

Abstract
Cerebromicrovascular rarefaction is believed to play a central role in cognitive impairment in patients receiving whole-brain irradiation therapy. To elucidate the mechanism underlying the deleterious effects of γ-irradiation on the cerebral microcirculation, rat primary cerebromicrovascular endothelial cells (CMVECs) were irradiated in vitro. We found that in CMVECs, γ-irradiation (2–8 Gy) elicited increased DNA damage, which was repaired less efficiently in CMVECs compared with neurons, microglia, and astrocytes. Increased genomic injury in CMVECs associated with increased apoptotic cell death. In the surviving cells, γ-irradiation promotes premature senescence (indicated by SA-β-galactosidase positivity and upregulation of p16INK4a), which was associated with impaired angiogenic capacity (decreased proliferation and tube-forming capacity). γ-Irradiated CMVECs acquired a senescence-associated secretory phenotype, characterized by upregulation of proinflammatory cytokines and chemokines (including IL-6, IL-1α, and MCP-1). Collectively, increased vulnerability of γ-irradiated CMVECs and their impaired angiogenic capacity likely contribute to cerebromicrovascular rarefaction and prevent regeneration of the microvasculature postirradiation. The acquisition of a senescence-associated secretory phenotype in irradiated CMVECs is biologically highly significant as changes in the cytokine microenvironment in the hippocampus may affect diverse biological processes relevant for normal neuronal function (including regulation of neurogenesis and the maintenance of the blood brain barrier).
Key Words: Senescence, Endothelial, Inflammation, Oxidative stress, Angiogenesis, Vascular cognitive impairment, Capillary.
There is growing evidence of the deleterious neurocognitive effects of ionizing radiation documented from radiotherapy patients. Whole-brain radiation therapy (WBRT) is the main treatment for brain metastases located in brain regions that are difficult to reach for surgical removal, as well as for primary brain tumors following surgical resection (1,2). Although WBRT has proven to be effective in extending survival, previous studies have demonstrated that a relatively large proportion of patients who received WBRT develop deficits in their learning, memory, and spatial information processing abilities, which manifest months to years after treatment (3–5). Radiation-induced progressive decline in cognitive function has been confirmed in animal models, including rodents (6,7) and nonhuman primates (8). However, the etiology of radiation-induced cognitive decline is not fully understood.
Recent studies on experimental animals suggest that ionizing radiation induces dose-dependent damage to the cerebrovasculature, which importantly contributes to the impairment in brain function (6,7). To sustain neuronal activity and normal brain function, the structural and functional integrity of the cerebrovasculature has to be preserved to maintain a consistent supply of oxygen, nutrients, and growth factors and to provide efficient removal of waste products from the brain cells (6,7). We have recently shown that whole-brain irradiation results in capillary rarefaction in subregions of the hippocampus, which is believed to play a causal role in impairment of learning and memory (6). Cerebral microvascular rarefaction was also shown to cause cognitive dysfunction in the absence of, or preceding, neurodegeneration in various other pathophysiological conditions (9,10). In that regard, it is highly significant that prevention of microvascular rarefaction in mice after exposure to ionizing radiation was shown to significantly improve cognitive function (7).
The mechanisms underlying capillary rarefaction observed after whole-brain irradiation likely involve radiation-induced apoptosis of the microvascular endothelial cells. Accordingly, endothelial apoptosis has been demonstrated within 24 hours after x-ray irradiation in the central nervous system (11), leading to ~15% loss of cerebromicrovascular endothelial cells (CMVECs) (12). Interestingly, the massive irradiation-induced loss of CMVECs does not seem to be accompanied by a similar loss in other cell types present in the brain. Yet, the mechanisms underlying the differences in relative susceptibility of CMVECs, neurons, and other cell types to the harmful effect of ionizing radiation remain elusive.
Previous studies suggest that microvascular rarefaction after WBRT induces local tissue hypoxia, but the hypoxic state within the tissue cannot stimulate an angiogenic response in the surviving endothelial cells to increase capillary density (6,7). It is believed that the lack of compensatory angiogenesis in the irradiated brain is a key mechanism that contributes to the long-term side effects of WBRT, promoting persisting microvascular rarefaction, cerebral hypoxia, and consequential cognitive decline. Despite these advances, the mechanisms underlying the impairment of the angiogenic response after whole-brain irradiation are not well understood. There is increasing evidence that upon irradiation, proliferating cells can initiate a complex cellular response by adopting a state of permanent cell-cycle arrest, termed “cellular senescence” (13,14). On the basis of the aforementioned studies, we hypothesized that in CMVECs, ionizing radiation elicits replicative senescence, impairing their angiogenic capacity.
This study was designed to test the hypotheses that (a) CMVECs are uniquely sensitive to DNA damage and cellular injury caused by ionizing radiation and that (b) even low doses of ionizing radiation impair the angiogenic capacity of CMVECs by inducing cellular senescence. To test these hypotheses, using primary rat CMVECs, we assessed the effects of γ-irradiation on DNA damage, apoptosis, DNA repair capacity, proliferation, colony-forming capacity, the ability to form capillary-like structures, the expression of cellular markers of replicative senescence, and cellular production of reactive oxygen species.
Materials and Methods
Establishment and Characterization of Primary CMVEC Cultures
Three-month-old male F344×BN rats were obtained from the National Institute on Aging. The rats were housed in an environmentally controlled vivarium under pathogen-free conditions with unlimited access to food and water and a controlled photoperiod (12 hours light; 12 hours dark). All rats were maintained according to National Institutes of Health guidelines and all animal use protocols were approved by the Institutional Animal Care and Use Committees of the participating institutions. The animals were euthanized with CO2 to establish primary CMVEC cultures according to our published protocols (15–17). In brief, the brains were removed aseptically, rinsed in ice-cold phosphate-buffered saline (PBS), and minced into ≈1mm squares. The tissue was washed twice in ice-cold 1×PBS by low-speed centrifugation (50g, 2–3 minutes). The diced tissue was digested in a solution of collagenase (800 U/g tissue), hyaluronidase (2.5 U/g tissue), and elastase (3 U/g tissue) in 1 mL PBS/100 mg tissue for 45 minutes at 37°C in rotating humid incubator. The digested tissue was passed through a 100-µm cell strainer to remove undigested blocks. The single cell lysate was centrifuged for 2 minutes at 70g. After removing the supernatant, the pellet was washed twice in cold PBS supplemented with 2.5% fetal calf serum, and the suspension was centrifuged at 300g, for 5 minutes at 4°C.
To create an endothelial cell–enriched fraction, the cell suspension was gradient centrifuged using OptiPrep solution (Axi-Shield, PoC, Norway). Briefly, the cell pellet was resuspended in Hanks’ balanced salt solution and mixed with 40% iodixanol (final concentration: 17% (w/v) iodixanol solution; ρ = 1.096g/mL). Hanks’ balanced salt solution (2mL) was layered on top and centrifuged at 400g for 15 minutes at 20°C. Endothelial cells, which banded at the interface between Hanks’ balanced salt solution and the 17% iodixanol layer, were collected. The endothelial cell–enriched fraction was incubated for 30 minutes at 4°C in the dark with anti-CD31/PE (BD Biosciences, San Jose, CA) and anti-MCAM/FITC (BD Biosciences). After washing the cells twice with MACS buffer (Milltenyi Biotech, Cambridge, MA), anti-FITC magnetic bead–labeled and anti-PE magnetic bead–labeled secondary antibodies were used for 15 minutes at room temperature. Endothelial cells were collected by magnetic separation using MACS LD magnetic separation columns according to the manufacturer’s guidelines (Milltenyi Biotech). The endothelial fraction was cultured on fibronectin-coated plates in endothelial growth medium (Cell Application, San Diego, CA) for 10 days. Endothelial cells were phenotypically characterized by flow cytometry (GUAVA 8HT, Merck Millipore, Billerica, MA). Briefly, antibodies against five different endothelial specific markers were used (anti-CD31-PE, anti-erythropoietin receptor-APC [allophycocyanin], antivascular endothelial growth factor R2-PerCP, anti-intercellular adhesion molecule-fluorescein, anti-CD146-PE), and isotype-specific antibody-labeled fractions served as negative controls. All antibodies were purchased from R&D Systems (Minneapolis, MN).
Cell Cultures and γ-Irradiation Protocol
For each experiment, CMVECs were cultured in rat brain endothelial growth medium (Cell Applications No. R819-500) according to the manufacturer’s protocol. Rat brain hippocampus astrocytes were purchased from Lonza (www.lonza.com; Lonza No. R-HiAs-521, passage 1) and cultured in astrocyte basal medium (Lonza No. CC-3187) supplemented with AGM-SingleQuots (Lonza No. CC-4123) according to the vendor’s guidelines. Rat brain hippocampal neuronal cells were purchased from Lonza (Lonza No. R-Hi-501) and were cultured according to the vendor’s guidelines in primary neuron basal medium (PNBMl Lonza No. CC-3256) supplemented with PNGM-SingleQuots (Lonza No. CC4462; which contains 2 mM l-glutamine, 50 μg/mL gentamicin, 37 μg/mL amphotericin, and 2% NSF-1). Rat microglia (Lonza No. R-G-535) were purchased from Lonza and cultured according to the vendor’s guidelines in Dulbecco’s Modified Eagle Medium (Lonza No. 12-604F) supplemented with 20% heat-inactivated fetal bovine serum (Lonza No. 14-503E), 1% penicillin/streptomycin (Lonza No. 17-602E), and 4.5g/L d-glucose (Sigma No. G-8769). γ-Irradiation to the cells was administered using a 137Cs gamma irradiator (GammaCell 40, Nordion International). The irradiator was then activated for a time calculated to deliver 2–8 Gy, which correspond to clinically relevant doses of radiation used for WBRT, depending on the protocol. Dosimetry was performed as previously described (6,7) to confirm the dose received.
Assessment of γ-Irradiation-Induced Cell Death
To compare susceptibility of cultured CMVECs, microglia, and neurons to cell death induced by γ-irradiation, the cells were irradiated (2–8 Gy) and the ratio of dead cells as a percentage of total cell number was assessed by flow cytometry (at 24 hours postirradiation) using the Guava ViaCount Assay (Millipore) according to the manufacturer’s protocol, as we previously reported (18). The ViaCount Assay distinguishes viable and nonviable cells based on differential permeabilities of two proprietary DNA-binding dyes, including a nuclear dye, which stains only nucleated cells, and a viability dye, which stains dying cells. To demonstrate that γ-irradiation elicits primarily apoptosis in CMVECs, the time course for changes in caspase-3/7 activity (a useful measure of apoptotic cell death) was assessed in irradiated CMVECs using the Caspase-Glo 3/7 assay kit (Promega, Madison, WI) as previously reported (19–22). In 96-well plates, a 50-µL sample was mixed for 30 seconds with 50 µL Caspase-Glo 3/7 reagent and incubated for 2 hours at room temperature. Lysis buffer with the reagent served as blank. Luminescence of the samples was measured using an Infinite M200 plate reader (Tecan, Research Triangle Park, NC). Luminescent intensity values were normalized to the sample protein concentration.
DNA Damage Analysis by Alkaline Single-Cell Gel Electrophoresis
To compare susceptibility of cultured CMVECs, microglia, neurons, and astrocytes to DNA damage induced by γ-irradiation, single cell gel electrophoresis was performed following our published protocol (23–25). In brief, 105 cells in 100 μL PBS were mixed with 100 µL of 1.5% low-melting agarose and 90 μL of this mixture spotted on CometAssay slides (Trevigen, Gaithersburg, MD) between two layers of 1% low-melting agarose. DNA damage was induced by exposure of the slides to γ-irradiation (4 Gy). The extent of DNA fragmentation was examined by single-cell electrophoresis (“comet assay”), as reported (23–25). The comet assay is based on the alkaline lysis of labile DNA at sites of damage. The slides were treated with a lysis solution (NaCl 2.5M, Na2EDTA 100mM, Triton X-100 1%, dimethylsulfoxide 10%, and Trizma base 10mM; pH 10; for 1 hour at 4°C in the dark), rinsed with a neutralization buffer (3 × 5 minutes, 0.4M Tris, pH 7.5) to remove detergents and salts, and then submerged in a high pH buffer (NaOH 300mM, Na2EDTA 1mM, pH > 13) for 20 minutes to allow for unwinding of the DNA and the expression of alkali-labile damage. Electrophoresis was performed using the same buffer at 25V (~0.74V/cm) and 300 mA for 20 minutes. At the end of the run, the slides were neutralized in Tris–HCl 0.4M, pH 7.5, submerged in absolute ethanol for 3 minutes, and air dried, and DNA was stained with SYBR green (Invitrogen, Carlsbad, CA). Fluorescent images of the nuclei were captured using a Nikon Eclipse TE300 fluorescent microscope (Nikon Corporation, Tokyo, Japan) at ×20 magnification. DNA damage was quantified by measuring the tail DNA content (as a percentage of total DNA) using the Comet Assay-IV software (Perceptive Instruments, Haverhill, Suffolk, United Kingdom). For assessment of DNA repair efficiency, the cells were irradiated with 4Gy, and the extent of DNA damage was assessed by the comet assay at 10, 20, 40, 60, and 120 minutes postirradiation (24). The percentage of residual DNA damage was plotted as a function of time. The time constant calculated from this plot and the residual DNA damage at 120 minutes postirradiation were used as indices of DNA repair capacity.
Tube Formation Assay
To investigate the influence of γ-irradiation on angiogenic capacity of endothelial cells, the tube formation ability of γ-irradiated CMVECs (7 days postirradiation) was assessed. In brief, CMVECs were irradiated (0, 2, 4, 6, or 8 Gy), washed with PBS and resuspended in complete growth media, and cultured in T75 flasks for 7 days (media was partially changed on Days 3 and 6). On Day 7, CMVECs were plated on Geltrex Reduced Growth Factor Basement Membrane Matrix (Invitrogen) in Medium 200PRF (Invitrogen). Briefly, 150 μL/well of Geltrex was distributed in 24-well plates. The gel was allowed to solidify while incubating the plates for 30 minutes at 37°C. CMVECs were then seeded at a density of 2.5 × 104 cells/well in 0.8mL OPTI-MEM, treated with vascular endothelial growth factor (VEGF 100ng/mL), and placed in the incubator for 24 hours. Microscopic images were captured using a Nikon Eclipse Ti microscope equipped with a ×10 phase-contrast objective (Nikon Instruments Inc., Melville, NY). The extent of tube formation was quantified by measuring total tube length in five random fields per well using NIS-Elements microscope imaging software (Nikon Instruments Inc.), as recently reported (15,17,26). The mean of the total tube length per total area imaged (µm tube/mm2) was calculated for each well. Experiments were run in quadruplicate. The experimenter was blinded to the groups throughout the period of analysis.
Colony Formation Assay
The colony formation assay has been used as gold standard for determining the cellular effects of ionizing radiation in vitro since first described by Puck and Marcus (27) in 1956. To further investigate the impact of γ-irradiation on the vasculogenic potential of primary endothelial cells, a colony-forming assay was performed with irradiated CMVECs. In brief, CMVECs were irradiated (0, 2, 4, 6, and 8 Gy), washed with PBS and kept for 7 days in T75 flasks in the presence of complete growth media (media was partially changed on Days 3 and 6). On Day 7 postirradiation, the cells were counted and 2.5 × 103 cells were plated into each well of six-well plates in 4 mL of growth media. Triplicate wells were set up for each condition. After an additional 7 days of incubation (without media change), CMVECs were rinsed twice with PBS and fixed with 70% ethanol for 10 minutes and then plates were air dried. Coomassie Blue Staining (Life Technology) was performed according to manufacture protocols. Excess of stain was removed by gentle wash with PBS. To assess the impact of γ-irradiation on total CMVEC cell mass, the optical image of each stained well was obtained using a gel documentation system (AlphaImager), and relative background-corrected optical densities (used as an index of cell mass) were compared. To analyze average colony size, microscopic images of colonies were captured (at ×4 magnification, 30 random fields per group). The number of colonies in each image and the size of each individual colony in each image were measured by a blinded observer using the NIS-Elements microscope imaging software.
Cell Proliferation Assay
To assess the impact of γ-irradiation on cell proliferation, capacity was also assessed in CMVECs, using the flow cytometry–based Guava CellGrowth assay (EMD Millipore) as recently reported (15,17). Briefly, on Day 7 postirradiation (0 or 2 Gy), cells were collected, resuspended in PBS containing 0.1% bovine serum albumin, and stained with 16 µmol/L carboxyfluorescein diacetate succinimidyl ester (CFSE) for 15 minutes at 37°C. This dye diffuses into cells and is cleaved by intracellular esterases to form an amine-reactive product that produces a detectable fluorescence and binds covalently to intracellular lysine residues and other amine sources. Upon cell division, CFSE divides equally into the daughter cells halving the CFSE concentration of the mother cell; therefore, there is an inverse correlation between the fluorescence intensity and the proliferation capacity of the cells. After incubation, unbound dye was quenched with serum-containing medium. Then, cells were washed three times and incubated for 24 hours with 100ng/mL VEGF. Finally, cells were collected, washed, stained with propidium iodide (to gate out dead cells), and analyzed with a flow cytometer (Guava EasyCyte 8HT; Millipore). The inverse of the fluorescence intensity was used as an index of proliferation.
Senescence-Associated β-Galactosidase Activity Assay
Cellular senescence is characterized by the appearance of senescence-associated β-galactosidase (SA-β-gal) activity, associated with the senescent phenotype in vitro. To assess the sensitivity of primary CMVECs to γ-irradiation-induced senescence, SA-β-gal activity was assessed in irradiated CMVECs. In brief, CMVECs were irradiated (0, 2, 4, 6, and 8 Gy) and cultured in six-well plates for 7 days in the presence of complete growth media. On Day 7 postirradiation, histochemical staining for SA-β-gal activity was performed using the Sigma-Senescence Cells Histochemical Staining Kit (Sigma, No. CS0030, St. Louis, MO), following the manufacturer’s guidelines. To analyze the ratio of senescent cells in each well, microscopic images of the stained CMVEC cultures were captured (at ×10 magnification, 30 random fields per group). The percentage of β-galactosidase-positive cells (blue cytoplasmic staining) was calculated by a naïve observer.
Quantitative Real-Time Reverse Transcription PCR
A quantitative real-time reverse transcription (RT) PCR technique was used to analyze mRNA expression of p16INK4a and other senescence markers in irradiated and control CMVECS. In brief, after 8 days in culture postirradiation (6 Gy), the cells were harvested. Total RNA was isolated with a Mini RNA Isolation Kit (Zymo Research, Orange, CA) and reverse transcribed using Superscript III RT (Invitrogen). The mRNA expression of p16INK4a and other senescence markers was analyzed using a Stratagen MX3000 platform, as reported (17,28–32). Amplification efficiencies were determined using a dilution series of a standard sample. Quantification was performed using the efficiency-corrected ΔΔCq method. The relative quantities of the reference genes B2m, Hprt, and Ywhaz were determined, and a normalization factor was calculated based on the geometric mean for internal normalization. Oligonucleotides used for quantitative real-time RT-PCR are listed in Table 1.
Table 1.
Oligonucleotides for Real-Time Reverse Transcription PCR
| mRNA Targets | Description | Sense | Antisense |
|---|---|---|---|
| Cdkn2a | Cyclin-dependent kinase inhibitor 2A; p16INK4a | TAGTCTCGCGTTGCCAGAA | TGCGGTATTTGCGGTATCT |
| Tp53 | Tumor protein p53 | CGTTGCTCTGATGGTGAC | ACTTGTAGTGGATAGTGGTATAG |
| Cdkn1a | Cyclin-dependent kinase inhibitor 1A (p21, CIP1) | TGGACTGTTTATCTTTAGCC | CGTGGGAGGTTTACAATC |
| Cdkn2d | Cyclin-dependent kinase inhibitor 2D (p19, inhibits CDK4) | TGAAGTGTGAGCCATCTA | TACAACAAAACTGGAAACCT |
| Mmp9 | Matrix metallopeptidase 9 (gelatinase B, 92kDa gelatinase, and 92kDa Type IV collagenase) | CTGCTCCAACTGCTGTAT | GGTGTCCTCCGATGTAAG |
| VEGF A | Vascular endothelial growth factor A | GCTTGTCCTGAGAAGATA | GTATGTAAATGTATATTGAAACCT |
| MCP-1 | Chemokine (C–C motif) ligand 2 | GCATCAACCCTAAGGACTTCAG | GGCATCACATTCCAAATCACAC |
| IL-1a | Interleukin-1α | CAGCAATGGTCGGGAC | ATAGGTAAGTGGTTGCCT |
| IL-6 | Interleukin-6 | TACCCCAACTTCCAATGC | GATACCCATCGACAGGAT |
| Hprt | Hypoxanthine phosphoribosyltransferase 1 | AAGACAGCGGCAAGTTGAATC | AAGGGACGCAGCAACAGAC |
| B2M | Beta-2 microglobulin | ATTCACACCCACCGAGAC | GGATCTGGAGTTAAACTGGTC |
| YWHAZ | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein | AACTGCCTACATATTGGT | CACACAGACTACACTCAT |
Analysis of Secreted Cytokines in Conditioned Media
Profiling of cytokines secreted by irradiated and control CMVECs was conducted using a magnetic multiplex bead array (EMD Millipore). In brief, after 8 days in culture postirradiation (6 Gy), the medium was changed to serum-free Minimum Essential Medium for 24 hours and then the conditioned media were collected for analysis of secreted cytokines and growth factors. The cells were harvested, and sample protein content was determined for normalization purposes by a spectrophotometric quantitation method using BCA reagent (Pierce Chemical Co., Rockford, IL). The concentration of a range of cytokines and growth factors involved in neurovascular pathophysiology, including IL-1α, IL-6, GRO/KC (the mouse KC is considered the homolog of the human GROs), granulocyte-macrophage colony-stimulating factor (CSF), G-CSF, MIP-1α, MCP-1, eotaxin, and IL-1β, was measured in the conditioned media as reported (30,33).
Measurement of Cellular Reactive Oxygen Species Production
To investigate the influence of γ-irradiation on cellular peroxide production, we used the cell-permeant oxidative fluorescent indicator dye 5(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA, Invitrogen) as we previously reported (18,22). In brief, CMVECs were irradiated (0, 2, 4, 6, and 8 Gy) as previously described. On Day 1 or Day 14 postirradiation, the cells were washed with warm PBS and incubated with CM-H2DCFDA (10 µM, at 37°C, for 30 minutes). CM-H2DCFDA fluorescence was assessed by flow cytometry as previously described (18,22). In separate experiments, mitochondrial 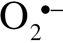 production in CMVECs was measured using MitoSOX Red (Invitrogen), a mitochondrion-specific hydroethidine-derivative fluorescent dye, as previously reported (23,30,34–36).
production in CMVECs was measured using MitoSOX Red (Invitrogen), a mitochondrion-specific hydroethidine-derivative fluorescent dye, as previously reported (23,30,34–36).
Data Analysis
Statistical analyses were performed using one-way ANOVA. P value less than .05 was considered statistically significant. Data are expressed as means ± standard error mean.
Results
CMVECs Exhibit Increased Radiosensitivity
Cultured endothelial cells, neurons, and microglia showed cellular morphology that is characteristic of respective cell types (Figure 1A). The ratio of viable and apoptotic/dead cells was compared using the ViaCount Assay. We found that γ-irradiation significantly increased cell death in CMVEC cultures in a dose-dependent manner (Figure 1B). Microglia and neuronal cells exhibited decreased sensitivity to γ-irradiation-induced cell death, as shown by the lower ratio of dead cells at each dose studied (Figure 1B). γ-Irradiation elicited significant increases in caspase-3/7 activity in CMVECs demonstrating that it elicits cell death primarily by inducing apoptosis in CMVECs (Figure 1C).
Figure 1.

Morphology of CMVECs (A), neurons (B), and microglia (C) in culture. (D) CMVECs exhibit increased radiosensitivity. Shown are γ-irradiation-induced increases in the ratio of dead cells in CMVECs, neurons, and microglia cultures at 24h postirradiation. The ratio of viable and apoptotic/dead cells was compared using the ViaCount Assay (Materials and Methods section). Data are means ± standard error mean. *p < .05 CMVECs vs neurons. # p < .05 CMVECs vs microglia. (E) γ-Irradiation induces apoptosis in CMVECs. Shown is the time course of increases in caspase-3/7 activity in CMVECs postirradiation. CMVECs = cerebromicrovascular endothelial cells.
CMVECs Exhibit Increased γ-Irradiation-Induced DNA Damage and Decreased DNA Repair Capacity
To assess DNA damage induced by γ-irradiation, a random sample of 200 cells was analyzed from each slide. Figure 2A shows examples of images of the comet assay. In each cell type without γ-irradiation, all the DNA was confined to the nuclei, as indicated by the percentage of DNA in the tail less than 5%. In CMVECs with γ-irradiation-induced DNA strand breaks, a bright fluorescent tail along the electric field was observed because small DNA fragments migrated out of the nuclei (Figure 2A). In neurons, microglia, and astrocytes, the tail momentum was significantly less indicating lower amount of DNA strand breaks at each dose of irradiation tested. Figure 2B, showing the DNA in the tail (indicating the severity of DNA damage) as a function of the dose of γ-irradiation in each cell type, indicates that CMVECs were significantly more sensitive to the DNA-damaging effects of γ-irradiation studied than neurons, microglia, and astrocytes.
Figure 2.

(A) CMVECs exhibit increased γ-irradiation-induced DNA damage and decreased DNA repair capacity. Cultured CMVECs, neurons, astrocytes, and microglia were γ irradiated, and DNA damage was assessed by single-cell electrophoresis (“comet assay”) at 10, 20, 40, 60, and 120 min postirradiation. Damaged DNA migrates during electrophoresis from the nucleus toward the anode, forming the shape of a “comet” with a head (cell nucleus with intact DNA) and a tail (relaxed and broken DNA). Panel A shows representative images of damaged DNA in irradiated (4 Gy) CMVECs, neurons, astrocytes, and microglia. Note the marked decline in tail DNA content at 60 min postirradiation due to DNA repair processes. (B) γ-Irradiated CMVECs exhibit increased DNA damage. Shown are γ-irradiation-induced increases in tail DNA content in CMVECs, neurons, astrocytes, and microglia cultures as a function of dose received. *p < .05 CMVECs vs neurons. (C) CMVECs exhibit decreased DNA repair capacity. For assessment of DNA repair efficiency, the percentage of residual DNA damage was plotted as a function of time postirradiation. Median values of tail DNA content at each time point are shown. The time constant calculated from this plot was the longest and the residual DNA damage at 120 min postirradiation was the greatest in CMVECs, indicating a less efficient DNA repair capacity in this cell type. CMVECs = cerebromicrovascular endothelial cells.
For assessment of DNA repair efficiency, the percentage of residual DNA damage was plotted as a function of time postirradiation. We found that the percentage of residual DNA damage was the highest in CMVECs at each time-point postirradiation (Figure 2C). The time constant calculated from this plot (Figure 2C, inset) was also the longest in CMVECs, indicating that CMVECs exhibit decreased DNA repair capacity.
γ-Irradiation Impairs Formation of Capillary-Like Structures by CMVECs
To elucidate the functional consequences of increased γ-irradiation-induced DNA damage in CMVECs, we assessed endothelial angiogenic capacity. When seeded onto Geltrex matrices, control CMVECs formed elaborated capillary networks in the presence of VEGF (Figure 3A). We found that γ-irradiation, in a dose-dependent manner, significantly inhibited the formation of capillary-like structures by CMVECs (Figure 3B–F).
Figure 3.

(A–E) γ-Irradiation impairs the ability of CMVECs to form capillary-like structures. CMVECs were γ irradiated (0–8 Gy). Cells were plated on Geltrex matrix–coated wells on Day 7 postirradiation, and tube formation was induced by treating cells with VEGF (100ng/mL, for 24h). Representative examples of capillary-like structures formed by cells irradiated with 0, 2, 4, 6, and 8 Gy are shown on Panels A, B, C, D, and E, respectively. Samples were run in quadruplicate. (F) Shown are summary data, expressed as percentage change in total tube length per total area scanned (µm tube/mm2). Data are means ± standard error mean. *p < .05 vs control. CMVECs = cerebromicrovascular endothelial cells; VEGF = vascular endothelial growth factor.
γ-Irradiation Impairs Colony-Forming Capacity and Proliferative Capacity of CMVECs
Proliferation represents a key step in angiogenesis. Using a colony formation assay at Day 14 after irradiation of CMVECs, the features, numbers, and size of adherent colonies were examined via light microscopy. We found that control CMVECs divided and gave rise to various sized colonies (Figure 4A–C). γ-Irradiation, in a dose-dependent manner, significantly inhibited total cell mass (Figure 4B), the average number of colonies (Figure 4D), and the average size of colonies (Figure 4E). We also noted that γ-irradiation increased the presence of cells with abnormal morphology, which were not associated with large colonies.
Figure 4.

Previous γ-irradiation inhibits colony formation by CMVECs. (A) Representative dishes of CMVECs by colony-forming assay. (B) The influence of previous γ-irradiation on the optical density of the images of the CMVEC cultures, indicating total cell mass. Data are mean of four independent determinations. *p < .05 vs control. (C) Representative micrographs showing the effects of CMVEC colonies. Note that γ-irradiation, in a dose-dependent manner, resulted in a significant reduction in individual colony size and the increased presence of cells with abnormal morphology (arrowheads; original magnification: ×10). Summary data showing the influence of γ-irradiation on the number of colonies (D) and the average colony size (E), as evaluated by clonogenic assay. *p < .05 vs control. CMVECs = cerebromicrovascular endothelial cells.
In separate studies, proliferative capacity of control and γ-irradiated CMVECs was compared after incubation with VEGF for 24 hours using a CFSE-based assay. We found that γ-irradiation significantly increased CFSE fluorescence in CMVECs, indicating that proliferation capacity is impaired by ionizing radiation (Figure 5).
Figure 5.

γ-Irradiation significantly impairs proliferation capacity of CMVECs. Cell proliferation capacity was assessed in CMVECs stimulated with VEGF (100ng/mL) using the flow cytometry based Guava CellGrowth assay (Materials and Methods section). As an index of proliferation capacity, the inverse of the mean fluorescence intensity of the indicator dye carboxyfluorescein diacetate succinimidyl ester was used. Data are means ± standard error mean (n = 5 in each group), *p < .05 vs control. CMVECs = cerebromicrovascular endothelial cells; VEGF = vascular endothelial growth factor.
γ-Irradiation Induces Premature Senescence in CMVECs
The impaired colony-forming and proliferative capacity of irradiated CMVECs, as well as the presence of cells with abnormal morphology, suggested that γ-irradiation of CMVECs induces premature senescence. To collect initial evidence that irradiation induces senescence in CMVECs, we performed SA-β-gal staining of irradiated and nonirradiated CMVEC cultures. We found that γ-irradiation, in a dose-dependent manner, increased the ratio of CMVECs with positive SA-β-gal staining. At the highest dose tested (8 Gy), ~30% of irradiated CMVECs were β-galactosidase positive on Day 14 postirradiation (Figure 6).
Figure 6.

(A–E) Representative micrographs showing that γ-irradiation, in a dose-dependent manner (0–8 Gy), increases the number of senescent cerebromicrovascular endothelial cells with positive senescent-associated β-galactosidase (SA-β-gal, blue) activity staining (original magnification: ×10). (F) Summary data showing the ratio of SA-β-gal-positive cells. Data are the median from four separate experiments.
γ-Irradiation Increases Expression of p16INK4a and Other Senescence Markers in CMVECS
Previous studies demonstrate that the senescence response induced by DNA damage in fibroblasts is controlled by the potent tumor suppressor p53, as well as by cyclin-dependent kinase inhibitors p21 (Cdkn1A, a direct target of p53 transactivation) and p16INK4a (Cdkn2A, a mediator of pRB-regulated growth arrest) (13,37–40). Here, we report that induction of premature senescence in CMVECs by γ-irradiation is also associated with upregulation of p53 and p16INK4a, as well as other molecular markers of senescence, whereas expression of p21/Cdkn1a did not change significantly (Figure 7).
Figure 7.

Quantitative reverse transcription PCR data showing expression of p16INK4a and other senescence markers in control CMVECs and γ-irradiated CMVECs (6 Gy, on Day 8 postirradiation). Data are means ± standard error mean (n = 6 in each group); *p < .05. CMVECs = cerebromicrovascular endothelial cells.
CMVECs Acquire a Senescence-Associated Secretory Phenotype in Response to γ-Irradiation
Previous studies show that irradiated senescent fibroblasts acquire a complex phenotype that includes the secretion of many inflammatory cytokines and chemokines, termed “senescence-associated secretory phenotype” (SASP) (38,40). Here, we report that γ-irradiated CMVECs also acquire an SASP, which include upregulation of IL-6, IL-1α, GM-CSF, G-CSF, MIP-1α, MCP-1, eotaxin, and IL-1β (Figure 8).
Figure 8.

Senescence-associated secretory phenotype in γ-irradiated CMVECs. Secretory profiles of control CMVECs and γ-irradiated CMVECs (6 Gy, on Day 8 postirradiation) were detected by a fluorescent multiplex bead array as described in Materials and Methods section. Data are means ± standard error mean (n = 4 in each group). *p < .05 vs control. CMVECs = cerebromicrovascular endothelial cells.
Effects of γ-Irradiation on Reactive Oxygen Species Production in CMVECs
Using CM-H2DCFDA and MitoSox fluorescence-based methods, we demonstrated that acute exposure to γ-irradiation results in increased cellular peroxide production and increased mitochondrial oxidative stress in CMVECs in a dose-dependent manner (Figure 9A and B, respectively). Interestingly, increased cellular peroxide production and increased mitochondrial oxidative stress were not evident on Day 14 postirradiation (Figure 9A and B).
Figure 9.

Flow cytometry data showing the effects of γ-irradiation on cellular peroxide production (A, assessed by measuring H2DCFDA fluorescence) and mitochondrial 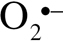 production (B, assessed by measuring MitoSox fluorescence) in CMVECs. Data are means ± standard error mean. *p < .05 vs control. Acute exposure to γ-irradiation (on Day 1 postirradiation) resulted in increased cellular reactive oxygen species production and increased mitochondrial oxidative stress in CMVECs in a dose-dependent manner. Increased cellular peroxide production and increased mitochondrial oxidative stress were not evident on Day 14 postirradiation. CMVECs = cerebromicrovascular endothelial cells.
production (B, assessed by measuring MitoSox fluorescence) in CMVECs. Data are means ± standard error mean. *p < .05 vs control. Acute exposure to γ-irradiation (on Day 1 postirradiation) resulted in increased cellular reactive oxygen species production and increased mitochondrial oxidative stress in CMVECs in a dose-dependent manner. Increased cellular peroxide production and increased mitochondrial oxidative stress were not evident on Day 14 postirradiation. CMVECs = cerebromicrovascular endothelial cells.
Discussion
The principal new findings of this study are that ionizing radiation in CMVECs (a) elicits increased DNA damage, which is repaired less efficiently than in other cell types and (b) promotes the acquisition of an SASP. Major steps of the angiogenic process, including proliferation and formation of capillary-like structures, are significantly inhibited in irradiated CMVECs, whereas the rate of apoptosis is significantly increased.
Our previous studies provide evidence that whole-brain irradiation results in significant microvascular rarefaction in the hippocampus, which appears to play a causal role in irradiation-induced cognitive decline (6,7). Here, we have devised an in vitro approach to determine the sensitivity of CMVECs to the harmful effects of γ-irradiation. We demonstrate that CMVECs exhibit increased radiosensitivity, manifested as an increased rate of acute apoptotic cell death in response to γ-irradiation (Figure 1). It is likely that γ-irradiation induced loss of endothelial cells in the brain contributes directly to cerebromicrovascular rarefaction after WBRT (6,7). The increased radiosensitivity of endothelial cells also appears to contribute to other harmful consequences of ionizing radiation as well. Previous studies on peripheral tissues confirm that microvascular networks, which control the delivery of oxygen and nutrients and the removal of metabolic waste, are the most sensitive parts of the vascular system to ionizing radiation (41). Indeed, previous studies reported that microvascular endothelial apoptosis also contributes to irradiation-induced pneumonitis and the gastrointestinal syndrome (42,43). Early studies have documented that peripheral skin capillaries are markedly radiosensitive (44). Interestingly, the synergistic effect of radiation-induced apoptosis of tumor endothelial cells and treatment with antiangiogenic drugs has been considered for clinical trials (45). Endothelial cell apoptosis induced by ionizing irradiation was reported to be mediated by a ceramide-dependent pathway (46–48).
To our knowledge, this is the first study demonstrating that CMVECs exhibit increased susceptibility to γ-irradiation-induced DNA damage (Figure 2A and B), which likely underlies their increased radiosensitivity compared with other cell types present in the brain (ie, neurons, microglia, and astrocytes). Most of the DNA-damaging effects of ionizing radiation are induced by 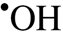 radicals (indirect effects) and by one-electron oxidation (direct effects). At present, it is unknown which of these mechanisms are more important for the observed tissue-specific differences in cellular sensitivity to γ-irradiation. We propose that the differences in the extent of DNA damage immediately postirradiation likely reflect differences in the nuclear DNA organization or antioxidant defenses because the irradiation was performed at 0°C, which effectively prevents enzymatic DNA repair processes. In addition, CMVECs also exhibit lower efficiency of DNA repair in response to the same dose of γ-irradiation (Figure 2C). It is thought that the spectrum of damages caused by ionizing radiation is mainly repaired by the base excision repair pathway, homolog/nonhomolog recombination, and end-joining repair (double-strand breaks), but the exact molecular mechanisms underlying cell type–dependent differences in repair of γ-irradiation-induced DNA damage are presently unknown.
radicals (indirect effects) and by one-electron oxidation (direct effects). At present, it is unknown which of these mechanisms are more important for the observed tissue-specific differences in cellular sensitivity to γ-irradiation. We propose that the differences in the extent of DNA damage immediately postirradiation likely reflect differences in the nuclear DNA organization or antioxidant defenses because the irradiation was performed at 0°C, which effectively prevents enzymatic DNA repair processes. In addition, CMVECs also exhibit lower efficiency of DNA repair in response to the same dose of γ-irradiation (Figure 2C). It is thought that the spectrum of damages caused by ionizing radiation is mainly repaired by the base excision repair pathway, homolog/nonhomolog recombination, and end-joining repair (double-strand breaks), but the exact molecular mechanisms underlying cell type–dependent differences in repair of γ-irradiation-induced DNA damage are presently unknown.
The regulation of cerebral capillary density is a dynamic process, which depends on the balance between capillary regression and angiogenesis. Here, we demonstrate that in addition to promoting endothelial cell loss, γ-irradiation also impairs the angiogenic capacity and proliferation of CMVECs (Figures 3–5) preventing regeneration of the microvasculature postirradiation. Early studies also documented that peripheral capillary damage induced by low doses of γ-radiation (from radium) is not associated with a marked degree of repair (44) and that radiation inhibits the replication of cultured endothelial cells derived from the peripheral circulation (49,50). The acute suppression of angiogenic capacity by irradiation has also been observed in cancer endothelial cells (51) and human umbilical vein endothelial cells (52,53). The mechanisms that underlie the impaired angiogenic capacity of irradiated CMVECs are likely multifaceted. Our findings suggest that increased sensitivity of CMVECs to γ-irradiation-induced DNA damage promotes the execution of the senescence program (termed “stress-induced premature senescence”) in a significant fraction of these cells (Figure 6). Senescent cells often express p16INK4a, a cyclin-dependent kinase inhibitor, which renders the senescence growth arrest irreversible. Here, growth arrest of γ-irradiated CMVECs is associated with upregulation of p16INK4a but not of another cyclin-dependent kinase inhibitor, p21 (CIP1/WAF1; Figure 7). The induction of p16INK4a was also associated with the upregulation of other markers of senescence (Figure 7). We found that irradiation induces the expression of p53 in CMVECs (Figure 7), but its function in growth arrest is uncertain as p53 deficiency was previously reported not to prevent radiation-induced inhibition of endothelial proliferation (52). Previous studies by the laboratory of Campisi and colleagues (40,54) have demonstrated that a hallmark trait of cellular senescence is the acquisition of an SASP, which entails a significant increase in the secretion of proinflammatory cytokines and chemokines. Here, we demonstrate that γ-irradiation results in the acquisition of an SASP in CMVECs (Figures 7 and 8), which shares many features of the SASP induced by γ-irradiation in cultured fibroblasts (38,39) and the age-associated arterial secretory phenotype (55). SASP factors that are upregulated by γ-irradiation both in CMVECs (Figure 8) and fibroblasts include the cytokines, IL-6 and IL-8, GM-CSF, and the chemokine MCP-1 (39). On the basis of data from the literature, we predict that SASP factors secreted from senescent CMVECs will impair the function of neighboring cells in a paracrine manner. Previous studies suggest that IL-6 plays a central role in regulation of senescence, but its function in CMVECs and after WBRT has not been explored. Interestingly, recent data suggest that the expression of the SASP in fibroblasts is not causally linked to p16INK4a activation, but rather it is an integral constituent of the cellular DNA damage response (40). Importantly, the presence of senescent cells is also likely to affect the function (including angiogenic capacity) of neighboring nonsenescent cells as well. (The name “senescence-messaging secretome” was proposed (56) to highlight that senescence-associated factors communicate the senescent state to the local microenvironment.) Further studies are clearly warranted to determine whether acquisition of an SASP by γ-irradiated CMVECs is causally linked to the growth arrest of these cells induced by ionizing radiation and whether SASP factors are able to impair the angiogenic capacity of nonsenescent CMVECs as well. The functional relevance of the SASP concept was first proposed by Campisi and colleagues (54), demonstrating that secreted factors from senescent fibroblasts promote the transformation of premalignant mammary epithelial cells. There is now growing evidence that senescence-associated activation of the inflammatory transcriptome regulates the extracellular environment and contributes to a number of age-related pathologies, including development of cardiovascular disease (for a recent review, see Wang and Bennett (57)). It is likely that γ-irradiation-induced SASP in CMVECs in vivo will promote neuroinflammation and contribute to the impairment of neuronal function by altering the cellular microenvironment. Importantly, the microvascular endothelium in the dentate gyrus of hippocampus is thought to play an important role in neurogenesis (58). Because microvascular endothelial cells secrete a number of trophic factors, including brain-derived neurotrophic factor, it is hypothesized that healthy endothelial function is required to maintain a microenvironment for optimal growth, survival, and differentiation of neuronal precursor cells (58). Inhibition of microvascular angiogenesis by ionizing radiation was reported to be associated with marked alterations in the neurogenic microenvironment and dysfunction of neural precursor cells (59). In addition to the increased secretion of cytokines and chemokines, senescent cells are also likely to secrete matrix metalloproteinases, which may also impair neuronal function. Endothelial cell senescence was also reported to be associated with disrupted cell–cell junctions and increased vascular permeability (60). These findings suggest that radiation-induced acquisition of an SASP in CMVECs may represent a novel target for future therapeutic interventions. Because the aging process itself is associated with endothelial senescence and cerebromicrovascular rarefaction, future studies should determine whether advanced age and γ-irradiation-induced acquisition of an SASP interact to exacerbate cognitive decline.
In conclusion, ionizing radiation elicits increased DNA damage in CMVECs, which is repaired less efficiently than in neurons, microglia, or astrocytes. Increased genomic injury in CMVECs likely exacerbates apoptotic cell death (which is a likely cause for microvascular rarefaction) and, in the surviving cells, promotes premature senescence. The resulting inhibition of angiogenesis prevents regeneration of the microvasculature postirradiation. The acquisition of an SASP in irradiated CMVECs is biologically highly significant as changes in the cytokine microenvironment in the hippocampus may affect diverse biological processes relevant for normal neuronal function (from regulation of neurogenesis to the maintenance of the blood–brain barrier).
Funding
This work was supported by grants from the American Federation for Aging Research (to A.C.), the Oklahoma Center for the Advancement of Science and Technology (to A.C., Z.U., and W.E.S.), the American Heart Association (to A.C. and Z.U.), the National Institutes of Health (AG031085 to A.C.; AT006526 to Z.U.; AG038747, NS056218, and P01 AG11370 to W.E.S.), the Ellison Medical Foundation (to W.E.S.), and the Hungarian National Development Agency (TÁMOP/SROP-4.2.1/b-10/2/KONV-2010-0012, to Z.U.).
Acknowledgments
The authors would like to express their gratitude for the support of the Donald W. Reynolds Foundation, which funds aging research at the University of Oklahoma Health Sciences Center under its Aging and Quality of Life Program.
References
- 1. Gaspar LE, Mehta MP, Patchell RA, et al. The role of whole brain radiation therapy in the management of newly diagnosed brain metastases: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2010;96:17–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Patil CG, Pricola K, Sarmiento JM, Garg SK, Bryant A, Black KL. Whole brain radiation therapy (WBRT) alone versus WBRT and radiosurgery for the treatment of brain metastases. Cochrane Database Syst Rev. 2012;9:CD006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Butler RW, Haser JK. Neurocognitive effects of treatment for childhood cancer. Ment Retard Dev Disabil Res Rev. 2006;12:184–191 [DOI] [PubMed] [Google Scholar]
- 4. Li J, Bentzen SM, Li J, Renschler M, Mehta MP. Relationship between neurocognitive function and quality of life after whole-brain radiotherapy in patients with brain metastasis. Int J Radiat Oncol Biol Phys. 2008;71:64–70 [DOI] [PubMed] [Google Scholar]
- 5. Welzel G, Fleckenstein K, Mai SK, Hermann B, Kraus-Tiefenbacher U, Wenz F. Acute neurocognitive impairment during cranial radiation therapy in patients with intracranial tumors. Strahlenther Onkol. 2008;184:647–654 [DOI] [PubMed] [Google Scholar]
- 6. Warrington JP, Csiszar A, Johnson DA, et al. Cerebral microvascular rarefaction induced by whole brain radiation is reversible by systemic hypoxia in mice. Am J Physiol Heart Circ Physiol. 2011;300:H736–H744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Warrington JP, Csiszar A, Mitschelen M, Lee YW, Sonntag WE. Whole brain radiation-induced impairments in learning and memory are time-sensitive and reversible by systemic hypoxia. PLoS One. 2012;7:e30444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Robbins ME, Bourland JD, Cline JM, Wheeler KT, Deadwyler SA. A model for assessing cognitive impairment after fractionated whole-brain irradiation in nonhuman primates. Radiat Res. 2011;175:519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Troen AM, Shea-Budgell M, Shukitt-Hale B, Smith DE, Selhub J, Rosenberg IH. B-vitamin deficiency causes hyperhomocysteinemia and vascular cognitive impairment in mice. Proc Natl Acad Sci U S A. 2008;105:12474–12479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lynch CD, Cooney PT, Bennett SA, et al. Effects of moderate caloric restriction on cortical microvascular density and local cerebral blood flow in aged rats. Neurobiol Aging. 1999;20:191–200 [DOI] [PubMed] [Google Scholar]
- 11. Peña LA, Fuks Z, Kolesnick RN. Radiation-induced apoptosis of endothelial cells in the murine central nervous system: protection by fibroblast growth factor and sphingomyelinase deficiency. Cancer Res. 2000;60:321–327 [PubMed] [Google Scholar]
- 12. Ljubimova NV, Levitman MK, Plotnikova ED, Eidus LKh. Endothelial cell population dynamics in rat brain after local irradiation. Br J Radiol. 1991;64:934–940 [DOI] [PubMed] [Google Scholar]
- 13. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740 [DOI] [PubMed] [Google Scholar]
- 14. Rodier F, Coppé JP, Patil CK, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ungvari Z, Tucsek Z, Sosnowska D, et al. Aging-induced dysregulation of dicer1-dependent microrna expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J Gerontol A Biol Sci Med Sci. 2012. 10.1093/gerona/gls242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tucsek Z, Gautam T, Sonntag WE, et al. Aging exacerbates microvascular endothelial damage induced by circulating factors present in the serum of septic patients. J Gerontol A Biol Sci Med Sci. 2012. 10.1093/gerona/gls232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Valcarcel-Ares MN, Gautam T, Warrington JP, et al. Disruption of Nrf2 signaling impairs angiogenic capacity of endothelial cells: implications for microvascular aging. J Gerontol A Biol Sci Med Sci. 2012;67:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Csiszar A, Podlutsky A, Podlutskaya N, et al. Testing the oxidative stress hypothesis of aging in primate fibroblasts: is there a correlation between species longevity and cellular ros production? J Gerontol A Biol Sci Med Sci. 2012;67(8):841–52. 10.1093/gerona/glr216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z, Ungvari Z. Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am J Pathol. 2007;170:388–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pepe M, Mamdani M, Zentilin L, et al. Intramyocardial VEGF-B167 gene delivery delays the progression towards congestive failure in dogs with pacing-induced dilated cardiomyopathy. Circ Res. 2010;106:1893–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ungvari Z, Ridgway I, Philipp EE, et al. Extreme longevity is associated with increased resistance to oxidative stress in arctica islandica, the longest-living non-colonial animal. J Gerontol A Biol Sci Med Sci. 2011;66(7):741–50. 10.1093/gerona/glr044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ungvari Z, Sosnowska D, Podlutsky A, Koncz P, Sonntag WE, Csiszar A. Free radical production, antioxidant capacity, and oxidative stress response signatures in fibroblasts from lewis dwarf rats: effects of life span-extending peripubertal GH treatment. J Gerontol A Biol Sci Med Sci. 2011;66(5):501–10. 10.1093/gerona/glr004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Csiszar A, Labinskyy N, Podlutsky A, et al. Vasoprotective effects of resveratrol and SIRT1: attenuation of cigarette smoke-induced oxidative stress and proinflammatory phenotypic alterations. Am J Physiol Heart Circ Physiol. 2008;294:H2721–H2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Csiszar A, Labinskyy N, Zhao X, et al. Vascular superoxide and hydrogen peroxide production and oxidative stress resistance in two closely related rodent species with disparate longevity. Aging Cell. 2007;6:783–797 [DOI] [PubMed] [Google Scholar]
- 25. Ungvari Z, Orosz Z, Rivera A, et al. Resveratrol increases vascular oxidative stress resistance. Am J Physiol Heart Circ Physiol. 2007;292:H2417–H2424 [DOI] [PubMed] [Google Scholar]
- 26. Csiszar A, Sosnowska D, Tucsek Z, et al. Circulating factors induced by caloric restriction in the nonhuman primate Macaca mulatta activate angiogenic processes in endothelial cells. J Gerontol A Biol Sci Med Sci. 2013;68:235–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Puck TT, Marcus PI. Action of x-rays on mammalian cells. J Exp Med. 1956;103:653–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bailey-Downs LC, Mitschelen M, Sosnowska D, et al. Liver-specific knockdown of igf-1 decreases vascular oxidative stress resistance by impairing the nrf2-dependent antioxidant response: a novel model of vascular aging. J Gerontol A Biol Sci Med Sci. 2012;67(4):313–29. 10.1093/gerona/glr164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bailey-Downs LC, Sosnowska D, Toth P, et al. Growth hormone and IGF-1 deficiency exacerbate high-fat diet-induced endothelial impairment in obese Lewis dwarf rats: implications for vascular aging. J Gerontol A Biol Sci Med Sci. 2012;67:553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Csiszar A, Sosnowska D, Wang M, Lakatta EG, Sonntag WE, Ungvari Z. Age-associated proinflammatory secretory phenotype in vascular smooth muscle cells from the non-human primate macaca mulatta: reversal by resveratrol treatment. J Gerontol A Biol Sci Med Sci. 2012;67(8):811–20. 10.1093/gerona/glr228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ungvari Z, Bailey-Downs L, Gautam T, et al. Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-{kappa}B activation in the nonhuman primate Macaca mulatta. J Gerontol A Biol Sci Med Sci. 2011;66:866–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ungvari Z, Gautam T, Koncz P, et al. Vasoprotective effects of life span-extending peripubertal GH replacement in Lewis dwarf rats. J Gerontol A Biol Sci Med Sci. 2010;65:1145–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bailey-Downs LC, Tucsek Z, Toth P, et al. Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: a paracrine mechanism contributing to vascular redox dysregulation and inflammation. J Gerontol A Biol Sci Med Sci. 2012. 10.1093/gerona/gls238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Csiszar A, Labinskyy N, Perez V, et al. Endothelial function and vascular oxidative stress in long-lived GH/IGF-deficient Ames dwarf mice. Am J Physiol Heart Circ Physiol. 2008;295:H1882–H1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ungvari Z, Labinskyy N, Mukhopadhyay P, et al. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H1876–H1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ungvari Z, Orosz Z, Labinskyy N, et al. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am J Physiol Heart Circ Physiol. 2007;293:H37–H47 [DOI] [PubMed] [Google Scholar]
- 37. Coppé JP, Kauser K, Campisi J, Beauséjour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006;281:29568–29574 [DOI] [PubMed] [Google Scholar]
- 38. Coppé JP, Patil CK, Rodier F, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One. 2010;5:e9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Coppé JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Coppé JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396–36403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roth NM, Sontag MR, Kiani MF. Early effects of ionizing radiation on the microvascular networks in normal tissue. Radiat Res. 1999;151:270–277 [PubMed] [Google Scholar]
- 42. Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155–1159 [DOI] [PubMed] [Google Scholar]
- 43. Fuks Z, Persaud RS, Alfieri A, et al. Basic fibroblast growth factor protects endothelial cells against radiation-induced programmed cell death in vitro and in vivo. Cancer Res. 1994;54:2582–2590 [PubMed] [Google Scholar]
- 44. Takahashi T. The action of radium upon the formation of blood capillaries and connective tissue. British Journal of Radiology (1930). 1930; 439–445 [Google Scholar]
- 45. Truman JP, García-Barros M, Kaag M, et al. Endothelial membrane remodeling is obligate for anti-angiogenic radiosensitization during tumor radiosurgery. PLoS One. 2010;5:e12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haimovitz-Friedman A, Kan CC, Ehleiter D, et al. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med. 1994;180:525–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haimovitz-Friedman A, Balaban N, McLoughlin M, et al. Protein kinase C mediates basic fibroblast growth factor protection of endothelial cells against radiation-induced apoptosis. Cancer Res. 1994;54:2591–2597 [PubMed] [Google Scholar]
- 48. Lozano J, Menendez S, Morales A, et al. Cell autonomous apoptosis defects in acid sphingomyelinase knockout fibroblasts. J Biol Chem. 2001;276:442–448 [DOI] [PubMed] [Google Scholar]
- 49. De Gowin RL, Lewis LJ, Hoak JC, Mueller AL, Gibson DP. Radiosensitivity of human endothelial cells in culture. J Lab Clin Med. 1974;84:42–48 [PubMed] [Google Scholar]
- 50. DeGowin RL, Lewis LJ, Mason RE, Borke MK, Hoak JC. Radiation-induced inhibition of human endothelial cells replicating in culture. Radiat Res. 1976;68:244–250 [PubMed] [Google Scholar]
- 51. Park MT, Oh ET, Song MJ, et al. The radiosensitivity of endothelial cells isolated from human breast cancer and normal tissue in vitro. Microvasc Res. 2012;84:140–148 [DOI] [PubMed] [Google Scholar]
- 52. Imaizumi N, Monnier Y, Hegi M, Mirimanoff RO, Rüegg C. Radiotherapy suppresses angiogenesis in mice through TGF-betaRI/ALK5-dependent inhibition of endothelial cell sprouting. PLoS One. 2010;5:e11084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ahmad M, Khurana NR, Jaberi JE. Ionizing radiation decreases capillary-like structure formation by endothelial cells in vitro. Microvasc Res. 2007;73:14–19 [DOI] [PubMed] [Google Scholar]
- 54. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072–12077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang M, Monticone RE, Lakatta EG. Arterial aging: a journey into subclinical arterial disease. Curr Opin Nephrol Hypertens. 2010;19:201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81–94 [DOI] [PubMed] [Google Scholar]
- 57. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111:245–259 [DOI] [PubMed] [Google Scholar]
- 58. Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–494 [DOI] [PubMed] [Google Scholar]
- 59. Monje ML, Mizumatsu S, Fike JR, Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nat Med. 2002;8:955–962 [DOI] [PubMed] [Google Scholar]
- 60. Krouwer VJ, Hekking LH, Langelaar-Makkinje M, Regan-Klapisz E, Post JA. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc Cell. 2012;4:12. [DOI] [PMC free article] [PubMed] [Google Scholar]