


Abstract
Type IV effectors (T4Es) are proteins produced by pathogenic bacteria to manipulate host cell gene expression and processes, divert the cell machinery for their own profit and circumvent the immune responses. T4Es have been characterized for some bacteria but many remain to be discovered. To help biologists identify putative T4Es from the complete genome of α- and γ-proteobacteria, we developed a Perl-based command line bioinformatics tool called S4TE (searching algorithm for type-IV secretion system effectors). The tool predicts and ranks T4E candidates by using a combination of 13 sequence characteristics, including homology to known effectors, homology to eukaryotic domains, presence of subcellular localization signals or secretion signals, etc. S4TE software is modular, and specific motif searches are run independently before ultimate combination of the outputs to generate a score and sort the strongest T4Es candidates. The user keeps the possibility to adjust various searching parameters such as the weight of each module, the selection threshold or the input databases. The algorithm also provides a GC% and local gene density analysis, which strengthen the selection of T4E candidates. S4TE is a unique predicting tool for T4Es, finding its utility upstream from experimental biology.
INTRODUCTION
Bacterial pathogens have evolved specific effector proteins to exploit host cell machinery and hijack the immune responses during infection (1). Dedicated multiprotein complexes, known as secretion systems, secrete these effectors. Type IV secretion systems (T4SS) are specialized ATP-dependent protein complexes used by many bacterial pathogens for the delivery of type IV effector (T4E) proteins into eukaryotic cells to subvert host cell processes during infection. Some T4Es have been identified in α-proteobacteria (Agrobacterium tumefaciens, Bartonella henselae, Brucella abortus, Anaplasma spp. and Ehrlichia chaffeensis) and γ-proteobacteria (Coxiella burnetii and Legionella pneumophila) and shown to be critical for pathogenicity making them first choice targets to understand bacterial virulence (1–12). Our group was initially interested in identifying T4Es in Ehrlichia ruminantium, which is the causative agent of heartwater, a fatal tropical disease of ruminants. This α-proteobacterium belong to the Anaplasmataceae family and is transmitted by ticks of genus Amblyomma (13).
Ehrlichia spp. and Anaplasma spp. of the Anaplasmataceae family are obligate intracellular pathogens of humans and animals capable of infecting various cell types, including endothelial cells, granulocytes, monocytes and macrophages (14). Once inside the host cell, Ehrlichia spp and Anaplasma spp. reside inside a membrane-bound vacuole where they replicate (14). The replicative vacuole interacts with cholesterol and autophagosome pathways for maturation (15,16). The biogenesis of this replicative niche depends on the function of T4SS and the related secretion of T4Es (16). However, only two T4Es have been described so far in Anaplasmataceae family and shown to play an important role in invasion and pathogenesis. The first effector, AnkA, was identified in Anaplasma phagocytophilum, based on sequence homology with tandemly repeated ankyrin motifs (17). AnkA is secreted by T4SS, is tyrosine phosphorylated and is then addressed into the nucleus to silence the CYBB gene expression of the host cell (18–20). This effector is part of the emerging family of the nucleomodulins that hijack nuclear processes to facilitate infection (21). The other known Anaplasmataceae effector, Ats-1, was identified in A. phagocytophilum and shown to be targeted by T4SS to the cytoplasm of infected cells. Ats-1 interacts with the host autophagosome initiation complex to recruit autophagosomes to the bacterial intracellular vacuole (16). Another portion of Ats-1 targets host cell mitochondria to exert antiapoptotic activity (12,22)
To facilitate the identification of putative T4Es in the whole genome of E. ruminantium, we explored bioinformatics for sequence motif search. Bacterial effectors can be classified into two main groups: those mimicking endogenous cell proteins and those modifying host cell proteins (1,23,24). Because of their various functions in mammalian cells, most of these effectors have characteristic eukaryotic-like domains involved in protein–protein interactions, localization signals and C-terminal features like positive charge, basicity or hydrophobicity that interfere with host cellular processes to promote bacterial replication (12,14,16,25). We first looked at the literature for bioinformatics tools developed for such motif searches and tested successfully for T4Es prediction (4,6,9–11,25–28). However, all the algorithms used in these works were based on the identification of a limited number of sequence motifs and were designed for specific α- or γ-proteobacteria. To circumvent the risk of missing important T4Es, we decided to develop a new algorithm combining searches for all motifs that previously predicted T4Es of α- and γ-proteobacteria. In addition, new sequence motifs were derived from the analysis of the extensive repertoire of T4Es characterized in L. pneumophila (8) and included in the algorithm.
In this article, we present ‘S4TE’ (Searching Algorithm for Type-IV secretion system Effectors), a tool for in silico screening of proteobacteria genomes and T4Es prediction based on the combined use of 13 distinctive features. This software was first probed against the comprehensive T4E dataset of L. pneumophila, strain Philadelphia (8) and subsequently tested on several genomes of α- and γ-proteobacteria. S4TE is both memory- and time-efficient. Although advanced users will be capable of modifying searching parameters of S4TE (e.g. exclusion of modules, change in module weighting, selection threshold or input databases), the common user can easily run the program with default settings. Installation process and basic command lines to launch and run S4TE are detailed in the user guide. S4TE package is freely available to non-commercial users at http://sate.cirad.fr/.
MATERIALS AND METHODS
Overview
We propose an easy-to-use and customizable algorithm for the prediction of candidate effector proteins secreted by T4SS. The algorithm can be used as a standard pre-selection technique for T4 effectors in genomes of any size. Its modularity will offer a simple and robust alternative to machine learning approach for less-studied pathogenic bacteria. In this section, we describe the algorithm used by S4TE, how the parameters of this software were estimated from the literature and how S4TE performs on different genomes. The essential features of the S4TE program, as depicted in Figure 1, are the following: (i) genome-wide screening based on 13 different criteria including homology to known T4Es, occurrence of eukaryotic-like domains or motifs and subcellular localization signals; (ii) T4Es prediction and ordering output based on criteria scoring; (iii) information on prediction performance compared with the reference L. pneumophila; (iv) analysis of G + C content and of space clustering of S4TE hits and (v) analysis of genome architecture and distribution of S4TE hits depending on local gene density.
Figure 1.

Flowchart of the bioinformatics search by S4TE to identify putative effector proteins (PEs). This bioinformatics pipeline is composed of 15 steps delimited by color boxes. Steps 1–13 look for T4 effector features in a given bacterial genome. Step 14 (Files compilation/Ranking) ranks and classifies the predicted T4 effectors based on their number of features to provide the best candidates for experimental validation (Supplementary Figures S2 and S3). Step 15 analyses the genome architecture and G + C content and shows the distribution of predicted effectors. Programs used are indicated in italics. Euk-like, Eukaryotic-like; Prok-like, Prokaryotic-like; NLS, nuclear localization signal; MLS, mitochondrial localization signal; C-ter, C-terminal.
S4TE search modules
S4TE is a modular program written in Perl that screens bacterial genomes. The 13 search parameters are listed in Table 1 and are run in 10 modules detailed below.
Table 1.
Description of the 13 features used in S4TE to screen a bacterial genome
| Feature number | Feature name | Description | References |
|---|---|---|---|
| 1 | De novo motif search | RRRSNTTTTY motif in the −300 bp (Supplementary Figure S1) | This work, http://meme.sdsc.edu/meme/cgi-bin/meme.cgi |
| 2 | Homology | Sequence identity to a known effector molecule; Blastp against effector database (e-value = 10−2) | (6,8) |
| 3 | Euk-like domains | Presence of eukaryotic domain: 58 eukaryotic domains | This work, Supplementary Table S1 |
| 4 | Prok-like domains | 3617 Domain of Unknown Function (DUF) domains | (29), http://pfam.sanger.ac.uk/search/ |
| 5 | NLS (nuclear localization signal) | Monopartite NLS; [KR]-[KR]-[KR]-[KR]-[KR] and bipartite NLS; K-[KR]-X(6,20)-[KR]-[KR]-X-[KR] | (30,31) |
| 6 | MLS (mitochondrial localization signal) | Probability of a sequence containing a mitochondrial targeting peptide (P > 0.95) | (32), http://www.bioperl.org/ |
| 7 | Prenylation domain | CaaX at the C-terminal; ‘C’ represents a cysteine residue, ‘a’ denotes an aliphatic amino acid and ‘X’ is one of four amino acids | (1,33,34) |
| 8 | Secondary structure | Probability of a coiled-coil structure for windows of 28 residues through a protein sequence (P > 0.95) | (35,36), http://emboss.bioinformatics.nl/cgi-bin/emboss/pepcoil |
| Coiled coils | |||
| 9 | C-ter basicity | ≤3 [HRK] in the C-terminal 25 amino acids | (3,7,22) |
| 10 | C-ter charges | Charge of C-terminal 25 amino acids ≥2; C-ter charge = number of [HRK]-number of [ED]-1 (COO-) | (3,7,22) |
| 11 | C-ter hydrophobicity | Hydropathy of C-terminal 25 amino acids; Hydrophobic residue at the −3rd or −4th position | (9,11,37,38) |
| 12 | Global hydrophilicity | Hydropathy of total protein <−200 | (9,11,37) |
| 13 | E-block | EEXXE in the C-terminal 30 amino acids | (39) |
(1) De novo regulatory motif search. In Legionella, the PmrA and CpxR response regulators regulate numerous T4Es (40). Because cis-acting regulatory sequences have not been described so far in α-proteobacteria, a motif search was performed in the promoter region of the 19 known T4Es of this class of bacteria in Anaplasma, Ehrlichia, Brucella and Bartonella genera. Enriched DNA motifs were searched in a window of 300 nt placed upstream of the start codon, using MEME (41) (http://meme.nbcr.net/meme/). A consensus motif of 10 nt was identified in 14 promoters. This motif, termed RS-TY, consists of 3 purines (R), 1 strong base G or C (S), any nucleotide (A, T, G, C), 4 thymines (T) and 1 pyrimidine (Y) (Supplementary Figure S1). Interestingly, this motif is reminiscent of the cis-regulatory elements characterized in L. pneumophila that are required for expression of T4SS-encoding genes (42). Also, for other pathogenic bacteria, the expression of genes encoding secretion systems and those scattered in the genome encoding their substrates is co-regulated by one master regulatory protein that binds a consensus or imperfect cis-regulatory element (43). Although the biological significance (if any) of this motif needs to be tested experimentally, it was included in S4TE algorithm. The corresponding RS_TY.pl module will extract the 300 nt upstream from the START codon and searches for the RS-TY motif thanks to a position-specific scoring matrix generated from multiple sequence alignments with the promoters of known T4Es of α-proteobacteria. Only alignments with a score >130 are selected.
(2) Homology. Effectors were shown not only to share local sequence similarities but also to diverge rapidly (8). BLAST 2.2 (44) was used for protein comparisons to look for homologies with known T4Es. The command line application can be downloaded from the NCBI web page. The blastp software allows the search of local similarity regions between two protein sequences. S4TE compares the database containing all known T4Es with the query proteome and returns all proteins containing a region that has local similarity with a cutoff of expected value (E) <0.01. This E-value cutoff of 0.01 was selected to show similarities between phylogenetically distant bacterial species (6). The majority of known T4Es are from γ-proteobacteria, and S4TE was expected to work also with α-proteobacteria. The blastcut.pl program was written to reformat the output file of blastp program for user-friendly reading and for S4TE compiler. The blastcut.pl returns only blastp positive alignments.
(3) and (4) Eukaryotic- and prokaryotic-like domains. The search for eukaryotic- and prokaryotic-like domains is done by Pfam-scan.pl in the PfamScan package (http://pfam.sanger.ac.uk) (45). To run properly, Pfam-scan.pl needs several softwares: the Moose module in CPAN (http://search.cpan.org/∼ether/Moose-2.0801/lib/Moose.pm), hmmer3.0rc2 and the BioPerl-1.6.1.tar.gz package. In addition, Pfam-scan.pl needs motif database Pfam-A.hmm (http://pfam.sanger.ac.uk). For file size purpose and memory saving, hmmcut.pl was designed to generate a motif database with the Pfam ID of each motif of interest. This motif database contains 58 eukaryotic-like domains previously found in effectors (Supplementary Table S1) and 3617 prokaryotic-like DUF domains (29). PfamManager.pl will reformat the output file of Pfam program for user convenience by separating search results for eukaryotic-like domains and for DUF domains.
(5) Nuclear localization signals (NLS). NLS are protein sequences that target proteins in the nucleus of eukaryotic cells (1). We assumed that the occurrence of NLS in a bacterial protein sequence would be a good indicator of secretion. There are two classes of NLS. Monopartite NLS consist of the PKKKRKV motif (30). Bipartite NLS are more complex and consist of two alkaline clusters (K and R) separated by a variable spacer (31). We wrote NLS.pl to search for monopartite NLS with the [KR]-[KR]-[KR]-[KR]-[KR] motif and for bipartite NLS with the K-[KR]-X(6,20)-[KR]-[KR]-X-[KR] motif. The latter motif was derived from multiple alignments of known eukaryotic protein sequences containing NLS (31).
(6) Mitochondrial localization signals (MLS). MLS are signal sequences located in the N-terminus of proteins that are targeted to mitochondria. This sequence is cleaved after translocation of the protein inside the mitochondria (1,22). To predict MLS and extract the predicted signal, we used the Mitoprot.pm package of Bioperl (32). Mitoprot.pl and Mitocut.pl were developed to use the Perl module Mitoprot.pm and to format the output file, respectively. Only MLS with P > 0.95 are selected by S4TE.
(7) Prenylation domains. Prenylation is a permanent post-translational modification that is required for protein stability (1). Prenylation involves the covalent addition of a 15-carbon farnesyl or a 20-carbon geranylgeranyl isoprenoid group to a Cys residue within the conserved C-terminal CaaX motif (in which ‘a’ represents an aliphatic residue and ‘X’ is one of the four amino acids) (33). Prenylation increases protein hydrophobicity, facilitating protein anchorage to membranes and targeting effector proteins to membrane-bound organelles (34). S4TE module CaaX.pl will search for prenylation domain in the C-terminal of proteins.
(8) Coiled coils. Coiled coils are structural motifs in proteins in which at least two α-helices are coiled together (35). Coiled-coil domains are protein interaction domains and have a role in the regulation of gene expression by stabilizing transcription factors (36). Coiled-coil–type proteins have similarities with pore-forming proteins in gram-negative pathogens and seem to be important for the delivery of effectors into host cells (46). Most proteins containing validated coiled-coil domains are of eukaryotic origin (47). Interestingly, coiled-coil domains are frequently found in secreted virulence effector proteins (47). To search for coiled-coil domains, we used pepcoil software of Emboss package (http://emboss.bioinformatics.nl/cgi-bin/emboss/pepcoil). The module pepcoilcut.pl of S4TE extracts coiled-coil domains with P > 0.95 and formats the output file.
(9) and (10) C-terminal basicity and charge. T4Es often have a positive charge and a large number of alkaline amino acids (HRK) in the 25 C-terminal amino acids (3,7,22). In S4TE, these two features are investigated with charge.pl module. In α-proteobacteria, all known T4Es have three or more alkaline amino acids (HRK) in the 25 C-terminal amino acids. This feature was used as a threshold to select positives. Charge is calculated by summing the positively charged amino acids (HRK) and by subtracting the number of negatively charged amino acids (ED) and the negative C-terminal charge (COO−). In α-proteobacteria, most known T4Es have a C-terminal charge of at least 2. This value was set as a threshold in charge.pl.
(11) and (12) C-terminal and global hydropathy. We calculated hydropathy profiles of proteins using Kyte–Doolittle scale, for which the more hydrophobic the residue, the higher its hydropathy value (>0) (37). Most known T4Es are hydrophilic, having total negative hydropathy scores, negative average hydropathy and highly hydrophilic C-termini (9,11,38). Negative hydropathy at the C-terminus and negative global hydropathy with scores of <200 were used in S4TE with hydro.pl for screening in bacterial proteomes. Moreover, hydro.pl looks for a hydrophobic residue at the third or fourth C-terminal positions (9,48).
(13) E-block. The E-block domain consists of a glutamate-rich sequence (EEXXE) in the C-terminal 30 amino acids and is associated with T4Es translocation in L. pneumophila. Huang et al. showed that an E-block motif is also important for the translocation of T4SS substrates (39). This motif is searched by S4TE with the E-block.pl module.
All search modules return a score of 0 or 1, based on the absence or presence of the parameter, or a parameter quantitative value over the threshold. The individual scores, weighted or not according to user decision, are summed, and the global score is compared with a threshold set by the user. All hits higher than the threshold are returned by S4TE. Each search module was individually designed and refined with dedicated T4Es datasets of different origins to achieve the best specificity (i.e. probability to find a true negative [TN]). We then adjusted S4TE (module weights and threshold) to have the best positive predictive value (PPV; proportion of true positives [TP] detected among all positives) and sensitivity (probability to find a TP) on the dataset of 275 T4Es characterized in L. pneumophila, strain Philadelphia (see Results section). S4TE is also designed to provide two performance indicators attached to any combination of searched parameters. The first indicator is the Sensitivity Index for Legionella (SIL) of a given combination: SIL is calculated by 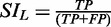 and ranges from 0 to 1. SIL gives information on the proportion of TP found in L. pneumophila with the same combination of parameters. The second indicator is defined as a Positivity Index for Legionella (PIL) and is generated by the formula
and ranges from 0 to 1. SIL gives information on the proportion of TP found in L. pneumophila with the same combination of parameters. The second indicator is defined as a Positivity Index for Legionella (PIL) and is generated by the formula 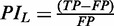 . Therefore, this indicator integrates the net yield of TP given by a combination of parameters, in proportion to the FP number. PIL ranges from −1 to 10 (Supplementary Table S2). When the FP number equals 0, PIL indicator is 0. By using S4TE on different genomes, the user will be able to look at the L. pneumophila performances of all the combinations that picked T4Es hits. For instance, if four hits are selected with combinations that give the following values for {TP, FP} in L. pneumophila; {25, 9}, {6, 1}, {0, 2} and {0, 0}; the corresponding SIL and PIL will be {0.74, 1.78}; {0.86, 5}; {0, −2} and {0, 0}, respectively. In this situation, to select the first target for further biological validation, the hit with SIL = 0.86 and PIL = 5 could be considered as more promising than the one with SIL = 0.74 and PIL = 1.78 (increased risk of FP selection with the latter). In contrast, a hit obtained with a combination that does not exist in L. pneumophila (SIL = 0 and PIL = 0) could be interesting to test because it could serve to identify an effector with an original association of features.
. Therefore, this indicator integrates the net yield of TP given by a combination of parameters, in proportion to the FP number. PIL ranges from −1 to 10 (Supplementary Table S2). When the FP number equals 0, PIL indicator is 0. By using S4TE on different genomes, the user will be able to look at the L. pneumophila performances of all the combinations that picked T4Es hits. For instance, if four hits are selected with combinations that give the following values for {TP, FP} in L. pneumophila; {25, 9}, {6, 1}, {0, 2} and {0, 0}; the corresponding SIL and PIL will be {0.74, 1.78}; {0.86, 5}; {0, −2} and {0, 0}, respectively. In this situation, to select the first target for further biological validation, the hit with SIL = 0.86 and PIL = 5 could be considered as more promising than the one with SIL = 0.74 and PIL = 1.78 (increased risk of FP selection with the latter). In contrast, a hit obtained with a combination that does not exist in L. pneumophila (SIL = 0 and PIL = 0) could be interesting to test because it could serve to identify an effector with an original association of features.
Data compilation
The main program S4TE.pl executes the 10 previously described modules to generate 13 result files. Afterward, a compiler (compilation.pl) will collect all information from result files generated during the pipeline execution and will highlight important data (Figure 1). For each feature, compilation.pl will search the identified proteins and will count the number of positive hits. Then, compilation.pl will sort the results by top/down scoring. For each analysis, the compiled results are written in CompilationFile.txt with the list of identified proteins, the number of hits, the score and the combination of positive features for each protein.
S4TE graphical outputs
G + C content and space clustering analysis
In L. pneumophila, T4Es have atypical G + C content that could result from horizontal gene transfers (HGT) acquired during evolution (26). In addition, effectors encoding genes are often clustered in specific regions, indicating possible HGT events and suggesting possible co-regulation (27,49). With the GC.pl program, S4TE calculates the G + C content (GC%) in a 10-kb window sliding every 200 nt across the genome and plots the GC%, the mean GC% and candidate effectors: effectors in regions with high G + C content are plotted in red, whereas others are plotted in green (Figure 3). This representation allows the user to easily see whether the hits are clustered or scattered in the genome and whether they are rich or low in G + C content.
Figure 3.

Schematic representation of putative T4 effectors in the L. pneumophila genome according to G + C content. This representation is an output file automatically generated by S4TE. The mean GC% is indicated by the blue line. Putative effectors in genomic regions with low G + C content are in green and those in regions with high G + C content are in red.
Genome architecture analysis
S4TE also proposes to analyse the genome architecture and its hit content through the visualization of the length and distribution of intergenic regions and the distribution of the hits according to local gene density (50–52). For every gene, we used two-dimensional data binning to visualize the distance to its closest coding gene neighbors on five prime and three prime (designated as 5′ and 3′ flanking intergenic regions [FIRs]) in a single representation (50). With FIR.pl and matrix.pl, S4TE sorts genes (or predicted effectors) into two-dimensional bins defined by the length of their 5′ and 3′ FIRs. Then, the gene (or effector) density distribution is represented in R by a color-coded heat map with filled.contour.pl. We used the median length of FIRs to distinguish between gene-dense regions (GDRs) and gene-sparse regions (GSRs). This method offers the opportunity to visualize the position of predicted effectors relative to the whole genome architecture (52).
Databases
To analyse a given genome, S4TE needs a genome database with four distinct files: (i) Genome.nucl containing the FASTA genome sequence; (ii) Genome.an, a csv file (with; as separator) containing the gene ID; the position of the first nucleotide of coding sequence; the position of the last nucleotide; the sense or antisense status; before use in S4TE, this file needs to be sorted in ascending order from the first nucleotide position; (iii) Genome.prot, a fasta file containing all the protein sequences of the genome; and (iv) Genome.csv, a file constructed from Genome.prot with nomprot.pl in the folder ∼/S4TE/DataBases/Genome/Tools/
A database of validated effectors derived from the literature was constructed in a protein sequence file of effectors (effector_db.txt) and formatted by makeblastdb -in effector_db.txt -dbtype prot
The database assembling the eukaryotic Pfam domains found in T4Es was constructed from the downloaded original wget ftp://ftp.sanger.ac.uk/pub/databases/Pfam/releases/Pfam26.0/Pfam-A.hmm*.gz
The tool hmmcut.pl will construct the Pfam-A.hmm database with the motifs used by S4TE only. The Pfam-A.hmm database is formatted by.hmmpress Pfam-A.hmm.
Software availability
S4TE is freely available to non-commercial users at http://sate.cirad.fr/. Programming was done using Perl 5.12 and BioPerl 1.6. The software runs on Linux platforms (Ubuntu 11.10 and Mac OS X). All required packages and the installation process are described in the user guide included in Supplementary Methods S1. The user guide also details S4TE options for running S4TE. By default, the command line to launch S4TE is.S4TE.pl -g “name of the genome” from the S4TE folder (cd way_to_S4TE/S4TE/). Some options are available for the user to launch S4TE: -c, suppression of a module in the pipeline; -w, modification of the weight of each module in the pipeline; -t, imposition of a threshold for effector selection. Each S4TE module creates an .txt file in the folder way_to_S4TE/S4TE/Jobs/job<Name_of_genome_folder><year><month><day><hour><min>
All the results are compiled in CompilationFile.txt and Results.txt in the same folder.
RESULTS
After the initial configuration of the different modules of the S4TE algorithm, achieved on all known T4Es, the whole program was run against the 275 known L. pneumophila T4Es and different parameter weightings were tested for optimizing the prediction. The next section details this optimization. The last section shows the outcome of S4TE on different representative bacteria genomes.
Effectors prediction and validation of the S4TE algorithm
For S4TE adjustment and validation, we used the extensive repertoire of 275 experimentally confirmed T4Es of L. pneumophila, strain Philadelphia (8,9). The work was carried out in three major steps consisting of the adjustment of parameter weighting for optimized prediction of L. pneumophila T4Es, the analysis of the relative importance of the different parameters in the optimized configuration and the link between the GC% and gene density in the genome and T4Es localization.
Optimization of S4TE forT4Es prediction on L. pneumophila
The genome of L. pneumophila, strain Philadelphia, contains the most extensive repertoire of T4Es ever identified, with 275 confirmed effector proteins encoded by 9.3% of the genome (8). These 275 T4Es were considered as TP, whereas the 2666 other proteins were included in the analysis as TN. By default, S4TE sets the selection threshold cutoff at 5. This threshold cutoff was first defined as the minimal score minus 1, necessary to allow the selection of all known T4Es of two α-proteobacteria: A. tumefaciens and B. henselae. The default threshold was selected to offset the overrepresentation of L. pneumophila effectors and to be less stringent for candidate effectors with no homology to known effectors. However, the user can adjust the threshold and S4TE will return all candidates scoring over the new threshold. On the L. pneumophila dataset, the algorithm with unweighted parameters and the default threshold at 5 selected 151 TP and 2428 TN. However, 238 hits were not T4Es (false positive [FP]), and 124 T4Es were not found by S4TE (false negative [FN]). This led to a sensitivity (Se) of 55%, a specificity (Sp) of 91%, PPV [PPV = TP/(TP + FP)] of 38.8% and a negative predictive value [NPV = TN/(TN + FN)] of 95.1%. To improve Se and PPV, different parameter weightings were tested. The best combination (i.e. 1311111111111) led to the selection of 223 TP of the 275 effectors of L. pneumophila (Se = 81%, PPV = 72.8%); it led to only 83 FP (Sp = 98.8%, NPV = 98%). The weighting code in brackets is a 13-digit code corresponding to the 13 features in the S4TE scheme and is reported in the head of the compilation file and the results file (Supplementary Figures S2 and S3).
All parameters in S4TE are relevant for the prediction of L. pneumophila T4Es
Beyond the final outcome of S4TE, we sought to determine the relative importance of each searching feature in the algorithm prediction of L. pneumophila T4Es. A variable distribution of the effectors across features was observed; some of them were highly selective and specific, whereas others were less efficient (Table 2). For Legionella, we confirmed the importance of the hydrophilic profile in the overall length of the protein and its C-terminus (PPV = 69% for feature 12), the charge of the C-terminus (PPV = 70% for feature 10) and the presence eukaryotic domains (PPV = 71% for feature 3), coiled-coil domains (PPV = 76% for feature 8) and E-block motif (PPV = 72% for feature 13) (Table 2). We then enumerated TP and FP identified by S4TE in L. pneumophila according to the number of matching features. TP and FP were well discriminated for combinations of 3 and 4 features (Figure 2A). Even with a slight increase in FP, combinations of 5, 6 and 7 features remained discriminant (Figure 2A). Although accuracy increased from 93% with a combination of 3 features to 95% with 7 features, specificity decreased from 99 to 97% (Figure 2B). The constant rise of the sensitivity from 26% with 3 features to 81% with 7 features shows the importance of our multi-criterion approach to identify a majority of candidate T4Es (Figure 2B). The complete list of feature combinations that generated hits for L. pneumophila was used to propose two performance indicators, SIL and PIL (see Materials and Methods section and Supplementary Table S2). These indicators are included in the result file appended to each predicted effector and will advise the user on the prediction efficacy of the same combination of features on L. pneumophila, thus providing additional help to select the right candidates for further biological evaluation.
Table 2.
Enumeration of L. pneumophila effectors predicted by individual features implemented in S4TE
| S4TE feature | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| True positives | 38 | 223 | 30 | 5 | 21 | 5 | 1 | 96 | 33 | 185 | 107 | 152 | 13 |
| False positives | 19 | 48 | 12 | 11 | 23 | 3 | 0 | 31 | 40 | 81 | 28 | 69 | 5 |
| PPV (%) | 67 | 82 | 71 | 31 | 48 | 63 | 100 | 76 | 45 | 70 | 79 | 69 | 72 |
The number of true positives (TP), false positives (FP) and the positive predictive value (PPV, expressed in %) is indicated.
Figure 2.
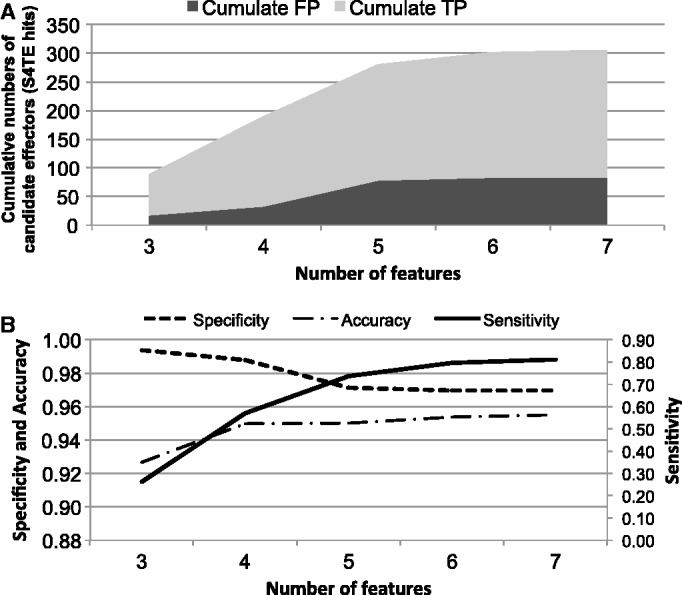
Distribution of the number of features that detected effector candidates in L. pneumophila. (A) Cumulated numbers of effectors correctly detected (TPs) and called by error (false positives, FP) by S4TE L. pneumophila genome. (B) Accuracy, sensitivity and specificity of S4TE analysis on L. pneumophila genome with combinations of 3, 4, 5, 6 and 7 features.
Genomic localization of T4Es depends on the G + C content, space clustering analysis and local gene density
The distribution of predicted effectors in the genome of L. pneumophila was analysed in detail. We first compared the G + C content of effector-containing regions with the mean G + C content (Figure 3). Effectors were found to be mainly in regions with low G + C content (in green; 60% of the predicted effectors), which agrees with the literature (26). Figure 3 also presents the spatial distribution of predicted effectors and indicates that some genomic regions are enriched in clustered effectors. Such clusters give meaningful information about putative gene co-regulation. The low G + C% content and spatial clustering support the hypothesis that the evolutionary origin of effectors was HGT (53,54). In prokaryotes, the genome architecture refers to the relative position of genetic elements on a genome, including gene order and operons (55). The evolution of genome architecture is largely compelled by specific lifestyles, such as intracellular replication of pathogenic bacteria (55). In some lineages, the active duplication of repeated sequences was associated with adaptation to the host (56), and the presence of insertion elements and transposons is a typical feature of pathogenicity islands (57). We therefore hypothesized that some predicted effectors would be associated with regions of low gene density in the genome of pathogenic bacteria. S4TE serves to visualize the distribution of predicted effectors relative to whole genome architecture (Figure 4). As a case study, we compared the FIRs of predicted effector genes with the architecture of the whole genome of L. pneumophila (Figures 4A and B). We found that predicted effectors frequently have both FIRs above the genome median value in L. pneumophila genome (Figure 4B and C, Table 3). Although 28% of L. pneumophila genes reside in GSRs, this percentage peaks at 52.8% for T4Es genes (162 of the 311 predicted) (Figure 4C). Furthermore, 92.3% of predicted T4Es had at least one FIR longer than the genome median and only 7.7% of the predicted effectors had both FIRs below the genome median (Figure 4B). These observations support the view that plastic genome regions with low gene density frequently harbor pathogenicity genes and may play a role in bacterial adaptation.
Figure 4.

Distribution of L. pneumophila genes and predicted T4 effectors according to local gene density (measured as length of flanking intergenic regions, FIRs). (A) Distribution of L. pneumophila genes according to their FIRs. Genes were sorted in two-dimensional bins according to the length of their 5′ (y-axis) and 3′ (x-axis) FIR lengths. The number of genes in bins is represented by a color-coded density graph. Genes with both FIRs longer than the median length of FIRs were considered as gene-sparse region (GSR) genes. Genes with both FIRs below the median value were considered as gene-dense region (GDR) genes. In between genes are genes with a long 5′ FIR and short 3′ FIR, and inversely. For L. pneumophila, this median value is 109-bp for 5′ FIRs and 65 bp for 3′ FIRs. The dotted line for the median length of FIR delimits the genes in GSR, GDR and in between. (B) Distribution of predicted T4 effectors according to their FIRs. The number of hits per T4 effectors in bins is indicated by a color scale. (C) Distribution of predicted T4 effectors in the GSRs and GDRs of L. pneumophila. The proportion of T4 effectors in GSRs, in between and GDRs is shown in red, yellow and blue, respectively, with percentage indicated.
Table 3.
Predicted T4 effectors in various genomes of α- and γ-proteobacteria
| Genome | ORFa | Known T4Es (%)b | Predicted T4Es (%)c | Predicted TP (%)d | Mean GC (%)e | High GC (%)f | Low GC (%)g | GDRs (%)h | IB (%)i | GSRs (%)j |
|---|---|---|---|---|---|---|---|---|---|---|
| Ehrlichia ruminantium, strain Gardel | 950 | NA | 22 (2,32) | NA | 28 | 68 | 32 | 23 | 36 | 41 |
| Anaplasma marginale | 963 | 4 (0,42) | 26 (2,70) | 100 | 50 | 62 | 38 | 15 | 46 | 38 |
| Brucella abortus chr I | 2000 | 3 (0,15) | 53 (2,65) | 100 | 57 | 62 | 38 | 25 | 40 | 34 |
| Brucella abortus chr II | 1034 | 1 (0,10) | 17 (1,64) | 100 | 57 | 41 | 59 | 6 | 65 | 29 |
| Coxiella burnetii | 2085 | 43 (2,06) | 126 (6,04) | 77 | 43 | 50 | 50 | 15 | 41 | 44 |
| Coxiella burnetii pl | 36 | 1 (2,78) | 4 (11,11) | 100 | 39 | 50 | 50 | 25 | 25 | 50 |
| Legionella pneumophila | 2943 | 275 (9,34) | 311 (10,57) | 81 | 38 | 40 | 60 | 8 | 40 | 53 |
aNumber of ORFs in the genome.
bNumber and proportion of known T4 effectors in the genome.
cNumber and proportion of predicted T4 effectors.
dProportion of true positives in S4TE prediction.
eMean G + C content of the genome.
fProportion of predicted T4 effectors in genomic regions with high G + C content.
gProportion of predicted T4 effectors in genomic regions with low G + C content.
hProportion of predicted T4 effectors in gene-dense regions.
iProportion of predicted T4 effectors in ‘in between’ regions.
jProportion of predicted T4 effectors in gene-sparse regions.
NA, not applicable; TP, true positives; GDRs, gene dense regions; GSRs, gene sparse regions; T4Es, type IV effectors.
S4TE successfully predicts T4Es in other bacterial pathogens
S4TE was used to analyse the genome of four representative α- and γ-proteobacteria. Because our validation analysis on L. pneumophila suggested that the software had high accuracy (>90%), we next focused on bacterial pathogens of α- and γ-classes having various genome sizes and for which some T4Es were already confirmed experimentally. To evaluate the computational requirements of our algorithm, the different runs on these datasets were timed. It took a maximum of 40 min for the bigger genomes (2–3 Mbp). In the annotation of each genome, S4TE indicated the total number of candidate T4Es, and their position relative to the G + C content and to their FIRs (Table 3). All known T4Es in Anaplasma marginale and B. abortus were picked up by S4TE, whereas 77% of known T4Es were identified for C. burnetii (Table 3). No T4Es of E. ruminantium are yet characterized; however, S4TE was able to detect all known T4Es of the closely related E. chaffeensis (not shown) and predicted 22 T4Es for E. ruminantium (Supplementary Table S3). As expected, orthologs of known T4Es of Anaplasmataceae such as AnkA and ECH_0825 were identified. Moreover, among other putative T4Es, 48% showed a global hydropathy lower than −200, 68% had a C-ter charge >2, 95% had at least three alkaline amino acids in the C-terminal 25 amino acids and 90% harbored characteristic eukaryotic-like domains (Supplementary Table S3). Interestingly, and in contrast to Legionella, predicted T4Es in the α-proteobacteria (E. ruminantium, A. marginale and B. abortus) were mainly localized in genome regions with high G + C content, except for the small chromosome of B. abortus (Table 3). However, we noticed a clear exclusion of predicted effectors from GDRs (Table 3). This could be an interesting predicting characteristic to explore in the future. Concerning C. burnetii, another γ-proteobacteria-like Legionella, the G + C content analysis revealed an equal distribution of predicted effectors throughout the genome but a strong prevalence in GSRs. Although effector prediction by S4TE is still accurate for plasmids (e.g. plasmid of C. burnetii) or for small replicons (e.g. chromosome II of B. abortus), one has to consider that analyses of the G + C content and genome architecture become meaningless.
DISCUSSION
Before this study, bioinformatics tools for genome-wide annotation of T4Es encoding genes have been developed in a limited number of studies for L. pneumophila, C. burnetii, B. abortus or A. marginale (6,10,11,27). However, these in silico screening tools search for only few criteria like a combination of homology hits to known effectors and occurrence of eukaryotic-like domains or motifs and coiled-coil domains (10). In another study, candidate T4Es were essentially selected on the basis of their hydropathy profiles and by eliminating proteins with known housekeeping functions and/or with predicted localization signals (11). Finally, Chen et al. investigated the genome of C. burnetii only for L. pneumophila paralogs before experimental validation (6). Our purpose with S4TE was to develop a more complete bioinformatics solution that not only looked for a wider range of sequence features for T4Es prediction but also proposed several visualization interfaces for the outputs. By investigating independently a set of 13 features, combining the different outcomes in a single score and comparing the score with a pre-defined threshold, we provide a computational method that can help biologists in selecting the best potential targets for subsequent experimental validation. S4TE also offers useful services for decision-makers like the representation of genome G + C content and gene density linked to the localization of T4Es and finally performance indicators calculated on L. pneumophila. The performances of S4TE, which uses 13 different criterions for T4Es prediction and three complementary analyses of genome contents linked to the localization of predicted effectors, were compared with the performances of other algorithms. C. burnetii and L. pneumophila are exceptional cases for which a broad repertoire of T4 effectors is identified and widely characterized. In silico screening with an accurate machine learning prediction algorithm is possible only when sets of negative and positive effector proteins are known, such as for L. pneumophila (27). However, for the vast majority of pathogenic bacteria with a T4SS, only few T4 effectors are known. The identification of novel effectors and the subsequent characterization of their function and their targets is a major step to understand how T4SS contributes to bacterial virulence. However, direct biological screening for T4Es can be a tough task, especially for obligate intracellular bacteria that are difficult to cultivate.
In this context, S4TE was designed as an easy-to-use, versatile and customizable algorithm for the prediction of putative effector proteins secreted by the T4SS of proteobacteria whatever the genome size. The high PPV obtained in the large T4Es repertoire of Legionella illustrates the relevance of the features combined by S4TE. These features were selected from independent searches on all known T4 effectors on L. pneumophila, C. burnetii, A. tumefaciens, B. abortus, Bartonella spp., Anaplasma spp. and Ehrlichia spp. The strength of S4TE relies on the compilation of these features to find TPs. However, user awareness must be raised of the fact that S4TE remains only a useful step toward the identification of T4Es, as non-effector proteins with characteristics similar to T4Es can also be selected, resulting in false positives. Experimental validation of T4SS-dependent translocation is therefore required to establish the effector status of the predicted proteins. Given that the S4TE algorithm is based on characteristics of known T4Es from different bacterial species, genera and even classes, we showed that it might be applicable to other distant pathogenic bacteria. This agrees with a growing number of studies suggesting conserved mode of action or targets for effectors across bacteria classes (21,24,58). The high number of true effectors picked with a potential C-terminal secretion signal seems in line with a T4SS biological function. Also, a previous study has localized the secretion signal at the effector C-terminus of T4Es (38).
Regarding E. ruminantium, our main study model, S4TE was able to identify 22 putative T4SS substrates that may contribute to modulation or evasion of host cellular processes. However, further biological testing of their T4SS-dependent secretion is required. Predicting the function of the identified Ehrlichia T4SS substrates based solely on their domains is a difficult task. Functional characterization of these candidate T4Es will provide valuable information about the molecular mechanisms underlying the pathogenesis of Ehrlichia. Finally, an integrated comprehension of the regulation of T4SS expression and translocation events during infection of host cells will establish the interaction of Ehrlichia with its environment.
Future directions
Depending on the availability of biologically validated effectors in α-proteobacteria and confirmation of a strong link between T4Es positions in the genome and the density of genes at these positions, the dense/sparse-gene feature will be integrated in the S4TE algorithm as a predictive value, as done previously for plant pathogenic fungi and oomycetes (51,52,59). We could also refine cutoffs and weighting, as well as add new features when new effectors are discovered. Finally, our approach could be applicable in the identification of candidate effectors in other pathosystems dealing with eukaryotic cells.
CONCLUSION
We have developed a computational tool, S4TE, dedicated to the prediction of candidate bacterial T4 effectors. Our software was designed to identify T4SS effector proteins in α- and γ-proteobacteria. First, the evaluation of S4TE performances demonstrated that the algorithm has a high specificity and high PPVs and NPVs for T4Es. Second, S4TE is time-efficient. Third, S4TE has a very high NPV by default. Yet, a few adjustments can be made by the user to improve confidence in the outcomes. The future-validated bona fide T4 effectors will help to refine the S4TE algorithm. In addition, we provide an automated pipeline to analyse effector space clustering and distribution in the genome according to the G + C content and local gene density. The algorithm can be used with default settings, but manual adjustment of the parameters is available. S4TE will be updated when new information (e.g. new validated effectors, new functional domains and new bacterial genomes) becomes available.
S4TE has been registered with the Agency for the Protection of Programs for version 1.0 under registration number IDDN.FR.001.310023.000.S.P.2012.000.31230, filed in June 2012.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [60].
FUNDING
UE, FEDER 2007–2013 [FED 1/1.4-30305] ‘Risk in animal and plant health’. Funding for open access charge: UE, FEDER 2007–2013 [FED 1/1.4-30305] ‘Risk in animal and plant health’.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank T. Lefrançois and D. Pleydell for useful suggestions, L. Bournez for her aid with Figure 1 and S. Gonzalez-Rizzo for her support and fruitful discussions. D.F.M also thanks P.L. and M.J. Meyer for inspiration. The authors are grateful to G. Burkhart and anonymous reviewers for critical reading and improvement of the manuscript.
REFERENCES
- 1.Hicks SW, Galan JE. Exploitation of eukaryotic subcellular targeting mechanisms by bacterial effectors. Nat. Rev. Micro. 2013;11:316–326. doi: 10.1038/nrmicro3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vergunst AC, Schrammeijer B, den Dulk-Ras A, de Vlaam CM, Regensburg-Tuïnk TJ, Hooykaas PJ. VirB/D4-dependent protein translocation from Agrobacterium into plant cells. Science. 2000;290:979–982. doi: 10.1126/science.290.5493.979. [DOI] [PubMed] [Google Scholar]
- 3.Vergunst AC, van Lier MC, den Dulk-Ras A, Stüve TA, Ouwehand A, Hooykaas PJ. Positive charge is an important feature of the C-terminal transport signal of the VirB/D4-translocated proteins of Agrobacterium. Proc. Natl Acad. Sci. USA. 2005;102:832–837. doi: 10.1073/pnas.0406241102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulein R, Guye P, Rhomberg TA, Schmid MC, Schröder G, Vergunst AC, Carena I, Dehio C. A bipartite signal mediates the transfer of type IV secretion substrates of Bartonella henselae into human cells. Proc. Natl Acad. Sci. USA. 2005;102:856–861. doi: 10.1073/pnas.0406796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Backert S, Meyer TF. Type IV secretion systems and their effectors in bacterial pathogenesis. Curr. Opin. Microbiol. 2006;9:207–217. doi: 10.1016/j.mib.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 6.Chen C, Banga S, Mertens K, Weber MM, Gorbaslieva I, Tan Y, Luo ZQ, Samuel JE. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc. Natl Acad. Sci. USA. 2010;107:21755–21760. doi: 10.1073/pnas.1010485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rikihisa Y, Lin M. Anaplasma phagocytophilum and Ehrlichia chaffeensis type IV secretion and Ank proteins. Curr. Opin. Microbiol. 2010;13:59–66. doi: 10.1016/j.mib.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gomez-Valero L, Rusniok C, Cazalet C, Buchrieser C. Comparative and functional genomics of legionella identified eukaryotic like proteins as key players in host-pathogen interactions. Front. Microbiol. 2011;2:208. doi: 10.3389/fmicb.2011.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo Z-Q. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One. 2011;6:e17638. doi: 10.1371/journal.pone.0017638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marchesini MI, Herrmann CK, Salcedo SP, Gorvel JP, Comerci DJ. In search of Brucella abortus type IV secretion substrates: screening and identification of four proteins translocated into host cells through VirB system. Cell. Microbiol. 2011;13:1261–1274. doi: 10.1111/j.1462-5822.2011.01618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lockwood S, Voth DE, Brayton KA, Beare PA, Brown WC, Heinzen RA, Broschat SL. Identification of Anaplasma marginale type IV secretion system effector proteins. PLoS One. 2011;6:e27724. doi: 10.1371/journal.pone.0027724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu H, Bao W, Lin M, Niu H, Rikihisa Y. Ehrlichia type IV secretion effector ECH0825 is translocated to mitochondria and curbs ROS and apoptosis by upregulating host MnSOD. Cell Microbiol. 2012;14:1037–1050. doi: 10.1111/j.1462-5822.2012.01775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allsopp BA. Natural history of Ehrlichia ruminantium. Vet. Parasitol. 2010;167:123–135. doi: 10.1016/j.vetpar.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 14.Dumler JS, Barbet AF, Bekker CP, Dasch GA, Palmer GH, Ray SC, Rikihisa Y, Rurangirwa FR. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and ‘HGE agent' as subjective synonyms of Ehrlichia phagocytophila. Int. J. Syst. Evol. Microbiol. 2001;51:2145–2165. doi: 10.1099/00207713-51-6-2145. [DOI] [PubMed] [Google Scholar]
- 15.Xiong Q, Rikihisa Y. Subversion of NPC1 pathway of cholesterol transport by Anaplasma phagocytophilum. Cell Microbiol. 2011;14:560–576. doi: 10.1111/j.1462-5822.2011.01742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niu H, Xiong Q, Yamamoto A, Hayashi-Nishino M, Rikihisa Y. Autophagosomes induced by a bacterial Beclin 1 binding protein facilitate obligatory intracellular infection. Proc. Natl Acad. Sci. USA. 2012;109:20800–20807. doi: 10.1073/pnas.1218674109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caturegli P, Asanovich KM, Walls JJ, Bakken JS, Madigan JE, Popov VL, Dumler JS. ankA: an Ehrlichia phagocytophila group gene encoding a cytoplasmic protein antigen with ankyrin repeats. Infect. Immun. 2000;68:5277–5283. doi: 10.1128/iai.68.9.5277-5283.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park J, Kim KJ, Choi K-S, Grab DJ, Dumler JS. Anaplasma phagocytophilum AnkA binds to granulocyte DNA and nuclear proteins. Cell Microbiol. 2004;6:743–751. doi: 10.1111/j.1462-5822.2004.00400.x. [DOI] [PubMed] [Google Scholar]
- 19.Lin M, Den Dulk-Ras A, Hooykaas PJ, Rikihisa Y. Anaplasma phagocytophilum AnkA secreted by type IV secretion system is tyrosine phosphorylated by Abl-1 to facilitate infection. Cell. Microbiol. 2007;9:2644–2657. doi: 10.1111/j.1462-5822.2007.00985.x. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Garcia JC, Rennoll-Bankert KE, Pelly S, Milstone AM, Dumler JS. Silencing of host cell CYBB gene expression by the nuclear effector AnkA of the intracellular pathogen Anaplasma phagocytophilum. Infect. Immun. 2009;77:2385–2391. doi: 10.1128/IAI.00023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bierne H, Cossart P. When bacteria target the nucleus: the emerging family of nucleomodulins. Cell Microbiol. 2012;14:622–633. doi: 10.1111/j.1462-5822.2012.01758.x. [DOI] [PubMed] [Google Scholar]
- 22.Niu H, Kozjak-Pavlovic V, Rudel T, Rikihisa Y. Anaplasma phagocytophilum Ats-1 is imported into host cell mitochondria and interferes with apoptosis induction. PLoS Pathog. 2010;6:e1000774. doi: 10.1371/journal.ppat.1000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ham H, Sreelatha A, Orth K. Manipulation of host membranes by bacterial effectors. Nat. Rev. Micro. 2011;9:635–646. doi: 10.1038/nrmicro2602. [DOI] [PubMed] [Google Scholar]
- 24.Anderson DM, Frank DW. Five mechanisms of manipulation by bacterial effectors: a ubiquitous theme. PLoS Pathog. 2012;8:e1002823. doi: 10.1371/journal.ppat.1002823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ninio S, Roy CR. Effector proteins translocated by Legionella pneumophila: strength in numbers. Trends Microbiol. 2007;15:372–380. doi: 10.1016/j.tim.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 26.De Felipe KS, Glover RT, Charpentier X, Anderson OR, Reyes M, Pericone CD, Shuman HA. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. 2008;4:e1000117. doi: 10.1371/journal.ppat.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burstein D, Zusman T, Degtyar E, Viner R, Segal G, Pupko T. Genome-scale identification of Legionella pneumophila effectors using a machine learning approach. PLoS Pathog. 2009;5:e1000508. doi: 10.1371/journal.ppat.1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carey KL, Newton HJ, Lührmann A, Roy CR. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog. 2011;7:e1002056. doi: 10.1371/journal.ppat.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prakash A, Yogeeshwari S, Sircar S, Agrawal S. Protein domain of unknown function 3233 is a translocation domain of autotransporter secretory mechanism in gamma proteobacteria. PLoS One. 2011;6:e25570. doi: 10.1371/journal.pone.0025570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell. 1984;39:499–509. doi: 10.1016/0092-8674(84)90457-4. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen Ba AN, Pogoutse A, Provart N, Moses AM. NLStradamus: a simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinformatics. 2009;10:202. doi: 10.1186/1471-2105-10-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Claros MG. MitoProt, a Macintosh application for studying mitochondrial proteins. Comput. Appl. Biosci. 1995;11:441–447. doi: 10.1093/bioinformatics/11.4.441. [DOI] [PubMed] [Google Scholar]
- 33.Al-Quadan T, Price CT, London N, Schueler-Furman O, Abukwaik Y. Anchoring of bacterial effectors to host membranes through host-mediated lipidation by prenylation: a common paradigm. Trends Microbiol. 2011;19:573–579. doi: 10.1016/j.tim.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 34.Ivanov SS, Charron G, Hang HC, Roy CR. Lipidation by the host prenyltransferase machinery facilitates membrane localization of Legionella pneumophila effector proteins. J. Biol. Chem. 2010;285:34686–34698. doi: 10.1074/jbc.M110.170746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 36.Boysen RI, Jong AJ, Wilce JA, King GF, Hearn MT. Role of interfacial hydrophobic residues in the stabilization of the leucine zipper structures of the transcription factors c-Fos and c-Jun. J. Biol. Chem. 2002;277:23–31. doi: 10.1074/jbc.M104556200. [DOI] [PubMed] [Google Scholar]
- 37.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 38.Nagai H, Cambronne ED, Kagan JC, Amor JC, Kahn RA, Roy CR. A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc. Natl Acad. Sci. USA. 2005;102:826–831. doi: 10.1073/pnas.0406239101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang L, Boyd D, Amyot WM, Hempstead AD, Luo Z-Q, O'Connor TJ, Chen C, Machner M, Montminy T, Isberg RR. The E Block motif is associated with Legionella pneumophila translocated substrates. Cell. Microbiol. 2011;13:227–245. doi: 10.1111/j.1462-5822.2010.01531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altman E, Segal G. The response regulator CpxR directly regulates expression of several Legionella pneumophila Icm/Dot components as well as new translocated substrates. J. Bacteriol. 2008;190:1985–1996. doi: 10.1128/JB.01493-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gal-Mor O, Zusman T, Segal G. Analysis of DNA regulatory elements required for expression of the Legionella pneumophilaicm and dot virulence genes. J. Bacteriol. 2002;184:3823–3833. doi: 10.1128/JB.184.14.3823-3833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cunnac S, Occhialini A, Barberis P, Boucher C, Genin S. Inventory and functional analysis of the large Hrp regulon in Ralstonia solanacearum: identification of novel effector proteins translocated to plant host cells through the type III secretion system. Mol. Microbiol. 2004;53:115–128. doi: 10.1111/j.1365-2958.2004.04118.x. [DOI] [PubMed] [Google Scholar]
- 44.Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic Acids Res. 2011;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barta ML, Dickenson NE, Patil M, Keightley A, Wyckoff GJ, Picking WD, Picking WL, Geisbrecht BV. The structures of coiled-coil domains from type III secretion system translocators reveal homology to pore-forming toxins. J. Mol. Biol. 2012;417:395–405. doi: 10.1016/j.jmb.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delahay RM, Frankel G. Coiled-coil proteins associated with type III secretion systems: a versatile domain revisited. Mol. Microbiol. 2002;45:905–916. doi: 10.1046/j.1365-2958.2002.03083.x. [DOI] [PubMed] [Google Scholar]
- 48.Lifshitz Z, Burstein D, Peeri M, Zusman T, Schwartz K, Shuman HA, Pupko T, Segal G. Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type-IVB secretion signal. Proc. Natl Acad. Sci. USA. 2013;110:E707–E715. doi: 10.1073/pnas.1215278110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kirzinger MW, Stavrinides J. Host specificity determinants as a genetic continuum. Trends Microbiol. 2012;20:88–93. doi: 10.1016/j.tim.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 50.Haas BJ, Kamoun S, Zody MC, Jiang RH, Handsaker RE, Cano LM, Grabherr M, Kodira CD, Raffaele S, Torto-Alalibo T, et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature. 2009;461:393–398. doi: 10.1038/nature08358. [DOI] [PubMed] [Google Scholar]
- 51.Raffaele S, Farrer RA, Cano LM, Studholme DJ, Maclean D, Thines M, Jiang RH, Zody MC, Kunjeti SG, Donofrio NM, et al. Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science. 2010;330:1540–1543. doi: 10.1126/science.1193070. [DOI] [PubMed] [Google Scholar]
- 52.Raffaele S, Win J, Cano LM, Kamoun S. Analyses of genome architecture and gene expression reveal novel candidate virulence factors in the secretome of Phytophthora infestans. BMC Genomics. 2010;11:637. doi: 10.1186/1471-2164-11-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Degtyar E, Zusman T, Ehrlich M, Segal G. A Legionella effector acquired from protozoa is involved in sphingolipids metabolism and is targeted to the host cell mitochondria. Cell. Microbiol. 2009;11:1219–1235. doi: 10.1111/j.1462-5822.2009.01328.x. [DOI] [PubMed] [Google Scholar]
- 54.Moliner C, Raoult D, Fournier PE. Evidence that the intra-amoebal Legionella drancourtii acquired a sterol reductase gene from eukaryotes. BMC Res. Notes. 2009;2:51. doi: 10.1186/1756-0500-2-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koonin EV, Wolf YI. Constraints and plasticity in genome and molecular-phenome evolution. Nat. Rev. Genet. 2010;11:487–498. doi: 10.1038/nrg2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collins NE, Liebenberg J, de Villiers EP, Brayton KA, Louw E, Pretorius A, Faber FE, van Heerden H, Josemans A, van Kleef M, et al. The genome of the heartwater agent Ehrlichia ruminantium contains multiple tandem repeats of actively variable copy number. Proc. Natl Acad. Sci. USA. 2005;102:838–843. doi: 10.1073/pnas.0406633102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Juhas M, Crook DW, Hood DW. Type IV secretion systems: tools of bacterial horizontal gene transfer and virulence. Cell. Microbiol. 2008;10:2377–2386. doi: 10.1111/j.1462-5822.2008.01187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shames SR, Finlay BB. Bacterial effector interplay: a new way to view effector function. Trends Microbiol. 2012;20:214–219. doi: 10.1016/j.tim.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 59.Saunders DG, Win J, Cano LM, Szabo LJ, Kamoun S, Raffaele S. Using hierarchical clustering of secreted protein families to classify and rank candidate effectors of rust fungi. PLoS One. 2012;7:e29847. doi: 10.1371/journal.pone.0029847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.