


Abstract
The effect of a cationic-neutral diblock polypeptide on the conformation of single DNA molecules confined in rectangular nanochannels is investigated with fluorescence microscopy. An enhanced stretch along the channel is observed with increased binding of the cationic block of the polypeptide to DNA. A maximum stretch of 85% of the contour length can be achieved inside a channel with a cross-sectional diameter of 200 nm and at a 2-fold excess of polypeptide with respect to DNA charge. With site-specific fluorescence labelling, it is demonstrated that this maximum stretch is sufficient to map large-scale genomic organization. Monte Carlo computer simulation shows that the amplification of the stretch inside the nanochannels is owing to an increase in bending rigidity and thickness of bottlebrush-coated DNA. The persistence lengths and widths deduced from the nanochannel data agree with what has been estimated from the analysis of atomic force microscopy images of dried complexes on silica.
INTRODUCTION
Advances in nanofabrication have made it possible to fabricate quasi one-dimensional devices with cross-sectional diameters on the order of tens to hundreds of nanometers. These nanochannels serve as a platform for studying, among others, single DNA molecules (1–8). Furthermore, confinement in a nanospace result in significant modification of certain biophysical phenomena, such as the knotting probability of circular DNA and the effect of macromolecular crowding on the conformation and folding of DNA (9–12). Nanochannels are also important for single-molecule technologies for mapping of large-scale genomic organization, including restriction enzyme cutting, nick labelling and denaturation mapping (13–18). Advantages of the nanochannel platform are that the DNA molecules are in an equilibrium conformation, high throughput can be achieved by using parallel arrays of channels and integration with lab-on-chip devices. Future devices might involve nonequilibrium translocation of DNA molecules through a pore or by a sensor, a process that can be facilitated by a nanochannel platform (19). The molecules of interest are usually visualized with fluorescence microscopy.
The resolution of the genome mapping technologies relies on the ability to stretch DNA to a length close to its contour length. Stretching of single DNA molecules to >60% of the contour length can be achieved by confinement in channels with cross-sectional diameters below 100 nm (3,14,15). Alternatively, DNA molecules can be stretched by working at low ionic strength of <1 mM (4–6). At such low ionic strength, the persistence length of the duplex exceeds 100 nm, and is significantly larger than the physiological value of around 50 nm. Following the latter strategy of amplifying the stretch by an increase in bending rigidity, we here propose to apply a polymer coating to the double-stranded DNA molecule. The advantage of this approach is that wider channels can be used to achieve a similar stretch. In particular, the less stringent channel diameter allows the use of nanofluidic devices produced by soft lithography and made of elastomer such as polydimethylsiloxane (PDMS). Elastomer chips have some advantages, including the possibility of obtaining multiple replicas by casting on a master stamp (3). Furthermore, a wider nanofluidic channel system allows in situ changes in environmental solution conditions, which is required for DNA binding assays. Using a novel device based on a cross-channel configuration, recently we have shown that DNA molecules trapped inside wider channels (250 nm) remain accessible for binding proteins and/or ligands (20). The wider channels also allow loading of DNA molecules by electrophoresis.
Recently, a cationic-neutral diblock polypeptide has been developed that qualifies as a suitable candidate for coating of DNA (21). The cationic DNA binding block is relatively short and consist of 12 lysine residues (K12). The K12 block is coupled to four repeats of a 100-amino acid long polypeptide (C4) (22). The C4 block has an amino acid composition similar to that of collagen. Furthermore, it is hydrophilic, net electroneutral and behaves as a flexible polymer in aqueous solution. The entire C4K12 diblock polypeptide is produced using recombinant DNA technology by large-scale expression with high yields in yeast. The diblock copolymers are monodisperse with a total molecular weight of 38.4 kDa. The polypeptide concentration is conveniently expressed as the N/P ratio, that is, the number of positively charged amino groups on the lysine K12 binding block divided by the number of phosphate groups on the DNA. As shown in previous work, complexation of DNA with C4K12 does not result in aggregates consisting of multiple DNA molecules (21). Instead, single DNA molecules are uniformly coated by a dense bottlebrush as illustrated in Figure 1. We have verified that DNA metabolism is not inhibited by coating with the polypeptide (see ‘Materials and Methods’ section). Two effects are expected to enhance stretching of the DNA–C4K12 complex in nanochannels: a larger cross-sectional diameter and a larger bending rigidity or persistence length. These effects will be gauged from nanofluidics experiments, atomic force microscopy and Monte Carlo simulation.
Figure 1.
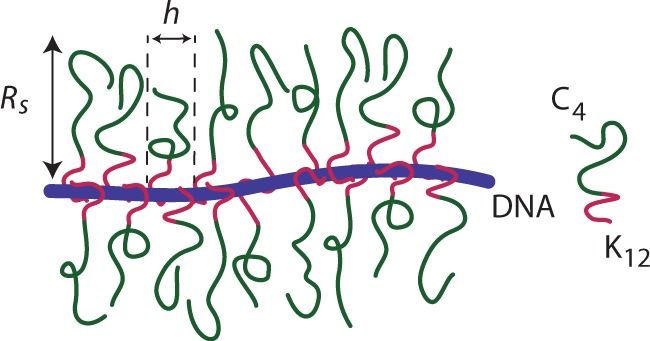
Illustration of a bottlebrush formed by binding of a diblock polypeptide copolymer to DNA. The binding block of the copolymer contains 12 cationic lysine residues.
MATERIALS AND METHODS
Sample preparation
C4K12 polypeptide (Mw = 38.4 kDa) was expressed in the yeast Pichia Pastoris as an exocellular protein and purified by NH4SO4 precipitation of the supernatant (21,22). The purity and absence of degradation of the polypeptides have been verified with sodium dodecyl sulphate-polyacrylamide gel electrophoresis and matrix-assisted laser desorption ionization time-of-flight mass spectrometer (MALDI-TOF). To verify the effect of polypeptide coating on DNA metabolism, we have done a digestion assay (21). As shown by the image of the gel in Figure 2, bare DNA is degraded by DNAaseI in <1 min. In the same condition, polypeptide-coated DNA with N/P = 7.5 is degraded in about an hour. DNA metabolism is hence slowed down, but not inhibited by the polypeptide coating. T4-DNA was purchased from Nippon Gene, Tokyo, and used without further purification. λ-DNA, Nb.BbvCI nicking endonuclease and VentR (exo-) DNA polymerase were purchased from New England Biolabs, Ipswich, MA, USA. YOYO-1 and ChromaTide Alexa Fluor 546-14-dUTP were purchased from Invitrogen, Carlsbad, CA, USA. λ-DNA was sequence-specifically stained with Alexa Fluor 546 (15). T4-DNA was stained with YOYO-1 with an intercalation ratio of 100 bp per dye molecule. For nanofluidics, samples were prepared by dialysing solutions of T4-DNA against 10 mM Tris/HCl and 1 mM EDTA in micro-dialysers. The Tris/HCl concentration is 10 mM Tris adjusted with HCl to pH 8.5, i.e. 2.9 mM Tris/Cl and 7.1 mM Tris. The ionic strength of the buffer was calculated with the Davies equation for estimating the activity coefficients of the ions and a dissociation constant pK = 8.08 for Tris. The polypeptide was dissolved in the same buffer. Solutions of polypeptide and DNA were subsequently mixed in equal volumes and incubated for 24 h at 277 K. The final DNA concentration is 3 mg/l. No anti-photo bleaching agent was used. Atomic force microscopy was done with pUC18 plasmid (2686 bp). pUC18 was purchased and linearized with HindIII restriction endonuclease (New England Biolabs, Ipswich, MA, USA). Solutions of plasmid and polypeptide were mixed in equal volumes to a final concentration of 0.2 mg of DNA/l and incubated for 24 h at 277 K.
Figure 2.
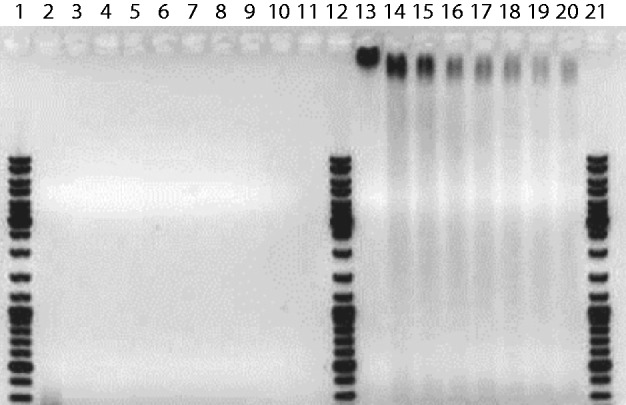
Degradation of pMTL23 (3.7 kb) by DNAaseI. The plasmid and DNAseI concentrations are 7.2 nM and 8.8 U/ml, respectively. Lanes 1, 12 and 21: DNA molar weight marker. Lanes 2–11: reaction product of bare DNA after 1, 5, 8, 15, 25, 35, 45, 60 min of incubation, respectively. Lanes 13–20: as lanes 2–11, but for C4K12-coated DNA with N/P = 7.5.
Chip fabrication
The nanofluidic devices were fabricated by replication in PDMS of patterned master stamps (5,23). The stamps were made in hydrogen silsesquioxane (HSQ) resist (Dow Corning, Midland, MI, USA) using a lithography process with proton beam writing (24,25). The  and
and  nm heights of the positive channel structures on the stamps were measured with atomic force microscopy (Dimension 3000, Veeco, Woodbury, NY, USA). The stamps were replicated in PDMS followed by curing with a curing agent (Sylgard, Dow Corning) at 338 K for 24 h (26). After both substrates were plasma oxidized (Harrick, Ossining, NY, USA), the PDMS replicas were sealed with glass slides. The widths of the channels in the replicas were measured with atomic force microscopy and the values agreed with those obtained from the scanning electron microscopy images of the master stamps. The nanochannels have a length of 60 μm and rectangular cross-sections of
nm heights of the positive channel structures on the stamps were measured with atomic force microscopy (Dimension 3000, Veeco, Woodbury, NY, USA). The stamps were replicated in PDMS followed by curing with a curing agent (Sylgard, Dow Corning) at 338 K for 24 h (26). After both substrates were plasma oxidized (Harrick, Ossining, NY, USA), the PDMS replicas were sealed with glass slides. The widths of the channels in the replicas were measured with atomic force microscopy and the values agreed with those obtained from the scanning electron microscopy images of the master stamps. The nanochannels have a length of 60 μm and rectangular cross-sections of 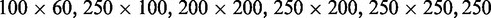
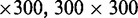 and
and 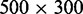 nm2. A fresh chip was made for every experiment.
nm2. A fresh chip was made for every experiment.
To check for possible adhesion of polypeptide to PDMS, we have prepared a flat PDMS chip. The chip was plasma oxidized and covered with glass slides. A square 2 × 2 mm2 window of the PDMS surface was left uncovered. The chip was exposed to a solution of polypeptide (1 g/l) for the typical duration of a nanofluidic experiment (30 min). The glass slides were removed, the chip was N2-dried and the surface was imaged with atomic force microscopy. We did not observe a difference in surface roughness or a transition in height level from the previously covered to the exposed PDMS surface area of the chip. Accordingly, there is no appreciable adhesion of polypeptide to PDMS.
Fluorescence imaging
The stained DNA molecules dispersed in the relevant solution were loaded into one of the two reservoirs connected by the nanochannels. The DNA molecules were subsequently driven into the channels by electrophoresis. For this purpose, two platinum electrodes were immersed in the reservoirs and connected to an electrophoresis power supply with a relatively low voltage in the range 0.1–10 V (Keithley, Cleveland, OH, USA). Once the DNA molecules were localized inside the nanochannels, the electric field was switched off and the molecules were allowed to relax to their equilibrium state for at least 60 s. The stained DNA molecules were visualized with a Nikon Eclipse Ti inverted fluorescence microscope equipped with a 200 W metal halide lamp, a filter set and a 100× oil immersion objective. A ultraviolet light illumination shutter controlled the exposure time. Images were collected with an electron multiplying charge coupled device camera (Andor iXon X3), and the extension of the DNA molecules inside the channels was measured with imageJ software (http://rsb.info.nih.gov/ij/).
DNA combing
Polystyrene (Mw = 280 kDa) is dissolved in toluene (Fisher Scientific, Pittsburgh, PA, USA) at a concentration of 100 g/l. Glass cover slips were cleaned by sonication in 70% ethanol for 30 min. The cover slips were spin-coated with the solution of polystyrene for 30 s at 2000 rpm. A 5-μl droplet was spotted onto the cover slip and sheared with the help of a pipette tip along the surface. The DNA molecules were visualized with fluorescence microscopy (27).
Atomic force microscopy
All imaging experiments were carried out at room temperature in air with a Dimension 3000 atomic force microscope, Veeco, Woodbury, NY, USA. Images were acquired in the tapping mode with silicon (Si) cantilevers (spring constant of 20–100 N/m) and operated below their resonance frequency (typically 230–410 kHz). The images were flattened, and the contrast and brightness were adjusted for optimum viewing conditions. A 20-μl droplet was spotted onto a fleshly cleaved mica or silica surface. Mica was used for bare DNA, whereas polypeptide-coated DNA was also adsorbed on silica. After 10 min to allow for DNA adsorption onto the surface, the specimens were developed by immersing them in ultrapure water for 30 min followed by drying in a stream of N2 gas.
Monte Carlo simulation
In the Monte Carlo protocol, the DNA chain is modelled as a string of 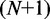 beads, which are connected by N inextensible bonds of length lb (28,29). The simulation model consists of bending energy between adjacent bonds and two interaction terms. The bending rigidity is set to reproduce persistence length of 50 nm. The interactions are hard-sphere repulsion between DNA beads and hard-wall repulsion between the beads and the wall. If the centre of a bead is beyond the channel wall, the potential becomes infinitely large and the configuration is rejected. The effective channel diameter is hence the real diameter minus the diameter of the bead. The contour length of the DNA chain was fixed at 8 μm. The diameter of the bead was set equal to the bond length lb, which is equivalent to the chain width w. Accordingly, for chains with a fixed contour length of 8 μm, the number of beads is 1601, 1068 or 801 for
beads, which are connected by N inextensible bonds of length lb (28,29). The simulation model consists of bending energy between adjacent bonds and two interaction terms. The bending rigidity is set to reproduce persistence length of 50 nm. The interactions are hard-sphere repulsion between DNA beads and hard-wall repulsion between the beads and the wall. If the centre of a bead is beyond the channel wall, the potential becomes infinitely large and the configuration is rejected. The effective channel diameter is hence the real diameter minus the diameter of the bead. The contour length of the DNA chain was fixed at 8 μm. The diameter of the bead was set equal to the bond length lb, which is equivalent to the chain width w. Accordingly, for chains with a fixed contour length of 8 μm, the number of beads is 1601, 1068 or 801 for 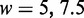 , or 10 nm, respectively. In each Monte Carlo cycle, we carried out one crankshaft and one reptation move. The simulation started from a random conformation and usually reached equilibrium after 107 cycles. In the production run, we generated
, or 10 nm, respectively. In each Monte Carlo cycle, we carried out one crankshaft and one reptation move. The simulation started from a random conformation and usually reached equilibrium after 107 cycles. In the production run, we generated 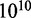 cycles and recorded the conformation every other 105 cycles. For each conformation, we calculated the extension as the maximum span of the DNA molecule along the channel axis. Finally, the extension was averaged over the ensemble of 105 conformations. Results for different persistence lengths and widths were obtained from the scale invariance of the Monte Carlo results for the relative extension. This implies that the maximum span divided by the contour length of a chain with persistence length P, width w, contour length L and confined inside a channel of diameter D is the same as the one of a chain with scaled parameters
cycles and recorded the conformation every other 105 cycles. For each conformation, we calculated the extension as the maximum span of the DNA molecule along the channel axis. Finally, the extension was averaged over the ensemble of 105 conformations. Results for different persistence lengths and widths were obtained from the scale invariance of the Monte Carlo results for the relative extension. This implies that the maximum span divided by the contour length of a chain with persistence length P, width w, contour length L and confined inside a channel of diameter D is the same as the one of a chain with scaled parameters 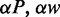 and
and 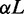 inside a channel of diameter
inside a channel of diameter  . We have verified that effects of finite contour length on the relative stretch are unimportant for the relevant range of channel diameters.
. We have verified that effects of finite contour length on the relative stretch are unimportant for the relevant range of channel diameters.
RESULTS AND DISCUSSION
Amplification of stretch
Our nanofluidic devices are made of PDMS casted on a high-quality master stamp, which was obtained by proton beam writing and ultraviolet optical lithography (5,23,24). T4-DNA (166 kb) was stained with a ratio of one YOYO-1 molecule per 100 bp. With this intercalation ratio, the YOYO-1 corrected contour length of T4-DNA amounts 57 μm. Furthermore, the concomitant minimal reduction in DNA charge has no effect on the electrostatic binding of the cationic block of the polypeptide on DNA. The stained molecules were subsequently incubated in solutions of various concentrations of polypeptide for at least 24 h. The molecules are accordingly coated with various amounts of C4K12, as indicated by the lysine to DNA N/P ratio (the DNA concentration is 3 mg/l).
In the nanofluidic experiment, the coated and stained DNA molecules were electrophoresed into an array of 60 μm long and rectangular nanochannels and imaged with fluorescence microscopy. A montage of images of single T4-DNA molecules in 10 mM Tris/HCl (2.9 mM Tris/Cl, 7.1 mM Tris, pH 8.5) and confined in channels with a cross-section of 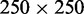 nm2 is shown in Figure 3A. The images refer to well-equilibrated conformations. After the electric field has been switched off, the molecules relax to their equilibrium state within 60 s. Video imaging was started 5–10 min after the molecules were brought into the channels and lasted for another 10 min. With increasing C4K12 to DNA ratio, the equilibrated molecules stretch in the longitudinal direction of the channel. Notice that the stretch of the bottlebrush with a 2-fold excess of polypeptide to DNA charge (N/P = 2.0) reaches ∼85% of the contour length. An excess of polypeptide is needed to achieve maximum stretch owing to the dynamic equilibrium of the binding process (law of mass action). A relative stretch of 85% has also been reported for DNA in narrow, 45-nm channels as well as for DNA at low ionic strength of 0.12 mM in 250-nm channels (6,17). An advantage of the present technology is the less stringent conditions in terms of channel diameter and ionic strength of the supporting medium.
nm2 is shown in Figure 3A. The images refer to well-equilibrated conformations. After the electric field has been switched off, the molecules relax to their equilibrium state within 60 s. Video imaging was started 5–10 min after the molecules were brought into the channels and lasted for another 10 min. With increasing C4K12 to DNA ratio, the equilibrated molecules stretch in the longitudinal direction of the channel. Notice that the stretch of the bottlebrush with a 2-fold excess of polypeptide to DNA charge (N/P = 2.0) reaches ∼85% of the contour length. An excess of polypeptide is needed to achieve maximum stretch owing to the dynamic equilibrium of the binding process (law of mass action). A relative stretch of 85% has also been reported for DNA in narrow, 45-nm channels as well as for DNA at low ionic strength of 0.12 mM in 250-nm channels (6,17). An advantage of the present technology is the less stringent conditions in terms of channel diameter and ionic strength of the supporting medium.
Figure 3.
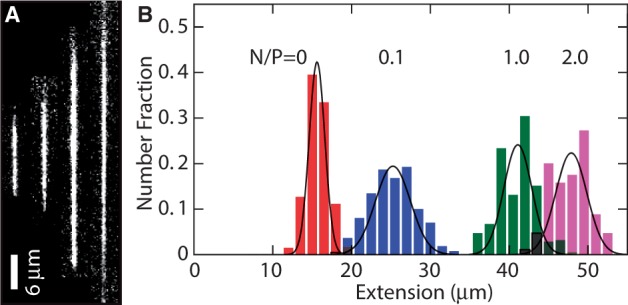
(A) Montage of fluorescence images of T4-DNA in 250 × 250 nm2 channels and in 10 mM Tris/HCl (pH 8.5). From left to right, N/P = 0 (polypeptide-free), 0.1, 1.0 and 2.0. The scale bar denotes 6 μm and the YOYO-1 intercalation ratio is 100 bp per dye molecule. (B) Distribution in extension of a population of ∼50 T4-DNA molecules with indicated N/P ratio. Gaussian fits give mean extensions of 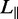 = 16 ± 2, 25 ± 3, 41 ± 3 and 48 ± 3 μm for N/P = 0, 0.1, 1.0 and 2.0, respectively.
= 16 ± 2, 25 ± 3, 41 ± 3 and 48 ± 3 μm for N/P = 0, 0.1, 1.0 and 2.0, respectively.
We have measured the extension of the T4-DNA molecules confined in the nanochannels of various cross-sectional diameters. With increasing N/P ratio, it becomes more difficult to electrophorese DNA molecules into channels of smaller cross-sectional diameter. For the highest used N/P ratio (2.0), we could not insert a large enough number of molecules into channels with a diameter <200 nm. For each experimental condition, we have used a fresh PDMS replica and measured ∼50 molecules. The distributions in extension are close to Gaussian. Examples pertaining to different N/P ratios are shown in Figure 3B. DNA fragments can easily be discerned because their extensions fall below the values pertaining to the intact molecules. For the cut-off, we have used the mean value minus two times the standard deviation. Resolution broadening can be neglected because the optical resolution of 200 nm is one order of magnitude smaller than the variance. The relative extensions 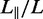 , that is, the mean stretch divided by the contour length of the DNA molecule, are set out in Figure 4. With decreasing channel diameter and/or increasing C4K12 to DNA ratio, the relative extension increases. For N/P = 0 (polypeptide-free) and 0.1, the extensions are in the range 0.2–0.5 times the contour length. In the case of N/P ratios of 1.0 and 2.0, the relative extensions are in the range 0.5–0.85. Furthermore, the extensions level off at a plateau value for smaller channel diameters and higher N/P ratios. The increase in relative extension is owing to an increase in bending rigidity and cross-sectional diameter of the bottlebrush-coated DNA. These effects will be gauged below with atomic force microscopy. We will, however, first demonstrate that large-scale genomic organization can be mapped with our channel platform in tandem with polypeptide coating.
, that is, the mean stretch divided by the contour length of the DNA molecule, are set out in Figure 4. With decreasing channel diameter and/or increasing C4K12 to DNA ratio, the relative extension increases. For N/P = 0 (polypeptide-free) and 0.1, the extensions are in the range 0.2–0.5 times the contour length. In the case of N/P ratios of 1.0 and 2.0, the relative extensions are in the range 0.5–0.85. Furthermore, the extensions level off at a plateau value for smaller channel diameters and higher N/P ratios. The increase in relative extension is owing to an increase in bending rigidity and cross-sectional diameter of the bottlebrush-coated DNA. These effects will be gauged below with atomic force microscopy. We will, however, first demonstrate that large-scale genomic organization can be mapped with our channel platform in tandem with polypeptide coating.
Figure 4.
Relative extension 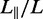 of T4-DNA (open symbols) and λ-DNA (closed symbols) in 10 mM Tris/HCl versus channel diameter D. The N/P ratios are 0 (▵), 0.1 (▿), 1.0 (□) and 2.0 (○). The solid curves represent Monte Carlo results with noted values of persistence length P and width w. The dashed curve represents deflection theory for narrow channels.
of T4-DNA (open symbols) and λ-DNA (closed symbols) in 10 mM Tris/HCl versus channel diameter D. The N/P ratios are 0 (▵), 0.1 (▿), 1.0 (□) and 2.0 (○). The solid curves represent Monte Carlo results with noted values of persistence length P and width w. The dashed curve represents deflection theory for narrow channels.
Large-scale genome mapping
The aim of large-scale genome mapping is not to determine the genetic code at the single base level, but rather to provide a map for assembling the genome at the larger scale and to sort out large-scale variations such as insertions, inversions and translocations. For the demonstration of genome mapping, we have site-specifically labelled nicks of λ-DNA (48.5 kb) with Alexa Fluor 546 (15,17). The YOYO-1 and Alexa labelled DNA was coated with C4K12 and electrophoresed into an array of nanochannels with cross-sectional diameters of 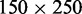 nm2. From an analysis of the YOYO-1 fluorescence, we have verified that the relative extensions of λ-DNA are the same as for T4-DNA (see Figure 4). There are four resolvable Alexa labelling sites at 8, 18.3 (average of two sites at 18.1 and 18.5 kb), 31.3 (average of 30.9, 31.2 and 31.8 kb) and 35.8 kb. Furthermore, the sticky ends of the λ-DNA molecule can also be labelled. As can be seen in panel A of Figure 5, individual Alexa labels are discernible with an N/P ratio of 2.0. Individual labels can also be discerned for a stoichiometric ratio of N/P = 1.0, albeit with decreased separation. In the case of polypeptide-free DNA, individual Alexa labelled sites cannot be discerned. This confirms that an excess of polypeptide to DNA charge is needed to achieve the highest possible stretch. For comparison, in panel B of Figure 5 we have displayed fluorescence images of combed, bare DNA molecules. In the combing experiment, the molecules are stretched to almost their full contour length. In both the nanofluidic and combing experiments, occasionally some Alexa labels are missing (in particular the ones at the end). Out of a pool of 100 molecules, 2, 10 and 40% of the molecules show 6, 5 and 4 discernible labels, respectively. Because we did not observe a difference in labelling efficiency for combed (bare) and channel-confined (coated) molecules, missing labels are due to incomplete labelling rather than caused by the coating procedure.
nm2. From an analysis of the YOYO-1 fluorescence, we have verified that the relative extensions of λ-DNA are the same as for T4-DNA (see Figure 4). There are four resolvable Alexa labelling sites at 8, 18.3 (average of two sites at 18.1 and 18.5 kb), 31.3 (average of 30.9, 31.2 and 31.8 kb) and 35.8 kb. Furthermore, the sticky ends of the λ-DNA molecule can also be labelled. As can be seen in panel A of Figure 5, individual Alexa labels are discernible with an N/P ratio of 2.0. Individual labels can also be discerned for a stoichiometric ratio of N/P = 1.0, albeit with decreased separation. In the case of polypeptide-free DNA, individual Alexa labelled sites cannot be discerned. This confirms that an excess of polypeptide to DNA charge is needed to achieve the highest possible stretch. For comparison, in panel B of Figure 5 we have displayed fluorescence images of combed, bare DNA molecules. In the combing experiment, the molecules are stretched to almost their full contour length. In both the nanofluidic and combing experiments, occasionally some Alexa labels are missing (in particular the ones at the end). Out of a pool of 100 molecules, 2, 10 and 40% of the molecules show 6, 5 and 4 discernible labels, respectively. Because we did not observe a difference in labelling efficiency for combed (bare) and channel-confined (coated) molecules, missing labels are due to incomplete labelling rather than caused by the coating procedure.
Figure 5.
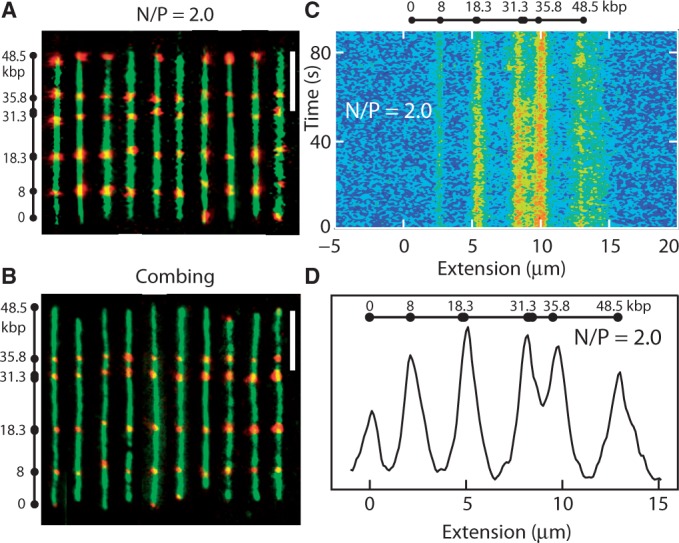
(A) Montage of fluorescence images of λ-DNA. The images obtained with YOYO-1 (green) and Alexa Fluor 541 (red) staining are superposed. The molecules are confined in 150 × 250 nm2 channels with N/P = 2.0. Labelling sites are noted. The scale bar denotes 5 μm. (B) As in panel (A), but for combed λ-DNA. (C) Time dependence of the Alexa Fluor intensity profile pertaining to N/P = 2.0 and in a 150 × 250 nm2 channel. The exposure time is 500 ms. (D) Average Alexa profile over a pool of 50 molecules with N/P = 2.0 and in 150 × 250 nm2 channels.
In the case of maximum stretch for N/P = 2.0 in  nm2 channels, we have analysed the Alexa fluorescence profiles of a pool of 50 DNA molecules. An example of the time-dependence of the Alexa fluorescence is shown in panel C of Figure 5. On the time scale of the experiment (90 s), the molecules are trapped inside the channel and there is no net displacement. Single molecule Alexa profiles were aligned and corrected for a minor variation in overall stretch of <5%. The locations of corresponding peaks for different molecules are in register. As shown by the fluorescence images and the average profile in panels A and D of Figure 5, respectively, the locations of the labels along the molecule are in good agreement with the locations of the nicking sites. Accordingly, the molecules are uniformly stretched along the direction of the channel without ever folding back. The averaged Alexa fluorescence intensity profile confirms a relative stretch of ∼85% for N/P = 2.0, as determined by the YOYO-1 fluorescence. The resolution in peak position is ∼2 kb, which is similar to the one reported for bare DNA in 45-nm channels (17).
nm2 channels, we have analysed the Alexa fluorescence profiles of a pool of 50 DNA molecules. An example of the time-dependence of the Alexa fluorescence is shown in panel C of Figure 5. On the time scale of the experiment (90 s), the molecules are trapped inside the channel and there is no net displacement. Single molecule Alexa profiles were aligned and corrected for a minor variation in overall stretch of <5%. The locations of corresponding peaks for different molecules are in register. As shown by the fluorescence images and the average profile in panels A and D of Figure 5, respectively, the locations of the labels along the molecule are in good agreement with the locations of the nicking sites. Accordingly, the molecules are uniformly stretched along the direction of the channel without ever folding back. The averaged Alexa fluorescence intensity profile confirms a relative stretch of ∼85% for N/P = 2.0, as determined by the YOYO-1 fluorescence. The resolution in peak position is ∼2 kb, which is similar to the one reported for bare DNA in 45-nm channels (17).
Persistence length and width
The persistence lengths were obtained from analysis of atomic force microscopy images of bottlebrush-coated DNA on silica. For this purpose, linearized pUC18 plasmid (2686 bp) was used at a concentration of 0.2 mg/l. The C4 blocks of the diblock polypeptide weakly adsorb to silica such that no additions to the buffer are necessary to promote adhesion. The C4-block mediated adsorption of bottlebrush-coated DNA is relatively weak because it is found that the molecules are more easily removed by flushing with water as compared with DNA molecules that have been adsorbed in the presence of either MgCl2 or high molecular weight polylysine. Accordingly, the bottlebrush is not kinetically trapped in a 3D conformation (30). For the reference case of bare DNA, molecules were bound to a mica surface by the use of a buffer comprising 10 mM MgCl2. Excess polypeptide and MgCl2 were removed from the interface by immersion of the specimens in ultrapure water for 30 min. During this development time, the molecules equilibrate on the surface in a 2D conformation. Subsequently, all specimens were N2-dried. A series of typical images for increasing C4K12 to DNA ratios are shown in Figure 6A–D. The relatively small plasmids are imaged in their entirety with a field of view of 3 × 3 μm2. We have also imaged polypeptide-coated 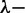 DNA with N/P = 2.0. However, as shown in panels E and F of Figure 6, only segments of these relatively long molecules can be visualized. Nevertheless, their appearance in terms of thickness and rigidity is similar to the one of the shorter plasmid molecules.
DNA with N/P = 2.0. However, as shown in panels E and F of Figure 6, only segments of these relatively long molecules can be visualized. Nevertheless, their appearance in terms of thickness and rigidity is similar to the one of the shorter plasmid molecules.
Figure 6.
(A–D) Tapping mode atomic force microscopy images of linearized pUC18 DNA complexed with increasing amounts of polypeptide. The N/P ratios are 0, 0.1, 1.0 and 2.0, in panels A, B, C and D, respectively. (E, F) As in panels (A–D), but for 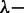 DNA with N/P = 2.0. The scale bar denotes 1 μm. Panels B–D are a montage.
DNA with N/P = 2.0. The scale bar denotes 1 μm. Panels B–D are a montage.
To obtain the persistence length from the atomic force microscopy images, we traced the centreline of a population of 10–30 individual pUC18 molecules, with the smaller number pertaining to the highest polypeptide concentrations. These were used to obtain the tangent vector correlation function 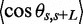 , where θ is the angle between tangent vectors at points s and s + L, by averaging s along the contour. For weakly adsorbed DNA molecules equilibrated in 2D conformation, this correlation follows:
, where θ is the angle between tangent vectors at points s and s + L, by averaging s along the contour. For weakly adsorbed DNA molecules equilibrated in 2D conformation, this correlation follows:
| (1) |
Experimental and fitted tangent correlation functions are shown in Figure 7. With increasing C4K12 to DNA ratio, fitted values of the persistence length P increase from 60 (bare DNA) to 240 nm at the highest polypeptide concentration. The value obtained for bare DNA agrees with the commonly accepted value of ∼50 nm (31). We have measured the length of the molecules by tracing the contour. No change in contour length induced by the binding of the polypeptide on DNA was observed.
Figure 7.
Orientation correlation of the tangent vectors at a pair of points separated by distance L along the contour. The lines represent exponential fits, and the polypeptide to DNA N/P ratios are noted. The fitted values of the persistence lengths P = 60 ± 5, 95 ± 5, 190 ± 10 and 240 ± 10 nm for N/P = 0, 0.1, 1.0 and 2.0, respectively.
The atomic force microscopy images show an increase in thickness of the bottlebrushes with increased polypeptide concentration. We analysed the cross-sectional profiles taken at 10 different and randomly chosen positions of the molecules. Gaussian fits to these profiles gives variances of 10, 15, 20 and 30 nm for N/P = 0, 0.1, 1.0 and 2.0, respectively. These values should, however, be taken as indicative of the thickness of the brushes because the profiles are broadened by the width of the tip and the brushes are dried and spread out on the silica surface.
The experimental values of the stretch can be compared with Monte Carlo results that we have obtained for the stretching of nanoconfined semiflexible polymers varying in persistence length P and effective thickness w. The computational results are also displayed in Figure 4. In comparing the experimental values to the Monte Carlo simulations, we have used the values of the persistence length P as obtained from atomic force microscopy, without any adjustment. Only for bare DNA, a slightly better fit was obtained with the nominal persistence length of 50 nm. For each N/P, the width w used in the Monte Carlo simulations was adjusted to obtain the best agreement with the experimental stretching data. The values of w obtained in this way show an increase with increased polypeptide coating and are in reasonable agreement with the variances of the cross-sectional profiles obtained from atomic force microscopy.
Theoretical considerations
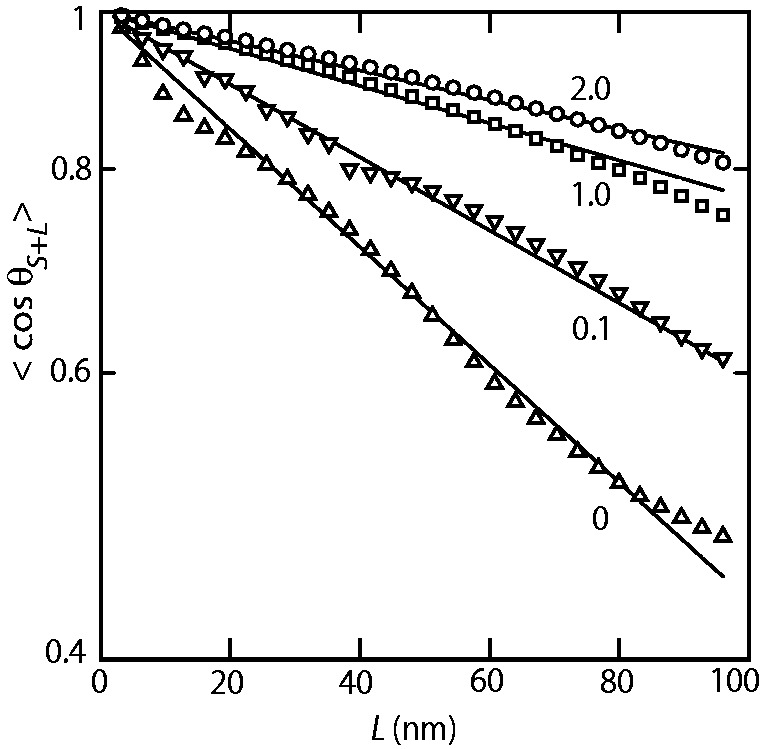
For  nm and N/P = 2.0, the data are close to the prediction of Odijk’s deflection model. Here, the bottlebrush is undulating along the channel by deflections from the wall (32). For larger values of D, the data fall below the mark set by deflection theory owing to back-folding and looping of the undulating wormlike bottlebrush conformation (28).
nm and N/P = 2.0, the data are close to the prediction of Odijk’s deflection model. Here, the bottlebrush is undulating along the channel by deflections from the wall (32). For larger values of D, the data fall below the mark set by deflection theory owing to back-folding and looping of the undulating wormlike bottlebrush conformation (28).
Existing theories support our findings of increasing effective thickness and persistence lengths for bottlebrush-coated DNA (21). Assuming that saturated binding corresponds to full charge neutralization of the DNA phosphates by the lysines on the K12 binding block, a single C4K12 molecule has a binding site size of 6 bp, and saturated binding corresponds to a distance h between bound polypeptides along the DNA contour of 2 nm. Recall that this requires an excess of polypeptide to DNA charge (N/P > 1), owing to the dynamic equilibrium of the binding process. Side chain stretching in bottlebrushes is controlled by the dimensionless parameter
| (2) |
where N is the number of segments in a side chain, l is the length of a side chain segment and h is the distance between grafted side chains along the main chain. If Rs is the mean-square end-to-end distance of the stretched side chain (in the bottlebrush) and R is the mean-square end-to-end distance of an unstretched side chain (in free solution), the scaling prediction for the degree of side chain stretching takes the form (33)
| (3) |
Assuming 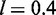 nm and N = 400, we find
nm and N = 400, we find 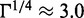 for the charge neutral complex with
for the charge neutral complex with 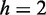 nm. Taking the hydrodynamic radius RH = 6 nm for the chain in free solution as a measure for R (21), the scaling estimate suggests that the side chains stretch to a value on the order of 20 nm. A precise value of Rs cannot be given because the proportionality factor in Equation (3) is unknown. Nevertheless, a maximum cross-sectional diameter of the bottlebrush of ∼40 nm at saturated binding is consistent with our experimental estimate for N/P = 2.0. The persistence length P of bottlebrush polymers can be estimated based on scaling theory, which was verified by self-consistent field calculations (21,34)
nm. Taking the hydrodynamic radius RH = 6 nm for the chain in free solution as a measure for R (21), the scaling estimate suggests that the side chains stretch to a value on the order of 20 nm. A precise value of Rs cannot be given because the proportionality factor in Equation (3) is unknown. Nevertheless, a maximum cross-sectional diameter of the bottlebrush of ∼40 nm at saturated binding is consistent with our experimental estimate for N/P = 2.0. The persistence length P of bottlebrush polymers can be estimated based on scaling theory, which was verified by self-consistent field calculations (21,34)
| (4) |
with a small numerical constant 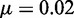 . For
. For 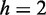 nm and the commonly accepted value of P0 = 50 nm for the DNA main chain intrinsic stiffness, the above theory predicts
nm and the commonly accepted value of P0 = 50 nm for the DNA main chain intrinsic stiffness, the above theory predicts 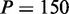 nm, or a 3-fold increase as compared with bare DNA. Hence, theoretical estimates confirm the expected order of magnitude of the stiffening effect, even though the predicted increase is <4- to 5-fold increase at N/P = 2.0 that we have estimated from the analysis of the atomic force microscopy images and the extensions of the complexes confined inside the nanochannels.
nm, or a 3-fold increase as compared with bare DNA. Hence, theoretical estimates confirm the expected order of magnitude of the stiffening effect, even though the predicted increase is <4- to 5-fold increase at N/P = 2.0 that we have estimated from the analysis of the atomic force microscopy images and the extensions of the complexes confined inside the nanochannels.
CONCLUSIONS
We have shown that we can amplify the stretch of DNA confined in nanochannels through a coating of the duplex with a diblock polypeptide. For channels with cross-sectional diameters of 200 nm we can achieve a uniform stretch of 85% of the contour length. Owing to the less stringent cross-sectional dimension of a few hundred nm, the channels are relatively easy to make out of elastomer with soft lithography. Furthermore, the DNA molecules can be loaded within the nanochannel using electrophoresis. Monte Carlo computer simulation shows that the stretch is due to an increase in bending rigidity and thickness of the bottlebrush-coated DNA. The persistence lengths and widths deduced from the stretch agree with what has been observed with atomic force microscopy. The highest degree of stretching corresponds to the deflection limit, in which the bottlebrush undulates within the nanochannel without ever folding back. We have demonstrated that the amplification of the stretch in tandem with site-specific fluorescence labelling allows the investigation of large-scale genomic organization of 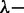 phage DNA. The limiting factor of the nanochannel platform is the length of the channels. With present-day technology it is possible to fabricate linear channels with a length of around a few millimetres, so that DNA molecules of a few mega base pairs can be stretched. For longer DNAs, one has to use fragments and/or nanofluidic devices with a back-folded channel layout.
phage DNA. The limiting factor of the nanochannel platform is the length of the channels. With present-day technology it is possible to fabricate linear channels with a length of around a few millimetres, so that DNA molecules of a few mega base pairs can be stretched. For longer DNAs, one has to use fragments and/or nanofluidic devices with a back-folded channel layout.
FUNDING
Singapore-MIT Alliance for Research and Technology, Singapore National Science Foundation [CBET-0852235]; Singapore MOE Academic Research Fund [MOE2009-T2-2-005]; Faculty Research Grant [R-144-000-312-112]; Dutch Polymer Institute, project [SynProt # 698]; Consejo Nacional de Ciencia y Tecnologia, Mexico (to A.H.G.). Funding for open access charge: National Science Foundation.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Sarabjit Singh and Lingjun Zhang for experimental support.
REFERENCES
- 1.Reisner W, Morton KJ, Riehn R, Wang YM, Yu Z, Rosen M, Sturm JC, Chou SY, Frey E, Austin RH. Statics and dynamics of single DNA molecules confined in nanochannels. Phys. Rev. Lett. 2005;94:196101. doi: 10.1103/PhysRevLett.94.196101. [DOI] [PubMed] [Google Scholar]
- 2.Mannion JT, Reccius CH, Cross JD, Craighead HG. Conformational analysis of single DNA molecules undergoing entropically induced motion in nanochannels. Biophys J. 2006;90:4538–4545. doi: 10.1529/biophysj.105.074732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reisner W, Beech JP, Larsen NB, Flyvbjerg H, Kristensen A, Tegenfeldt JO. Nanoconfinement-enhanced conformational response of single DNA molecules to changes in ionic environment. Phys. Rev. Lett. 2007;99:058302. doi: 10.1103/PhysRevLett.99.058302. [DOI] [PubMed] [Google Scholar]
- 4.Jo K, Dhingra DM, Odijk T, de Pablo JJ, Graham MD, Runnheim R, Forrest D, Schwartz DC. A single-molecule barcoding system using nanoslits for DNA analysis. Proc. Natl Acad. Sci. USA. 2007;104:2673–2678. doi: 10.1073/pnas.0611151104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang C, Zhang F, van Kan JA, van der Maarel JRC. Effects of electrostatic screening on the conformation of single DNA molecules confined in a nanochannel. J. Chem. Phys. 2008;128:225109. doi: 10.1063/1.2937441. [DOI] [PubMed] [Google Scholar]
- 6.Kim Y, Kim KS, Kounovsky KL, Chang R, Jung GY, dePablo JJ, Jo K, Schwartz DC. Nanochannel confinement: DNA stretch approaching full contour length. Lab. Chip. 2011;11:1721–1729. doi: 10.1039/c0lc00680g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Persson F, Utko P, Reisner W, Larsen NB, Kristensen A. Confinement spectroscopy: probing single DNA molecules with tapered nanochannels. Nano Lett. 2009;9:1382–1385. doi: 10.1021/nl803030e. [DOI] [PubMed] [Google Scholar]
- 8.Su T, Das SK, Xiao M, Purohit PK. Transition between two regimes describing internal fluctuation of DNA in a nanochannel. PLoS One. 2011;6:e16890. doi: 10.1371/journal.pone.0016890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai L, van der Maarel JRC, Doyle PS. Effect of nanoslit confinement on the knotting probability of circular DNA. ACS Macro Lett. 2012;1:732–736. doi: 10.1021/mz3001622. [DOI] [PubMed] [Google Scholar]
- 10.Zhang C, Shao PG, van Kan JA, van der Maarel JRC. Macromolecular crowding induced elongation and compaction of single DNA molecules confined in a nanochannel. Proc. Natl Acad. Sci. USA. 2009;106:16651–16656. doi: 10.1073/pnas.0904741106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones JJ, van der Maarel JRC, Doyle PS. Effect of nanochannel geometry on DNA structure in the presence of macromolecular crowding agent. Nano Lett. 2011;11:5047–5053. doi: 10.1021/nl203114f. [DOI] [PubMed] [Google Scholar]
- 12.Zhang C, Gong ZY, Guttula D, Malar PP, van Kan JA, Doyle PS, van der Maarel JRC. Nanouidic compaction of DNA by like-charged protein. J. Phys. Chem. B. 2012;116:3031–3036. doi: 10.1021/jp2124907. [DOI] [PubMed] [Google Scholar]
- 13.Riehn R, Lu M, Wang YM, Lim SF, Cox EC, Austin RH. Restriction mapping in nanofluidic devices. Proc. Natl Acad. Sci. USA. 2005;102:10012–10016. doi: 10.1073/pnas.0503809102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levy SL, Craighead HG. DNA manipulation, sorting, and mapping in nanofluidic systems. Chem. Soc. Rev. 2010;39:1133–1152. doi: 10.1039/b820266b. [DOI] [PubMed] [Google Scholar]
- 15.Das SK, Austin MD, Akana MC, Deshpande P, Cao H, Xiao M. Single molecule linear analysis of DNA in nano-channel labelled with sequence specific fluorescent probes. Nucleic Acids Res. 2010;38:e177. doi: 10.1093/nar/gkq673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reisner W, Larsen NB, Silahtaroglu A, Kristensen A, Tommerup N, Tegenfeldt JO, Flyvbjerg H. Single-molecule denaturation mapping of DNA in nanofluidic channels. Proc. Natl Acad. Sci. USA. 2010;107:13294–13299. doi: 10.1073/pnas.1007081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 2012;30:771–776. doi: 10.1038/nbt.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reisner W, Pedersen JN, Austin RH. DNA Confinement in nanochannels: physics and biological applications. Rep. Prog. Phys. 2012;75:106601. doi: 10.1088/0034-4885/75/10/106601. [DOI] [PubMed] [Google Scholar]
- 19.Min SK, Kim WY, Cho Y, Kim KS. Fast DNA sequencing with a graphene-based nanochannel device. Nature Nanotech. 2011;6:162–165. doi: 10.1038/nnano.2010.283. [DOI] [PubMed] [Google Scholar]
- 20.Zhang C, Jiang K, Liu F, Doyle PS, van Kan JA, van der Maarel JRC. A nanofluidic device for single molecule studies with in situ control of environmental solution conditions. Lab. Chip. 2013;13:2821–2826. doi: 10.1039/c3lc50233c. [DOI] [PubMed] [Google Scholar]
- 21.Hernandez-Garcia A, Werten MW, Stuart MC, de Wolf FA, de Vries R. Coating of single DNA molecules by genetically engineered protein diblock copolymers. Small. 2012;8:3491–3501. doi: 10.1002/smll.201200939. [DOI] [PubMed] [Google Scholar]
- 22.Werten MW, Wisselink WH, Jansen-van den Bosch TJ, de Bruin EC, de Wolf FA. Secreted production of a custom-designed, highly hydrophilic gelatin in Pichia pastoris. Protein Eng. 2001;14:447–454. doi: 10.1093/protein/14.6.447. [DOI] [PubMed] [Google Scholar]
- 23.van Kan JA, Zhang C, Malar PP, van der Maarel JRC. High throughput fabrication of disposable nanofluidic lab-on-chip devices for single molecule studies. Biomicrofluidics. 2012;6:036502. doi: 10.1063/1.4740231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Kan JA, Bettiol AA, Watt F. Proton beam writing of three-dimensional nanostructures in hydrogen silsesquioxane. Nano Lett. 2006;6:579–582. doi: 10.1021/nl052478c. [DOI] [PubMed] [Google Scholar]
- 25.van Kan JA, Bettiol AA, Watt F. Three-dimensional nanolithography using proton beam writing. Appl. Phys. Lett. 2003;83:1629–1631. [Google Scholar]
- 26.Shao PG, van Kan JA, Ansari K, Bettiol AA, Watt F. Poly (dimethyl siloxane) micro/nanostructure replication using proton beam written masters. Nucl. Instrum. Methods Phys. Res. B. 2007;260:479–482. [Google Scholar]
- 27.Allemand JF, Bensimon D, Jullien L, Bensimon A, Croquette V. pH-dependent specific binding and combing of DNA. Biophys. J. 1997;73:2064–2070. doi: 10.1016/S0006-3495(97)78236-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dai L, Ng SY, Doyle PS, van der Maarel JRC. Conformation model of back-folding and looping of a single DNA molecule confined inside a nanochannel. ACS Macro Lett. 2012;1:1046–1050. doi: 10.1021/mz300323a. [DOI] [PubMed] [Google Scholar]
- 29.Dai L, Jones JJ, van der Maarel JRC, Doyle PS. A systematic study of DNA conformation in slit-like confinement. Soft Matter. 2012;8:2972–2982. [Google Scholar]
- 30.Rivetti C, Guthold M, Bustamante C. Scanning force microscopy of DNA deposited onto mica: equilibrium versus kinetic trapping studied by statistical polymer chain analysis. J. Mol. Biol. 1996;264:919–932. doi: 10.1006/jmbi.1996.0687. [DOI] [PubMed] [Google Scholar]
- 31.Wiggins PA, van der Heijden T, Herrero FM, Spakowitz A, Phillips R, Widom J, Dekker C, Nelson PC. High flexibility of DNA on short length scales probed by atomic force microscopy. Nat. Nanotechnol. 2006;1:137–141. doi: 10.1038/nnano.2006.63. [DOI] [PubMed] [Google Scholar]
- 32.Odijk T. On the statistics and dynamics of confined or entangled stiff polymers. Macromolecules. 1983;16:1340–1344. [Google Scholar]
- 33.Hsu HP, Paul W, Binder K. Understanding the multiple length scales describing the structure of bottle brush polymers by Monte Carlo simulation methods. Macromol. Theory Simul. 2011;20:510–525. [Google Scholar]
- 34.Feuz L, Leermakers FAM, Textor M, Borisov O. Bending rigidity and induced persistence length of molecular bottle brushes: a self-consistent field theory. Macromolecules. 2005;38:8891–8901. [Google Scholar]