


Abstract
Chromatin modifications have been comprehensively illustrated to play important roles in gene regulation and cell diversity in recent years. Given the rapid accumulation of genome-wide chromatin modification maps across multiple cell types, there is an urgent need for computational methods to analyze multiple maps to reveal combinatorial modification patterns and define functional DNA elements, especially those are specific to cell types or tissues. In this current study, we developed a computational method using differential chromatin modification analysis (dCMA) to identify cell-type-specific genomic regions with distinctive chromatin modifications. We then apply this method to a public data set with modification profiles of nine marks for nine cell types to evaluate its effectiveness. We found cell-type-specific elements unique to each cell type investigated. These unique features show significant cell-type-specific biological relevance and tend to be located within functional regulatory elements. These results demonstrate the power of a differential comparative epigenomic strategy in deciphering the human genome and characterizing cell specificity.
INTRODUCTION
All human cells share the same genetic information encoded by genomic DNA sequences, regardless of the cells’ type. However, cells exhibit dramatically diverse phenotypes (1). In eukaryotic cells, genomic DNA is modulated by numerous chemical modifications, thus adding an extra layer of information to the genome sequence. These modifications enable genomic DNA to encode a vast and complex program of gene regulation (2–4), giving rise to diverse protein expression patterns and subsequent tissue-specific phenotypic diversity (5). Discovering functional elements and understanding how diverse modifications regulate these elements are central challenges to elucidate global gene regulation in humans.
Cooperative binding of chromatin modifications, including histone modifications, DNA methylation and regulatory proteins shape the macro-environment of DNA and affect context-dependent interpretation of it. With the advent of chromatin immunoprecipitation coupled with tilling arrays (ChIP-chip) or parallel DNA sequencing (ChIP-seq), people have come genome-wide profiling of binding sites (6). More and more data sets are being generated for various chromatin features in multiple cell types, providing abundant resources for decoding chromatin modification patterns.
One popular approach used to interpret epigenomic data is to identify and functionally characterize combinatorial patterns and systematically define DNA regulatory elements. For example, methylation of both lysine 4 and lysine 27 on histone H3 is an epigenetic signature characteristic of embryonic stem cells, which keeps silenced developmental genes poised for activation (7,8). Another example is, by applying supervised regression framework, histone modification intensities around promoters were shown to be predictive for gene expression (3,9). Clustering approaches, used in early signature detection work, grouped well-annotated promoters on the basis of specific histone modification patterns (10). This research has recently been adopted into two data analysis platforms: seqMINER (11) and Cistrome (12). Hon et al. (13) developed a probabilistic method, ChromaSig, to identify histone modification signatures de novo that are repeated across the genome, without using any existing annotations. Jascheck and Tanay (14) proposed a spatial clustering algorithm to identify sets of common combinatorial modification patterns, defined over contiguous genomic regions. More recently, the hidden Markov model (HMM) and Bayesian network approaches have been used to uncover recurrent chromatin states by segmenting the epigenome into regions defined by characteristic histone mark combinations (15,16). In contrast to these clustering-type methods, Ucar et al. (17) developed a biclustering algorithm, CoSBI, to search combinatorial patterns involving only subsets of histone marks. Teng and Tan (18) further described a semi-supervised version of the CoSBI algorithm to incorporate existing knowledge of combinatorial patterns into the mining procedure.
Although these methods facilitated the identification and characterization of chromatin modification patterns and functional genomic elements in the human genome, we are far from understanding the underlying modification patterns and biological mechanisms that dictate phenotypes of cells and tissues. In a recent pioneering study, Ernst et al. (19) applied the HMM method (15) to define chromatin states of nine cell types. Furthermore, cell-type-specific changes, which go toward elucidating cell-type specificities and predict regulatory mechanisms that drive gene regulation, were investigated. To date, cell-type-specific functional regions of the genome, which have key roles in cell diversity, have not been analyzed or defined directly.
In this article, we aim to identify cell-type-specific regulatory elements (CSREs) in the human genome, by differential Chromatin Modification Analysis (dCMA). To this end, we developed a computational framework capable of identifying genomic regions, containing distinctive chromatin modifications that are unique to each cell type. We applied this method to the public ChIP-seq data set used in (19) to demonstrate its effectiveness. We identified CSREs for each cell type related to various genomic features, which showed significant, cell-type-specific biological relevance and tended to be regulatory elements. Moreover, a large majority of CSREs are located in non-coding regions lacking annotations. These results can shed light on experimental human genome investigations. The proposed framework demonstrates the power of a differential comparative epigenomics strategy in deciphering aspects of the human genome and characterizing cell specificity.
MATERIALS AND METHODS
Materials
Genome-wide maps of nine chromatin marks and a control case in nine cell types have been generated using ChIP-seq in a recent study (19). These nine cell types include embryonic stem cells (H1 ES or H1), erythrocytic leukemia cells (K562), B-lymphoblastoid cells (GM12878), hepatocellular carcinoma cells (HepG2), umbilical vein endothelial cells (HUVEC), skeletal muscle myoblasts (HSMM), normal lung fibroblasts (NHLF), normal epidermal keratinocytes (NHEK) and mammary epithelial cells (HMEC). In each cell type, nine chromatin marks consisting of CCCTC-binding factor (CTCF), H3K27me3, H3K36me3, H4K20me1, H3K4me1, H3K4me2, H3K4me3, H3K27ac and H3K9ac were profiled, and a whole-cell extract (WCE) sequencing was conducted as control. RNA expression profiles of the nine cell types were also generated using Affymetrix GeneChip arrays. In this article, we downloaded these data for the following studies.
The raw ChIP-seq data were preprocessed as previously described in (19). Whole-genome data were divided into non-overlapping 200 bp bins. All sequencing reads were extended by 200 bp in the 3′ direction, to capture actual binding sites and then assigned a unique 200 bp bin according to their middle points. We had an integer for each bin, which summaries neighboring read counts. Next, the integers were binarized using a Poisson null model. Specifically, the null hypothesis of a ChIP-seq experiment is that all reads distribute uniformly across the genome; therefore, the mean of the Poisson distribution can be calculated by dividing the total read counts by the number of 200 bp bins in the human genome. A threshold of 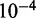 was chosen to transform the integers into binary values. After binarization, we observed that many 200 bp bins have no signals in all cell types, which may consist of sequences with low ‘mappability’ (20). Those consecutive regions with length >10 kb were removed from the genome (20.7% of the whole genome) in the analyses. Finally, we obtained nine binary matrices of size 10 (nine chromatin marks and WCE) by N (the number of 200 bp bins in human genome). Cytosolic RNA was isolated and quantified using Affymetrix GeneChip arrays (19). The raw data stored in CEL files were processed with RMA, and replicate expression values were averaged. The resulting expression profiles were processed using quantile normalization (21) across the nine cell types.
was chosen to transform the integers into binary values. After binarization, we observed that many 200 bp bins have no signals in all cell types, which may consist of sequences with low ‘mappability’ (20). Those consecutive regions with length >10 kb were removed from the genome (20.7% of the whole genome) in the analyses. Finally, we obtained nine binary matrices of size 10 (nine chromatin marks and WCE) by N (the number of 200 bp bins in human genome). Cytosolic RNA was isolated and quantified using Affymetrix GeneChip arrays (19). The raw data stored in CEL files were processed with RMA, and replicate expression values were averaged. The resulting expression profiles were processed using quantile normalization (21) across the nine cell types.
Methods
After data preprocessing, we obtained binary modification profiles for all cell types (Figure 1A). We introduce the following steps to identify CSREs. We have implemented this method and made a package called dCMA, which can be easily used for other researchers (Supplementary Methods).
Figure 1.

Illustration of the framework used in identifying CSREs. (A) The data profiles of nine cell types as characterized by nine marks and one control. The raw ChIP-seq reads were mapped to 200 bp bins and the signals were binarized using a Poisson null model. (B) For each bin, the dissimilarity of the resulting binary vectors between two different cell types was measured using hamming distance. For each cell type, the DMS of a bin was the summation of pairwise hamming distance (sPHD) computed between it and other cell types. (C) The DMS profile of each cell type was normalized across the genome. Then, each column of the matrix was multiplied by the corresponding Z-scores to consider the variance in the column. (D) Wavelets smoothing strategy was adopted to smooth the resulting differential profile of each cell type. CSREs were extracted by selecting suitable height and length parameters. Statistical significance (P-value) of each CSRE was calculated by a non-parametric test.
Step 1: Calculation of the differential modification score. For each genomic position (a 200 bp bin), hamming distance was used to measure differences of the modification characteristics between each pair of cell types (Figure 1B). Suppose there were K cell types and M chromatin marks and the human genome was divided into N 200 bp bins. Let 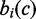 denote the mark occurrence profile at genomic bin coordinate c in cell type i (a binary vector with M components). Then, the Differential Modification Score (DMS) for a particular cell type t at genomic coordinate c was the summation of hamming distances between this cell type and others, which can be represented as
denote the mark occurrence profile at genomic bin coordinate c in cell type i (a binary vector with M components). Then, the Differential Modification Score (DMS) for a particular cell type t at genomic coordinate c was the summation of hamming distances between this cell type and others, which can be represented as 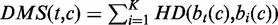 , where HD represents the hamming distance operator. After these calculations, we had a DMS profile across the human genome in each cell type.
, where HD represents the hamming distance operator. After these calculations, we had a DMS profile across the human genome in each cell type.
Step 2: Normalization and correction of the DMSs. First, we normalized the sum of squares of each cell type’s DMS profile to a constant. We further calculated the Z-score of DMSs for each bin among all cell types: 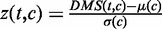 , where
, where 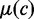 and
and 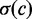 are the mean and the standard deviation of the DMS at genomic position c, respectively. The normalized DMS were then multiplied by the corresponding Z-scores to get the corrected scores.
are the mean and the standard deviation of the DMS at genomic position c, respectively. The normalized DMS were then multiplied by the corresponding Z-scores to get the corrected scores.
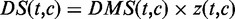 (Figure 1C). The corrected scores reflect both the scale and relative size of the original scores.
(Figure 1C). The corrected scores reflect both the scale and relative size of the original scores.
Step 3: Wavelet smoothing and CSRE extraction. The wavelet transform is a widely used filtering and smoothing technique, which has been comprehensively applied in computational biology (Figure 1D). It has been used in applications such as noise removal from microarray data (22) and normalization of diverse data types to a common scale in integrative analysis (23). We applied the maximal overlap discrete wavelet transform (24), a modification of discrete wavelet transform, to the corrected DMS profiles to further reduce noises and enhance the signal-to-noise ratio (Figure 1D). The genomic regions corresponding to smoothed DSs peaks in each cell type are defined as CSREs and can be directly extracted with given height and length parameters. Finally, the results with height = 1.5 and length = 12 were used to illustrate the strong biological relevance of CSREs. As to the selection of these two parameters, we performed enrichment analysis on CSREs identified from four other groups of parameter settings to show their robustness (Supplementary Methods).
Step 4: Measuring the statistical significance of CSREs. The null hypothesis in defining CSREs is chromatin marks occupy the 200 bp bins with equal probability in all nine cell types. We estimated the probability of occurrence for the marks in each bin and simulate data under null hypothesis, according to the estimated probabilities. The summation of the DMS, within the CSRE, was chosen as the testing statistic, and then the right-sided probability in the null distribution (fitted by a Gaussian distribution) was computed as P-value. P-values were corrected for multiple hypothesis testing by Benjamini–Hochberg correction (25).
Mapping CSRE bins to various genomic features
The CSREs were identified in a comparative manner, and we examined their functional potential roles by mapping them to known genomic features. All base pairs of the human genome were categorized into six classes: promoter, 5′ UTR, exon, intron, 3′ UTR and intergenic. Specifically, base pairs within 2 kb from a known RefSeq transcription start site (TSS) were labeled as promoter, and those located further than 2 kb from any RefSeq gene were labeled as intergenic. We purposefully defined intergenic regions in this loose fashion to allow us to cover the majority of the human genome using these six categories. The CSRE bins were then classified into six feature categories according to their bins’ feature annotations, as described earlier in the text. The CSREs bins were classified in a hierarchical fashion, i.e. if the bins of a CSRE belong to more than one features, it was processed according to the order: promoter >5′ UTR >3′ UTR > exon > intron > intergenic. For each kind of genomic region, the corresponding proportion in CSREs was divided by the genome-wide proportion to determine the fold enrichment. The statistical significance was evaluated with Fisher’s exact test.
Overlap analysis of the CSREs between cell types
To test whether the CSREs of each cell type are cell-type specific, we calculate the overlap of CSREs between any two cell types. Two CSREs A and B are defined to be overlapped if 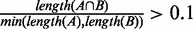 , where length(X) represents the number of bins in fragment X. Then, for each pair of cell types, namely, i and j, we can determine the number of CSREs in cell type i overlapped by those in cell type j. To test the statistical significance of the overlap, Fisher’s exact tests were applied (left-sided and right-sided P-values for significant less and more overlaps, respectively).
, where length(X) represents the number of bins in fragment X. Then, for each pair of cell types, namely, i and j, we can determine the number of CSREs in cell type i overlapped by those in cell type j. To test the statistical significance of the overlap, Fisher’s exact tests were applied (left-sided and right-sided P-values for significant less and more overlaps, respectively).
Enrichment analysis
To investigate the biological relevance of CSREs, we conducted housekeeping gene, biological function and network enrichment analysis. CSREs were mapped to RefSeq genes first (version hg18, downloaded from UCSC Genome Browser). Specifically, a CSRE was mapped to a gene when it overlaps the gene region. Here, the gene region starts from the TSS and ends at the transcription end site. We call the overlapping gene as the neighboring gene of this CSRE.
To assess whether the CSREs are cell-type-specific regulatory elements, we calculated the overlap between CSRE neighboring genes and housekeeping genes. We downloaded the housekeeping gene list generated in (26), and 3218 genes were obtained after mapping to the RefSeq gene list (version hg18). Fold enrichments of the overlaps were obtained, and the corresponding statistical significance was evaluated using Fisher’s exact test.
Gene Ontology (GO) terms enrichment analysis was performed using STEM (27) where Fisher’s exact test was used and the Bonferroni corrected q-values were reported. For each cell type, the top five enriched GO terms associating 5–500 genes were selected (Figure 3E). A human protein–protein interaction network consisting of 13 207 proteins and 64 549 interactions was downloaded from the BioGRID website (28). For each cell type, a sub-network was determined by mapping CSRE neighboring genes to the protein–protein interaction, and we let m be the number of interactions observed therein. The expected number of interactions in the sub-network was 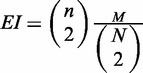 , where N and n are the numbers of nodes in the whole network and the sub-network, respectively, and M is the number of observed interactions in the whole network. The fold enrichment was
, where N and n are the numbers of nodes in the whole network and the sub-network, respectively, and M is the number of observed interactions in the whole network. The fold enrichment was 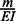 . The statistical significance of the fold enrichment was calculated using right-sided Fisher’s exact test.
. The statistical significance of the fold enrichment was calculated using right-sided Fisher’s exact test.
Figure. 3.

Functional relevance and cell-type specificity of CSREs. (A) The proportion of CSREs belonging to single, two or more cell types. (B) Overlaps of CSREs between each pair of cell types. The values in the diagonal correspond to the number of identified CSREs in nine cell types and the value in row i column j records the number of CSREs in cell type i overlapped by those in cell type j. (C) CSRE neighboring genes tended to be more significantly connected than was expected, with 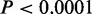 indicated by (asterisk). (D) CSRE neighboring genes are more likely to be non-housekeeping genes with the exception of those in K562 and GM12878. Dashed vertical line on the left side represent the P-value threshold (
indicated by (asterisk). (D) CSRE neighboring genes are more likely to be non-housekeeping genes with the exception of those in K562 and GM12878. Dashed vertical line on the left side represent the P-value threshold (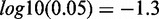 ). (E) CSRE neighboring genes show distinct functional enrichments highly relevant to the corresponding cell type contexts. We chose the top five enriched GO terms in each cell type, and
). (E) CSRE neighboring genes show distinct functional enrichments highly relevant to the corresponding cell type contexts. We chose the top five enriched GO terms in each cell type, and 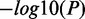 was used to generate the heat map. (F) Enriched phenotypes of SNPs located in the CSREs in three cell types.
was used to generate the heat map. (F) Enriched phenotypes of SNPs located in the CSREs in three cell types.
Relationship between CSREs and disease-associated variants
The NHGRI’s collection of trait/disease-associated SNPs from published genome-wide association studies were downloaded from UCSC genome browser (July 12, 2012) (29,30). There were 7899 SNPs associated with 575 traits in total. For each cell type, we extract a subset of SNPs corresponding to CSREs. The traits of the set of SNPs are used to investigate their characteristics. We aimed to investigate the relevance between the selected SNPs’ traits and each cell type, based on the identified CSREs. To this end, we tested whether SNPs associated with a trait are significantly located in CSREs of a cell type through the Fisher’s exact test. P-values were corrected for multiple hypotheses testing by Benjamini–Hochberg correction.
Relationship between CSREs and DNase I hypersensitive and EP300 binding sites
DNase I hypersensitive sites (DHSs) peaks in eight of the nine studied cell types (except HepG2) and EP300 binding peaks for three cell types (H1 ES, GM12 878 and HepG2) were obtained from the UCSC browser (http://genome.ucsc.edu). Cell-type specific peaks are defined by filtering out those peaks appearing in other cell types. Considering the chromatin modifications happened on nucleosomes, while the DHSs and transcription factor binding sites are usually located in nucleosome depleted regions, we extended these cell-type-specific peaks by 2.5 kb in each direction to their upstream and downstream, respectively. (We have also tested this with a more stringent criterion and get similar results, see Supplementary Methods and Supplementary Figure S1.) CSREs that overlapped, by at least 1 bp, the extended peaks were considered as DHS or EP300 proximal. For genome-wide background, we randomly selected 1000 sets of CSREs for each cell type with length and chromosome attributes reserved and calculated the corresponding number of DHS or EP300 proximal ones. One-sample Wilcoxon test was used to evaluate the statistical significance of the real number.
RESULTS
We have developed a multi-stage method to identify CSREs, which are likely to be cell-selective regulatory regions (Supplementary Table S1). We applied our method to the data set generated in (19) for nine cell types. Analysis of these data sets defined, on average, 4110.8 CSREs per cell type (ranging from 1701 in NHLF to 6659 in GM12878; Supplementary Figure S2A) spanning an average 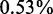 of the genome. On average, 92% of CSREs in each cell type are statistical significant with q < 0.0001. The median lengths of CSREs across the nine cell types was similar (∼3 Kb), except two of them (K562 and GM12878) are slightly longer than the others (Supplementary Figure S2B). The total numbers of base pairs, found in cell-type specific CSREs, varies from ∼7.5 (NHLF) to 28.5 Mb (GM12878) (Supplementary Figure S2C). The number of genes near to CSREs also varies, from ∼1546 (NHLF) to 4907 (GM12 878) (Supplementary Figure S2D). These diverse distributions may imply the functional complexity of these cell types (Supplementary Figure S2).
of the genome. On average, 92% of CSREs in each cell type are statistical significant with q < 0.0001. The median lengths of CSREs across the nine cell types was similar (∼3 Kb), except two of them (K562 and GM12878) are slightly longer than the others (Supplementary Figure S2B). The total numbers of base pairs, found in cell-type specific CSREs, varies from ∼7.5 (NHLF) to 28.5 Mb (GM12878) (Supplementary Figure S2C). The number of genes near to CSREs also varies, from ∼1546 (NHLF) to 4907 (GM12 878) (Supplementary Figure S2D). These diverse distributions may imply the functional complexity of these cell types (Supplementary Figure S2).
CSREs relate to various genomic features
We explored the relations between CSREs and various genomic features to illustrate their potential functional roles. The proportion of CSREs in different genomic regions varied across the cell types (Figure 2A). The CSREs were significantly enriched at well-known regulatory regions, such as promoters, 5′ and 3′ UTRs (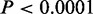 , Fisher’s exact tests) (Figure 2B). Exon and intron regions were also enriched in CSREs, suggesting that part of a gene body may serve as regulatory elements, such as enhancers, for its own expression (31). The strong enrichments of CSREs in promoter regions (
, Fisher’s exact tests) (Figure 2B). Exon and intron regions were also enriched in CSREs, suggesting that part of a gene body may serve as regulatory elements, such as enhancers, for its own expression (31). The strong enrichments of CSREs in promoter regions (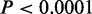 , Fisher’s exact tests) demonstrates underlying modifications acting in promoter regions play critical roles in regulating gene expression. In particular, CSREs in the promoter regions of H1 ES cells were substantially enriched when compared to those found in the other cell types. These results imply more promoter regions are under epigenetic regulation in embryonic stem cells. As discussed later in the text, many promoters in H1 ES are marked by H3K27me3, a repressive chromatin modification, but these regions are tuned in poised status. These characteristics are consistent with the unique cellular context of pluripotent cells.
, Fisher’s exact tests) demonstrates underlying modifications acting in promoter regions play critical roles in regulating gene expression. In particular, CSREs in the promoter regions of H1 ES cells were substantially enriched when compared to those found in the other cell types. These results imply more promoter regions are under epigenetic regulation in embryonic stem cells. As discussed later in the text, many promoters in H1 ES are marked by H3K27me3, a repressive chromatin modification, but these regions are tuned in poised status. These characteristics are consistent with the unique cellular context of pluripotent cells.
Figure 2.

Relationship between CSREs and various genomic features. (A) The distribution of CSREs in six different genomic regions, including promoter, 5′ UTR, 3′ UTR, exon, intron and intergenic regions. (B) The fold enrichments of the CSREs in the six different genomic regions. (C) Box plot of the distance between the intergenic CSREs and the nearest TSSs, compared with those of randomly generated ones. For each intergenic CSRE, the random one was an arbitrarily selected genomic element from the same chromosome with the same length. Then, the distances between the random regions to their nearest TSS were computed. (D) The normalized proportion of CSREs in each chromosome (1–22, X) in all cell types. (The bar of chromosome 22 in K562 was truncated to 0.03 for visualization, and its real number is 0.056) (E) Bar plot of the CV (defined as the ratio of the standard deviation to the mean) of normalized proportion of CSREs in each cell type.
Although CSREs were not enriched in intergenic regions, this group constitute ∼36.4% (averaged across the nine cell types) of total CSREs. Moreover, CSREs in intronic regions made up ∼25.5% (averaged across the nine cell types) of the total CSREs. These results highlight the potential regulatory roles of non-coding regions and, also, the power of a comparative epigenetics strategy in investigating functional roles of the human genome. Intuitively, we hypothesized most of the CSREs target their neighboring genes. The distances are significantly shorter than those found in the genomic background, which, to some extent, verifies our assumption (Figure 2C and Supplementary Figure S3).
We further explored the distribution of CSREs in all 23 chromosomes (1–22, X) in each cell type (Figure 2D). Specifically, we calculated the normalized proportion of CSREs, with respect to chromosome length, for each cell type. We used the coefficient of variation (CV) to quantify the dispersion of the proportions and found that the CVs of two cancer cells (K562 and HepG2) are apparently larger than those of the others (Figure 2E). We observed significantly more CSREs in K562’s chromosome 22 and in HepG2’s chromosomes 16, 17 and 20. Interestingly, a reciprocal translocation between chromosome 9 and 22 in leukemia cells (32) has been well studied. The translocation results in the oncogenic BCR-ABL gene fusion, which is a highly sensitive marker for leukemia (33). For HepG2, chromosome translocations involving chromosomes 16 and 17 have been observed with spectral karyotyping. Moreover, chromosomes 2, 14 and 20 were found to be amplified (34). These previous studies confirm the informative nature of CSREs. In this case, it is non-uniform distribution of CSREs, in K562 and HepG2 cells. These observations demonstrate that the CSREs defined by chromatin modifications may relate to the structural abnormity of chromatin, which contributes to cancer progression, as well as other disorders.
In total, we identified 34 721 distinct CSREs in the human genome (collectively spanning 4.62%), most of which (94.1%) were specific to a single cell type with only 5.9% being found in two or more cell types (Figure 3A). Next, we calculated the number of overlapping CSREs between cell types (Figure 3B). As expected, most pairs of cell types exhibit significantly less overlaps (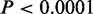 , left-sided Fisher’s exact tests). In contrast, significant overlaps (
, left-sided Fisher’s exact tests). In contrast, significant overlaps (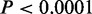 , Fisher’s exact tests) of CSREs occurred between NHEK and HMEC cell lines. This observation is not surprising, given NHEK are keratinizing epithelial cells, and their functions are more similar with HMEC than other cell types.
, Fisher’s exact tests) of CSREs occurred between NHEK and HMEC cell lines. This observation is not surprising, given NHEK are keratinizing epithelial cells, and their functions are more similar with HMEC than other cell types.
We observed protein products of genes near to CSREs tend to interact with each other in each cell type (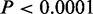 , Fisher’s exact tests) (Figure 3C and Supplementary Figure S4). This result suggests genes near CSREs are more likely to work in a cooperative fashion to carry out biological functions. We also analyzed any overlaps between CSRE neighboring genes in each cell type and housekeeping genes. As expected, in seven cell types, CSRE neighboring genes tend not to be housekeeping genes (
, Fisher’s exact tests) (Figure 3C and Supplementary Figure S4). This result suggests genes near CSREs are more likely to work in a cooperative fashion to carry out biological functions. We also analyzed any overlaps between CSRE neighboring genes in each cell type and housekeeping genes. As expected, in seven cell types, CSRE neighboring genes tend not to be housekeeping genes (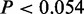 ) (Figure 3D and Supplementary Figure S5). This observation implies genes near to CSREs are likely to execute cell-type-specific biological functions.
) (Figure 3D and Supplementary Figure S5). This observation implies genes near to CSREs are likely to execute cell-type-specific biological functions.
CSREs are defined as genomic regions exhibiting distinctive modification patterns, relative to those in other cell types. It is a reasonable assumption that these CSRE neighboring genes are involved in cell-type-specific biological processes. We mapped CSREs to RefSeq genes, and a gene set was obtained for each cell type. Using GO enrichment analysis, we found overrepresented GO terms were highly relevant to the functions of corresponding cell types, showing distinct cell-type specificity (Figure 3E and Supplementary Tables S2–S5). For example, terms related to development such as ‘brain development’ and ‘regulation of nervous system development’ are enriched in embryonic stem cells (H1 ES); terms related to immune response such as ‘lymphocyte activation’ and ‘immune response-regulating signaling pathway’ are enriched in B-lymphoblastoid cells (GM12 878). As shown in the overlap analysis, NHEK and HMEC are highly related cell types; this is also seen in the functional enrichment analysis data. Functional categories enriched in NHEK are likely to be enriched in HMEC. These results suggest that CSREs act in the regulation of cell-type-specific biological processes, which highlights the role of chromatin modifications in controlling and maintaining differential cell-type gene expression patterns.
Numerous genome-wide association studies have provided a plethora of links between common genetic variants to their resulting traits and diseases. However, the mechanisms behind these links are ill defined, and research needs to be done to explore the downstream effects of disease-associated SNPs (30). To this end, we identified overrepresented traits in each cell type where their associated SNPs were located in the CSREs. We found characteristics of identified traits are consistent with their associated cellular contexts (Figure 3F and Supplementary Table S6). For example, SNPs with the trait ‘hematological and biochemical traits’ and ‘soluble levels of adhesion molecules’ are significantly located in CSREs in erythrocytic leukemia cells (K562). We also found variants relating to immunological diseases, such as ‘primary biliary cirrhosis’ (35), ‘systemic lupus erythematosus’ (36) and ‘Rheumatoid arthritis’ (37), are significantly located in CSREs in B-lymphoblastoid cells (GM12 878). These observations suggest SNPs may change the binding surface of nucleic acids in CSREs altering gene expression patterns, and ultimately disturbing downstream transcriptional regulation. These findings demonstrate that chromatin modifications charactering CSREs help define underlying mechanisms of downstream functional effects of DNA variants including those in non-coding regions.
CSRE neighboring genes reveal diverse transcriptional behaviors
Next, we investigated the transcriptional behavior of CSRE neighboring genes in the nine cell types studied. Each cell type was divided into two groups: ‘CSRE neighboring’ group denotes CSRE neighboring genes, and ‘other’ group denotes genes that are not in a proximal location to CSREs. Only genes that could be mapped to Affymetrix probes are involved in this analysis. Transcription levels were compared between these two groups in all nine cell types. All ‘CSRE neighboring’ groups except that of H1 ES had significantly higher transcriptional levels than the corresponding ‘other’ groups (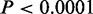 , two-sample Wilcoxon tests) (Figure 4A). We also found that the neighboring genes for all cell types expect H1 ES contains significantly more highly expressed genes than expectation (P < 0.001) (Supplementary Figure S6). In comparison, we checked the overlaps between CSRE neighboring gene set and downregulated gene set in each cell type. We found they did not show significance in any cell type (Supplementary Figure S7). These results indicate most chromatin modifications defining CSREs play a role in gene activation to accomplish specific biological functions. In contrast, CSRE neighboring genes in H1 ES exhibit significantly lower transcription level than the ‘other’ group (
, two-sample Wilcoxon tests) (Figure 4A). We also found that the neighboring genes for all cell types expect H1 ES contains significantly more highly expressed genes than expectation (P < 0.001) (Supplementary Figure S6). In comparison, we checked the overlaps between CSRE neighboring gene set and downregulated gene set in each cell type. We found they did not show significance in any cell type (Supplementary Figure S7). These results indicate most chromatin modifications defining CSREs play a role in gene activation to accomplish specific biological functions. In contrast, CSRE neighboring genes in H1 ES exhibit significantly lower transcription level than the ‘other’ group (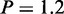 e–7, two-sample Wilcoxon tests), suggesting that chromatin modification defining CSREs in H1 ES have a repressive role in neighboring gene transcription. As expected, H3K27me3, a repressive chromatin modification mark, had a stronger mean intensity in H1 ES CSREs than in the other cell types (Figure 4B). Furthermore, we analyzed the extent of transcription diversity of genes labeled as ‘CSRE neighboring’ in at least one cell type. These genes behaved in a more diverse fashion, based on RNA expression levels, than the remaining genes did (Figure 4C), supporting the proposed potential cell-type-specific roles of these genes.
e–7, two-sample Wilcoxon tests), suggesting that chromatin modification defining CSREs in H1 ES have a repressive role in neighboring gene transcription. As expected, H3K27me3, a repressive chromatin modification mark, had a stronger mean intensity in H1 ES CSREs than in the other cell types (Figure 4B). Furthermore, we analyzed the extent of transcription diversity of genes labeled as ‘CSRE neighboring’ in at least one cell type. These genes behaved in a more diverse fashion, based on RNA expression levels, than the remaining genes did (Figure 4C), supporting the proposed potential cell-type-specific roles of these genes.
Figure 4.

Transcription levels of CSRE neighboring genes. (A) Boxplots comparison of ‘CSRE neighboring’ and ‘Other’ genes. The significance of the difference was calculated using two-sample Wilcoxon tests with 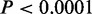 . The ‘CSRE neighboring’ gene group had higher expression levels, with exception of the H1 ES cell line. (B) Bar plot of the mean intensity of H3K27me3, a repressive chromatin modification, among all CSREs. (C) The comparison of transcription diversity of all CSRE neighboring genes and remaining genes. For each gene, the CV was used to measure the diversity of expression levels across the nine cell types examined. Boxplot comparison of the CV was shown, and the statistical significance was evaluated using two-sample Wilcoxon test.
. The ‘CSRE neighboring’ gene group had higher expression levels, with exception of the H1 ES cell line. (B) Bar plot of the mean intensity of H3K27me3, a repressive chromatin modification, among all CSREs. (C) The comparison of transcription diversity of all CSRE neighboring genes and remaining genes. For each gene, the CV was used to measure the diversity of expression levels across the nine cell types examined. Boxplot comparison of the CV was shown, and the statistical significance was evaluated using two-sample Wilcoxon test.
CSREs have proximity to DHSs and EP300 binding sites
DHSs are marks of regulatory DNA and have been extensively used to map regulatory DNA regions in diverse cell lines (38). Moreover, it was reported that differential DNase I hypersensitivity is predictive for perturbation-induced transcription factor-binding sites (39), highlighting the role of DHSs in investigating regulatory DNA. Nucleosomes flanking DHSs have been shown to acquire various covalent modifications, such as methylation and acetylation, facilitating the binding of regulators. We expected CSREs were most likely to lie adjacent to DHSs. Surprisingly, we found most CSREs to be DHS proximal in all eight cell types we tested, which further demonstrates CSREs, as well as their underlying modifications, could play important regulatory roles (Figure 5A). Moreover, ∼61.9% of CSREs are located in intronic and intergenic regions, and they are more likely to act as enhancers or facilitate enhancer activities. EP300 is an enhancer-binding transcriptional co-activator and has been used to identify enhancers, genome-wide, through chromatin immunoprecipitation experiments (40). We found that the CSREs overlap EP300 ChIP peaks significantly more than random ones do (Figure 5B), indicating that many CSREs are adjacent to enhancers.
Figure 5.

Relationship of CSREs to DHSs and EP300 binding sites. The CSREs overlap (A) DHSs in eight cell types and (B) EP300 ChIP peaks, in three cell types, significantly more than randomly simulated ones. The histograms showing the overlap distributions are calculated based on random simulated CSREs, and the red curves are plotted based on kernel density estimate. The black arrows indicate the true number of overlaps. Statistical significance is measured by one-sample Wilcoxon test with 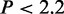 e–16 for all cases.
e–16 for all cases.
CSREs reveal epigenetic mechanisms of cellular dysfunction in cells: two case studies
The first case study we will explore is known as the Philadelphia translocation. The Philadelphia translocation is a chromosome abnormality involving chromosomes 9 and 22, which results in an oncogenic fusion gene BCR-ABL, and is a hallmark of chronic myelogenous leukemia (41,42). In the erythrocytic leukemia cells (K562), both pieces of the BCR-ABL fusion gene (BCR on chromosomes 22 and ABL1 on chromosomes 9) contain CSREs, demonstrating distinctly different epigenetic profile from the other cell types (Figure 6A). Specifically, BCR contains a CSRE consisting of 766 200 bp bins and ABL1 contains five adjacent CSREs. We found three marks including H3K27me3, H3K36me3 and H3K20me1 have distinctive characteristics at these regions (Figure 6A and B). We observed that H3K27me3, a histone modification associated with Polycomb-repressed regions (43), appears upstream of the BCR transcriptional start site and downstream of the ABL1 transcriptional termination site; however, this modification is almost absent in both gene bodies. In contrast, H3K36me3, a histone modification related to transcribed regions (6), had strong signals across the gene body of both genes, exhibiting a mutually exclusive occupancy pattern to that of H3K27me3. These observations imply that there is precise epigenetic control on the transcriptional boundaries of this fusion gene. Another histone modification, H3K20me1, also related to transcribed gene regions (6), is present in the region ranging from upstream of BCR to downstream of ABL1; moreover, it does not demonstrate any exclusivity with H3K27me3 or H3K36me3, suggesting a different functional role. Finally, CTCF, H3K27ac and H3K9ac exhibit stronger signals across the BCR-ABL regions in K562 cells than those observed in the other cell types examined. These distinctive chromatin modification patterns highlight specialized epigenetic regulation of these two genes; any dysfunction in them may be directly related to transcription of the fusion gene.
Figure 6.

Illustration of two distinctive modification patterns revealed by CSREs: the BCR-ABL fusion gene and the INSIG1 gene. (A) The left two plots show the binary modification profiles of the two component genes that make up the BCR-ABL fusion gene in all nine cell types investigated. BCR and ABL gene are covered by one and five CSRE, respectively. Five red bottom lines indicate those five adjacent CSREs covering ABL gene. The top right two plots show the corresponding detailed modification patterns in K562, where the Philadelphia translocation results in the fusion gene. (B) The average intensity comparison of three marks before and after the BCR gene TSS. (C) Binary modification patterns of the CSRE encompassing the promoter region of the INSIG1 gene. (D) The dramatic loss of five active marks including H3K4me1/2/3, H3K27ac and H3K9ac in HepG2 (note that the heights of corresponding bars are almost zero). The modification intensity in the ‘others’ group was calculated based on the combination profiles of the other eight cell types.
Our second case study is insulin-induced gene 1 (INSIG-1), a membrane protein, which plays a critical role in cholesterol homeostasis; it is expressed in almost all tissues and highly expressed in the liver (44). Previously reported lower INSIG-1 expression levels in HepG2 were confirmed here (Supplementary Figure S8), and it correlates to dysregulation of sterol regulatory element-binding protein (45). As we expected, the INSIG-1 promoter region contains a CSRE, consisting of 24 200 bp bins, in which there are almost no modification signals (Figure 6C and D). In the other cell types examined, this corresponding region is marked by active histone modifications including H3K4me1/2/3, H3K27ac and H3K9ac. The dramatic loss of necessary epigenetic modifications may result in lower INSIG-1 expression in HepG2 cells. Both of these examples illustrate the identified CSREs are relevant to biological processes with respect to cell types, and the chromatin modifications characterizing the CSREs may provide clues to explain their underlying mechanisms.
DISCUSSION
The wealth of accumulated ChIP-seq data regarding various chromatin marks (e.g. DNA methylation, histone modifications), in diverse cellular contexts (e.g. cell types, tissues, conditions), provide a unique opportunity to investigate the intrinsic regulatory layer of the human genome. Here, we propose genomic regions, exhibiting distinctive chromatin characteristics, across different cell types contribute to cell-type-specific gene regulation. In this article, we developed a powerful method to identify CSREs within the human genome using dCMA. As a proof of principle, we used it to analyze public data consisting of nine chromatin marks in nine different cell types. Extensive analyses revealed that identified CSREs demonstrate distinct and cell-type specific roles. Their underlying modification patterns may be a key to understanding their potential role in cell-type-specific regulatory mechanisms. These results suggest comparative epigenomics is a promising strategy in deciphering aspects of the human genome.
Compared with related methods (14,15,17), our method specifically looks at differential modifications, which were not addressed by previous ones. These previous methods were designed for jointly analyzing chromatin maps of a single cell type or cell line. However, our method was designed for jointly analyzing chromatin maps of multiple cell types directly. Second, among previous methods, only Ernst et al. (19) have further applied their method onto chromatin maps of multiple cell types. However, the results of these two studies are different. The study of Ernst et al. identified 15 chromatin states, whereas ours discovered ∼4111 cell-type regulatory elements for each cell type on average. Third, the previous methods may potentially be applied for the task we are approaching here. However, it cannot be used directly; we have to compare the state of genomics elements of a cell type with those of others (see Supplementary Methods and Supplementary Figures S9–S12). This is not a trivial issue. How to determine the number of hidden states to control the model complexity for the HMM-based method is difficult too. Moreover, the data matrices corresponding to different cell types are simply concatenated, which may ignore the heterogeneity among cellular contexts. In contrast, our method is essentially non-parametric, requiring no model assumption, and hence the results are data-driven. Lastly, the application of our method on chromatin maps of multiple cell types does have novel biological findings, which have not been recovered by previous studies. For example, in the two case studies, the identified CSREs are corresponding to interesting biological phenomena. The HMM-based method by Xu et al. (46) was designed to find differential histone modification sites between ChIP libraries, and it was limited to do comparison between two cell types for a single mark. However, our method was designed for finding genomic elements that are specific to a single cell type by comparing its modification profiles of multiple marks to the same modification information of other multiple cell types. It is not limited by the numbers of cell types and epigenetic marks. We note that the study by Xu et al. cannot be directly extended to address our task.
Here, we found the chromatin modifications of different marks defining CSREs demonstrate interesting properties. For example, H3K27me3 is highly represented in H1 ES CSREs, but not in any other cell type examined. In contrast, the H3K27ac modification had strong signals in all cell types except for H1 ES. Taking into account that H3K27ac is an active regulatory mark and H3K27me3 is a repressive regulatory mark, these findings may indicate that most of the CSREs play active roles in gene regulation, except in H1 ES. This is consistent with H1 ES cellular characteristics, in which most genes carrying out specific biological processes or that are active in the early steps of embryogenesis are poised for activation (47). Moreover, H3K9ac, another chromatin mark associated with active regulatory regions (48), exhibits an intensity pattern resembling that of H3K27ac.
The accuracy of identifying CSREs relies on the number of known chromatin marks and the number of cell types used in the comparison study. We evaluated the importance of each chromatin mark in defining CSREs by removing them from the data sets and then testing how many CSREs were recovered (Supplementary Table S7). As expected, removal of the control sequencing experiment, WCE, almost has no effect on the results (average recovery = 98.8%), suggesting that our method did not rely on controls. Removal of CTCF marginally affected the results (average recovery = 97.6%), implying that CTCF binding sites are relative stable across all cell types, which was confirmed in a recent study by directly comparing the binding sites of CTCF in 11 different human cell lines (49). In contrast, removal of four marks related to enhancer regions, H3K4me1, H3K4me2, H3K27ac and H3K9ac, resulted in dramatic changes to the results (average recovery = 79.4, 85.6, 73.8, 86.1%, respectively). Thus, we speculate enhancer activity dynamics control cell-type-specific gene regulation.
In this study, we have preprocessed the data using a binarization way. We note that real-value and binarization ways both have their own advantages and disadvantages. The real-value way can keep more information, but it may not be robust to noise. It is difficult to model real-valued signals using common probability distributions such as Gaussian because there are many extremely high values in the signals, causing distorting effects. In a recently published nature article aiming at genome segmentation (16), raw real-valued signals are first transformed with the inverse hyperbolic sine function to reduce the distorting effects and then modeled by Gaussians. Moreover, as the authors pointed that the real-valued model (16) requires much more training time, the authors only trained the model in 1% of the genome. Although the binarization process may loss some information, it does have some advantages. First, the binarization procedure implicitly normalizes the ChIP libraries. As for the raw read counts, cross experiments normalization is still a challenging problem. Second, the binary representation is more robust, insensitive to outliers in the raw read counts. Third, the binarized representation of the data enables the use of a simple and computational tractable method to solve this problem, and the results are more interpretable. In this study, we used Poisson as null model to transform the raw real-valued signals into binary values. It has been argued that the Poisson distribution is not perfect to model sequencing reads distribution for its incapability in describing over-dispersion. As it is still debatable, we will investigate the effects of different null models. More specifically, we are trying locally null model fitting, considering that the chromosome structures are more stable within some small regions.
We expect the promising method presented here to become a useful tool in analyzing complex chromatin modification data across multiple cell types. Moreover, the method also has the potential to identity distinct, biologically relevant, functional DNA elements in the genome, as more genome-wide epigenetic data become available and more cell types are systematically profiled. Specifically, with the progresses of several large-scale epigenome efforts [e.g. ENCODE (50), modENCODE (51) and Epigenome Roadmap project (52)], our dCMA strategy can play a valuable role in deciphering the human epigenome and its implications in human disease. We should also note that the chromatin states of a single cell type are changing over the lifetime of a cell, which may lead to some variations in the current analyses. Further, as more time-series epigenetic data are available, our method can be extended to explore the regulatory elements that are specific to the development stages or conditions of a single cell type in the same way.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China, No. [11001256 and 11131009]; ‘Special Presidential Prize’ - Scientific Research Foundation of the CAS, the Special Foundation of President of AMSS at CAS for ‘Chen Jing-Run’ Future Star Program and the Foundation for Members of Youth Innovation Promotion Association, CAS. Funding for open access charge: National Natural Science Foundation of China, No. [11001256 and 11131009].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Prof. Zhiping Weng from University of Massachusetts Medical School for help discussions. They greatly appreciate the anonymous reviewers and editor for their very useful comments and suggestions.
REFERENCES
- 1.Maniatis T, Goodbourn S, Fischer JA. Regulation of inducible and tissue-specific gene expression. Science. 1987;236:1237–1245. doi: 10.1126/science.3296191. [DOI] [PubMed] [Google Scholar]
- 2.Dong X, Greven MC, Kundaje A, Djebali S, Brown JB, Cheng C, Gingeras TR, Gerstein M, Guigo R, Birney E, et al. Modeling gene expression using chromatin features in various cellular contexts. Genome Biol. 2012;13:R53. doi: 10.1186/gb-2012-13-9-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karlic R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc. Natl Acad. Sci. USA. 2010;107:2926–2931. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crick F. Central dogma of molecular biology. Nature. 1970;227:561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- 6.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Azuara V, Perry P, Sauer S, Spivakov M, Jorgensen HF, John RM, Gouti M, Casanova M, Warnes G, Merkenschlager M, et al. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006;8:532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 9.Cheng C, Yan KK, Yip KY, Rozowsky J, Alexander R, Shou C, Gerstein M. A statistical framework for modeling gene expression using chromatin features and application to modENCODE datasets. Genome Biol. 2011;12:R15. doi: 10.1186/gb-2011-12-2-r15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 11.Ye T, Krebs AR, Choukrallah MA, Keime C, Plewniak F, Davidson I, Tora L. seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 2011;39:e35. doi: 10.1093/nar/gkq1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hon G, Ren B, Wang W. ChromaSig: a probabilistic approach to finding common chromatin signatures in the human genome. PLoS Comput. Biol. 2008;4:e1000201. doi: 10.1371/journal.pcbi.1000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaschek R, Tanay A. Spatial clustering of multivariate genomic and epigenomic information. Res. Comput. Mol. Biol. 2009;5541:170–183. [Google Scholar]
- 15.Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods. 2012;9:473–476. doi: 10.1038/nmeth.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ucar D, Hu Q, Tan K. Combinatorial chromatin modification patterns in the human genome revealed by subspace clustering. Nucleic Acids Res. 2011;39:4063–4075. doi: 10.1093/nar/gkr016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teng L, Tan K. Finding combinatorial histone code by semi-supervised biclustering. BMC Genomics. 2012;13:301. doi: 10.1186/1471-2164-13-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rozowsky J, Euskirchen G, Auerbach RK, Zhang ZD, Gibson T, Bjornson R, Carriero N, Snyder M, Gerstein MB. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat. Biotechnol. 2009;27:66–75. doi: 10.1038/nbt.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 22.Klevecz RR, Murray DB. Genome wide oscillations in expression. Wavelet analysis of time series data from yeast expression arrays uncovers the dynamic architecture of phenotype. Mol. Biol. Rep. 2001;28:73–82. doi: 10.1023/a:1017909012215. [DOI] [PubMed] [Google Scholar]
- 23.Thurman RE, Day N, Noble WS, Stamatoyannopoulos JA. Identification of higher-order functional domains in the human ENCODE regions. Genome Res. 2007;17:917–927. doi: 10.1101/gr.6081407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Percival DB, Walden AT. Wavelet Methods for Time Series Analysis. Cambridge; New York: Cambridge University Press; 2000. [Google Scholar]
- 25.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Methodol. 1995;57:289–300. [Google Scholar]
- 26.Chang CW, Cheng WC, Chen CR, Shu WY, Tsai ML, Huang CL, Hsu IC. Identification of human housekeeping genes and tissue-selective genes by microarray meta-analysis. PLoS One. 2011;6:e22859. doi: 10.1371/journal.pone.0022859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ernst J, Bar-Joseph Z. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinformatics. 2006;7:191. doi: 10.1186/1471-2105-7-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 32.Rowley JD. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 33.Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–3356. [PubMed] [Google Scholar]
- 34.Wong N, Lai P, Pang E, Leung TW, Lau JW, Johnson PJ. A comprehensive karyotypic study on human hepatocellular carcinoma by spectral karyotyping. Hepatology. 2000;32:1060–1068. doi: 10.1053/jhep.2000.19349. [DOI] [PubMed] [Google Scholar]
- 35.Juran BD, Hirschfield GM, Invernizzi P, Atkinson EJ, Li Y, Xie G, Kosoy R, Ransom M, Sun Y, Bianchi I, et al. Immunochip analyses identify a novel risk locus for primary biliary cirrhosis at 13q14, multiple independent associations at four established risk loci and epistasis between 1p31 and 7q32 risk variants. Hum. Mol. Genet. 2012;21:5209–5221. doi: 10.1093/hmg/dds359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koffler D, Agnello V, Thoburn R, Kunkel HG. Systemic lupus erythematosus: prototype of immune complex nephritis in man. J. Exp. Med. 1971;134:169–179. [PMC free article] [PubMed] [Google Scholar]
- 37.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 38.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He HH, Meyer CA, Chen MW, Jordan VC, Brown M, Liu XS. Differential DNase I hypersensitivity reveals factor-dependent chromatin dynamics. Genome Res. 2012;22:1015–1025. doi: 10.1101/gr.133280.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorkin DU, Lee D, Reed X, Fletez-Brant C, Bessling SL, Loftus SK, Beer MA, Pavan WJ, McCallion AS. Integration of ChIP-seq and machine learning reveals enhancers and a predictive regulatory sequence vocabulary in melanocytes. Genome Res. 2012;22:2290–2301. doi: 10.1101/gr.139360.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 42.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer. 2005;5:172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 43.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 45.Janowski BA. The hypocholesterolemic agent LY295427 up-regulates INSIG-1, identifying the INSIG-1 protein as a mediator of cholesterol homeostasis through SREBP. Proc. Natl Acad. Sci. USA. 2002;99:12675–12680. doi: 10.1073/pnas.202471599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu H, Wei CL, Lin F, Sung WK. An HMM approach to genome-wide identification of differential histone modification sites from ChIP-seq data. Bioinformatics. 2008;24:2344–2349. doi: 10.1093/bioinformatics/btn402. [DOI] [PubMed] [Google Scholar]
- 47.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, 3rd, Gingeras TR, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 49.Lee BK, Bhinge AA, Battenhouse A, McDaniell RM, Liu Z, Song L, Ni Y, Birney E, Lieb JD, Furey TS, et al. Cell-type specific and combinatorial usage of diverse transcription factors revealed by genome-wide binding studies in multiple human cells. Genome Res. 2012;22:9–24. doi: 10.1101/gr.127597.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Celniker SE, Dillon LA, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, et al. Unlocking the secrets of the genome. Nature. 2009;459:927–930. doi: 10.1038/459927a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.