

A developmental program termed epithelial–mesenchymal transition (EMT) plays a critical role in promoting metastasis in epithelial-derived carcinomas. Recent studies also implicate its reverse program, mesenchymal–epithelial transition (MET), in this metastatic process. In this review, Tsai and Yang discuss the functional requirement of EMT and/or MET during the individual steps of tumor metastasis and the potential for targeting these programs when treating metastatic diseases.
Keywords: epithelial–mesenchymal transition (EMT), mesenchymal–epithelial transition (MET), carcinoma metastasis, extravasation, intravasation, invasion, tumor dormancy
Abstract
Tumor metastasis is a multistep process by which tumor cells disseminate from their primary site and form secondary tumors at a distant site. Metastasis occurs through a series of steps: local invasion, intravasation, transport, extravasation, and colonization. A developmental program termed epithelial–mesenchymal transition (EMT) has been shown to play a critical role in promoting metastasis in epithelium-derived carcinoma. Recent experimental and clinical studies have improved our knowledge of this dynamic program and implicated EMT and its reverse program, mesenchymal–epithelial transition (MET), in the metastatic process. Here, we review the functional requirement of EMT and/or MET during the individual steps of tumor metastasis and discuss the potential of targeting this program when treating metastatic diseases.
Epithelial and mesenchymal cell types have long been recognized by their unique cell morphology and organization in tissues. Epithelial cells form polarized sheets or layers of cells that are connected laterally via several types of cellular junctions, including adherens junctions, desmosomes, and tight junctions. In addition, epithelial cells anchor themselves to the underlying basement membrane via hemidesmosomes to maintain apical–basal polarity. Both desmosomes and hemidesmosomes further connect with the epithelial-specific cytokeratin intermediate filaments. In contrast, mesenchymal cells embed themselves inside the extracellular matrix (ECM) and rarely establish tight contact with neighboring cells. During specific embryonic morphogenesis processes such as mesoderm formation and neural crest development, epithelial cells can exhibit enormous plasticity and transit into a mesenchymal state by activating the epithelial–mesenchymal transition (EMT) program. After EMT, these cells lose their epithelial junctions and switch to producing vimentin filaments. The functional hallmark of the EMT program is to allow stationary epithelial cells to gain the ability to migrate and invade during developmental morphogenesis (Boyer and Thiery 1993; Hay 1995).
Although epithelial cells convert into the mesenchymal state during developmental EMT, entering the EMT program is not necessarily an irreversible commitment, as evident during kidney tubule formation. These epithelial cells can activate a transitory EMT program and then undergo a reverse process called mesenchymal–epithelial transition (MET) to continue their differentiation paths (Thiery et al. 2009; Lim and Thiery 2012). In many instances, the identification of an epithelial versus a mesenchymal state can be relatively fluid, and a partial EMT/MET frequently occurs to fulfill unique developmental tasks. These dynamic EMT/MET events highlight the enormous flexibility of presumably differentiated cells during morphogenesis.
In the past decade, an increasing number of studies have provided strong evidence for the reinitiation of the EMT developmental program in carcinoma progression and metastasis. This EMT program in tumor metastasis possesses many morphological and molecular features similar to those of the developmental EMT program. Importantly, due to the heterogeneity and constantly evolving microenvironment in human tumors, the EMT program in metastasis adapts to these conditions to allow tumor cells to successfully metastasize.
While EMT has been accepted as a critical program during embryogenesis, its role in carcinoma metastasis has been under debate (Tarin et al. 2005; Thomson et al. 2005; Garber 2008; Ledford 2011; Chui 2013). Many cell culture and mouse tumor model studies have clearly demonstrated the importance of EMT in tumor progression. However, the EMT process in human cancer, if present, remains difficult to identify, since carcinoma cells undergoing EMT share many similar morphological and molecular features with surrounding stromal fibroblasts. Furthermore, although primary tumors and circulating tumor cells (CTCs) present EMT features, distant metastases are generally epithelial in morphology, suggesting that, if functional, the EMT program is likely to be dynamically regulated during metastasis. In 2002, Jean Paul Thiery (2002), proposed the reversible EMT metastasis model in which primary epithelial tumor cells activate EMT to invade and disseminate throughout the body, while, upon arriving at distant sites, disseminated tumor cells (DTCs) undergo a reversion process, or MET, to form epithelial metastases. This intriguing hypothesis has brought EMT research to the forefront of carcinoma metastasis study.
The molecular program of EMT
Given that a number of recent reviews have focused on the molecular pathways regulating EMT/MET (Thiery et al. 2009; De Craene and Berx 2013; Zheng and Kang 2013), this section aims to provide an overview of the cellular and molecular definition for EMT/MET and establish a foundation for detailed discussions of these pathways in the context of tumor metastasis. The complex morphological and cellular changes during EMT require the cooperation of a large number of molecular signaling pathways and regulators. Based on their functionalities during EMT, we categorize them into three groups: the effector molecules that execute the EMT program (EMT effectors), the transcription factors that orchestrate the EMT program (EMT core regulators), and the extracellular cues that activate the EMT program (EMT inducers).
EMT effectors
Many of the hallmark EMT effector molecules are subcellular structure proteins that demarcate the epithelial or mesenchymal identity of a cell. During EMT, key molecular components of these structures are subjected to various levels of regulation. For example, the genes encoding various epithelial junction proteins, such as E-cadherin, α-catenin, and γ-catenin, are down-regulated at the mRNA and protein levels. Among them, E-cadherin is regarded as a gatekeeper of the epithelial state in various epithelial cell types. During EMT, E-cadherin gene transcriptional repression (Batlle et al. 2000; Cano et al. 2000; Hajra et al. 2002), promoter methylation (Graff et al. 1995; Kanai et al. 1997; Saito et al. 1998), and protein phosphorylation and degradation (Zhou et al. 2004; Bachelder et al. 2005; Lester et al. 2007) have all been observed in response to various inducing signals. Furthermore, intermediate filaments are shown to switch from cytokeratin to vimentin during EMT. Increased vimentin levels are also a consistent marker during various EMT events, while cytokeratin subtype switches tend to be variable and tissue type-specific.
Some additional key EMT effector molecules are proteins that promote cell migration and invasion during EMT. Fibronectin, an extracellular protein required for mesenchymal cell migration, is frequently induced upon activation of EMT. To promote cellular invasion through the ECM during EMT, a PDGF/PDGF receptor (PDGFR) autocrine loop is activated to promote invadopodia-mediated ECM degradation (Eckert et al. 2011). A number of nonepithelial cadherins, such as N-cadherin, and cell surface proteins, such as CD44 (Kuo et al. 2009) and integrin β6 (Bates et al. 2005), are induced and thought to be critical for proper cell migration. All of these proteins have been used to define the occurrence of EMT in tumors.
EMT core regulators
Execution of the EMT program involves the transcriptional alteration of many genes regulating cell adhesion, mesenchymal differentiation, cell migration, and invasion. In general, three core groups of transcriptional regulators have been consistently shown to be critical during various EMT events, thus being regarded as the core EMT regulators.
The first group is the transcription factors of the Snail zinc finger family, including Snail1 and Snail2, both of which are capable of directly binding to the E-boxes of the E-cadherin promoter to repress its transcription (Batlle et al. 2000; Cano et al. 2000; Hajra et al. 2002). The second group is the distantly related zinc finger E-box-binding homeobox family proteins Zeb1 and Zeb2, which are also able to suppress E-cadherin transcription (Comijn et al. 2001; Eger et al. 2005) via a double-negative feedback loop controlling Zeb1/Zeb2 and miRNA-200 family expression (Christoffersen et al. 2007; Bracken et al. 2008; Burk et al. 2008; Gregory et al. 2008; Korpal and Kang 2008; Korpal et al. 2008; Park et al. 2008; Kim et al. 2011b). Both the Snail and Zeb families of transcription factors have also been shown to repress the expression of other cellular junction proteins, such as claudins and ZO-1 (Ohkubo and Ozawa 2004; Vandewalle et al. 2005). The third group is the basic helix–loop–helix (bHLH) family of transcription factors, including Twist1 (Yang et al. 2004), Twist2 (Fang et al. 2011), and E12/E47 (Perez-Moreno et al. 2001), all of which can induce EMT alone or cooperatively. For example, Twist1 can not only repress E-cadherin through induction of Snail transcription factors (Li et al. 1995; Yang et al. 2004; Casas et al. 2011) but also activate programs associated with tumor invasion (Eckert et al. 2011), thus coordinating two major aspects of the EMT program.
EMT inducers
During tumor progression, EMT induction in tumor cells has not been associated with genetic alterations of the EMT core transcription factors, perhaps due to their essential roles in embryonic morphogenesis. Instead, carcinoma cells are thought to undergo EMT in response to a combination of extracellular signals in the tumor microenvironment. Many EMT-inducing signals tend to be cell type- or tissue type-specific and probably require the cooperation between multiple pathways. All major developmental signaling pathways, including TGF-β, Wnt, Notch, and growth factor receptor signaling cascades, have been implicated in some aspect of the EMT program. Most notably, the TGF-β pathway appears to be a primary inducer of EMT (Katsuno et al. 2013). For example, TGF-β and BMPs have been shown to induce the EMT core transcription factors Snail1/2, Zeb1/2, and Twist1 (Thiery et al. 2009; Eckert et al. 2011). Interestingly, the ability of TGF-β/Smad signaling to induce EMT depends on the cooperation of several other pathways, including activation of the Ras kinase cascade via activated receptor tyrosine kinases (RTKs) or Ras mutations (Grunert et al. 2003) and cooperation from the Wnt/β-catenin/LEF-1 signaling pathway (Nawshad et al. 2005). One of the major sources of TGF-β in tumors is the stromal fibroblast cells in the tumor microenvironment (Hanahan and Weinberg 2011).
In addition to growth factor signaling, inflammatory cytokines and hypoxia in the tumor microenvironment have also been shown to promote EMT. The inflammatory cytokine TNFα can stabilize Snail1 via NF-κB activation (Wu et al. 2009) and induce Twist1 expression via IKK-β and NF-κB p65 activation (Li et al. 2012). Cytokines in the tumor microenvironment can also activate Stat3 via JAK kinases to induce Twist1 expression (Lo et al. 2007; Cheng et al. 2008). Hypoxic responses mediated by HIF-1 were also shown to induce the expression of Twist1 and Snail1 to promote EMT (Yang et al. 2008; Mak et al. 2010). Together, these studies indicate that extracellular cues from the tumor microenvironment play a critical role in activating EMT.
In summary, the EMT program involves a large number of cellular and molecular alterations. Since EMT-inducing signals are diverse and often context-dependent, EMT effectors and core transcription regulators are most widely used as molecular markers of EMT in human cancers. Further analysis of how individual EMT-inducing signals impinge on the EMT core regulators and effectors will provide a more comprehensive inventory of key players in EMT.
EMT/MET in tumor metastasis
The metastatic process is thought to consist of several steps. The initial escape from the primary site requires the epithelial tumor cells to become motile and degrade the underlying basement membrane and ECM; breakdown of these barriers initiates invasion into the nearby tissue parenchyma (step I: invasion). The next step of metastasis is termed intravasation, during which tumor cells invade across the endothelial lamina, penetrate into the blood or lymphatic vessels, and thereby enter the systemic circulation (step II: intravasation). Once in the circulation, only a small number of the disseminated neoplastic cells appear to be capable of surviving various insults within the circulation (step III: systemic transport). Eventually, some of the surviving cells may arrest in the vascular lumen and extravasate through the capillary endothelium into the parenchyma of distant organs (step IV: extravasation). In the new stromal environment, an even smaller subset of tumor cells establish micrometastases with the potential to proliferate into fully malignant, secondary tumors that are clinically detectable and eventually life-threatening (step V: colonization) (Thiery 2002; Fidler 2003; Kalluri and Weinberg 2009).
To clearly define the role of EMT in metastasis, we discuss both experimental and clinical evidence of EMT and its reversion program, MET, during the appropriate individual steps of tumor metastasis (Fig. 1).
Figure 1.
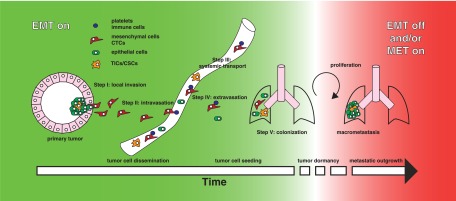
Model for reversible EMT over time. Epithelial cells undergo genetic transformation to become carcinoma in situ. Microenvironmental and genetic factors can promote the malignant conversion of these cells to activate the EMT program. During these early stages of tumor development, tumor cells that have undergone EMT can invade the local matrix (step I) and intravasate into the vasculature (step II). These epithelial–mesenchymal-transitioned cells are then transported in the circulation and survive via various prosurvival mechanisms (step III). At the distant tissue site, maintenance of the EMT program is required to help tumor cells extravasate into the parenchyma (step IV) to establish micrometastases. This initial seeding of tumor cells at distant sites can occur rapidly, after which cells may remain “dormant” for a long period of time. Subsequent colonization in distant organs requires the reversion of EMT and/or activation of the MET program to establish secondary tumors (step V).
Step I: EMT in malignant conversion and local invasion
Malignant conversion
Tumor initiation is often characterized by both genetic changes intrinsic to the tumor cells and alterations in the local microenvironment that promote tumor progression. Activation of the EMT program is classically thought to be a late stage event in malignant cancer to promote metastasis. However, several studies have implicated a possible role of EMT core transcription factors in the initial malignant transformation. Expression of Twist1 mRNA was detected in atypical ductal hyperplasia, a very early stage of primary breast tumor development in the MMTV-Neu mouse tumor model (Husemann et al. 2008). Twist1 was also shown to override oncogene-induced senescence and apoptosis by binding to p53 and promoting its degradation (Valsesia-Wittmann et al. 2004; Ansieau et al. 2008; Lee and Bar-Sagi 2010; Piccinin et al. 2012). By inhibiting p53, Twist1 was able to cooperate with oncogenes such as Her2 and H-ras to promote malignant transformation (Valsesia-Wittmann et al. 2004; Ansieau et al. 2008; Morel et al. 2012; Piccinin et al. 2012), suggesting a potential role of EMT genes in tumor initiation.
Activation of EMT is considered essential to allow carcinoma cells to lose cell–cell junctions and dissociate from each other for single-cell migration and invasion. For example, TGF-β pathway activation in EpH4-Ras mouse mammary carcinoma cells resulted in the loss of E-cadherin-mediated adherens junctions and gain of mesenchymal markers in cell culture and mouse tumor xenografts (Oft et al. 1996, 1998; Janda et al. 2002). Intercrossing the mouse pancreatic β-cell tumor (RIP-Tag2) model with transgenic mice that maintain E-cadherin expression in β-cell tumors arrested tumor development at the early adenoma stage, whereas expression of a dominant-negative form of E-cadherin induced early invasion and metastasis (Perl et al. 1998). Furthermore, mice carrying a genetic deletion of the E-cadherin gene on a mammary-specific p53-null background developed invasive lobular carcinomas, a subtype of breast cancer that presents individual migrating tumor cells (Derksen et al. 2006). Together, these studies strongly support a role of EMT in promoting single-cell invasion in primary tumors.
Degradation of the ECM
Carcinoma invasion requires tumor cells to gain the ability to degrade the underlying basement membrane and ECM. The EMT program is involved in this process through up-regulation of various matrix degradation enzymes by the EMT core regulators. Snail1 expression in MDCK epithelial cells and MCF-7 breast carcinoma cells increased MT1-MMP, MT2-MMP, and MMP9 expression (Olmeda et al. 2007a; Ota et al. 2009) and facilitated the breakdown of the basement membrane (Hotary et al. 2006; Ota et al. 2009). Conversely, Snail1 inhibition in epidermal and breast carcinoma cells decreased MMP9 expression and tumor growth and metastasis (Olmeda et al. 2007a,b). Consistent with these results, Snail2 was found to play an essential role in regulating tumor metastasis through induction of MT4-MMP and MMP2 (Shih et al. 2005; Huang et al. 2009). Interestingly, proteases have also been implicated in activating EMT by disrupting cell–cell junctions. Radisky et al. (2005) showed that MMP3 induces expression of Rac1, which increases reactive oxygen species (ROS) production and Snail1 expression to promote EMT (Orlichenko and Radisky 2008). Together, these results suggest a possible interplay between the loss of cellular junctions and the induction of proteases during EMT to promote tumor invasion.
More recently, there is emerging evidence that EMT transcription factors can also induce the formation of specialized subcellular structures called invadopodia to invade local ECMs (Eckert et al. 2011; Murphy and Courtneidge 2011). Invadopodia are actin-based protrusions that recruit various proteases such as membrane-tethered proteases (MT-MMPs), ADAMs, and MMPs, etc. to cell–matrix contact points to degrade ECM (Murphy and Courtneidge 2011). The EMT core transcription factor Twist1 was found to promote invadopodia formation through induction of PDGFRα expression and Src activation (Eckert et al. 2011). TGF-β was also shown to induce invadopodia formation in EpH4 and MCF10A mammary epithelial cells through up-regulation of Twist1 (Eckert et al. 2011) and the focal adhesion protein Hic-5 (Pignatelli et al. 2012) to promote matrix degradation and invasion. Furthermore, Zeppo1, a novel metastasis promoter that can repress E-cadherin expression, was found to induce EpH4.9 cells to form invadopodia-like structures in three dimensions (Slorach et al. 2011). Together, these studies suggest that the EMT program not only allows carcinoma cells to dissociate from each other but also provides them the ability to degrade ECM for single-cell invasion to initiate the metastatic cascade (Fig. 2).
Figure 2.
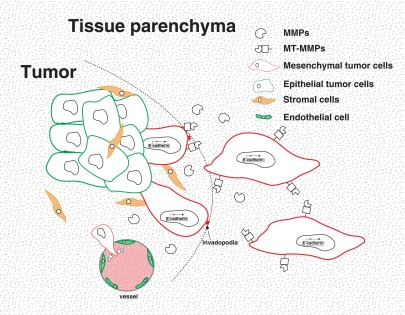
EMT in local invasion and intravasation. Activation of the EMT program is mostly characterized by the loss of E-cadherin expression. In order to invade through local basement membrane (surrounding the tumor or the tumor vasculature), these mesenchymal tumor cells up-regulate several secreted (MMPs) and membrane-tethered (MT-MMPs) proteases to break down ECM components. In addition, EMT factors can up-regulate specialized cellular structures such as invadopodia to promote local invasion. Expression of proteases can further induce EMT by breaking down cell–cell junctions, resulting in a positive feedback loop during malignant transformation of these cells.
Clinical evidence of EMT in primary tumor invasion
In the past, identification of carcinoma cells undergoing EMT in human tumor tissues has largely relied on histological analysis of E-cadherin expression. A partial loss of E-cadherin is associated with carcinoma progression and poor prognosis in various human tumor types, consistent with the role of E-cadherin as a caretaker of the epithelial state in carcinomas (Hirohashi 1998; Vincent-Salomon and Thiery 2003). In a few carcinoma subtypes, E-cadherin is lost at an early stage of the disease, so the tumor types present a permanent EMT phenotype. For example, a portion of lobular breast carcinomas (Berx et al. 1995, 1998) and diffuse gastric cancers (Becker et al. 1994; Oda et al. 1994) contain E-cadherin gene nonsense or frameshift mutations, while transcriptional suppression or post-translational modification of E-cadherin also contributes to the lack of E-cadherin expression in these tumors (Droufakou et al. 2001). Furthermore, in many late stage human carcinomas, E-cadherin expression appears to be heterogeneous, with E-cadherin-negative tumor cells interspersed within foci of E-cadherin-positive areas in the tumor (Bukholm et al. 1998, 2000), suggesting that some carcinoma cells might have undergone EMT.
Recent progress in gene expression profiling has shed more light on the prevalence of EMT in human cancer. Most notably, microarray analysis classified a claudin-low subtype of breast ductal carcinoma with low expression of E-cadherin and found that this subtype was enriched with an EMT gene signature including EMT core transcription factors Snail1, Twist1/2, and Zeb2 (Prat et al. 2010). In addition, immunohistochemical analysis of 28 molecular markers in 479 invasive breast carcinomas revealed clustering of EMT markers in tumors with a basal-like phenotype (Sarrio et al. 2008). Further analysis of these invasive basal/triple-negative subgroups found tumor cells that are double-positive for keratin and vimentin at the invasive front (Sorlie et al. 2001; Livasy et al. 2006; Rakha et al. 2006; Bonnomet et al. 2012). In colorectal carcinoma, ZEB1- and Snail1-positive cells with a mesenchymal morphology at the invasive front showed strong nuclear β-catenin signals (due to an APC mutation), suggesting that colon carcinoma cells, but not stromal cells, have undergone EMT (Brabletz et al. 2001, 2005; Spaderna et al. 2006). Recently, Celesti et al. (2013) performed Twist1 immunostaining in combination with fluorescent in situ hybridization (FISH) analysis of chromosome translocation unique to human colorectal tumor cells and found that 17 out of 20 human colon tumors contained Twist1+ tumor cells with a mesenchymal phenotype. Together, these studies provide convincing clinical evidence of the occurrence of EMT (or at least partial EMT) in primary human carcinomas.
Step II: EMT in tumor cell intravasation
Following local invasion, tumor cells need to undergo an intravasation process to enter into the vasculature (lymphatic or blood vessels) for systemic dissemination. The precise mechanism of how tumor cells cross the endothelial barrier is largely unknown. Because of their size, tumors cells may require additional machinery to intravasate, unlike the smaller leukocytes that rely on diapedesis to migrate between endothelial cell (EC) junctions (Miles et al. 2008). The EMT program is thought to modulate the migratory and invasive properties of carcinoma cells to promote entry into the vasculature.
Technological advances in transendothelial assays, chick chorioallantoic membrane (CAM) assays, and intravital live imaging have facilitated the investigation of EMT during the intravasation process. Using a transendothelial migration assay, Drake et al. (2009) found that Zeb1 expression in PC-3 human prostate cancer cells was required for enhanced migration through the EC barrier and increased metastatic colonization. Using a modified CAM assay that allows visualization of chorionic epithelium-derived vascular basement membrane, Ota et al. (2009) found that MCF-7 breast cancer cells expressing Snail1 could transmigrate through the underlying chorionic basement membrane and intravasate into the host vasculature. More interestingly, they showed that overexpression of Snail1 promoted intravasation through activation of membrane-bound MT1-MMP and MT2-MMP but not secreted MMPs, suggesting that direct contact between tumor cells and the endothelium is likely to be required for intravasation (Ota et al. 2009). Although further advances in in vivo imaging techniques are needed to carefully investigate the mechanisms regulating intravasation, these studies support a role of EMT in promoting carcinoma cells to breach through endothelium during intravasation (Fig. 2).
Step III: EMT in systemic transport
Upon entering systemic circulation, surviving tumor cells must possess the proper machinery to survive anoikis and then attach onto the blood vessel wall to prepare for extravasation from the circulation. Many recent studies in human cancer patients and mouse tumor models have identified the presence of EMT molecular markers in CTCs using various methods, including immunostaining, in situ hybridization, RNA sequencing, real-time quantitative PCR, and expression analysis (Yu et al. 2011). In general, most studies detected expression of EMT markers in CTCs; however, the functional significance of EMT in CTCs still awaits further evaluation.
Experimental evidence for EMT in CTCs
Currently, only a limited number of reports using mouse tumor models directly examine the involvement of EMT in producing CTCs. This may be due in part to the challenges in capturing CTCs that have fully undergone EMT, given that current isolation methods rely primarily on using epithelial markers, such as EpCam, to capture CTCs from the blood or bone marrow (Yu et al. 2011). To investigate tumor cell dissemination in vivo, Husemann et al. (2008) used the MMTV-Her2 mouse mammary tumor model and found increased Twist1 expression in hyperplastic lesions during early primary tumor development. Concomitantly, these mice presented increased DTCs in the bone marrow at this stage, suggesting that EMT may be partially responsible for the induction of CTCs/DTCs (Husemann et al. 2008). Consistent with these findings, in a K-Ras-driven mouse pancreatic tumor model, Rhim et al. (2012) detected circulating pancreatic tumor cells at the premalignant stage of tumor progression. The majority of these CTCs presented a mesenchymal phenotype and expressed Zeb2, indicating activation of the EMT program in these cells (Rhim et al. 2012). To demonstrate that activation of EMT directly promotes the production of CTCs, Tsai et al. (2012) examined the number of CTCs in a squamous cell carcinoma mouse tumor model in response to Twist1 induction. Indeed, Twist1 induction dramatically increased the number of CTCs compared with control mice, and these CTCs presented an EMT phenotype with loss of E-cadherin and expression of vimentin (Tsai et al. 2012). In the MDA-MB-468 breast tumor xenograft model, expression of Snail1 and Snail2 also increased the presence of vimentin-positive CTCs (Bonnomet et al. 2012). In these experimental studies, an increase in CTCs was associated with an increase in metastasis incidence, suggesting that EMT-induced CTCs directly contribute to effective metastasis formation.
Recent insightful studies have also revealed a potential mechanism for maintaining the mesenchymal state of CTCs. Labelle et al. (2011) found that CTCs were preferentially associated with platelet cells, which are a major source of TGF-β production in the blood. Importantly, depletion of platelets or inhibition of TGF-β secretion solely in platelets drastically reduced distant metastases (Labelle et al. 2011). Consistent with these experimental data, mesenchymal CTCs isolated from breast cancer patients were found clustered with platelet cells, and gene expression profiling of these CTCs found an enrichment of the TGF-β pathway (Yu et al. 2013). Together, these studies suggest that successful metastasis may depend on the maintenance of a mesenchymal state in CTCs.
One potential hypothesis for why CTCs need to maintain the EMT program is that EMT can prevent single tumor cells from detachment-induced anoikis while in circulation. For example, CTC survival may be aided by microtubule-based membrane protrusions called microtentacles that are thought to allow CTC aggregation and/or cell attachment to the blood wall. These structures have previously been shown to form in detached breast tumor circulating cells (Matrone et al. 2010a,b; Whipple et al. 2010). Expression of Twist1 or Snail1 in human mammary epithelial cells promotes microtentacle formation in detached cells, suggesting that EMT could aid CTC survival via microtenacle-based attachment of CTCs to leukocytes, platelets, and endothelium (Fig. 3; Matrone et al. 2010a).
Figure 3.

EMT in systemic transport and extravasation. Experimental and clinical samples have revealed an EMT signature in CTCs. This signature provides a possible biomarker to monitor tumor progression and/or therapeutic response. Experimental evidence suggests that platelets play a critical role in maintaining EMT activation in CTCs by providing the TGF-β signal. Furthermore, studies suggest that activation of EMT promotes microtentacle formation that allows tumor cell attachment to the endothelium and promotes cell survival. In order to extravasate at distant sites, tumor cells maintain an EMT phenotype and express cellular protrusions that allow extravasation, which is mediated by β1 integrin signaling.
Clinical evidence for EMT in CTCs
Many recent clinical studies have highlighted the use of CTCs as a prognostic marker for cancer progression and an indicator of therapeutic response. Studies in patients with metastatic breast and colorectal cancer showed that CTCs serve as an independent predictor of progression-free survival and overall survival (Christoffersen et al. 2007; Cohen et al. 2008). Interestingly, many clinical studies also detected the presence of EMT molecular markers in the CTCs. For example, CTCs from breast cancer patients showed reduced expression of epithelial markers and/or increased mesenchymal markers (Aktas et al. 2009; Kallergi et al. 2011; Raimondi et al. 2011; Mego et al. 2012). Expression of Twist1 in DTCs isolated from bone marrows of breast cancer patients was correlated with early distant relapse (Watson et al. 2007). Using a similar approach, CTCs isolated from hepatocellular carcinoma (HCC) patients with metastasis expressed almost 20 times more Snail1 transcripts when compared with patients with no metastasis, demonstrating a potential functional role of Snail1 in HCC metastasis (Min et al. 2009). A recent study by Yu et al. (2013) quantified the proportions of CTCs with epithelial or mesenchymal phenotypes and found an association of mesenchymal CTCs with disease progression. One caveat of many studies involving CTCs is that, as previously discussed, the current CTC isolation methods rely primarily on using epithelial markers to capture CTCs. Thus, CTCs in the mesenchymal state may be missed during isolation (Paterlini-Brechot and Benali 2007; Pantel and Alix-Panabieres 2010; Kang and Pantel 2013). New technical advances are required to explore the potential utility of CTCs in both monitoring therapeutic responses and predicting cancer patient survival.
Step IV: EMT in tumor cell extravasation
Many classic studies of EMT regulators have used experimental metastasis assays, such as tail vein injection and intracardiac injection, to investigate tumor cell extravasation in the organ of interest. Studies have suggested that extravasation is a relatively rapid process (i.e., tumor cells extravasate 1–2 d following tail vein injection) (Cameron et al. 2000; Mendoza et al. 2010). However, tail vein assays involve injecting high numbers of tumor cells directly into the circulation. These injected tumor cells arrive at the lung microvasculature in such a large quantity that this process often results in intravessel growth rather than the sequential step of tumor growth in the tissue parenchyma following tumor cell extravasation. Thus, experimental models that more closely mimic physiological dissemination are needed to analyze the extravasation step.
Recently, new experimental systems have been developed to investigate how EMT is involved in the extravasation step. Stoletov et al. (2010) established an extravasation assay in zebrafish, which are optically transparent and allow real-time imaging of cell movement. Using this elegant system, they found that Twist1 expression in breast tumor cells promoted tumor cell extravasation through a β1 integrin-independent mechanism. Furthermore, they showed that Twist1-expressing cells formed large dynamic membrane protrusions during extravasation (Stoletov et al. 2010). Interestingly, Shibue et al. (2012) found that upon arriving at the distant tissue parenchyma, tumor cells present filopodium-like protrusions (FLPs) that contain integrin β1 to interact with the ECM. Not only was FLP formation found to be essential for successful metastasis, but the ability of various breast tumor cells to generate FLPs was correlated with their mesenchymal states, and their formation can be induced by the expression of Twist1 and Snail1 (Shibue et al. 2012). Together, these data indicate that the EMT program may promote extravasation and the initial lodging of tumor cells in distant organs (Fig. 3).
Step V: Reversion of EMT in metastatic colonization
As discussed, primary carcinomas and CTCs show strong cellular and molecular signatures of EMT; in contrast, resulting macrometastases are largely epithelial, which suggests that the involvement of EMT during metastasis is likely to be dynamic. Indeed, Chao et al. (2010) showed that tail vein injection of mesenchymal MDA-MB-231 cells into the secondary organ environment resulted in re-expression of E-cadherin through the passive loss of methylation in the E-cadherin promoter. Using vimentin as the mesenchymal marker, Bonnomet et al. (2012) found that primary MDA-MB-468 tumor xenografts and the resulting lung metastases showed a heterogeneous expression pattern of vimentin, while CTCs expressed high levels of vimentin, Snail1, and Snail2. This suggests that vimentin-positive CTCs might have undergone MET to form vimentin-negative macrometastasis. Two recent studies have provided concrete experimental data to support such epithelial–mesenchymal plasticity during tumor metastasis. Using an inducible Twist1 mouse bearing skin tumors, it has been demonstrated that activation of EMT promotes the early steps of metastasis, including local invasion, intravasation, and extravasation. However, the loss of an EMT-inducing signal at the distant site was essential for cell proliferation and macrometastasis formation (Tsai et al. 2012). Consistent with these studies, Ocana et al. (2012) showed that down-regulation of a novel EMT inducer, Prrx1, in BT549 human breast cancer cells was required for lung metastasis colonization upon tail vein injection. Specifically, Prrx1 cooperated with Twist1 to promote a more invasive phenotype, while loss of Prrx1 was required to revert EMT (Ocana et al. 2012). Together, these studies strongly argue that reversion of EMT is essential for metastasis colonization.
Why do tumor cells need to revert to an epithelial state to grow into macrometastases? Studies in cell culture showed that induction of EMT by Snail1 and Zeb2 directly represses cell division by inhibiting Cyclin D activity (Vega et al. 2004; Mejlvang et al. 2007). In an in vivo skin tumor model, activation of Twist1 was found to be associated with reduced tumor cell proliferation (Tsai et al. 2012). Since colonization demands tumor cells to restart proliferation upon extravasation into a foreign microenvironment, reversion of EMT may be required to provide such growth advantage. While these studies suggest that the induction of proliferation likely plays a key role in the reversion of EMT during colonization, it remains unanswered which signaling pathways couple EMT with cell proliferation. Other studies suggest that EMT regulators might provide additional assistance for metastatic colonization. The miR-200 family members are negative regulators of the EMT inducer Zeb1 and vice versa (Christoffersen et al. 2007; Bracken et al. 2008; Burk et al. 2008; Gregory et al. 2008; Korpal and Kang 2008; Korpal et al. 2008; Park et al. 2008; Kim et al. 2011b). Interestingly, re-expression of miR-200 family members was shown to enhance colonization possibly by repressing Sec23a-mediated secretion of metastasis-suppressive proteins, including Igfbp4 and Tinagl1 (Fig. 4; Korpal et al. 2011).
Figure 4.

Mechanisms of EMT reversion. Colonization at distant sites requires the reversion of EMT to promote tumor cell proliferation. The interplay between EMT activators and inhibitors (i.e., MET activators) plays a critical role in metastatic outgrowth. The loss of EMT activators such as Twist1 or Prrx1 appears to be required to promote EMT reversion. However, signals from the microenvironment in distant sites may also shift the balance from EMT activators to EMT inhibitors or MET activators. It is unknown when or how these factors are regulated during tumor progression, which may impact treatment of metastatic disease.
Another unanswered question is how the EMT reversion process occurs in distant organs. In other words: Is the absence of an EMT-inducing signal sufficient for EMT reversion? Are additional MET-inducing signals required to actively promote MET? Recent studies indicate that both scenarios are possible in promoting EMT reversion. In a skin tumor model, the absence of a Twist1 signal (or the withdrawal of Twist1-activating signal) in DTCs resulted in macrometastasis formation (Tsai et al. 2012). However, this study does not exclude the possibility that additional MET-inducing signals may also contribute to colonization. Gao et al. (2012) showed that versican expression by myeloid cells in the lung metastatic niche promoted lung colonization by inducing MET, thus supporting the notion that signal inputs from the metastatic niche could regulate epithelial–mesenchymal plasticity in distant sites. Although it is currently unknown how microenvironmental signals regulate MET, EMT core transcription factors are considered primary targets for such regulation. For example, a number of EMT core transcription factors are negatively regulated by miRNAs, including miR-200 family members that regulate Zeb2 as well as miR-34 family members that regulate Snail1 and Zeb1. It is possible that microenvironmental signals could impinge on these miRNAs to turn off EMT at distant organs (Fig. 4; Kim et al. 2011a; Siemens et al. 2011). Given that micrometastasis outgrowth is a key rate-limiting step in metastasis, more studies on the molecular regulators of MET could shed light on therapeutic approaches to inhibit tumor colonization.
Adding to the complexity of metastatic colonization is the clinical observation that DTCs can remain “dormant” for many years before regrowth. These cells are thought to reside in the secondary tissue parenchyma as a micrometastatic lesion or in the bone marrow and mobilize to secondary sites prior to regrowth (Hedley and Chambers 2009). Due to the lack of specific molecular markers to detect and isolate dormant micrometastases from epithelial organs in cancer patients and mouse tumor models, our current understanding of tumor dormancy is largely based on micrometastases isolated from the bone marrow. In one study, micrometastases from the bone marrow of breast cancer patients were isolated, and Twist1 expression was identified as a marker for early distant metastasis relapse (Watson et al. 2007). This result is consistent with a role of EMT in promoting tumor cell dissemination, as discussed earlier. More importantly, together with the notion that reversion of EMT is required for macrometastasis colonization, they suggest that the inability to revert EMT in DTCs might contribute to metastasis dormancy. Further studies using relevant experimental tumor models are needed to better evaluate the involvement of EMT in tumor cell dormancy.
While large numbers of studies have demonstrated a cell-autonomous role of EMT in promoting tumor cell dissemination, an alternative “cooperative” model proposes a supporting role for tumor cells that have undergone EMT to aid metastasis. By mixing uniquely labeled epithelial tumor cells and mesenchymal tumor cells to generate primary tumors, Tsuji et al. (2008) reported that epithelial cells require the cooperation of mesenchymal cells to lead the way for intravasation and CTC generation. However, only the epithelial tumor cells, but not the mesenchymal tumor cells, could generate macrometastases (Tsuji et al. 2008). In addition, the collaboration between tumor-initiating cells (TICs) and cells having undergone EMT accelerated metastatic colonization by the TICs (Celia-Terrassa et al. 2012). Both studies suggest that epithelial and mesenchymal tumor cells may cooperate to successfully form metastatic tumors (Tsuji et al. 2009). However, given that the mesenchymal tumor cells used in these studies have undergone a permanent EMT and cannot colonize in distant organs, it is plausible that the epithelial tumor cells still need to undergo a dynamic EMT/MET process to succeed in generating macrometastases.
Emerging frontiers of EMT
EMT and cancer stem cells (CSCs)
The role of CSCs or TICs in tumor progression has been at the forefront of cancer research in recent years (Nguyen et al. 2012). CSCs are generally characterized by their ability to initiate tumors in serial dilution transplantation assays. They express multiple cell surface markers, including CD44high, CD24low, and CD133high, depending on the tumor type. Interestingly, a number of studies have demonstrated that EMT activation can generate CSCs or disseminated cells that have tumor-initiating properties. Studies by the Weinberg group (Mani et al. 2008) and the Puisieux group (Morel et al. 2008) showed that activation of EMT by TGF-β, Snail1, Twist1, and Zeb1 in normal human mammary epithelial cells can promote a CSC-like phenotype with tumor-initiating properties. In mouse mammary epithelial cells, Snail2 was found to be a critical player in regulating normal mammary stem cells (Guo et al. 2012). Twist1 was also found to be capable of suppressing CD24 expression, providing a direct connection between an EMT transcription factor and CSC generation (Vesuna et al. 2009). In breast cancer cells, activation of urokinase-type plasminogen activator receptor (uPAR) was found to reversibly activate EMT, and activation of this pathway was capable of generating CSC-like properties (Lester et al. 2007; Jo et al. 2009, 2010). More interestingly, recent studies also found that basal breast cancer non-CSCs are populations that can generate CSCs de novo, and the CSC plasticity is controlled by the chromatin state of the Zeb1 promoter (Chaffer et al. 2011), further highlighting the critical role of EMT regulators in regulating CSC plasticity during tumor progression.
Consistent with these experimental results, numerous studies have provided correlative evidence relating EMT to the emergence of a CSC phenotype in human cancers. In breast cancer patients, disseminated breast cancer cells from pleural effusions, which likely have undergone EMT, are enriched for a CD44high, CD24low CSC-like population (Al-Hajj et al. 2003). In addition, stem cells isolated from normal breast tissue or breast cancers express a number of canonical EMT markers. Of clinical significance, studies have shown that the tumor immune response resulted in EMT-associated emergence of CD44high–CD24low CSCs (Santisteban et al. 2009).
Despite the strong evidence of a pro-CSC-forming role by EMT core regulators, there are experimental data that implicate EMT as having a negative impact on TICs. Celia-Terrassa et al. (2012) showed that cancer cells with a pronounced epithelial phenotype were enriched with highly metastatic TICs, whereas the mesenchymal-like cells lacked TICs. Furthermore, forced expression of Snail1 in the phenotypically epithelial cells suppressed their self-renewal and metastatic capabilities, suggesting that EMT activation may in fact suppress the tumor-initiating properties of CSCs (Celia-Terrassa et al. 2012). These contradictory results could be due to the difference in additional genetic/epigenetic alterations in the individual cell types studied. A recent study found that removing an EMT regulator, Prrx1, was required for the tumor-initiating ability of breast cancer cells expressing Twist1 (Ocana et al. 2012). Future studies to clearly define the difference in signaling pathways regulating EMT versus TICs will provide much-needed information on how the EMT program and TIC regulation are intervened.
EMT and drug resistance
One major obstacle in cancer therapy is that cancer patients can develop resistance to treatment over time. An increasing number of reports suggests a potential role of EMT in conferring drug resistance. Studies using colorectal cell lines have shown that expression of EMT inducers Snail1 or TGF-β in SMAD4-null cells increased resistance to chemotherapy (Hoshino et al. 2009; Papageorgis et al. 2011). Conversely, colorectal cell lines that were rendered oxaliplatin-resistant showed phenotypic and gene expression changes consistent with EMT (Yang et al. 2006; Kawamoto et al. 2012). Interestingly, tumor specimens taken from patients who had received 1 wk of preoperative chemotherapy prior to surgery resection displayed a more mesenchymal gene signature compared with prechemotherapy biopsy samples. Furthermore, recurrent tumors frequently exhibited an EMT gene signature (e.g., decreased E-cadherin and increased vimentin) (Kawamoto et al. 2012). Shintani et al. (2011) found that, in non-small-cell lung carcinoma (NSCLC) patients, tumor biopsy prior to chemotherapy treatment showed epithelial markers but that this phenotype shifted toward mesenchymal markers following treatment. Consequently, the disease-free survival rate was lower in patients whose tumors presented an EMT phenotype compared with EMT-negative tumors (Shintani et al. 2011).
Besides the evidence of drug resistance to chemotherapy, recent studies have also suggested a role of EMT in drug resistance toward targeted therapies. Using several human NSCLC lines, Thomson et al. (2005) demonstrated that cell lines expressing epithelial markers were more sensitive to epidermal growth factor receptor (EGFR) inhibition, whereas cells lines presenting mesenchymal markers were more resistant to treatment. In gefitinib-resistant PC9/AB2 lung cancer cells, Notch1 was found to promote EMT. Knockdown of Notch1 reverted the EMT phenotype and rendered these cells sensitive to gefitinib (Xie et al. 2012), implicating a strong correlation between EMT activation and drug resistance.
While it remains unknown whether and how the EMT program directly impinges on drug resistance, several possible mechanisms might be in play. First, since activation of EMT reduces cell proliferation (Vega et al. 2004; Mejlvang et al. 2007), slowing the cell cycle machinery will increase resistance to chemotherapy, which generally targets highly proliferative cells. Second, EMT core transcription factors, including TGF-β, Snail1, Snail2, and Twist1, have been shown to confer resistance to cell death via various pathways, most notably by antagonizing p53-mediated apoptosis (Inoue et al. 2002; Kajita et al. 2004; Vega et al. 2004; Gal et al. 2008), thus providing a potential survival advantage. Third, as discussed above, the EMT program can promote CSC properties. CSCs were found to be inherently more resistant to conventional cancer chemotherapies than rapidly proliferating progenitor cells and differentiated tumor cells. These CSCs were also found to be responsible for tumor reoccurrence and capable of establishing metastases (Phillips et al. 2006; Li et al. 2008). Indeed, conventional chemotherapy is shown to enrich CSCs in breast cancer patients. For example, mammospheres isolated from chemotherapy-treated breast tumors showed similar mammosphere-initiating capacity after eight passages in culture, whereas cells from untreated tumors vanished within three passages, suggesting again an increase in CSCs after chemotherapy (Yu et al. 2007). Li et al. (2008) further demonstrated that chemotherapy increased the number of CD44high–CD24low cell population and that these cells have stem-like features. Together, these studies strongly indicate that activation of EMT contributes to cancer therapy resistance.
Future directions in therapeutic targeting of EMT/MET
Metastatic diseases are responsible for >90% of carcinoma-related deaths. Given the strong evidence supporting a critical role of EMT in tumor metastasis, targeting this process is thought to be a promising approach to treat invasive cancer. However, current treatment modalities remain limited in their efficacy in targeting cells undergoing EMT. This may be due in part to potential drug resistance in this population of cells (as discussed above) and the lack of appropriate targets in the core EMT program. Because EMT core transcription factors remain technically challenging to target, targeting the activation or the functional consequence of EMT is perhaps the more effective approach. Several groups have performed high-content drug screens to identify potential inhibitors of EMT in response to various EMT-inducing signals. Interestingly, the majority of the compounds obtained appear to be involved in inhibiting the specific EMT-inducing signals used in the screen. For example, rapamycin and 17-AGG were identified as inhibitors of TGFβ-induced EMT by modifying the TGFβ pathway (Reka et al. 2011), while inhibitors of ALK5, MEK, and SRC could interfere with EMT in response to EGF, HGF, and IGF-1 (Reka et al. 2011; Chua et al. 2012). Salinomycin was also identified as inducing selective killing of E-cadherin-null breast epithelial cells compared with E-cadherin-positive cells (Gupta et al. 2009), although its molecular action toward EMT is unknown.
The plasticity of EMT in metastasis provides another level of complexity regarding the appropriate time window to target EMT in patients. Several studies suggest that targeting the TGFβ pathway to inhibit EMT or blocking tumor cell invasion through inhibiting PDGR signaling may be appropriate as metastasis prevention strategies in early stage carcinomas. Once tumor cells have disseminated from the primary tumor, inhibiting EMT could be counterproductive, since reversion of EMT appears to be beneficial for disseminated carcinoma cells to regain proliferation and colonize distant organs (Tsai et al. 2012). Instead, the EMT features in CTCs and disseminated dormant tumor cells present several unique opportunities for therapeutic intervention. First, since DTCs present molecular markers of EMT, unique EMT surface markers expressed in these cells could be ideal targets for T-cell-based immunotherapy. Second, since an increasing number of studies suggest a role of EMT in promoting chemoresistance, combining chemotherapies with EMT inhibitors holds promise to overcome chemoresistance in dormant tumor cells, thus providing a unique therapeutic approach to eradicate dormant tumor cells. Last, preventing the reversion of the EMT program in dormant micrometastases would be a novel approach to prevent resurrection of dormant tumor cells. In the near future, improving our understanding of the molecular regulation of the dynamic EMT/MET programs during tumor metastasis will help to provide much-needed effective treatment to eradicate metastatic diseases.
Acknowledgments
We apologize to the many researchers in this field whose work we were unable to cite due to space restrictions. We thank Helicia Paz, Navneeta Pathak, Danielle Murphy, and anonymous reviewers for their insightful comments. Our research on tumor metastasis is supported by grants from the National Institutes of Health (1DP2OD002420), National Cancer Institute (1RO1CA168689), American Cancer Society (grant RSG-09-282-01-CSM), The Hartwell Foundation, and DOD Breast Cancer Program (W81XWH-13-1-0132) to J.Y., and by the NIH (T32CA121938) and California Breast Cancer Program post-doctoral fellowship (16FB-0009) to J.H.T.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.225334.113.
References
- Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S 2009. Stem cell and epithelial–mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res 11: R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF 2003. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci 100: 3983–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, et al. 2008. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 14: 79–89 [DOI] [PubMed] [Google Scholar]
- Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM 2005. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: Implications for the epithelial–mesenchymal transition. J Cell Biol 168: 29–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates RC, Bellovin DI, Brown C, Maynard E, Wu B, Kawakatsu H, Sheppard D, Oettgen P, Mercurio AM 2005. Transcriptional activation of integrin β6 during the epithelial–mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J Clin Invest 115: 339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A 2000. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2: 84–89 [DOI] [PubMed] [Google Scholar]
- Becker KF, Atkinson MJ, Reich U, Becker I, Nekarda H, Siewert JR, Hofler H 1994. E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res 54: 3845–3852 [PubMed] [Google Scholar]
- Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, van Roy F 1995. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J 14: 6107–6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berx G, Becker KF, Hofler H, van Roy F 1998. Mutations of the human E-cadherin (CDH1) gene. Hum Mutat 12: 226–237 [DOI] [PubMed] [Google Scholar]
- Bonnomet A, Syne L, Brysse A, Feyereisen E, Thompson EW, Noel A, Foidart JM, Birembaut P, Polette M, Gilles C 2012. A dynamic in vivo model of epithelial-to-mesenchymal transitions in circulating tumor cells and metastases of breast cancer. Oncogene 31: 3741–3753 [DOI] [PubMed] [Google Scholar]
- Boyer B, Thiery JP 1993. Epithelium-mesenchyme interconversion as example of epithelial plasticity. APMIS 101: 257–268 [DOI] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T 2001. Variable β-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci 98: 10356–10361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T, Hlubek F, Spaderna S, Schmalhofer O, Hiendlmeyer E, Jung A, Kirchner T 2005. Invasion and metastasis in colorectal cancer: Epithelial–mesenchymal transition, mesenchymal–epithelial transition, stem cells and β-catenin. Cells Tissues Organs 179: 56–65 [DOI] [PubMed] [Google Scholar]
- Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ 2008. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial–mesenchymal transition. Cancer Res 68: 7846–7854 [DOI] [PubMed] [Google Scholar]
- Bukholm IK, Nesland JM, Karesen R, Jacobsen U, Borresen-Dale AL 1998. E-cadherin and α-, β-, and γ-catenin protein expression in relation to metastasis in human breast carcinoma. J Pathol 185: 262–266 [DOI] [PubMed] [Google Scholar]
- Bukholm IK, Nesland JM, Borresen-Dale AL 2000. Re-expression of E-cadherin, α-catenin and β-catenin, but not of γ-catenin, in metastatic tissue from breast cancer patients. J Pathol 190: 15–19 [DOI] [PubMed] [Google Scholar]
- Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T 2008. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 9: 582–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron MD, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC, Chambers AF, MacDonald IC 2000. Temporal progression of metastasis in lung: Cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res 60: 2541–2546 [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA 2000. The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2: 76–83 [DOI] [PubMed] [Google Scholar]
- Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J 2011. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res 71: 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celesti G, Di Caro G, Bianchi P, Grizzi F, Basso G, Marchesi F, Doni A, Marra G, Roncalli M, Mantovani A, et al. 2013. Presence of Twist1-positive neoplastic cells in the stroma of chromosome-unstable colorectal tumors. Gastroenterology 145: 647–657 [DOI] [PubMed] [Google Scholar]
- Celia-Terrassa T, Meca-Cortes O, Mateo F, de Paz AM, Rubio N, Arnal-Estape A, Ell BJ, Bermudo R, Diaz A, Guerra-Rebollo M, et al. 2012. Epithelial–mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest 122: 1849–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, et al. 2011. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci 108: 7950–7955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao YL, Shepard CR, Wells A 2010. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer 9: 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M, Xu LM, Costanzo C, Cheng JQ, Wang LH 2008. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem 283: 14665–14673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersen NR, Silahtaroglu A, Orom UA, Kauppinen S, Lund AH 2007. miR-200b mediates post-transcriptional repression of ZFHX1B. RNA 13: 1172–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua KN, Sim WJ, Racine V, Lee SY, Goh BC, Thiery JP 2012. A cell-based small molecule screening method for identifying inhibitors of epithelial–mesenchymal transition in carcinoma. PLoS ONE 7: e33183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chui MH 2013. Insights into cancer metastasis from a clinicopathologic perspective: Epithelial–mesenchymal transition is not a necessary step. Int J Cancer 132: 1487–1495 [DOI] [PubMed] [Google Scholar]
- Cohen SJ, Punt CJ, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, Picus J, Morse M, Mitchell E, Miller MC, et al. 2008. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol 26: 3213–3221 [DOI] [PubMed] [Google Scholar]
- Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F 2001. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 7: 1267–1278 [DOI] [PubMed] [Google Scholar]
- De Craene B, Berx G 2013. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 13: 97–110 [DOI] [PubMed] [Google Scholar]
- Derksen PW, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, van Beijnum JR, Griffioen AW, Vink J, Krimpenfort P, et al. 2006. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 10: 437–449 [DOI] [PubMed] [Google Scholar]
- Drake JM, Strohbehn G, Bair TB, Moreland JG, Henry MD 2009. ZEB1 enhances transendothelial migration and represses the epithelial phenotype of prostate cancer cells. Mol Biol Cell 20: 2207–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droufakou S, Deshmane V, Roylance R, Hanby A, Tomlinson I, Hart IR 2001. Multiple ways of silencing E-cadherin gene expression in lobular carcinoma of the breast. Int J Cancer 92: 404–408 [DOI] [PubMed] [Google Scholar]
- Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J 2011. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 19: 372–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R 2005. δEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 24: 2375–2385 [DOI] [PubMed] [Google Scholar]
- Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang Z, Yang CJ, Yuan L, Ouyang G 2011. Twist2 contributes to breast cancer progression by promoting an epithelial–mesenchymal transition and cancer stem-like cell self-renewal. Oncogene 30: 4707–4720 [DOI] [PubMed] [Google Scholar]
- Fidler IJ 2003. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3: 453–458 [DOI] [PubMed] [Google Scholar]
- Gal A, Sjoblom T, Fedorova L, Imreh S, Beug H, Moustakas A 2008. Sustained TGF βa exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene 27: 1218–1230 [DOI] [PubMed] [Google Scholar]
- Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, Sadik H, Argani P, Wagner P, Vahdat LT, et al. 2012. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res 72: 1384–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber K 2008. Epithelial-to-mesenchymal transition is important to metastasis, but questions remain. J Natl Cancer Inst 100: 232–233, 239 [DOI] [PubMed] [Google Scholar]
- Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, Isaacs WB, Pitha PM, Davidson NE, Baylin SB 1995. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 55: 5195–5199 [PubMed] [Google Scholar]
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ 2008. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10: 593–601 [DOI] [PubMed] [Google Scholar]
- Grunert S, Jechlinger M, Beug H 2003. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol 4: 657–665 [DOI] [PubMed] [Google Scholar]
- Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, et al. 2012. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148: 1015–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES 2009. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138: 645–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajra KM, Chen DY, Fearon ER 2002. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res 62: 1613–1618 [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA 2011. Hallmarks of cancer: The next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Hay ED 1995. An overview of epithelio–mesenchymal transformation. Acta Anat (Basel) 154: 8–20 [DOI] [PubMed] [Google Scholar]
- Hedley BD, Chambers AF 2009. Tumor dormancy and metastasis. Adv Cancer Res 102: 67–101 [DOI] [PubMed] [Google Scholar]
- Hirohashi S 1998. Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol 153: 333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino H, Miyoshi N, Nagai K, Tomimaru Y, Nagano H, Sekimoto M, Doki Y, Mori M, Ishii H 2009. Epithelial-mesenchymal transition with expression of SNAI1-induced chemoresistance in colorectal cancer. Biochem Biophys Res Commun 390: 1061–1065 [DOI] [PubMed] [Google Scholar]
- Hotary K, Li XY, Allen E, Stevens SL, Weiss SJ 2006. A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes Dev 20: 2673–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CH, Yang WH, Chang SY, Tai SK, Tzeng CH, Kao JY, Wu KJ, Yang MH 2009. Regulation of membrane-type 4 matrix metalloproteinase by SLUG contributes to hypoxia-mediated metastasis. Neoplasia 11: 1371–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmuller G, et al. 2008. Systemic spread is an early step in breast cancer. Cancer Cell 13: 58–68 [DOI] [PubMed] [Google Scholar]
- Inoue A, Seidel MG, Wu W, Kamizono S, Ferrando AA, Bronson RT, Iwasaki H, Akashi K, Morimoto A, Hitzler JK, et al. 2002. Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2: 279–288 [DOI] [PubMed] [Google Scholar]
- Janda E, Litos G, Grunert S, Downward J, Beug H 2002. Oncogenic Ras/Her-2 mediate hyperproliferation of polarized epithelial cells in 3D cultures and rapid tumor growth via the PI3K pathway. Oncogene 21: 5148–5159 [DOI] [PubMed] [Google Scholar]
- Jo M, Lester RD, Montel V, Eastman B, Takimoto S, Gonias SL 2009. Reversibility of epithelial–mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling. J Biol Chem 284: 22825–22833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo M, Eastman BM, Webb DL, Stoletov K, Klemke R, Gonias SL 2010. Cell signaling by urokinase-type plasminogen activator receptor induces stem cell-like properties in breast cancer cells. Cancer Res 70: 8948–8958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajita M, McClinic KN, Wade PA 2004. Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol Cell Biol 24: 7559–7566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallergi G, Papadaki MA, Politaki E, Mavroudis D, Georgoulias V, Agelaki S 2011. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res 13: R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA 2009. The basics of epithelial–mesenchymal transition. J Clin Invest 119: 1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Ushijima S, Hui AM, Ochiai A, Tsuda H, Sakamoto M, Hirohashi S 1997. The E-cadherin gene is silenced by CpG methylation in human hepatocellular carcinomas. Int J Cancer 71: 355–359 [DOI] [PubMed] [Google Scholar]
- Kang Y, Pantel K 2013. Tumor cell dissemination: Emerging biological insights from animal models and cancer patients. Cancer Cell 23: 573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno Y, Lamouille S, Derynck R 2013. TGF-β signaling and epithelial–mesenchymal transition in cancer progression. Curr Opin Oncol 25: 76–84 [DOI] [PubMed] [Google Scholar]
- Kawamoto A, Yokoe T, Tanaka K, Saigusa S, Toiyama Y, Yasuda H, Inoue Y, Miki C, Kusunoki M 2012. Radiation induces epithelial–mesenchymal transition in colorectal cancer cells. Oncol Rep 27: 51–57 [DOI] [PubMed] [Google Scholar]
- Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, Cha SY, Ryu JK, Yoon D, Fearon ER, et al. 2011a. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial–mesenchymal transition. J Cell Biol 195: 417–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S, Pineau P, Marchio A, Palatini J, Suh SS, et al. 2011b. p53 regulates epithelial–mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med 208: 875–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M, Kang Y 2008. The emerging role of miR-200 family of microRNAs in epithelial-mesenchymal transition and cancer metastasis. RNA Biol 5: 115–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M, Lee ES, Hu G, Kang Y 2008. The miR-200 family inhibits epithelial–mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 283: 14910–14914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celia-Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua Y, et al. 2011. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med 17: 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YC, Su CH, Liu CY, Chen TH, Chen CP, Wang HS 2009. Transforming growth factor-β induces CD44 cleavage that promotes migration of MDA-MB-435s cells through the up-regulation of membrane type 1-matrix metalloproteinase. Int J Cancer 124: 2568–2576 [DOI] [PubMed] [Google Scholar]
- Labelle M, Begum S, Hynes RO 2011. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20: 576–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledford H 2011. Cancer theory faces doubts. Nature 472: 273. [DOI] [PubMed] [Google Scholar]
- Lee KE, Bar-Sagi D 2010. Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell 18: 448–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester RD, Jo M, Montel V, Takimoto S, Gonias SL 2007. uPAR induces epithelial–mesenchymal transition in hypoxic breast cancer cells. J Cell Biol 178: 425–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Cserjesi P, Olson EN 1995. Dermo-1: A novel twist-related bHLH protein expressed in the developing dermis. Dev Biol 172: 280–292 [DOI] [PubMed] [Google Scholar]
- Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, et al. 2008. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 100: 672–679 [DOI] [PubMed] [Google Scholar]
- Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, Chao CH, Yamaguchi H, Yang NK, Ding Q, et al. 2012. Epithelial–mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res 72: 1290–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Thiery JP 2012. Epithelial–mesenchymal transitions: Insights from development. Development 139: 3471–3486 [DOI] [PubMed] [Google Scholar]
- Livasy CA, Karaca G, Nanda R, Tretiakova MS, Olopade OI, Moore DT, Perou CM 2006. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol 19: 264–271 [DOI] [PubMed] [Google Scholar]
- Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung MC 2007. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial–mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res 67: 9066–9076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA, Gouvin LM, Sharma VM, Mercurio AM 2010. ERβ impedes prostate cancer EMT by destabilizing HIF-1α and inhibiting VEGF-mediated snail nuclear localization: Implications for Gleason grading. Cancer Cell 17: 319–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. 2008. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell 133: 704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrone MA, Whipple RA, Balzer EM, Martin SS 2010a. Microtentacles tip the balance of cytoskeletal forces in circulating tumor cells. Cancer Res 70: 7737–7741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrone MA, Whipple RA, Thompson K, Cho EH, Vitolo MI, Balzer EM, Yoon JR, Ioffe OB, Tuttle KC, Tan M, et al. 2010b. Metastatic breast tumors express increased tau, which promotes microtentacle formation and the reattachment of detached breast tumor cells. Oncogene 29: 3217–3227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mego M, Mani SA, Lee BN, Li C, Evans KW, Cohen EN, Gao H, Jackson SA, Giordano A, Hortobagyi GN, et al. 2012. Expression of epithelial–mesenchymal transition-inducing transcription factors in primary breast cancer: The effect of neoadjuvant therapy. Int J Cancer 130: 808–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejlvang J, Kriajevska M, Vandewalle C, Chernova T, Sayan AE, Berx G, Mellon JK, Tulchinsky E 2007. Direct repression of cyclin D1 by SIP1 attenuates cell cycle progression in cells undergoing an epithelial mesenchymal transition. Mol Biol Cell 18: 4615–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza A, Hong SH, Osborne T, Khan MA, Campbell K, Briggs J, Eleswarapu A, Buquo L, Ren L, Hewitt SM, et al. 2010. Modeling metastasis biology and therapy in real time in the mouse lung. J Clin Invest 120: 2979–2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles FL, Pruitt FL, van Golen KL, Cooper CR 2008. Stepping out of the flow: Capillary extravasation in cancer metastasis. Clin Exp Metastasis 25: 305–324 [DOI] [PubMed] [Google Scholar]
- Min AL, Choi JY, Woo HY, Kim JD, Kwon JH, Bae SH, Yoon SK, Shin SH, Chung YJ, Jung CK 2009. High expression of Snail mRNA in blood from hepatocellular carcinoma patients with extra-hepatic metastasis. Clin Exp Metastasis 26: 759–767 [DOI] [PubMed] [Google Scholar]
- Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A 2008. Generation of breast cancer stem cells through epithelial–mesenchymal transition. PLoS One 3: e2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel AP, Hinkal GW, Thomas C, Fauvet F, Courtois-Cox S, Wierinckx A, Devouassoux-Shisheboran M, Treilleux I, Tissier A, Gras B, et al. 2012. EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin-low tumors in transgenic mice. PLoS Genet 8: e1002723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DA, Courtneidge SA 2011. The ‘ins’ and ‘outs’ of podosomes and invadopodia: Characteristics, formation and function. Nat Rev Mol Cell Biol 12: 413–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawshad A, Lagamba D, Polad A, Hay ED 2005. Transforming growth factor-β signaling during epithelial–mesenchymal transformation: Implications for embryogenesis and tumor metastasis. Cells Tissues Organs 179: 11–23 [DOI] [PubMed] [Google Scholar]
- Nguyen LV, Vanner R, Dirks P, Eaves CJ 2012. Cancer stem cells: An evolving concept. Nat Rev Cancer 12: 133–143 [DOI] [PubMed] [Google Scholar]
- Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, Nieto MA 2012. Metastatic colonization requires the repression of the epithelial–mesenchymal transition inducer Prrx1. Cancer Cell 22: 709–724 [DOI] [PubMed] [Google Scholar]
- Oda T, Kanai Y, Oyama T, Yoshiura K, Shimoyama Y, Birchmeier W, Sugimura T, Hirohashi S 1994. E-cadherin gene mutations in human gastric carcinoma cell lines. Proc Natl Acad Sci 91: 1858–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E 1996. TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev 10: 2462–2477 [DOI] [PubMed] [Google Scholar]
- Oft M, Heider KH, Beug H 1998. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol 8: 1243–1252 [DOI] [PubMed] [Google Scholar]
- Ohkubo T, Ozawa M 2004. The transcription factor Snail downregulates the tight junction components independently of E-cadherin downregulation. J Cell Sci 117: 1675–1685 [DOI] [PubMed] [Google Scholar]
- Olmeda D, Jorda M, Peinado H, Fabra A, Cano A 2007a. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene 26: 1862–1874 [DOI] [PubMed] [Google Scholar]
- Olmeda D, Moreno-Bueno G, Flores JM, Fabra A, Portillo F, Cano A 2007b. SNAI1 is required for tumor growth and lymph node metastasis of human breast carcinoma MDA-MB-231 cells. Cancer Res 67: 11721–11731 [DOI] [PubMed] [Google Scholar]
- Orlichenko LS, Radisky DC 2008. Matrix metalloproteinases stimulate epithelial–mesenchymal transition during tumor development. Clin Exp Metastasis 25: 593–600 [DOI] [PubMed] [Google Scholar]
- Ota I, Li XY, Hu Y, Weiss SJ 2009. Induction of a MT1-MMP and MT2-MMP-dependent basement membrane transmigration program in cancer cells by Snail1. Proc Natl Acad Sci 106: 20318–20323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantel K, Alix-Panabieres C 2010. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol Med 16: 398–406 [DOI] [PubMed] [Google Scholar]
- Papageorgis P, Cheng K, Ozturk S, Gong Y, Lambert AW, Abdolmaleky HM, Zhou JR, Thiagalingam S 2011. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res 71: 998–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Gaur AB, Lengyel E, Peter ME 2008. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22: 894–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterlini-Brechot P, Benali NL 2007. Circulating tumor cells (CTC) detection: Clinical impact and future directions. Cancer Lett 253: 180–204 [DOI] [PubMed] [Google Scholar]
- Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A 2001. A new role for E12/E47 in the repression of E-cadherin expression and epithelial–mesenchymal transitions. J Biol Chem 276: 27424–27431 [DOI] [PubMed] [Google Scholar]
- Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G 1998. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 392: 190–193 [DOI] [PubMed] [Google Scholar]
- Phillips TM, McBride WH, Pajonk F 2006. The response of CD24−/low/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst 98: 1777–1785 [DOI] [PubMed] [Google Scholar]
- Piccinin S, Tonin E, Sessa S, Demontis S, Rossi S, Pecciarini L, Zanatta L, Pivetta F, Grizzo A, Sonego M, et al. 2012. A ‘twist box’ code of p53 inactivation: Twist box: p53 interaction promotes p53 degradation. Cancer Cell 22: 404–415 [DOI] [PubMed] [Google Scholar]
- Pignatelli J, Tumbarello DA, Schmidt RP, Turner CE 2012. Hic-5 promotes invadopodia formation and invasion during TGF-β-induced epithelial–mesenchymal transition. J Cell Biol 197: 421–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM 2010. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res 12: R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, et al. 2005. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 436: 123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi C, Gradilone A, Naso G, Vincenzi B, Petracca A, Nicolazzo C, Palazzo A, Saltarelli R, Spremberg F, Cortesi E, et al. 2011. Epithelial–mesenchymal transition and stemness features in circulating tumor cells from breast cancer patients. Breast Cancer Res Treat 130: 449–455 [DOI] [PubMed] [Google Scholar]
- Rakha EA, Putti TC, Abd El-Rehim DM, Paish C, Green AR, Powe DG, Lee AH, Robertson JF, Ellis IO 2006. Morphological and immunophenotypic analysis of breast carcinomas with basal and myoepithelial differentiation. J Pathol 208: 495–506 [DOI] [PubMed] [Google Scholar]
- Reka AK, Kuick R, Kurapati H, Standiford TJ, Omenn GS, Keshamouni VG 2011. Identifying inhibitors of epithelial–mesenchymal transition by connectivity map-based systems approach. J Thorac Oncol 6: 1784–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, et al. 2012. EMT and dissemination precede pancreatic tumor formation. Cell 148: 349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Takazawa H, Uzawa K, Tanzawa H, Sato K 1998. Reduced expression of E-cadherin in oral squamous cell carcinoma: Relationship with DNA methylation of 5′ CpG island. Int J Oncol 12: 293–298 [DOI] [PubMed] [Google Scholar]
- Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, Haluska P, Ingle JN, Hartmann LC, Manjili MH, et al. 2009. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res 69: 2887–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrio D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J 2008. Epithelial–mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res 68: 989–997 [DOI] [PubMed] [Google Scholar]
- Shibue T, Brooks MW, Inan MF, Reinhardt F, Weinberg RA 2012. The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov 2: 706–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih JY, Tsai MF, Chang TH, Chang YL, Yuan A, Yu CJ, Lin SB, Liou GY, Lee ML, Chen JJ, et al. 2005. Transcription repressor slug promotes carcinoma invasion and predicts outcome of patients with lung adenocarcinoma. Clin Cancer Res 11: 8070–8078 [DOI] [PubMed] [Google Scholar]
- Shintani Y, Okimura A, Sato K, Nakagiri T, Kadota Y, Inoue M, Sawabata N, Minami M, Ikeda N, Kawahara K et al. 2011. Epithelial to mesenchymal transition is a determinant of sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann Thorac Surg 92: 1794–1804 [DOI] [PubMed] [Google Scholar]
- Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U, Hermeking H 2011. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial–mesenchymal transitions. Cell Cycle 10: 4256–4271 [DOI] [PubMed] [Google Scholar]
- Slorach EM, Chou J, Werb Z 2011. Zeppo1 is a novel metastasis promoter that represses E-cadherin expression and regulates p120-catenin isoform expression and localization. Genes Dev 25: 471–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. 2001. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci 98: 10869–10874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaderna S, Schmalhofer O, Hlubek F, Berx G, Eger A, Merkel S, Jung A, Kirchner T, Brabletz T 2006. A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology 131: 830–840 [DOI] [PubMed] [Google Scholar]
- Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, Klemke R 2010. Visualizing extravasation dynamics of metastatic tumor cells. J Cell Sci 123: 2332–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarin D, Thompson EW, Newgreen DF 2005. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res 65: 5996–6000 [DOI] [PubMed] [Google Scholar]
- Thiery JP 2002. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer 2: 442–454 [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA 2009. Epithelial–mesenchymal transitions in development and disease. Cell 139: 871–890 [DOI] [PubMed] [Google Scholar]
- Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, Iwata KK, Gibson N, Haley JD 2005. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res 65: 9455–9462 [DOI] [PubMed] [Google Scholar]
- Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J 2012. Spatiotemporal regulation of epithelial–mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22: 725–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji T, Ibaragi S, Shima K, Hu MG, Katsurano M, Sasaki A, Hu GF 2008. Epithelial–mesenchymal transition induced by growth suppressor p12CDK2–AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res 68: 10377–10386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji T, Ibaragi S, Hu GF 2009. Epithelial–mesenchymal transition and cell cooperativity in metastasis. Cancer Res 69: 7135–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valsesia-Wittmann S, Magdeleine M, Dupasquier S, Garin E, Jallas AC, Combaret V, Krause A, Leissner P, Puisieux A 2004. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell 6: 625–630 [DOI] [PubMed] [Google Scholar]
- Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, Tulchinsky E, Van Roy F, Berx G 2005. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res 33: 6566–6578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA 2004. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev 18: 1131–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesuna F, Lisok A, Kimble B, Raman V 2009. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 11: 1318–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent-Salomon A, Thiery JP 2003. Host microenvironment in breast cancer development: Epithelial–mesenchymal transition in breast cancer development. Breast Cancer Res 5: 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson MA, Ylagan LR, Trinkaus KM, Gillanders WE, Naughton MJ, Weilbaecher KN, Fleming TP, Aft RL 2007. Isolation and molecular profiling of bone marrow micrometastases identifies TWIST1 as a marker of early tumor relapse in breast cancer patients. Clin Cancer Res 13: 5001–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whipple RA, Matrone MA, Cho EH, Balzer EM, Vitolo MI, Yoon JR, Ioffe OB, Tuttle KC, Yang J, Martin SS 2010. Epithelial-to-mesenchymal transition promotes tubulin detyrosination and microtentacles that enhance endothelial engagement. Cancer Res 70: 8127–8137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP 2009. Stabilization of snail by NF-κB is required for inflammation-induced cell migration and invasion. Cancer Cell 15: 416–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Zhang L, He CS, Xu F, Liu JL, Hu ZH, Zhao LP, Tian Y 2012. Activation of Notch-1 enhances epithelial–mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J Cell Biochem 113: 1501–1513 [DOI] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA 2004. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117: 927–939 [DOI] [PubMed] [Google Scholar]
- Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R, Gray MJ, Cheng H, Hoff PM, Ellis LM 2006. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res 12: 4147–4153 [DOI] [PubMed] [Google Scholar]
- Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ 2008. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat Cell Biol 10: 295–305 [DOI] [PubMed] [Google Scholar]
- Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, et al. 2007. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 131: 1109–1123 [DOI] [PubMed] [Google Scholar]
- Yu M, Stott S, Toner M, Maheswaran S, Haber DA 2011. Circulating tumor cells: Approaches to isolation and characterization. J Cell Biol 192: 373–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, et al. 2013. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339: 580–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Kang Y 2013. Multilayer control of the EMT master regulators. Oncogene doi: 10.1038/onc.2013.128 [DOI] [PubMed] [Google Scholar]
- Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC 2004. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial–mesenchymal transition. Nat Cell Biol 6: 931–940 [DOI] [PubMed] [Google Scholar]