

Abstract
Background
Bacterial genomes displaying a strong bias between the leading and the lagging strand of DNA replication encode two DNA polymerases III, DnaE and PolC, rather than a single one. Replication is a highly unsymmetrical process, and the presence of two polymerases is therefore not unexpected. Using comparative genomics, we explored whether other processes have evolved in parallel with each polymerase.
Results
Extending previous in silico heuristics for the analysis of gene co-evolution, we analyzed the function of genes clustering with dnaE and polC. Clusters were highly informative. DnaE co-evolves with the ribosome, the transcription machinery, the core of intermediary metabolism enzymes. It is also connected to the energy-saving enzyme necessary for RNA degradation, polynucleotide phosphorylase. Most of the proteins of this co-evolving set belong to the persistent set in bacterial proteomes, that is fairly ubiquitously distributed. In contrast, PolC co-evolves with RNA degradation enzymes that are present only in the A+T-rich Firmicutes clade, suggesting at least two origins for the degradosome.
Conclusion
DNA replication involves two machineries, DnaE and PolC. DnaE co-evolves with the core functions of bacterial life. In contrast PolC co-evolves with a set of RNA degradation enzymes that does not derive from the degradosome identified in gamma-Proteobacteria. This suggests that at least two independent RNA degradation pathways existed in the progenote community at the end of the RNA genome world.
Keywords: replication, degradosome, LUCA, phylogenetic profiling, nanoRNase
Background
Future developments of Synthetic Biology require that patterns of gene organization in genomes are carefully taken into account [1]. Following the pioneering work of Sueoka [2], Lobry and co-workers identified a replication-linked bias in the nucleotide distribution in bacterial chromosomes. Subsequently, a bias in favor of genes transcribed in the same direction as that of the movement of the replication fork was observed in most bacterial genomes [3,4]. The bias was correlated with the presence in the genome of a single origin of replication. Taken together, these observations led to the construction of algorithms meant to identify in silico the origins of replication [5]. The cause of the bias has been a matter of speculation until it was observed that Firmicutes displayed the strongest bias [6,7] reaching 87% in organisms such as Thermoanaerobacter tengcongensis [8]. A first hypothesis proposed that the bias was favoring genes requiring high expression [9]. Yet, there was no correlation between gene expressivity and transcription from the leading strand. Indeed, many genes of the replications machinery are expressed at a low level, and they are transcribed from the leading strand [10]. The leading strand bias had therefore to be accounted for by physical constraints: transcription of key genes must avoid head-on collision with the replication machinery to prevent formation of truncated transcripts. The latter are known to be toxic for the cell, in particular when they code for polypeptides belonging to protein complexes (see [11] for a general description of the process).
A further observation noticed that organisms that were strongly biased in the leading vs lagging strand replication encoded two DNA polymerases III, DnaE and PolC, rather than a single one [12]. DnaE was originally identified in Escherichia coli [13] whereas PolC was identified in Bacillus subtilis [14]. Yet, in contrast with the situation in Eukarya where the presence of two polymerases is the norm, most bacterial species listed in genome reference databases code for only one DNA polymerase III. In E. coli, the same structural type of DnaE replicase acts on both the leading and lagging strand. Two identical replicase molecules are held together in a complex with the replicative helicase and subunits with priming activities, allowing two identical alpha catalytic subunits to assume different functions on the two strands of the replication fork [15]. In contrast, in B. subtilis, asymmetric DNA synthesis requires replicative DNA polymerases with two distinct structures, DnaE and PolC. In contrast to PolC, DnaE, which replicates the lagging strand, is devoid of 3' --> 5'-proofreading exonuclease activity and has a low processivity (1-75 nucleotides), requiring additional factors to fulfill its role in replication [16].
DnaE and PolC differ both in structure and activity [15]. This prompted us to explore whether their genes co-evolved with consistent groups of genes, allowing us to propose scenarios of the origins of the replication machineries. In particular the cell manages compartmentalization via the cell's envelope, appendages, but also nanomachines such as the ribosome, ATP synthase, the degradosome and many others [17]. We present here a phylogenetic profile analysis focused on the bacterial dnaE and polC genes and show that proteins co-evolving with PolC have distinct features, and may form a specific kind of degradosome. The consequences in terms of the origin of bacteria are discussed.
Methods
To separate between the history of DnaE and that of PolC, we established a heuristics meant to identify genes that co-evolved in bacterial genomes. The approach is straightforward: we first identify orthologous genes by pairwise comparison to compute phylogenetic profiles; we subsequently compare them after statistical validation, taking into account the phylogenetic distances between organisms with co-occurring genes; finally, we combine phylogenetic profiling with other methods that take into account the genomic context.
Pairwise gene comparison and phylogenetic profiles
Phylogenetic profiling uses binary vectors that, taking genes one by one, identify in which organisms an ortholog is present (resp. absent). To this aim, Tatusov introduced the notion of "occurrence vector" for groups of orthologous proteins [18]. Here we used the complete RefSeq NCBI database of bacterial genomes [19] comparing the cognate proteomes using BlastP (all genes of a proteome against all proteomes). We subsequently identified orthologs using bidirectional best hits (BBH) as described by Koonin and co-workers [20]. Next, we retained orthologs according to the distribution of a gene similarity scores, s, designed to take into account biological constraints other than orthology, using relevant thresholds (see equation 4 for definition of the thresholds).
Lacking an evolutionary model common to all genomes, we chose the simplest model: the similarity score we used is the direct convolution of identity (i) and coverage (c: length of the BlastP alignment divided by the length of the longest protein) of the BlastP hits:
| (1) |
We computed the values of s for each gene ortholog present in the target organism. As an example, Figure 1 (left panel) displays the behavior of sorted s for 8 genes of the B. subtilis histidine pathway. The x axis indicates the number of orthologs of the target gene having a s score below x. The curves displayed in Figure 1 show that if we used a common threshold (score of the y axis) for all genes, we would find inconsistent levels of orthology. For example, fixing the threshold at 40 (arbitrary units, dashed black line) for the gene hisF retained almost all hisF orthologs but only some hisC orthologs (selected orthologs have similarity scores above the threshold). Ths is because there is no common molecular evolutionary clock [21] for the genes in the pathway: hisC orthologs genes maintain their function despite a high rate of mutation, generating BlastP hits with lower similarity scores. This constrasts with the evolution of hisF orthologs. Had we used the same threshold for hisC and hisF genes we would not have found them to be correlated.
Figure 1.
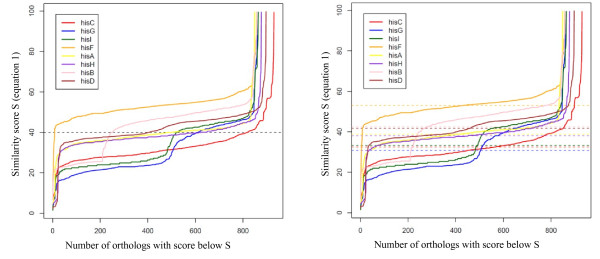
Curves of sorted similarity score s (equation 1) for 8 genes of the B. subtilis histidine pathway. The x axis represents the number of orthologs of the target gene having a s score below y. Left panel: The same threshold (dashed black line at 40) for all genes is used. Right panel: Different thresholds chosen according to the average of s are used for each gene.
We therefore selected orthologous genes verifying the following formula:
| (2) |
This allowed us to select orthologous genes with a similarity score s above the average of s, avg(s) (see Figure 1 right panel). Now, the number of orthologous genes selected for the genes hisF and hisC is almost the same. As a consequence, if these orthologs belong to the same organisms, the two genes will be found to be correlated, a functionally relevant observation.
Next, we included in the model the fact that orthologous genes must have a similar homology score because positive selection pressure tends to retain only a limited number of mutations among those that are constantly created. This implies that a high density of similarity score should correspond to families of organisms that keep the function of a gene with no alteration. To this aim, we first computed the distribution of s values, d(s), for each ortholog of target genes.
Figure 2 shows how d(s) varies for the histidine pathway's genes of B. subtilis. We subsequently retained the orthologs genes verifying the following formula:
Figure 2.

Distribution d(s) of orthologs s values (equation 1) for the histidine pathway's genes of B. subtilis.
| (3) |
Finally, we took into account the fact that proteins may keep their function while suffering different mutation rates (a different threshold for each gene) and that families of organisms that keep the function of a gene have similar homology scores because they undergo positive selection pressure (high densities of homology score). To this aim we selected orthologs with a similarity score s above the average and otherwise orthologs genes with a s density above the average, combining (2) and (3).
| (4) |
Figure 3 shows an example of this selection (selected orthologs genes have their s values indicated in grey) for hisI orthologs.
Figure 3.

Distribution d(s) of orthologs s values (equation 1) for gene hisI of B. subtilis. Orthologs of hisI which have their s values indicated in grey correspond to orthologs selected applying equation 4.
Relevant selection of orthologs has been computed for each gene of the target organism. This allowed the building up of binary vectors of presence/absence for these genes to explore the hypothesis that functionally linked genes have the same occurrence vectors. The next step was then to compare together these occurrence vectors to underscore functional relationships that group together genes of the target organism.
Comparison of phylogenetic profiles
As previous authors, we assumed that in the course of evolution functionally-related genes tend to be gained or lost together. This results in a correlation of their occurrence vectors. The first exploration of this hypothesis compared co-occurrence profiles using Hamming's distance [22]. Subsequently, many different statistical approaches to compare phylogenetic profiles have been used, such as mutual information [23], Pearson correlation coefficient [24] and Fisher's test [25]. Here we used the phi coefficient to compare phylogenetic profiles of two genes X and Y. This measure is similar to the Pearson correlation coefficient. In fact, a Pearson correlation coefficient estimated for two binary variables will return the phi coefficient:
| (5) |
With n11, the number of organisms in which X and Y are present; n00, the number of organisms in which X and Y are absent; n10, the number of organisms in which X is absent and Y is present; n01, the number of organisms in which X is present and Y is absent. The formula is symmetric:
| (6) |
Measure of phylogenetic distances
The Pearson correlation, as other statistical methods, ignores that organisms are phylogenetically related and that the phylogenetic kinships may be biased. This must be taken into account, as the genome samples that have been sequenced is considerably biased, in terms of relative phylogenetic proximity. This may have a negative influence on our predictions. To reduce the effect of this phylogenetic skew, we modified the formula, taking into account the phylogenetic distance between organisms in which the genes co-occur. The idea was to give a larger weight to genes co-occurring in distant organisms than to those present/absent simultaneously in closely related organisms. As we do not have a detailed model that would account for the sampling bias in genome data, we used a plausible straightforward phenomenological measure of the proximity between two organisms, A and B, D(A,B):
| (7) |
with N(A,B) the number of genes occurring in organisms A and B, N(A) the number of genes in organism A and N(B) the number of genes in organism B.
In the absence of a model describing genome evolution, and knowing that there is a considerable anthropomorphic bias in the choice of the organisms that have been sequenced we chose a somewhat arbitrary non-linear model to increase the weight of distant organisms. Our formula to measure the functional link between two genes X and Y is now:
| (8) |
with A and B the most distant organisms in which genes X and Y co-occur.
Coupling with genomic context methods
Further biologically-relevant factors must be taken into account to construct a plausible heuristics. A great many methods use the genomic context of genes to predict functional links between proteins. For example, functional links are suggested by conservation of gene neighbourhood and gene order [26], gene fusion events [27], correlation of the genes' evolutionary rate [28] and correlation of genes' occurrence in organisms (phylogenetic profiles). In [23] an evaluation of the methods that emphasized these factors showed that conservation of the gene neighborhood and the gene order covered 45% of the functional interaction between genes of Mycoplasma genitalium. In the present work, the gene neighborhood was measured using the Syntonizer software (implemented in the MicroScope platform [29]), which is based on an exact graph-theoretical approach to measure synteny [30]. A factor K was used: K(X,Y) = 1 if the two genes X and Y are found at least once in synteny and 0.9 otherwise.
The final formula to compute the co-evolution score C between two genes X and Y is then:
| (9) |
with ϕ(X,Y) the correlation score between phylogenetic profiles of the genes X and Y, D(A,B) the distance between the most distant organisms A and B in which the genes X and Y co-occur and K(X,Y) a factor measuring the conservation of genes neighborhood.
This phylogenetic profile method (PhyloProfile) has been integrated in the MicroScope platform [29]. It is directly available in the gene editor and allows the users to compute dynamically co-evolution scores of the target gene against all genes of the organism of interest.
Construction of clusters of co-evolution
Finally, following computation of the relevant phylogenetic profiles, we constructed co-evolution clusters. To this aim, we computed co-evolution scores, C, for all genes of B. subtilis, E. coli and Acinetobacter baylyi. If n is the number of genes of an organism, we obtained n*(n-1) scores. These scores were used to build up networks in which nodes correspond to genes and edges correspond to scores of co-evolution between genes. Applying a clustering method on these networks will allow construction of partitions of these genomes into clusters of co-evolving genes. Here we used as a clustering method the Markov Cluster algorithm (MCL; Van Dongen 2000; http://micans.org/mcl/) that is designed to cluster large numbers of relationships in a similarity space. The MCL algorithm is a fast and scalable unsupervised cluster algorithm for networks, based on simulation of flows in graphs. It has successfully been applied for clustering large sets of protein sequences [31,32]. In the present work, clusters corresponding to functionally relevant processes, such as metabolic pathways (histidine biosynthesis...) or global functions (degradosome ...) were obtained, depending on the threshold used in the clustering procedure.
As a validation of our approach we verified that molybdopterin biosynthesis and use have disappeared in a concerted way, as observed in Pseudoalteromonas haloplanktis [33].
Results
The contrasted replication-associated gene orientation bias uncovered between most bacterial clades and Firmicutes prompted us to explore the underlying phylogenetic constraints that support this discrepancy. To this aim we meant to uncover the functions that co-evolved with either DnaE or PolC.
To get a first crude view of the processes underlying this replication-associated bias we used the JMP® software (SAS Institute, Cary, NC) to compute a hierarchical clustering of the B. subtilis essential genes [34] according to their occurrence proportion in different Bacteria clades (Figure 4). Three main clusters were obtained. Unexpectedly, they were all related to DNA replication. This substantiated the conjecture that the way replication is organized was indeed at the core of some important functional variation specific to bacteria forming a given clade.
Figure 4.
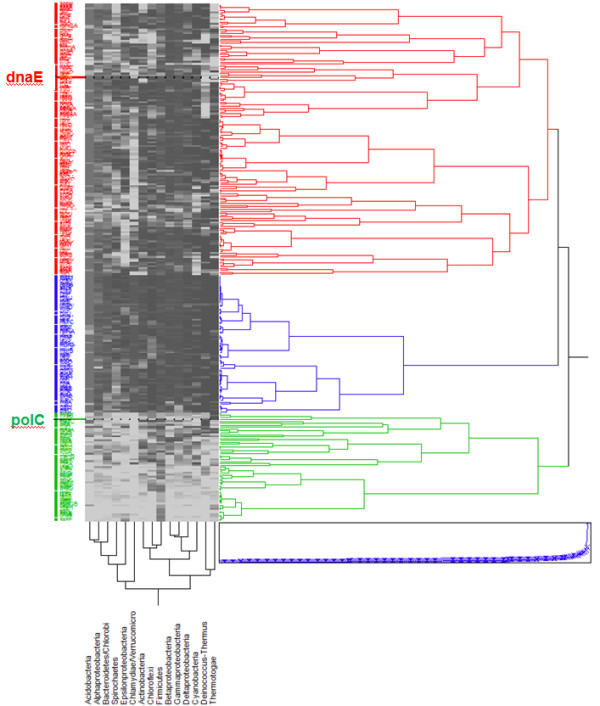
Hierarchical clustering of occurrence proportion vectors of B. subtilis essential genes in clades of the domain Bacteria. Squares show the proportion of a phylum (ascending from white to black) having an occurrence of a gene. The dendogram depicts distances between the proportion vectors. This dendogram separates B. subtilis essential genes into three clusters. The first one (red) contains dnaE and corresponds to genes occurring in the majority of bacterial clades. The second one (blue) contains dnaA and spans the whole domain of bacteria. The last one (green) contains polC and is composed of genes essentially specific to Firmicutes. The computing and visualization of the clusters was performed using the JMP® software (SAS Institute, Cary, NC).
We observed the following, from top to bottom in Figure 4: A first cluster corresponded to genes present in almost all bacterial clades. This cluster contains dnaE, which, however, seems to be absent from the Deinococcus-Thermus and Chlamydia-Verrucomicrobia clades. It also comprises the bulk of the translation machinery (ribosome protein genes), including a limited number of tRNA synthetases, RNA polymerase genes, core iron-sulfur metabolism, and the core genes required to synthesize an envelope (Additional file 1). The second cluster (in blue) corresponded to genes spanning the whole domain Bacteria. It comprizes most genes of tRNA synthetases and the remaining set of ribosomal protein genes. Remarkably it clusters with dnaA as well as the gene for DNA primase. Finally, the third cluster corresponded to genes mostly specific to Firmicutes. This cluster co-evolves with polC, genes involved in cell division and genes involved in RNA degradation.
While already revealing, this first analysis cannot give us a detailed view of gene co-evolution as it is based on a considerably biased sample of genomes. Indeed, genomes have been chosen to be sequenced as a function of the history of biological studies in the academic world (and particularly oriented towards bacteria of medical interest), and not based on a view spanning the whole tree of life, with equal weight for all extant species. In general, finding the same frequency of gene occurrence in a clade does not imply that these genes are simultaneously present or absent in the same organisms. This is particularly true when clades are made of only a few organisms (Thermotogae, Acidobacteria, Chloroflexi, Deinococcus have less than twelve organisms). Naturally, this is much less so in clades that are well represented (Firmicutes and gamma-Proteobacteria comprize more than 250 organisms). Furthermore this preliminary analysis was only based on the essential genes of B. subtilis, and it is well established that functional ubiquity does not equate to gene ubiquity: a same function could be essential in other clades, but performed by a gene of a completely different descent.
In order to investigate more accurately gene co-evolution we needed therefore to analyze the occurrence profiles of all B. subtilis genes at the level of several phylogenetically distant model organisms rather than clades. To this aim, we designed a specific phylogenetic profile heuristics (PhyloProfile, see Methods) and based our exploration on the recently re-sequenced and re-annotated genome of B. subtilis [35] in parallel with that of the reference sequence and annotation of E. coli [36], an organism phylogenetically distant from B. subtilis. The method allowed us to identify clusters of genes that specificallly co-evolved with dnaE and polC.
Genes co-evolving with dnaE
As a possibly ancestral protein, DnaE co-evolves with a very large number of genes. We used different co-evolution scores thresholds (0.50 to 0.90) to investigate how the proteins of the whole proteome clustered with DnaE. When the threshold was higher than 0.75, the number of genes co-evolving with DnaE stabilized to approximately 250 genes. As an example, using 0.77 as a threshold, we listed the genes co-evolving with DnaE from B. subtilis (Additional file 1). This list matched remarkably well with the key genes coding for the persistent functions that are required to allow reproduction and replication [37], substantiating the validity of the heuristics.
Most of the genes coding for the translation and transcription machineries, including factors and enzymes involved in modification and maturation of basic components of the machineries, were found in the list. This list also comprized the genes coding for the major metabolic pathways allowing construction of the cell: pyrimidine and purine biosynthesis and salvage, the core of the glycolytic/gluconeogenesis pathway (eno, gapA, pgk, tpiA...) and ATP synthase, as well as the secretion machinery. A number of components of the replication apparatus (DnaA, DnaC, DnaG, DnaN, GyrAB, LigA, PcrA, RnhB, SsbA) and genes involved in recombination and repair (Nth, Obg, PolA, RadA, RecA, RecG, RecN, RecO, RuvAB) were present as well in this set of proteins co-evolving with DnaE. Remarkably, a set of functions was missing from this list, that which corresponds to many components of RNA degradation (see below) [38]. Indeed, we found only a very limited set of RNA maturation and catabolism genes: rnc (coding for RNase III), rnhB (already mentioned, allowing degradation of RNA/DNA hybrid sequences during replication of the DNA lagging strand) and pnpA, a gene coding for an enzyme degrading RNA while preserving phosphate bond energy, polynucleotide phosphorylase.
Beside genes coding for known functions, a consistent set of genes coding for unknown functions was present in most bacterial clades. The corresponding list of the corresponding 33 « y » B. subtilis genes within the larger DnaE-related gene set is displayed in Table 1. These genes were further analyzed as they correspond to unknown or poorly identified functions that, if understood, would enhance considerably our understanding of bacteria. In the course of this exploration, we benefited from continuous re-annotation of the updated genome sequence [35]. Using the most recent publications, this limited the unknowns to at most 18 genes. Interestingly, the majority of the newly identified functions was involved in ribosomal RNA metabolism (rsmI(yabC), rsmD(ylbH), rlmN(yloN), rimP(ylxS), hflX(ynbA), cpgA(yloQ)), further substantiating the DnaE/translation connection. We also found in this set the hibernation promoting factor (hpf(yvyD)) and further genes involved in recombination and repair (uup(yfmR), recD(yrrC), rarA(yrvN)). All of these genes have a counterpart in E. coli. A gene, shorter in B. subtilis than in E. coli, codes for a function important for mRNA turnover, rpsA in E. coli and ypfD (renamed rpfA) in B. subtilis. This gene codes for ribosomal protein S1 in E. coli and there is a clear indication, in this case, that the ribosomal protein function has been superimposed on the general function of mRNA presentation to the degradosome [39]. This is a first indication that there is divergence between proteins functionally related to DnaE polymerase III in E. coli and B. subtilis, in particular at the level of RNA degradation.
Table 1.
Genes of unknown function co-evolving with Bacillus subtilis dnaE
| Label | B. subtilis | old name | E. coli | Function |
|---|---|---|---|---|
| BSU00200 | yaaK | yaaK | ybaB | DNA binding protein1 |
| BSU00360 | rsmI | yabC | yraL | ribosomal RNA small subunit methyltransferase I2 |
| BSU00390 | yabD | yabD | ycfH | metal-dependent DNase1 |
| BSU00480 | yabJ | yabJ | yjgF | putative enzyme resulting in alteration of gene expression |
| BSU05910 | ydiB | ydiB | yjeE | putative ATPase or kinase UPF0079 |
| BSU05920 | ydiC | ydiC | yeaZ | putative chaperone or protease |
| BSU05950 | ydiF | ydiF | ybiT | putative ABC transporter (ATP-binding protein)3 |
| BSU07370 | uup | yfmR | ycbH | putative ABC protein involved in RecA-independent precise excision of transposons1 |
| BSU07900 | rbn | yfkH | yihY | putative ribonuclease BN2 |
| BSU09240 | yhcW | yhcW | yniC | putative phosphoglycolate phosphatase |
| BSU14350 | yknX | yknX | ybjY (macA) | putative efflux permease3 |
| BSU15010 | rsmD | ylbH | yhhF | ribosomal RNA small subunit methyltransferase D2 |
| BSU15380 | ylmE | ylmE | yggS | conserved hypothetical protein |
| BSU15750 | rlmN | yloN | yfgB | 23S rRNA m2A2503 methyltransferase2 |
| BSU15780 | cpgA | yloQ | rsgA (yjeQ) | GTPase involved in ribosome and sacculus morphogenesis2 |
| BSU16590 | rimP | ylxS | yhbC | ribosome maturation factor2 |
| BSU17430 | hflX | ynbA | hflX | ribosome associating GTPase2 |
| BSU21450 | yolJ | yolJ | "-" | putative glycosyltransferase |
| BSU22880 | rpfA | ypfD | "-" rpsA | RNA degradation presenting factor (ribosomal protein S1 homolog)2 |
| BSU24790 | yqgX | yqgX | ycbL | putative metal-binding hydrolase |
| BSU24890 | folN | yqgN | ygfA | 5-formyltetrahydrofolate cyclo-ligase |
| BSU25320 | yqfG | yqfG | ybeY | putative metal-dependent hydrolase |
| BSU25620 | yqeL | yqeL | ybeB | putative ribosomal maturation protein2 |
| BSU27370 | yrrL | yrrL | yceG | conserved hypothetical protein |
| BSU27390 | yrrK | yrrK | yqgF | putative Holliday junction resolvase1 |
| BSU27480 | recD | yrrC | recD | exodeoxyribonuclease V alpha chain1 |
| BSU27490 | yrrB | yrrB | yciM | putative tetratricopeptide repeat family protein |
| BSU27530 | rarA | yrvN | ycaJ | DNA-dependent ATPase1 |
| BSU27700 | yrbF | yrbF | yajC | component of the preprotein translocase |
| BSU28360 | rdgB | ysnA | yggV (rdgB) | inosine/xanthosine triphosphate pyrophosphatase (subunit A) |
| BSU30680 | ytjA | ytjA | yidD | conserved hypothetical protein |
| BSU35310 | hpf | yvyD | yhbH | ribosome-associated sigma 54 modulation protein2 |
| BSU36950 | tamT | ywlC | yrdC | tRNA threonylcarbamoyladenosine biosynthesis protein2 |
1: DNA metabolism
2: RNA metabolism
3: transport
Starting with the B. subtilis nomenclature, we grouped the 18 remaining genes of unknown function according to common features, either extracted from the literature or using the neighborhood software STRING [40], with the hope to further uncover some of the associated functions. Despite its consistently valid outcomes we refrained from using our PhyloProfile to avoid circular validation. Interestingly, all but one (yolJ, a gene belonging to bacteriophage SPbeta, that disappeared from PhyloProfile when the threshold was increased) of these co-evolving genes have at least one clear ortholog in E. coli. yaaK belongs to an operon involved in DNA replication and repair, its STRING pattern further subtantiates this functional association. yabD and yrrK yrrL are connected together and relate to genes of the translation machinery (metG, ksgA, serS, rsmG) as well to genes involved in DNA metabolism and aromatic compounds biosynthesis. yabJ (yjgF in E. coli) codes for a putative enzyme with a 3D known structure conserved in all three domains of life [41]. Here, it is related to intermediary metabolism via aminotransferase PatB, the control of purine metabolism (purR, relA, gmk, rdgB), isoleucine biosynthesis and to proteolysis (via ClpX). In contrast, in E. coli, it is related to catabolism of threonine, pyrimidine metabolism and to a network of genes of unknown function. ydiB (coding for a putative shikimate dehydrogenase [42]), ydiC and ydiF connected to ribosome maturation and sulfur metabolism. yhcW connects to ilvA and translation; yknX, in an operon coding for a putative efflux permease, down regulated in the presence of benzoate [43]; ylmE to cell division and proline and purine/polyphosphate metabolism; yqgX to translation via aspartyl and histidyl tRNA synthetases, as well as D-tyrosine deacylase, transformylase and RelA; yqfG to translation and phosphate metabolism; yqeL and ytjA to translation and tRNA modification; finally, yrrB and yrbF are connected to several genes involved in tRNA modification.
In summary, DnaE co-evolves with most of the replication, recombination, translation, transcription and secretion machineries, and with some of the core metabolic biosynthetic pathways. We noted however the absence of a consistent RNA degradation pathway as well as most enzymes of the envelope biosynthetic pathways. This suggested that both RNA degradation and envelope biosynthesis, while functionally essential, might derive from different descent in different bacterial clades.
Genes co-evolving with polC
The list of the 162 genes co-evolving with polC differed considerably from that co-evolving with dnaE (Additional file 1). In particular, among those, 69 were genes of unknown function, a considerable proportion (Table 2). Interestingly, when analyzing their neighborhoods with STRING, we observed that there was seldom any connection with the dnaE-related networks, as if polC had evolved from a completely different origin. We also noticed that almost all genes in the list, while present in B. subtilis, do not have an ortholog in E. coli. This is exactly the opposite of what we found with genes co-evolving with dnaE. As for DnaE, the PolC-related proteins could be clustered into functionally significant groups (Tables 2 and 3). Remarkably, many of these clusters correspond to some aspect of RNA metabolism. Using STRING, these genes could be further clustered into seven groups (Table 3), the other ones remaining isolated.
Table 2.
Genes of unknown function co-evolving with Bacillus subtilis polC
| label | B. subtilis | E. coli | Function | Functional connection | String groups |
|---|---|---|---|---|---|
| BSU00030 | yaaA | ybcJ | putative RNase or phosphorylase; conserved in yeast; very weak in E. coli2 | RNA/DNA metabolism | group 1 |
| BSU00330 | yabA dnaH | "_ | subunit of the DNA replication complex1 | DNA polymerase and "y" network | group 1 |
| BSU00340 | yabB | "_ | putative RNA methyltransferase2 | RNA metabolism | group 1 |
| BSU00630 | yabR | "_ | putative RNA degradation protein; polyribonucleotide nucleotidyltransferase or phosphorylase2 | degradosome network with RfaA | group 2 |
| BSU31390 | yugI | "_ | putative RNA degradation protein or phosphorylase or nucleotidyl transferase; | degradosome network with RfaA | group 2 |
| BSU01750 | ybbP | "_ | homolog of YabR and RpfA2 DAC domain protein present in Archaea | group 3 | |
| BSU01760 | ybbR | "_ | substrate for Sfp phosphopantetheinyl transferase-catalyzed protein labeling by small molecule-CoA conjugates | group 3 | |
| BSU40510 | yybT | ". | phosphodiesterase acting on cyclic dinucleotides; possibly nanornase2 | RNA metabolism | group 3 |
| BSU15060 | ylbM | "_ | conserved hypothetical protein, found in a ribonuleoprotein complex in Mus musculus2 | RNA metabolism | group 3 |
| BSU25630 | yqeK | "_ | putative hydrolase | group 3 | |
| BSU25680 | yqeG | ". | putative hydrolase | group 3 | |
| BSU11030 | yitL | "_ | RNA-binding protein2 | RNA metabolism | group 3 |
| BSU19680 | yozE | ". | conserved hypothetical protein2 | RNA metabolism | group 3 |
| BSU24860 | yqgQ | "_ | putative single strand nucleic acid binding protein2 | RNA metabolism | group 3 |
| BSU14540 | ykzG | "_ | omega subunit of RNA polymerase2 | RNA metabolism | group 3 |
| BSU16610 | ylxR | "_ | putative RNA binding protein; putative new fold2 | RNA binding | group 3 |
| BSU27380 | yrzB | "_ | putative anti-sigma factor | group 3 | |
| BSU15830 | yloU | "_ | conserved hypothetical protein | group 3 | |
| BSU15840 | yloV | dhaL | putative dihydroxyacetone/glyceraldehyde kinase | group 3 | |
| BSU08680 | ygaC | "_ | putative factor, domain associated with ribonuclease E and G, possibly involved in Fe-S group formation2 | degradosome network | group 3 |
| BSU15970 | ylxM | "- | conserved hypothetical protein2 | RNA/DNA metabolism | group 3 |
| BSU27950 | ysxB | "- | conserved hypothetical protein with ribosomal function2 | ribosome | group 3 |
| BSU16000 | ylqC | "- | putative RNA binding protein2 | RNA binding | group 3 |
| BSU25670 | yqeH | "- | GTPase involved in ribosome 30S assembly2 | ribosome | group 3 |
| BSU16970 | ymdB | "- | putative phosphoesterase | group 3 | |
| BSU27400 | yrzL | "- | conserved hypothetical protein functionally linked to alanine tRNA loading2 | RNA metabolism | group 3 |
| BSU32310 | yutD | "- | conserved hypothetical protein2 | RNA metabolism | group 3 |
| BSU14640 | yktA | "- | conserved hypothetical protein | group 3 | |
| BSU15000 | ylbG | "- | conserved hypothetical protein | group 3 | |
| BSU17880 | ynzC | "- | conserved hypothetical protein | group 3 | |
| BSU22190 | ypsA | "- | conserved hypothetical protein | group 3 | |
| BSU07420 | yfmM | "- | putative polyphosphate-AMP phosphotransferase | group 3 | |
| BSU07430 | yfmL | "- | putative ATP-dependent RNA helicase2 | RNA metabolism | group 3 |
| BSU00970 | yacP | "- | putative ribonuclease with PIN and NYN domains; similar to eukaryotic RNases2 | RNA metabolism | group 4 |
| BSU23950 | yqjA | "- | conserved hypothetical protein putative | group 4 | |
| BSU32280 | yutG | "- | phosphatidyl-glycerophosphatas e A | group 4 | |
| BSU16845 | ymfF | "- | putative metalloprotease | protein metabolism | group 4 |
| BSU16860 | ymfH | "- | putative processing protease | protein metabolism | group 4 |
| BSU09800 | yheA | "- | conserved hypothetical protein | group 4 | |
| BSU09930 | yhaM | "- | 3'-5' exonuclease2 | DNA/RNA metabolism | group 4 |
| BSU01450 | ybxA ecfA | "- | energizing coupling factor ABC multiple influx transporter (ATP-binding protein)3 | specific transport | group 5 |
| BSU01460 | ybaE ecfB | "- | energizing coupling factor ABC multiple influx transporter (ATP-binding protein)3 | specific transport | group 5 |
| BSU01470 | ybaF ecfT | "- | permease component of the EcfAB influx transporters3 | specific transport | group 5 |
| BSU11590 | yjbL | "- | putative phosphatase | group 6 | |
| BSU11600 | yjbM | "- | (p)ppGpp synthetase | group 6 | |
| BSU11620 | yjbO | rluD | pseudouridylate synthase2 | RNA metabolism | group 6 |
| BSU29820 | ytpR | "- | putative protein with RNA binding domain2 | RNA binding | group 7 |
| BSU29840 | ytpP | "- | putative thiol-disulfide oxidoreductase with thioredoxin domain5 | sulfur metabolism | group 7 |
| BSU29860 | ytoP | frvX | glutamyl aminopeptidase; deblocking enzyme (wrong annotation in E. coli) | protein metabolism | group 7 |
| BSU13480 | ykrK | "_ | conserved hypothetical protein | ||
| BSU14100 | ykuJ | "_ | putative RNA-specific modification enzyme subunit2 | RNA metabolism network of "y" genes | |
| BSU14130 | ykuL | "_ | conserved hypothetical protein | ||
| BSU15410 | ylmH | "_ | factor involved in shape determination, RNA-binding fold2 | ||
| BSU17440 | ynbB | "_ | putative C-S lyase5 | sulfur metabolism | |
| BSU17699 | ynzK | "_ | putative membrane protein | ||
| BSU17910 | yneF | "_ | conserved hypothetical protein; methionine-rich5 | sulfur metabolism | |
| BSU19620 | yodJ | "_ | D-alanyl-D-alanine carboxypeptidase lipoprotein4 | ||
| BSU24260 | yqxC | "_ | putative methyltransferase with RNA binding domain2 | RNA binding | |
| BSU28650 | ysgA | "_ | putative RNA methylase2 | ||
| BSU29780 | ytxG | pqiB | homolog involved in DNA repair in Mus musculus1 | DNA repair | |
| BSU29980 | ytjP | "_ | putative dipeptidase | protein metabolism | |
| BSU30460 | ytrA | "_ | transcriptional regulator (GntR family) | ||
| BSU30490 | ytqB | "_ | putative RNA methylase2 | RNA metabolism | |
| BSU30870 | ytcB | "_ | putative UDP-glucose epimerase4 | ||
| BSU36340 | ywpE | "_ | putative sortase | ||
| BSU36910 | ywlG | "_ | conserved hypothetical protein; present in Archaea and Eukarya | ||
| BSU37340 | ywiB | "_ | putative RNA binding protein possibly involved in aminoacyl-tRNA editing2 | RNA binding | |
| BSU38499 | ywzH | "_ | conserved hypothetical protein | ||
| BSU40939 | yyzM | "_ | conserved hypothetical protein |
1: DNA metabolism
2: RNA metabolism
3: transport
4: envelope metabolism
5: sulfur metabolism
Table 3.
Genes groups identified using the STRING software [40]
| Groups | STRING genes |
|---|---|
| group 1 | yazA yabB yaaT yaaR yabC yabA yaaA yadB tmk holB dnaN dnaA dnaD recF |
| group 2 | yabR rpfA yugI yabMNOPQ hprT rpmE divIC rpfA(ypfD) alaRT ftsZ gatA ppiB acpS alaS aroD cbsA cca cotD csbA ctsR cysS defB dltC dltD dnaB dnaC dnaD ecsB ffh ftsK ftsL ftsY glcU glmM gluKP glyQS gpsB hcrA infB jag kapB lepA ltaSA lysA mecAB mtnN murB mutSB nadD nrnA nusA obg polC priA prmA rbfA recA recG recU recX rex ribC ribT rimM rmpA rnc rnhB rnjA rplI rplS rplU rpmB rpmF rpoZ rpsP rpsR rsiW |
| group 3 | scpAB sdaAA sdaAB sfp sigW smc speA spoIIIAD spoIVFA spoIVFB spoVS suhB sul tkt trmD truB ung uppS yaaQ yaaR yabF yabO yazC ybbP ybbR ydcK yebG yerH yetN yfhJ yfmKLMO ygaBCD yhaL yhcV yheA yisL yitKL yizA yktAB ykzG ylaH ylbFGMNP yloUV ylqC ylxMPQRS ymdAB yneABER ynzC yozE ypeQ ypsA yqeGHIKLM yqfC yqgNQY yqhP yqzF yrdA yrrKLM yrzBL yslB ysxB ytpI ytwI yuiB yutDEM yuzB yvcKL ywfO ywnH yybPRST yycHI |
| group 4 | yacOP yueI yazC yvcS ywhD ylaL yozC yetH yqjB artMQ sigH ispDF cysS gltX cysE radA yqjAB mecB yutGH yneR ftsL cinA veg ydaS ypjQ yheA yojF ymxH fabR ymfFGHIJ lgt mreC recFG ctsR ybaF yheAB yqgQ yhaMO ywbD aroCF tyrA cwlJ yhaMNO rpe tmk rsgA ksgA thiN smpB topA cca |
| group 5 | ksgA ecfAB ecfT ytlC cbiOA cbiOB rpsI rpoA truA rplQ rplM skfE ykoCD yuiG bioY |
| group 6 | ywpF ywhD ymfK ywzC ylaL ywjG ypjB bofC ppnKA ndk rpoZ rpoABCE relA pyk folE yjbLMO yaaC ytzF lspA prfB truAB rluB |
| group 7 | yrvN polA msrB pheST metG alaS ispG ytpPQRST ytoPQ ysdC ytzB comEA |
Group 1 clusters a subunit of the DNA replication complex, DnaH(YabA), together with putative RNA binding proteins (YaaA and YabB). Distant homologs exist outside of Firmicutes, in particular in Eukarya.
Group 2 comprizes proteins involved in the Firmicute degradosome network and possibly involved in stress management (YabR and YugI (general stress protein GSP13 [44], essential in Staphylococcus aureus [45]), both displaying a S1 motif [46]).
Group 3, the largest group, is organized around several proteins, most of which small or very small. They have often a known 3D structure but do not yet have an idenfied function. They are involved in RNA metabolism (transcription, RNA modification and turnover): YitL, an RNA binding protein, RnpZA(YkzG), a component of the omega subunit of RNA polymerase, YktA (possibly involved in polyamine metabolism, YlbG (possibly involved in activity of a conserved small RNA, CsfG [47]), YozE, of unknown function, YqgQ (putative single-strand nucleic acid binding protein involved in transcription [48]), and finally YrzL (essential in S. aureus [45]) and YutD, unknown proteins that are possibly hydrolases. With STRING we observed that this cluster is further connected to RnjA, the non-orthologous functional equivalent of RNase E in proteobacteria [49]. Many of these proteins are members of UniProt Unknown Protein Families (UPFs), most of which associated to Firmicutes and sometimes Archaea and Eukarya. This cluster further connects via YqgQ to DNA replication and recombination, to energy-dependent proteases, to divalent metal transporters and to proteins involved in shaping the cell. Via YloU (with a paralog, YqhY), YloV (dihydroxyacetone kinase-like), YlxM (conserved in Mycoplasma sp.), YnzC, YfmM (proposed to code for polyphosphate-AMP phosphotransferase in Staphylococcus epidermidis [50]), YmdB (putative phosphoesterase), YqeG (essential in S. aureus [45]), this cluster again comprises a large number of UPFs. It has some connection with the recombination machinery (RecA, RecG) and the envelope (PlsX, Ffh). YloU has similarity to yeast S-adenosylmethionine-dependent tRNA (uracil-5-)-methyltransferase. This may be significant, because in A+T-rich Firmicutes this activity differs from that in most other organisms, TrmFO, the methylating activity, depending on methylene-tetrahydrofolate rather than AdoMet, thus suggesting recruitment for another RNA modification activity [51]. Finally a sub-cluster of this large group associates YlbM (member of a ribonucleoprotein complex), YqeG (hydrolase), YqeK, YqeH (phosphohydrolase), and YybT (a putative phosphodiesterase, with motif GGDQV related to that of cyclic-diGMP synthesis and degradation). It is connected to Cca, RplI and YqeI, a putative RNA binding protein. Again, this cluster has a clear RNA metabolism flavour.
Group 4 comprises YmfF and YmfG, two peptidyl hydrolases of unknown substrates, perhaps associated to transport of peptidyl siderophores [52] and connects to YheA (related to metabolism of aromatics (YwbD, AroC, TyrA, AroF)). It contains proteins such as YqjA (a putative membrane bound protein), YacP (possibly involved in ribosomal RNA maturation), connected to a network with tRNA synthetases CysS and GltX and with sigma factor SigH. It also comprises YhaM (a 3'-5' oligonucleotidase) and YutG (a putative phosphatidyl-glycerophosphatase A).
Group 5 has now an identified function: it corresponds to the EcfABT energy-dependent activating part of a multi-substrate transporter [53].
Group 6 clusters together YjbL, a putative phosphatase, YjbO, similar to RluD pseudouridylate synthase and YjbM, connected to RelA and RNA polymerase (RpoABCZ) as expected from its putative function in ppGpp synthesis and turnover. This homolog of RelA/SpoT seems to be specific to A+T-rich Firmicutes. It is also connected to riboflavin and folate biosynthesis.
Group 7 associates YtpP and YtpR, connected to tRNA synthetases PheST, AlaS and MetG, as well as DNA polymerase PolA and methionine sulfoxide reductase MsrA. It contains also YtoP (FrvX, a glutamyl aminopeptidase in E. coli).
Finally, the 20 remaining genes do not belong to clear clusters and do not have obvious functions. Yet, six among them (ykuJ, ylmH, yqxC, ysgA, ytqB, ywlB) code for proteins having RNA binding properties. ynbB and yneF are connected to sulfur metabolism and possibly tRNA modification. Protein YtxG has an homolog involved in DNA repair in Mus musculus. YodJ and YtcB are protein likely to be involved in murein synthesis or turnover.
Discussion
Cells encode several DNA polymerases that fulfill a variety of reactions: genome replication, repair and recombination. Eukarya have two types of DNA replicases. In the same way, the genomes of the Firmicutes clade have a unique heterodimeric DNA polymerase III α-subunits, PolC and DnaE [54]. In contrast the E. coli replicase is made of two identical DnaE subunits. The difference between the replicase of E. coli and that of B. subtilis is further reflected by the composition of the other subunits of DNA polymerase III that differ from those in other clades. In E. coli, a single protein, the AAA+ ATPase DnaC (counterpart of DnaI in B. subtilis), is used to load helicase DnaB [55]. In contrast, in B. subtilis and other low-G+C Firmicutes, three different proteins, DnaD, DnaB (no counterpart in E. coli, used for loading to DnaC), and the AAA+ ATPase DnaI, are needed to load the replicative helicase (DnaC in B. subtilis is the counterpart of DnaB in E. coli). DnaA binds first, followed by DnaD and then DnaB, and finally the DnaI-mediated loading of helicase occurs [56].
The existence of two replicases implies a physical difference in the replication process that should be reflected in a bias in nucleotide strand composition. Analysis of compositional strand asymmetries of prokaryotic genome sequences in terms of the presence or absence of PolC has found not only a correlation with PolC [12], but also with a purine asymmetry [7]. This latter asymmetry is however probably not the result of the physical differences in the leading and lagging strands replication, but, rather, the consequence of the increase in the gene content of the leading strand in organisms with PolC, which alleviates conflicts between the transcription and replication machineries [10,57]. In particular it was noticed that G seems to favour the leading strand in most bacterial genomes, which fits with an over representation of genes in this strand in Firmicutes, as coding regions are overrepresenting GNN codons [58].
DNA composition analysis is of limited interest per se, as it does not provide much information about gene functions. The present study showed that while the functional connection of the DnaE subunit spans the whole domain Bacteria, with almost all functions that are expected to make a minimal genome, this is not so of the PolC co-evolving genes. Furthermore, the latter group of genes seems highly specific and related to original features of biological processes, RNA metabolism in particular.
PolC is the subunit of DNA replicase that replicates the leading strand of the DNA double helix in Firmicutes. Structural data on the PolC family of replicases shows that it evolved separately from DnaE [59]. This process requires in addition to PolC, ten other proteins. In addition to these 11 proteins, lagging strand replication requires DnaE and primase [56]. Progression of the replisome implies that DnaE uses Okasaki fragments as primers, and these fragments need to be removed, so that they do not interfere with progression of PolC-mediated leading strand replication progression [60]. It is even suspected that, because replication of the lagging strand must be slow, bacterial replicases are made of not of two, but three replication subunits, two of them involved in replication of the lagging strand [61].
In this context, management of the degradation of RNA derived from Okazaki fragments has a central importance. In particular degradation of nanoRNAs, fragments of size smaller than 5 nts, becomes crucial, as they can enter the replication bubble. It is established that PolC discriminates against RNA primers while DnaE uses RNA primers efficiently [54,56]. As a consequence it can be expected that PolC co-evolved with a particular type of RNA degradosome. Indeed, in the present work, many proteins co-evolving with PolC could be seen as associated to properties of a Firmicutes-specific degradosome (or downstream from it) [39], comprizing nanoRNases such as NrnA [62], an RNase which degrades short oligoribonucleotides (and which is present in the smallest genome of an autonomous living organism, M. genitalium) but also RnmV, MrnC(YazC), YacP, Rny(YmdA), YhaM, YkzG, YlbM, YlmH, RnhC(YsgB) and YybT, proteins that are not present in the E. coli degradosome (Additional file 1). The presence of a particular type of RNase H (RnhC), required to hydrolyze RNAs belonging to RNA-DNA hybrids, essential in B. subtilis [63], is particularly revealing, as it further supports the conjecture that the co-evolution we observed is related to RNA turnover. A further related observation is that there is no global counterpart of the degradosome proteins in those evolving with DnaE, except for the core phosphorylase, polynucleotide phosphorylase, and a specific RNase H (RnhB) of descent different from that of RnhC. NanoRNases (essential for degrading Okazaki fragments) have now been identified from three descents: Orn in gamma-Proteobacteria, NrnA in Firmicutes) and NrnC in alpha-Proteobacteria [64]. This indicates that there may exist more than two degradosomes in the Bacteria domain. Further work similar to the one presented here will tell.
Finally, we remarked that the genes that co-evolve with polC have often counterparts in the three domains of life. This suggested that, despite their absence from the genes co-evolving with the majority of the essential cellular processes, these genes are of very ancient descent. Interestingly, many of the corresponding functions are related to RNA metabolism but also to phosphate or polyphosphate metabolism.
Conclusion
Analysis of the genes co-evolving with the two forms of DNA replicase found in Bacteria, DnaE and PolC revealed that, while DnaE co-evolved with the translation and transcription machineries, PolC co-evolved with proteins that do not belong to the same group. In particular PolC co-evolved with a form of the RNA degradation machinery that is distinct from that characterized in gamma-Proteobacteria, the E. coli degradosome [38]. Among other possibilities, this observation suggests that, while there may exist a last common ancestor to the translation/transcription machinery, this was probably not so for the machineries leading to RNA turnover, which may have appeared independently on several occasions. The RNA world, that developed RNA-centered metabolism [65], predated the RNA genome world, in which RNA synthesis and turnover must have been essential. Our observations are consistent with the discovery of DNA and DNA replication at least twice [66] suggesting that the origin of present living organisms was a community of organisms developing more or less independently from one another (the progenote hypothesis [67,68]), splitting and fusing as time elapsed until the present domains were more stably defined.
Authors' contributions
SE and DV designed and implemented the PhyloProfile algorithm. SE wrote part of the manuscript. CM coordinated the implementation in the MicroScope platform. AD designed and coordinated this study, performed the biological interpretation of the results and wrote the bulk of the manuscript. All authors read, modified and approved the final manuscript.
Supplementary Material
Gene clusters obtained using PhyloProfile with a threshold of 0.77. The first two clusters comprise the dnaE and the polC clusters. At the end of the list many clusters contain only two genes that co-evolve significantly.
Contributor Information
Stefan Engelen, Email: stefan.engelen@genoscope.cns.fr.
David Vallenet, Email: david.vallenet@genoscope.cns.fr.
Claudine Médigue, Email: claudine.medigue@genoscope.cns.fr.
Antoine Danchin, Email: antoine.danchin@normalesup.org.
Acknowledgements
This work benefited from the support of the European Union Microme programme FP7-KBBE-2007-3-2-08-222886.
References
- Rocha EP, Guerdoux-Jamet P, Moszer I, Viari A, Danchin A. Implication of gene distribution in the bacterial chromosome for the bacterial cell factory. J Biotechnol. 2000;78:209–219. doi: 10.1016/S0168-1656(00)00197-8. [DOI] [PubMed] [Google Scholar]
- Sueoka N. On the genetic basis of variation and heterogeneity of DNA base composition. Proc Natl Acad Sci USA. 1962;48:582–592. doi: 10.1073/pnas.48.4.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobry JR. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 1996;13:660–665. doi: 10.1093/oxfordjournals.molbev.a025626. [DOI] [PubMed] [Google Scholar]
- Rocha EP, Danchin A, Viari A. Universal replication biases in bacteria. Mol Microbiol. 1999;32:11–16. doi: 10.1046/j.1365-2958.1999.01334.x. [DOI] [PubMed] [Google Scholar]
- Frank AC, Lobry JR. Oriloc: prediction of replication boundaries in unannotated bacterial chromosomes. Bioinformatics. 2000;16:560–561. doi: 10.1093/bioinformatics/16.6.560. [DOI] [PubMed] [Google Scholar]
- Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessieres P, Bolotin A, Borchert S. et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- Hu J, Zhao X, Yu J. Replication-associated purine asymmetry may contribute to strand-biased gene distribution. Genomics. 2007;90:186–194. doi: 10.1016/j.ygeno.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Bao Q, Tian Y, Li W, Xu Z, Xuan Z, Hu S, Dong W, Yang J, Chen Y, Xue Y. et al. A complete sequence of the T. tengcongensis genome. Genome Res. 2002;12:689–700. doi: 10.1101/gr.219302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton BR. Strand asymmetry and codon usage bias in the chloroplast genome of Euglena gracilis. Proc Natl Acad Sci USA. 1999;96:5123–5128. doi: 10.1073/pnas.96.9.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EP, Danchin A. Essentiality, not expressiveness, drives gene-strand bias in bacteria. Nat Genet. 2003;34:377–378. doi: 10.1038/ng1209. [DOI] [PubMed] [Google Scholar]
- Wagner E, Lykke-Andersen J. mRNA surveillance: the perfect persist. J Cell Sci. 2002;115:3033–3038. doi: 10.1242/jcs.115.15.3033. [DOI] [PubMed] [Google Scholar]
- Rocha E. Is there a role for replication fork asymmetry in the distribution of genes in bacterial genomes? Trends Microbiol. 2002;10:393–395. doi: 10.1016/S0966-842X(02)02420-4. [DOI] [PubMed] [Google Scholar]
- Gefter ML, Hirota Y, Kornberg T, Wechsler JA, Barnoux C. Analysis of DNA polymerases II and 3 in mutants of Escherichia coli thermosensitive for DNA synthesis. Proc Natl Acad Sci USA. 1971;68:3150–3153. doi: 10.1073/pnas.68.12.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love E, D'Ambrosio D, Brown NC. Mapping of the gene specifying DNA polymerase III of Bacillus subtilis. Mol Gen Genet. 1976;144:313–321. doi: 10.1007/BF00341730. [DOI] [PubMed] [Google Scholar]
- McHenry CS. Chromosomal replicases as asymmetric dimers: studies of subunit arrangement and functional consequences. Mol Microbiol. 2003;49:1157–1165. doi: 10.1046/j.1365-2958.2003.03645.x. [DOI] [PubMed] [Google Scholar]
- Le Chatelier E, Becherel OJ, d'Alencon E, Canceill D, Ehrlich SD, Fuchs RP, Janniere L. Involvement of DnaE, the second replicative DNA polymerase from Bacillus subtilis, in DNA mutagenesis. J Biol Chem. 2004;279:1757–1767. doi: 10.1074/jbc.M310719200. [DOI] [PubMed] [Google Scholar]
- Vendeville A, Lariviere D, Fourmentin E. An inventory of the bacterial macromolecular components and their spatial organization. FEMS Microbiol Rev. 2010;35:395–414. doi: 10.1111/j.1574-6976.2010.00254.x. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerkandl E. On the molecular evolutionary clock. J Mol Evol. 1987;26:34–46. doi: 10.1007/BF02111280. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynen M, Snel B, Lathe W, Bork P. Predicting protein function by genomic context: quantitative evaluation and qualitative inferences. Genome Res. 2000;10:1204–1210. doi: 10.1101/gr.10.8.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazko GV, Mushegian AR. Detection of evolutionarily stable fragments of cellular pathways by hierarchical clustering of phyletic patterns. Genome Biol. 2004;5:R32. doi: 10.1186/gb-2004-5-5-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker D, Pagel M. Predicting functional gene links from phylogenetic-statistical analyses of whole genomes. PLoS Comput Biol. 2005;1:e3. doi: 10.1371/journal.pcbi.0010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Jensen LJ, von Mering C, Bork P. Analysis of genomic context: prediction of functional associations from conserved bidirectionally transcribed gene pairs. Nat Biotechnol. 2004;22:911–917. doi: 10.1038/nbt988. [DOI] [PubMed] [Google Scholar]
- Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA. Protein interaction maps for complete genomes based on gene fusion events. Nature. 1999;402:86–90. doi: 10.1038/47056. [DOI] [PubMed] [Google Scholar]
- Pazos F, Valencia A. Similarity of phylogenetic trees as indicator of protein-protein interaction. Protein Eng. 2001;14:609–614. doi: 10.1093/protein/14.9.609. [DOI] [PubMed] [Google Scholar]
- Vallenet D, Engelen S, Mornico D, Cruveiller S, Fleury L, Lajus A, Rouy Z, Roche D, Salvignol G, Scarpelli C. et al. MicroScope: a platform for microbial genome annotation and comparative genomics. Database (Oxford) 2009;2009:bap021. doi: 10.1093/database/bap021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer F, Morgat A, Labarre L, Pothier J, Viari A. Syntons, metabolons and interactons: an exact graph-theoretical approach for exploring neighbourhood between genomic and functional data. Bioinformatics. 2005;21:4209–4215. doi: 10.1093/bioinformatics/bti711. [DOI] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medigue C, Krin E, Pascal G, Barbe V, Bernsel A, Bertin PN, Cheung F, Cruveiller S, D'Amico S, Duilio A. et al. Coping with cold: the genome of the versatile marine Antarctica bacterium Pseudoalteromonas haloplanktis TAC125. Genome Res. 2005;15:1325–1335. doi: 10.1101/gr.4126905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P. et al. Essential Bacillus subtilis genes. Proc Natl Acad Sci USA. 2003;100:4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbe V, Cruveiller S, Kunst F, Lenoble P, Meurice G, Sekowska A, Vallenet D, Wang T, Moszer I, Medigue C. et al. From a consortium sequence to a unified sequence: the Bacillus subtilis 168 reference genome a decade later. Microbiology. 2009;155:1758–1775. doi: 10.1099/mic.0.027839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley M, Abe T, Arnaud MB, Berlyn MK, Blattner FR, Chaudhuri RR, Glasner JD, Horiuchi T, Keseler IM, Kosuge T. et al. Escherichia coli K-12: a cooperatively developed annotation snapshot--2005. Nucleic Acids Res. 2006;34:1–9. doi: 10.1093/nar/gkj405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Rocha E, Danchin A. How essential are nonessential genes? Mol Biol Evol. 2005;22:2147–2156. doi: 10.1093/molbev/msi211. [DOI] [PubMed] [Google Scholar]
- Carpousis AJ. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu Rev Microbiol. 2007;61:71–87. doi: 10.1146/annurev.micro.61.080706.093440. [DOI] [PubMed] [Google Scholar]
- Danchin A. A phylogenetic view of bacterial ribonucleases. Prog Mol Biol Transl Sci. 2009;85:1–41. doi: 10.1016/S0079-6603(08)00801-5. [DOI] [PubMed] [Google Scholar]
- Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M. et al. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deriu D, Briand C, Mistiniene E, Naktinis V, Grutter MG. Structure and oligomeric state of the mammalian tumour-associated antigen UK114. Acta Crystallogr D Biol Crystallogr. 2003;59:1676–1678. doi: 10.1107/S0907444903014306. [DOI] [PubMed] [Google Scholar]
- Lutke-Eversloh T, Stephanopoulos G. Combinatorial pathway analysis for improved L-tyrosine production in Escherichia coli: identification of enzymatic bottlenecks by systematic gene overexpression. Metab Eng. 2008;10:69–77. doi: 10.1016/j.ymben.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Kitko RD, Cleeton RL, Armentrout EI, Lee GE, Noguchi K, Berkmen MB, Jones BD, Slonczewski JL. Cytoplasmic acidification and the benzoate transcriptome in Bacillus subtilis. PLoS One. 2009;4:e8255. doi: 10.1371/journal.pone.0008255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Yu B, Hu J, Xia W, Jin C, Xia B. 1H, 13C, and 15N resonance assignments of a general stress protein GSP13 from Bacillus subtilis. Biomol NMR Assign. 2008;2:163–165. doi: 10.1007/s12104-008-9111-y. [DOI] [PubMed] [Google Scholar]
- Chaudhuri RR, Allen AG, Owen PJ, Shalom G, Stone K, Harrison M, Burgis TA, Lockyer M, Garcia-Lara J, Foster SJ. et al. Comprehensive identification of essential Staphylococcus aureus genes using Transposon-Mediated Differential Hybridisation (TMDH) BMC Genomics. 2009;10:291. doi: 10.1186/1471-2164-10-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorski P, Proux F, Cheraiti C, Dreyfus M, Hermann-Le Denmat S. The deleterious effect of an insertion sequence removing the last twenty percent of the essential Escherichia coli rpsA gene is due to mRNA destabilization, not protein truncation. J Bacteriol. 2007;189:6205–6212. doi: 10.1128/JB.00445-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchais A, Duperrier S, Durand S, Gautheret D, Stragier P. CsfG, a sporulation-specific, small non-coding RNA highly conserved in endospore formers. RNA Biol. 2011;8 doi: 10.4161/rna.8.3.14998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshminarasimhan D, Eswaramoorthy S, Burley SK, Swaminathan S. Structure of YqgQ protein from Bacillus subtilis, a conserved hypothetical protein. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:8–11. doi: 10.1107/S1744309109047009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon C. What is the role of RNase J in mRNA turnover? RNA Biol. 2010;7:316–321. doi: 10.4161/rna.7.3.11913. [DOI] [PubMed] [Google Scholar]
- Wei W, Cao Z, Zhu YL, Wang X, Ding G, Xu H, Jia P, Qu D, Danchin A, Li Y. Conserved genes in a path from commensalism to pathogenicity: comparative phylogenetic profiles of Staphylococcus epidermidis RP62A and ATCC12228. BMC Genomics. 2006;7:112. doi: 10.1186/1471-2164-7-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbonavicius J, Brochier-Armanet C, Skouloubris S, Myllykallio H, Grosjean H. In vitro detection of the enzymatic activity of folate-dependent tRNA (Uracil-54,-C5)-methyltransferase: evolutionary implications. Methods Enzymol. 2007;425:103–119. doi: 10.1016/S0076-6879(07)25004-9. [DOI] [PubMed] [Google Scholar]
- Miethke M, Schmidt S, Marahiel MA. The major facilitator superfamily-type transporter YmfE and the multidrug-efflux activator Mta mediate bacillibactin secretion in Bacillus subtilis. J Bacteriol. 2008;190:5143–5152. doi: 10.1128/JB.00464-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Beek J, Duurkens RH, Erkens GB, Slotboom DJ. Quaternary structure and functional unit of energy coupling factor (ECF)-type transporters. J Biol Chem. 2011;286:5471–5475. doi: 10.1074/jbc.M110.199224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHenry CS. Breaking the rules: bacteria that use several DNA polymerase IIIs. EMBO Rep. 2011;12:408–414. doi: 10.1038/embor.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu RE, Yao NY, O'Donnell M. Single-molecule analysis of the Escherichia coli replisome and use of clamps to bypass replication barriers. FEBS Lett. 2010;584:2596–2605. doi: 10.1016/j.febslet.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders GM, Dallmann HG, McHenry CS. Reconstitution of the B. subtilis replisome with 13 proteins including two distinct replicases. Mol Cell. 2010;37:273–281. doi: 10.1016/j.molcel.2009.12.025. [DOI] [PubMed] [Google Scholar]
- Rocha EP, Danchin A. Gene essentiality determines chromosome organisation in bacteria. Nucleic Acids Res. 2003;31:6570–6577. doi: 10.1093/nar/gkg859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DJ, Fresco JR. Greater GNN pattern bias in sequence elements encoding conserved residues of ancient proteins may be an indicator of amino acid composition of early proteins. Gene. 2003;303:177–185. doi: 10.1016/s0378-1119(02)01176-9. [DOI] [PubMed] [Google Scholar]
- Evans RJ, Davies DR, Bullard JM, Christensen J, Green LS, Guiles JW, Pata JD, Ribble WK, Janjic N, Jarvis TC. Structure of PolC reveals unique DNA binding and fidelity determinants. Proc Natl Acad Sci USA. 2008;105:20695–20700. doi: 10.1073/pnas.0809989106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao NY, O'Donnell M. Replisome structure and conformational dynamics underlie fork progression past obstacles. Curr Opin Cell Biol. 2009;21:336–343. doi: 10.1016/j.ceb.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu RE, Kurth I, O'Donnell ME. Single-molecule studies reveal the function of a third polymerase in the replisome. Nat Struct Mol Biol. 2011. [DOI] [PMC free article] [PubMed]
- Mechold U, Fang G, Ngo S, Ogryzko V, Danchin A. YtqI from Bacillus subtilis has both oligoribonuclease and pAp-phosphatase activity. Nucleic Acids Res. 2007;35:4552–4561. doi: 10.1093/nar/gkm462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itaya M, Omori A, Kanaya S, Crouch RJ, Tanaka T, Kondo K. Isolation of RNase H genes that are essential for growth of Bacillus subtilis 168. J Bacteriol. 1999;181:2118–2123. doi: 10.1128/jb.181.7.2118-2123.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MF, Cescau S, Mechold U, Wang J, Cohen D, Danchin A, Boulouis HJ, Biville F. Identification of a new nanoRNase in Bartonella. Microbiology. 2012. in press . [DOI] [PubMed]
- Robertson MP, Joyce GF. The Origins of the RNA World. Cold Spring Harb Perspect Biol. 2010. in press . [DOI] [PMC free article] [PubMed]
- Leipe DD, Aravind L, Koonin EV. Did DNA replication evolve twice independently? Nucleic Acids Res. 1999;27:3389–3401. doi: 10.1093/nar/27.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giulio M. The last universal common ancestor (LUCA) and the ancestors of archaea and bacteria were progenotes. J Mol Evol. 2011;72:119–126. doi: 10.1007/s00239-010-9407-2. [DOI] [PubMed] [Google Scholar]
- Woese CR, Fox GE. The concept of cellular evolution. J Mol Evol. 1977;10:1–6. doi: 10.1007/BF01796132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene clusters obtained using PhyloProfile with a threshold of 0.77. The first two clusters comprise the dnaE and the polC clusters. At the end of the list many clusters contain only two genes that co-evolve significantly.