicheau D. Autophagy: a protective mechanism against nephrotoxicant-induced renal injury. Kidney Int. 2009;75:118–119. doi: 10.1038/ki.2008.537. author reply 9 doi:10.1038/ki.2008.537. [DOI] [PubMed] [Google Scholar]
- 20.Hartleben B, Godel M, Meyer-Schwesinger C, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084–1096. doi: 10.1172/JCI39492. doi:10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. doi:10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. doi:10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 23.Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 24.Korolchuk VI, Mansilla A, Menzies FM, et al. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. doi:10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie Z, Zhang J, Wu J, et al. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes. 2008;57:3222–3230. doi: 10.2337/db08-0610. doi:10.2337/db08-0610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Colombo SL, Moncada S. AMPKalpha1 regulates the antioxidant status of vascular endothelial cells. Biochem J. 2009;421:163–169. doi: 10.1042/BJ20090613. [DOI] [PubMed] [Google Scholar]
- 27.Mishra R, Cool BL, Laderoute KR, et al. AMP-activated protein kinase inhibits transforming growth factor-beta-induced Smad3-dependent transcription and myofibroblast transdifferentiation. J Biol Chem. 2008;283:10461–10469. doi: 10.1074/jbc.M800902200. doi:10.1074/jbc.M800902200. [DOI] [PubMed] [Google Scholar]
- 28.Morrow VA, Foufelle F, Connell JM, et al. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem. 2003;278:31629–31639. doi: 10.1074/jbc.M212831200. doi:10.1074/jbc.M212831200. [DOI] [PubMed] [Google Scholar]
- 29.Centeno-Baez C, Dallaire P, Marette A. Resveratrol inhibition of inducible nitric oxide synthase in skeletal muscle involves AMPK but not SIRT1. Am J Physiol Endocrinol Metab. 2011;301:E922–E930. doi: 10.1152/ajpendo.00530.2010. doi:10.1152/ajpendo.00530.2010. [DOI] [PubMed] [Google Scholar]
- 30.Vellai T. Autophagy genes and ageing. Cell Death Differ. 2009;16:94–102. doi: 10.1038/cdd.2008.126. doi:10.1038/cdd.2008.126. [DOI] [PubMed] [Google Scholar]
- 31.Matsui Y, Takagi H, Qu X, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. doi:10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 32.Lee IH, Cao L, Mostoslavsky R, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. doi:10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Narita M, Young AR, Arakawa S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332:966–970. doi: 10.1126/science.1205407. doi:10.1126/science.1205407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duran A, Amanchy R, Linares JF, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–146. doi: 10.1016/j.molcel.2011.06.038. doi:10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. doi: 10.1038/nature09076. doi:10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez A, Duran A, Selloum M, et al. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006;3:211–222. doi: 10.1016/j.cmet.2006.01.011. doi:10.1016/j.cmet.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Xiong X, Tao R, Depinho RA, et al. The Autophagy Related Gene 14 (Atg14) is regulated by Forkhead Box O Transcription Factors and Circadian Rhythms and lays a critical role in hepatic autophagy and Lipid Metabolism. J Biol Chem. 2012;287:39107–39114. doi: 10.1074/jbc.M112.412569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilkinson S, Ryan KM. Autophagy: an adaptable modifier of tumourigenesis. Curr Opin Genet Dev. 2010;20:57–64. doi: 10.1016/j.gde.2009.12.004. doi:10.1016/j.gde.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 39.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. doi:10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gustafsson AB. Bnip3 as a dual regulator of mitochondrial turnover and cell death in the myocardium. Pediatr Cardiol. 2011;32:267–274. doi: 10.1007/s00246-010-9876-5. doi:10.1007/s00246-010-9876-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bellot G, Garcia-Medina R, Gounon P, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. doi:10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawakami T, Inagi R, Takano H, et al. Endoplasmic reticulum stress induces autophagy in renal proximal tubular cells. Nephrol Dial Transplant. 2009;24:2665–2672. doi: 10.1093/ndt/gfp215. doi:10.1093/ndt/gfp215. [DOI] [PubMed] [Google Scholar]
- 43.Kouroku Y, Fujita E, Tanida I, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–239. doi: 10.1038/sj.cdd.4401984. doi:10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 44.Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. doi:10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. doi:10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 46.Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. doi:10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24:3052–3065. doi: 10.1096/fj.09-144519. doi:10.1096/fj.09-144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng J, Ohsaki Y, Tauchi-Sato K, et al. Cholesterol depletion induces autophagy. Biochem Biophys Res Commun. 2006;351:246–252. doi: 10.1016/j.bbrc.2006.10.042. doi:10.1016/j.bbrc.2006.10.042. [DOI] [PubMed] [Google Scholar]
- 49.Kovsan J, Bluher M, Tarnovscki T, et al. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab. 2011;96:E268–E277. doi: 10.1210/jc.2010-1681. doi:10.1210/jc.2010-1681. [DOI] [PubMed] [Google Scholar]
- 50.Ost A, Svensson K, Ruishalme I, et al. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med. 2010;16:235–246. doi: 10.2119/molmed.2010.00023. doi:10.1007/s00894-009-0539-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou L, Liu F. Autophagy: roles in obesity-induced ER stress and adiponectin downregulation in adipocytes. Autophagy. 2010;6:1196–1197. doi: 10.4161/auto.6.8.13478. doi:10.4161/auto.6.8.13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou L, Zhang J, Fang Q, et al. Autophagy-mediated insulin receptor down-regulation contributes to endoplasmic reticulum stress-induced insulin resistance. Mol Pharmacol. 2009;76:596–603. doi: 10.1124/mol.109.057067. doi:10.1124/mol.109.057067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, Goldman S, Baerga R, et al. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci USA. 2009;106:19860–19865. doi: 10.1073/pnas.0906048106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goldman S, Zhang Y, Jin S. Autophagy and adipogenesis: implications in obesity and type II diabetes. Autophagy. 2010;6:179–181. doi: 10.4161/auto.6.1.10814. doi:10.4161/auto.6.1.10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang L, Li P, Fu S, et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. doi:10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quan W, Hur KY, Lim Y, et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia. 2012;55:392–403. doi: 10.1007/s00125-011-2350-y. doi:10.1007/s00125-011-2350-y. [DOI] [PubMed] [Google Scholar]
- 57.Las G, Serada SB, Wikstrom JD, et al. Fatty acids suppress autophagic turnover in beta-cells. J Biol Chem. 2011;286:42534–42544. doi: 10.1074/jbc.M111.242412. doi:10.1074/jbc.M111.242412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem. 2011;286:32324–32332. doi: 10.1074/jbc.M111.254417. doi:10.1074/jbc.M111.254417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huber TB, Edelstein CL, Hartleben B, et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy. 2012;8:1009–1031. doi: 10.4161/auto.19821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kume S, Uzu T, Maegawa H, et al. Autophagy: a novel therapeutic target for kidney diseases. Clin Exp Nephrol. 2012;16:827–832. doi: 10.1007/s10157-012-0695-2. [DOI] [PubMed] [Google Scholar]
- 61.Tanaka Y, Kume S, Kitada M, et al. Autophagy as a therapeutic target in diabetic nephropathy. Exp Diabetes Res. 2012;2012:628978. doi: 10.1155/2012/628978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weide T, Huber TB. Implications of autophagy for glomerular aging and disease. Cell Tissue Res. 2011;343:467–473. doi: 10.1007/s00441-010-1115-0. doi:10.1007/s00441-010-1115-0. [DOI] [PubMed] [Google Scholar]
- 63.Inoki K, Mori H, Wang J, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121:2181–2196. doi: 10.1172/JCI44771. doi:10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mori H, Inoki K, Masutani K, et al. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem Biophys Res Commun. 2009;384:471–475. doi: 10.1016/j.bbrc.2009.04.136. doi:10.1016/j.bbrc.2009.04.136. [DOI] [PubMed] [Google Scholar]
- 65.Yang Y, Wang J, Qin L, et al. Rapamycin prevents early steps of the development of diabetic nephropathy in rats. Am J Nephrol. 2007;27:495–502. doi: 10.1159/000106782. doi:10.1159/000106782. [DOI] [PubMed] [Google Scholar]
- 66.Torras J, Herrero-Fresneda I, Gulias O, et al. Rapamycin has dual opposing effects on proteinuric experimental nephropathies: is it a matter of podocyte damage? Nephrol Dial Transplant. 2009;24:3632–3640. doi: 10.1093/ndt/gfp367. doi:10.1093/ndt/gfp367. [DOI] [PubMed] [Google Scholar]
- 67.Stallone G, Infante B, Pontrelli P, et al. Sirolimus and proteinuria in renal transplant patients: evidence for a dose-dependent effect on slit diaphragm-associated proteins. Transplantation. 2011;91:997–1004. doi: 10.1097/TP.0b013e318211d342. doi:10.1097/TP.0b013e318211d342. [DOI] [PubMed] [Google Scholar]
- 68.Godel M, Hartleben B, Herbach N, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest. 2011;121:2197–2209. doi: 10.1172/JCI44774. doi:10.1172/JCI44774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cina DP, Onay T, Paltoo A, et al. Inhibition of MTOR disrupts autophagic flux in podocytes. J Am Soc Nephrol. 2012;23:412–420. doi: 10.1681/ASN.2011070690. doi:10.1681/ASN.2011070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Till A, Lakhani R, Burnett SF, et al. Pexophagy: the selective degradation of peroxisomes. Int J Cell Biol. 2012;2012:512721. doi: 10.1155/2012/512721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kitada M, Kume S, Takeda-Watanabe A, et al. Sirtuins and renal diseases: relationship with aging and diabetic nephropathy. Clin Sci (Lond) 2013;124:153–164. doi: 10.1042/CS20120190. doi:10.1042/CS20120190. [DOI] [PMC free article] [PubMed] [Google Scholar]