

The RNA-binding protein RNPC1/Rbm38 is a p53 target and repressor of p53 mRNA translation. While the p53–RNPC1 loop is critical for modulating p53 tumor suppression, it is unclear how the loop is regulated. Here, Zhang et al. show that glycogen synthase kinase 3 regulates p53 mRNA translation via phosphorylation of RNPC1. Surprisingly, they find that phosphorylation of a specific site converts RNPC1 from a repressor to an activator of p53. This study may have important therapeutic implications for the modulation of p53 activity in cancer cells.
Keywords: p53, RNPC1, Rbm38, GSK3, eIF4E, eIF4G
Abstract
The RNPC1 RNA-binding protein, also called Rbm38, is a target of p53 and a repressor of p53 mRNA translation. Thus, the p53–RNPC1 loop is critical for modulating p53 tumor suppression, but it is not clear how the loop is regulated. Here, we showed that RNPC1 is phosphorylated at Ser195 by glycogen synthase kinase 3 (GSK3). We also showed that GSK3 promotes p53 mRNA translation through phosphorylation of RNPC1. Interestingly, we found that the phosphor-mimetic mutant S195D and the deletion mutant Δ189–204, which lacks the GSK3 phosphorylation site, are unable to repress p53 mRNA translation due to loss of interaction with eukaryotic translation factor eIF4E on p53 mRNA. Additionally, we found that phosphorylated RNPC1, RNPC1-S195D, and RNPC1(Δ189–204) promote p53 mRNA translation through interaction with eukaryotic translation factor eIF4G, which then facilitates the assembly of the eIF4F complex on p53 mRNA. Furthermore, we showed that upon inhibition of the phosphatidylinositol 3-kinase (PI3K)–Akt pathway, GSK3 is activated, leading to increased RNPC1 phosphorylation and increased p53 expression in a RNPC1-dependent manner. Together, we postulate that the p53–RNPC1 loop can be explored to increase or decrease p53 activity for cancer therapy.
A variety of stress signals, such as DNA damage, hypoxia, and oncogene activation, induce p53 for tumor suppression by blocking cell cycle progression and promoting apoptotic cell death and senescence (Levine and Oren 2009; Vousden and Prives 2009). As a transcription factor, p53 activates and represses a broad range of target genes for tumor suppression (Harms et al. 2004; Riley et al. 2008), and thus p53 activity needs to be fine-tuned in response to the type and level of stress (Kruse and Gu 2009; Vousden and Prives 2009). Recently, translational regulation has emerged as a key mechanism to control p53 activity (Zhang and Chen 2008). For example, ATM-dependent phosphorylation of Mdm2 enhances p53 mRNA–Mdm2 interaction and promotes p53 translation in response to cellular stresses (Gajjar et al. 2012). Additionally, several RNA-binding proteins (RBPs), including HuR, ribosomal protein L26 (RPL26), and nucleolin, are found to modulate p53 activity through mRNA translation (Mazan-Mamczarz et al. 2003; Takagi et al. 2005; Chen and Kastan 2010).
The RBP RNPC1, also called Rbm38, is a target of p53 and E2F1 (Shu et al. 2006; Feldstein et al. 2012). Interestingly, RNPC1 interacts with translation factor eIF4E on p53 mRNA and represses p53 translation (Zhang et al. 2011). RNPC1 is also capable of enhancing p21 and p73, but decreasing p63 and Mdm2, mRNA stability through binding to the AU/U-rich element in their 3′ untranslated regions (UTRs) (Shu et al. 2006; Zhang et al. 2010; Yan et al. 2012; Xu et al. 2013). In addition, RNPC1 is found to relieve microRNA repression of several p53 targets, including p21, DDIT4, LATS2, and RNPC1 itself (Leveille et al. 2011). Importantly, RNPC1 expression is found to be altered in breast and other cancer patients with poor prognosis (Jenssen et al. 2002; Chin et al. 2006; Jonsson et al. 2007; Leveille et al. 2011; Feldstein et al. 2012). Recently, we showed that in dog lymphoma, overexpression of RNPC1 is correlated with decreased expression of p53 (Zhang et al. 2011).
Glycogen synthase kinase 3 (GSK3) is a ubiquitously expressed proline-directed serine/threonine kinase and is encoded by two distinct but related genes: GSK3α and GSK3β (Doble and Woodgett 2003). GSK3 was identified as a critical regulator of the insulin signaling pathway (Cohen and Frame 2001). It is now known that GSK3 regulates numerous signaling pathways and cellular processes, including cell proliferation, apoptosis, differentiation, and neural development (Cohen and Frame 2001; Wu and Pan 2010). Due to its diverse functions, GSK3 is implicated in the pathogenesis of many human diseases, such as diabetes, neurodegenerative diseases, bipolar disorder, and cancer (Frame and Cohen 2001; Grimes and Jope 2001). As a multifunctional kinase, GSK3β is found to regulate p53 activity directly or indirectly via Mdm2 (Kulikov et al. 2005; Pluquet et al. 2005; Charvet et al. 2011). In the present study, we showed that GSK3β regulates p53 through a novel mechanism; i.e., GSK3β controls p53 mRNA translation via phosphorylation of RNPC1. We also provided evidence that Ser195 phosphorylation converts RNPC1 from a repressor to an activator of p53.
Results
RNPC1 is phosphorylated at Ser195
We showed previously that RNPC1, as a p53 target, represses p53 mRNA translation, and thus the mutual regulation of p53 and RNPC1 constitutes a novel feedback loop in the p53 pathway (Zhang et al. 2011). The RNPC1 gene encodes two isoforms, RNPC1a with 239 amino acids and RNPC1b with 121 amino acids, but only RNPC1a has an activity toward p53 expression. For simplicity, RNPC1 and RNPC1a are used interchangeably throughout this study. Interestingly, in an SDS-PAGE gel, the RNPC1a protein is expressed as two polypeptides (Shu et al. 2006; Zhang et al. 2011), suggesting that post-translational modifications of RNPC1 may modulate the p53–RNPC1 loop. Therefore, we examined whether RNPC1 is phosphorylated. To test this, cell extracts from MCF7 and HCT116 cells that were induced to express HA-tagged RNPC1 were mock-treated or treated with λ protein phosphatase (λ-PPase). We found that upon treatment with λ-PPase, the slow-migrating band of RNPC1 was diminished, accompanied by increased levels of the fast-migrating band, suggesting that the slow-migrating band is phosphorylated (p-RNPC1) (Fig. 1A, cf. lanes 1,3 and 2,4). Similarly, upon λ-PPase treatment, the slow-migrating band of endogenous RNPC1 was decreased along with an increased level of the fast-migrating band (Fig. 1B).
Figure 1.

RNPC1 is phosphorylated at Ser195. (A) The slow-migrating band of RNPC1 is a phosphorylated form of RNPC1 (p-RNPC1). MCF7 and HCT116 cells were induced to express HA-tagged RNPC1 for 24 h. Cell lysates were collected and treated with or without λ-PPase (400 U) for 1 h, followed by Western blot analysis using antibodies against HA or actin. (B) The slow-migrating band of endogenous RNPC1 is diminished upon λ-PPase treatment. The experiment was performed as described in A except that HCT116 cell lysates were used. (C) Schematic representations of RNPC1, RNPC1b, and various RNPC1 deletion mutants. The locations of RNP1 and RNP2 are shown in gray boxes, and the potential phosphorylation sites are indicated. (D) The region from amino acid 204 to 239 in RNPC1 is required for expression of p-RNPC1. MCF7 cells were transiently transfected with 1 μg of pcDNA3 vector expressing HA-tagged RNPC1, RNPC1b, or an individual RNPC1 deletion mutant for 24 h followed by Western blot analysis with antibodies against HA or actin. (E) Ser195 phosphorylation is responsible for expression of p-RNPC1. HCT116 cells were transiently transfected with 1 μg of pcDNA3 vector expressing HA-tagged RNPC1 or various mutants (S195A, T198A, T141A, and S201A) for 24 h followed by Western blot analysis using antibodies against HA or actin. (F) The slow-migrating but not the fast-migrating band is recognized by anti-phospho-S195 RNPC1 (α-p-RNPC1). MCF7 and HCT116 cells were mock-treated or treated with doxorubicin (250 ng/mL) for 18 or 24 h followed by Western blot analysis using antibodies against p53, RNPC1, p-RNPC1, and actin.
Previously, we showed that in an SDS-PAGE gel, RNPC1b is expressed as one polypeptide (Shu et al. 2006), suggesting that the phosphorylation site is located within the C-terminal region of RNPC1. To map the residue subjected to phosphorylation, a series of deletions was made in the C-terminal region of RNPC1 (Fig. 1C). We showed that like RNPC1, RNPC1(1–220) was expressed as two bands, whereas RNPC1(1–204) and three other mutants were expressed as one band (Fig. 1D), suggesting that a serine or threonine in the region surrounding residue 204 is likely to be the phosphorylation site. To test this, alanine substitution mutants were made at S195, S201, T141, and T198. We found that only the S195A mutant was expressed as one fast-migrating band, whereas T198A, T141A, and S201A were expressed as two bands (Fig. 1E).
To confirm that the slow-migrating band is phosphorylated at S195, we generated an antibody specifically recognizing S195-phosphorylated RNPC1, designated α-p-RNPC1. We found that the antibody recognized the slow-migrating band of ectopically expressed RNPC1 and the S195D mutant, in which Ser195 was substituted with phosphor-mimetic aspartic acid, but not S195A, which is nonphosphorylatable (Supplemental Fig. S1A). We also found that upon treatment with λ-PPase, the slow-migrating band of RNPC1 immunoprecipitated by α-p-RNPC1 was decreased, which was accompanied by an increased level of the fast-migrating band (Supplemental Fig. S1B). We note that RNPC1 can form a homodimer in solution. Consistently, a minute amount of the fast-migrating RNPC1 was detected in the α-p-RNPC1 immunoprecipitates (Supplemental Fig. S1B, lane 2). Furthermore, we showed that the slow-migrating band of endogenous RNPC1 was recognized by α-p-RNPC1, which was increased in MCF7 and HCT116 cells upon treatment with doxorubicin in a dose-dependent manner (Fig. 1F, lanes 4–6,10–12). Doxorubicin, a DNA-damaging agent, induced p53 accumulation, which then led to increased expression of RNPC1 (Fig. 1F), consistent with our previous reports (Shu et al. 2006). Together, we showed that RNPC1 is a phosphor-protein and that phosphorylation of Ser195 is responsible for the expression of the slow-migrating form of RNPC1.
GSK3β is a kinase responsible for Ser195 phosphorylation
Since Ser195 is followed by a proline, a proline-directed kinase, such as GSK3, is likely involved in Ser195 phosphorylation. To test this, the levels of p-RNPC1 were measured in RNPC1-expressing HCT116 and MCF7 cells treated with lithium, an inhibitor of GSK3. We found that the levels of p-RNPC1 were decreased in a dose-dependent manner along with increased levels of unphosphorylated RNPC1 (Fig. 2A). Similarly, the levels of p-RNPC1 were decreased in MCF7 and RKO cells upon treatment with another GSK3 inhibitor, 6-bromoindirubin-3′-oxime (BIO) (Fig. 2B). In addition, upon knockdown of GSK3β, the ratio of exogenous p-RNPC1 versus RNPC1 in HCT116 cells was decreased from 0.86 to 0.41 (Fig. 2C). Next, to examine whether GSK3β directly phosphorylates RNPC1, an in vitro kinase assay was performed using recombinant GSK3β and GST-tagged RNPC1, RNPC1b, and S195A. As shown in Figure 2D, wild-type RNPC1, but not S195A and RNPC1b, was found to be phosphorylated.
Figure 2.

GSK3β is a kinase responsible for S195 phosphorylation. (A,B) The level of p-RNPC1 is decreased by Li+ (A) or BIO (B) in a dose-dependent manner. Cells were induced to express RNPC1 for 12 h followed by mock treatment or treatment with 0–40 mM Li+ (A) or 0–1 μM BIO (B) for 12 h. Cell lysates were harvested for Western blot analysis with antibodies against HA or actin. (C) Knockdown of GSK3β reduces the level of p-RNPC1. HCT116 cells were induced to express RNPC1 for 24 h followed by transient transfection with a scrambled siRNA or an siRNA against GSK3β for 72 h. Cell lysates were harvested and subjected to Western blot analysis with antibodies against RNPC1, GSK3β, and actin. The relative level of RNPC1 and p-RNPC1 was measured by densitometry, and the ratio of p-RNPC1 versus RNPC1 is shown below. (D) GSK3β phosphorylates RNPC1 in vitro. In vitro kinase assay was performed as described in the Materials and Methods. The samples were subjected to autoradiography for 24 h (top panel) and subsequently subjected to Western blot analysis with antibody against GST (bottom panel). (E) GSK3β is required for phosphorylation of endogenous RNPC1. HCT116 and RKO cells were transduced with lentivirus particles expressing a control shRNA or an shRNA against GSK3β for 72 h followed by treatment with 250 ng/mL doxorubicin for 16 h. Cell lysates were harvested and subjected to Western blot analysis with antibodies against RNPC1, GSK3β, and actin. (F,G) p-RNPC1 interacts with GSK3β in vivo. HCT116 cell lysates were immunoprecipitated with a control IgG or antibodies against RNPC1 (F) or GSK3β (G) followed by Western blot analysis with antibodies against RNPC1 and GSK3β.
To determine whether GSK3β phosphorylates endogenous RNPC1, we measured the ratio of p-RNPC1 versus RNPC1. We found that under the condition of DNA damage induced by treatment with doxorubicin, the ratio of p-RNPC1 versus RNPC1 was markedly decreased by knockdown of GSK3β in both HCT116 (1.96 vs. 0.15) and RKO (1.98 vs. 0.23) cells (Fig. 2E). To avoid potential off-target effects, GSK3β was knocked down by another shRNA targeting a different region in GSK3β, and similar results were observed in MCF7 and MDCK cells (Supplemental Fig. S2A,B). Similarly, we found that the ratio of p-RNPC1 versus RNPC1 was decreased in GSK3β−/− mouse embryonic fibroblasts (MEFs) as compared with that in wild-type MEFs (Supplemental Fig. S2C). However, under a basal condition, knockdown of both GSK3β and GSK3α, but not knockdown of each individually, significantly suppressed RNPC1 phosphorylation (Supplemental Fig. S2D). Moreover, knockdown of GSK3β, but not GSK3α, had a major effect on RNPC1 phosphorylation under the condition of DNA damage (Supplemental Fig. S2E, cf. lanes 2 and 3).
GSK3 phosphorylates a priming substrate with a consensus sequence of Ser/Thr–Pro–X–X–p-Ser/Thr (where X represents any residue) (Cohen and Frame 2001). As RNPC1 (195SPATAAS201) is not a classical priming substrate, we examined whether an interaction between GSK3β and its substrate is used for docking GSK3β (Biondi and Nebreda 2003). Indeed, we found that GSK3β was detected in anti-RNPC1 but not IgG immunoprecipitates (Fig. 2F). Conversely, RNPC1, especially p-RNPC1, was detected in GSK3β but not IgG immunocomplexes (Fig. 2G).
Ser195 phosphorylation converts RNPC1 from a repressor to an activator of p53 mRNA translation via binding to the p53 3′ UTR
Previously, we showed that RNPC1 suppresses p53 mRNA translation (Zhang et al. 2011) and is necessary for the maintenance of p21 mRNA stability (Shu et al. 2006; Cho et al. 2010). Thus, we examined whether Ser195 phosphorylation has an effect on RNPC1 activity. To test this, we generated multiple cell lines that inducibly express HA-tagged wild-type RNPC1, S195D, or S195A. We found that like wild-type RNPC1, S195A and S195D were capable of inducing p21 expression in both HCT116 and p53-null HCT116 cells (Fig. 3A,B, p21 panels), suggesting that RNPC1 is capable of regulating p21 expression independent of Ser195 phosphorylation. In addition, we found that while wild-type RNPC1 and S195A suppressed p53 expression, S195D promoted p53 expression in both HCT116 and p21-null HCT116 cells (Fig. 3A,C), suggesting that Ser195 phosphorylation converts RNPC1 from a repressor to an activator of p53 expression independent of p21. Similarly, we found that S195D promoted p53 expression in MCF7 and RKO cells (Supplemental Fig. S3A,B). To rule out potential interference of the HA tag with RNPC1 activity, we generated a group of HCT116 cell lines that inducibly express wild-type RNPC1, S195D, and S195A, none of which was tagged with an HA epitope. We found that S195D enhanced, whereas wild-type and S195A inhibited, p53 expression (Supplemental Fig. S3C). Since RNPC1 is known to suppress p53 expression at both the basal and DNA damage conditions (Zhang et al. 2011), we examined whether DNA damage has an effect on S195D and S195A activity. As expected, p53 and p21 were accumulated upon treatment with camptothecin (Supplemental Fig. S3D, cf. lanes 1,5,9 and 3,7,11). We found that p53 expression was repressed, whereas p21 expression was enhanced, by wild-type RNPC1 and S195A at both the basal and DNA damage conditions (Supplemental Fig. S3D, cf. lanes 1,3,9,11 and 2,4,10,12). Consistent with the above studies, we also found that the levels of p53 and p21 were increased by S195D at both the basal and DNA damage conditions (Supplementa1 Fig. S3D, cf. lanes 5,7 and 6,8).
Figure 3.

S195 phosphorylation converts RNPC1 from a repressor to an activator of p53 expression. (A,C) The level of p53 protein is increased by S195D but decreased by wild-type RNPC1 or S195A. HCT116 (A) and p21-null HCT116 (C) cells were uninduced or induced to express HA-tagged wild-type RNPC1, S195D, and S195A for 48 h followed by Western blot analysis with antibodies against HA, p53, p21, or actin. The level of p53 protein was normalized to that of actin, and the relative-fold change is shown below each lane. The data are representative of three independent experiments. (B) The level of p21 protein is increased by wild-type RNPC1, S195D, and S195A. p53-null HCT116 cells were uninduced or induced to express wild-type RNPC1, S195D, or S195A for 48 h followed by Western blot analysis with antibodies against HA, p21, or actin. (D,E) HCT116 (D) and RKO (E) cells were uninduced or induced to express wild-type RNPC1, S195D, and S195A for 24 h and then subjected to 35S-labeling for 10 min. Cell lysates were collected and immunoprecipitated with 1 μg of p53 antibody. The immunocomplexes were resolved by SDS-PAGE, and p53 was visualized by autoradiography. The relative level of p53 was measured by densitometry, and the relative fold change is shown below each pair.
Since RNPC1 inhibits p53 mRNA translation (Zhang et al. 2011), we wanted to determine whether the increased expression of p53 by S195D is due to increased p53 mRNA translation. To test this, 35S metabolic labeling was performed to measure the level of the newly synthesized p53 protein with or without RNPC1 expression. We showed that the level of de novo synthesized p53 protein was decreased by wild-type RNPC1 and S195A but increased by S195D in both HCT116 and RKO cells (Fig. 3D,E). To rule out the possibility that p53 mRNA stability is regulated by Ser195 phosphorylation, the level of p53 transcript was measured in HCT116 cells. We showed that the level of p53 protein was increased by S195D and decreased by wild-type RNPC1 and S195A (Supplemental Fig. S3E), consistent with the above study (Fig. 3). In addition, we found that the level of p21 transcript was increased by wild-type RNPC1, S195A, and S195D (Supplemental Fig. S3F, p21 panel), consistent with our previous studies (Shu et al. 2006; Cho et al. 2010). However, the level of p53 transcript in HCT116 cells remained unchanged regardless of expression of wild-type RNPC1, S195A, or S195D (Supplemental Fig. S3F, p53 panel).
The ability of RNPC1 to specifically suppress p53 mRNA translation is dependent on the binding of RNPC1 to p53 5′ and/or 3′ UTRs (Zhang et al. 2011). This led us to investigate whether Ser195 phosphorylation alters the binding affinity of RNPC1 to the p53 5′ or 3′ UTR. To test this, cell extracts were isolated from H1299 cells that were cotransfected with a vector expressing HA-tagged RNPC1 or S195D along with a pGL3 luciferase reporter carrying either the p53 5′ UTR or 3′ UTR. The extracts were then subjected to immunoprecipitation with anti-HA antibody to capture HA-tagged RNPC1 and S195D or a control IgG followed by RT–PCR (RNA-ChIP [chromatin immunoprecipitation). We showed that like wild-type RNPC1, S195D was found to interact with both the p53 5′ and 3′ UTRs (Supplemental Fig. S4A,B), suggesting that Ser195 phosphorylation does not alter the RNA-binding activity of RNPC1 to p53 mRNA.
Next, we examined whether activation of p53 mRNA translation by S195D is dependent on the binding of RNPC1 to p53 5′ and/or 3′ UTRs. To test this, cell extracts were isolated from H1299 cells that were cotransfected with a vector expressing wild-type RNPC1, S195D, or S195A along with an expression vector that contains the p53 coding region alone or together with the p53 5′ UTR, 3′ UTR, or both. We found that p53 expression was inhibited by wild-type RNPC1 and S195A in a dose-dependent manner as long as the p53 transcript contains 5′ and/or 3′ UTRs (Fig. 4A,C), consistent with the previous report (Zhang et al. 2011). Interestingly, we found that S195D increased p53 expression in a dose-dependent manner from the p53 transcript that contains the 3′ UTR alone or both 5′ and 3′ UTRs but not the coding region alone or together with the 5′ UTR (Fig. 4B). To rule out potential interference from endogenous RNPC1 in H1299 cells, the experiment was performed with p53−/−; RNPC1−/− double-knockout MEFs. We showed that S195D increased, whereas wild-type RNPC1 and S195A decreased, p53 expression from p53 expression vectors that contain the 3′ UTR alone or together with the 5′ UTR (Fig. 4F,G, cf. lanes 1,3,5 and 2,4,6). In addition, S195D had no effect on p53 expression from p53 transcripts that contain the coding region alone or together with the 5′ UTR (Fig. 4D,E, cf. lanes 3 and 4). These results suggest that S195D increases p53 mRNA translation through binding to the p53 3′ UTR.
Figure 4.

The 3′ UTR, but not the 5′ UTR, of p53 mRNA is required for S195D to enhance p53 expression. (A–C) H1299 cells were transiently cotransfected with a control pcDNA3 vector or various amounts of pcDNA3 vector expressing HA-tagged wild-type RNPC1 (A), S195D (B), or S195A (C) along with a fixed amount of p53 expression vector that contains the coding region (ORF) alone or in combination with the 5′ UTR, 3′ UTR, or both. Twenty-four hours post-transfection, cell lysates were collected, and the levels of p53, RNPC1, and actin were analyzed by Western blot analysis. The level of p53 protein was normalized to that of actin, and the relative fold change is shown below each lane. (D–G) RNPC1−/−; p53−/− MEFs were transiently cotransfected with a p53 expression vector that contains the coding region (ORF) alone (D) or together with the 5′ UTR (E), 3′ UTR (F), or both (G) along with a control vector or a pcDNA3 vector expressing HA-tagged wild-type RNPC1, S195D, or S195A for 24 h. Cell lysates were collected and subjected to Western blot analysis to determine the level of p53, RNPC1, and actin. The level of p53 protein was normalized to that of actin, and the relative fold change is shown below each lane.
Ser195 phosphorylation abrogates physical interaction of RNPC1 with eIF4E on p53 mRNA but promotes the binding of eIF4E to p53 mRNA potentially via eIF4G
Upon binding to the p53 5′ and/or 3′ UTRs, RNPC1 and cap-binding protein eIF4E physically interact on p53 mRNA, which then weakens the binding of eIF4E to the p53 mRNA 5′ cap, resulting in specific inhibition of p53 mRNA translation (Zhang et al. 2011). To test whether Ser195 phosphorylation has an effect on the binding of eIF4E to the p53 mRNA 5′ cap, an RNA-ChIP assay was performed. We showed that upon expression of wild-type RNPC1 or S195A, the relative level of p53 transcripts associated with eIF4E was markedly decreased (Fig. 5A,C, lanes 5,6), consistent with the previous report (Zhang et al. 2011). In contrast, S195D markedly enhanced the binding of eIF4E to p53 mRNA (Fig. 5B, lanes 5,6). As a control, RNPC1 did not bind to actin mRNA and was not able to inhibit the binding of eIF4E to actin mRNA (Fig. 5A,C, actin panels, lanes 5–8).
Figure 5.
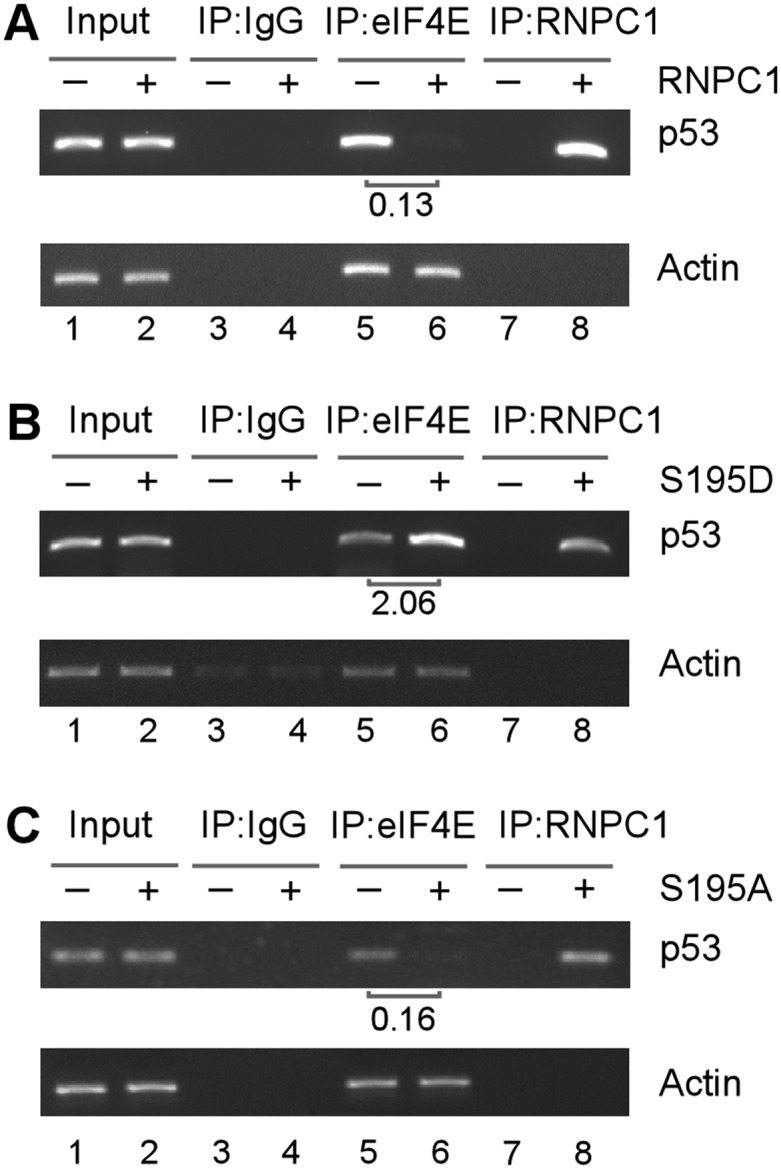
S195D enhances, whereas wild-type RNPC1 and S195A inhibit, eIF4E binding to p53 mRNA. HCT116 cells were uninduced or induced to express HA-tagged wild-type RNPC1 (A), S195D (B), and S195A (C) for 48 h followed by immunoprecipitation with a control IgG or anti-HA that recognizes RNPC1 or anti-eIF4E. Total RNAs were purified from immunocomplexes and subjected to RT–PCR analysis to measure the level of p53 and actin mRNA. The relative level of p53 mRNA was measured by densitometry, and the relative fold change is shown below each pair.
Next, we examined whether Ser195 phosphorylation has an effect on the interaction of RNPC1 with eIF4E. As expected, eIF4E was detected in anti-RNPC1 immunocomplexes (Fig. 6A), and RNPC1 was detected in anti-eIF4E immunocomplexes (Fig. 6B). However, the ratio of p-RNPC1 versus RNPC1 in anti-eIF4E immunocomplexes (0.12) was much lower than that in the input control (1.96), suggesting that p-RNPC1 has a weak affinity to eIF4E. To further test this, we measured the interaction of S195D and S195A with eIF4E. We showed that like wild-type RNPC1, S195A was able to interact with eIF4E (Fig. 6C,D, lanes 3,9). In contrast, the interaction of S195D with eIF4E was very weak or undetectable (Fig. 6C,D, lane 6). These observations led us to speculate that the region surrounding S195 is necessary for RNPC1 to interact with eIF4E. To test this, we delineated the regions in RNPC1 and eIF4E for their physical interaction. We showed that endogenous eIF4E was detected in immunocomplexes containing RNPC1, RNPC1(amino acids 1–220), and RNPC1(amino acids 1–204) but not the ones containing RNPC1(amino acids 1–189) and RNPC1(Δ189–204) (Supplemental Fig. S5A,B). In addition, a GST pull-down assay was performed and showed that His-tagged eIF4E fusion protein physically interacted with GST-tagged RNPC1 and RNPC1(amino acids 175–220) but not RNPC1(amino acids 1–189), RNPC1(amino acids 189–204), RNPC1(amino acids 220–239), and RNPC1(Δ189–204) (Supplemental Fig. S5C,D). These data suggest that the domain in RNPC1 for interaction with eIF4E is located within amino acids 175–220, which supports the idea that Ser195 phosphorylation abrogates physical interaction of RNPC1 with eIF4E. Conversely, the GST pull-down assay showed that His-tagged RNPC1 physically interacted with GST-tagged eIF4E, eIF4E(amino acids 141–218), and eIF4E(amino acids 194–218) but exhibited weak or no interaction with eIF4E(amino acids 1–70), eIF4E(amino acids 1–140), eIF4E(amino acids 141–167), and eIF4E(amino acids 168–193) (Supplemental Fig. S5E,F). These data suggest that the domain in eIF4E for interaction with RNPC1 is located within amino acids 194–218.
Figure 6.

S195 phosphorylation abrogates eIF4E, but promotes eIF4G, to interact with RNPC1. (A,B) S195 phosphorylation abrogates the interaction between endogenous eIF4E and RNPC1. HCT116 cell lysates were treated with RNase A and then immunoprecipitated with a control IgG or antibodies against RNPC1 (A) or eIF4E (B). Immunocomplexes were examined by Western blot analysis with anti-RNPC1 and anti-eIF4E. The relative level of RNPC1 and p-RNPC1 was measured by densitometry, and the ratio of p-RNPC1 versus RNPC1 is shown in B. (C,D) Wild-type RNPC1 and S195A, but not S195D, were capable of interacting with eIF4E. HCT116 cells were induced to express HA-tagged wild-type RNPC1, S195D, or S195A for 24 h. Cell lysates were collected and treated with RNase A followed by immunoprecipitation with a control IgG or anti-eIF4E (C) or anti-HA that recognizes RNPC1 (D). The immunocomplexes were examined by Western blot analysis with anti-RNPC1 and anti-eIF4E. (E,F) eIF4G preferentially interacts with p-RNPC1. HCT116 (E) or RKO (F) cell lysates were treated with RNase A and then immunoprecipitated with a control IgG or antibodies against RNPC1 (E) or eIF4G (F). Immunocomplexes were examined by Western blot analysis with anti-RNPC1 and anti-eIF4G. (G) HCT116 cells were induced to express HA-tagged wild-type RNPC1 for 24 h. Cell lysates were collected and treated with RNase A followed by immunoprecipitation with a control IgG, anti-RNPC1, or anti-p-RNPC1. The immunocomplexes were examined by Western blot analysis with anti-eIF4G, anti-eIF4E, and anti-HA (HA-RNPC1). The ratio of p-RNPC1 versus RNPC1 is shown below the lane. (H) Extracts were collected from HCT116 cells induced to express HA-tagged wild-type RNPC1, S195D, or S195A for 24 h and then immunoprecipitated with anti-RNPC1. The immunocomplexes were digested with V8 protease for 1 h at 37°C. The digested samples were subjected to Western blot analysis with anti-HA. (I,J) Deletion mutant Δ189–204, which is capable of interacting with eIF4G but not eIF4E, promotes p53 expression. H1299 cells were transiently transfected with a p53 expression vector (which contains either the 5′ UTR or 3′ UTR) along with a control vector or a pcDNA3 vector that expresses HA-tagged wild-type RNPC1, Δ189–204, or S195D for 24 h. Cell lysates were collected and subjected to Western blot analysis to determine the level of p53, RNPC1, and actin. The level of p53 protein was normalized to that of actin, and the relative fold change is shown below each pair. (K) Deletion mutant Δ189–204 promotes p53 expression via the p53 3′ UTR in a dose-dependent manner. The experiment was performed as in I and J except that two doses of the Δ189–204 expression vector were used.
The above studies suggest that Ser195 phosphorylation blocks physical interaction between RNPC1 and eIF4E on p53 mRNA and thus abrogates RNPC1 to inhibit p53 mRNA translation. However, it remains unclear how Ser195 phosphorylation promotes p53 mRNA translation. Hence, Ser195 phosphorylation must impact on other components of the translation machinery. eIF4G, a scaffold protein, recruits several translation initiation factors, including eIF4A and eIF4E, to form the eIF4F complex, a key component for the assembly of the translation machinery (Haghighat and Sonenberg 1997; Hentze 1997; von der Haar et al. 2000). Indeed, eIF4G is a common target for translational regulation (Kapasi et al. 2007; Rajyaguru et al. 2012). Thus, we examined whether RNPC1 interacts with eIF4G. We found that eIF4G was detected in anti-RNPC1 but not IgG immunoprecipitates (Fig. 6E). Conversely, we found that RNPC1, especially p-RNPC1, was detected in anti-eIF4G but not IgG immunoprecipitates (Fig. 6F). Similarly, the relative level of eIF4G was much higher in α-p-RNPC1 immunoprecipitates than in α-RNPC1 immunoprecipitates (Fig. 6G; Supplemental Fig. S6A). Due to strong interaction between eIF4E and eIF4G, a substantial level of eIF4E was detected in anti-RNPC1 and anti-p-RNPC1 immunoprecipitates (Fig. 6G; Supplemental Fig. S6A).
To examine whether RNPC1 directly interacts with eIF4G, GST pull-down assays were performed. We showed that RNPC1 interacted with eIF4G(amino acids 1–156) and eIF4G(amino acids 1046–1600) but not eIF4G(amino acids 157–667) and eIF4G(amino acids 668–1045) (Supplemental Fig. S6B,C). Conversely, we found that endogenous eIF4G interacted with full-length RNPC1 and RNPC1(Δ189–204) and weakly with RNPC1(amino acids 1–220) but not with RNPC1(amino acids 1–204) and RNPC1(amino acids 1–189) (Supplemental Fig. S6D,E). These data suggest that the region of amino acids 204–239 in RNPC1 is capable of interacting with two regions in eIF4G (amino acids 1–156 and 1046–1600).
Since S195D and p-RNPC1 have a stronger affinity to eIF4G than S195A, there is a possibility that the conformation of RNPC1 protein is altered by S195D substitution and/or S195 phosphorylation. To test this, a V8 protease sensitivity assay was performed. Indeed, we found that the protease sensitivity of S195D was quite different from that for wild-type RNPC1 and especially S195A (Fig. 6H).
Since RNPC1(Δ189–204) is capable of interacting with eIF4G but not eIF4E, we reasoned that, like S195D, Δ189–204 would increase p53 mRNA translation via the p53 3′ UTR. To test this, Δ189–204 was cotransfected in H1299 cells with a p53 expression vector along with wild-type RNPC1 and S195D as a control. We found that wild-type RNPC1 suppressed p53 expression from the p53 transcript that carries either the p53 5′ or 3′ UTR (Fig. 6I,J), consistent with the above study (Fig. 4) and our previous study (Zhang et al. 2011). In contrast, we found that, like S195D, Δ189–204 increased p53 expression from the p53 transcript that carries the p53 3′ UTR but not the 5′ UTR (Fig. 6I,J). In addition, p53 expression was increased by Δ189–204 in a dose-dependent manner from the p53 transcript that carries the p53 3′ UTR but not the 5′ UTR (Fig. 6K).
The phosphatidylinositol 3-kinase (PI3K)–Akt pathway regulates p53 expression via GSK3-mediated phosphorylation of RNPC1
Finally, we asked whether GSK3β-mediated phosphorylation of RNPC1 has an effect on p53 mRNA translation. To test this, GSK3β was knocked down in HCT116 cells by lentiviral shRNA along with treatment with doxorubicin (Fig. 7A) or camptothecin (Fig. 7B). We found that the level of p-RNPC1 was decreased upon knockdown of GSK3β, but the levels of p53 protein were not found to be significantly decreased (Fig. 7A,B, left panels). This is not surprising, since inhibition of GSK3β leads to decreased activity of Mdm2 (Kulikov et al. 2005) and increased stability of p53 (Qu et al. 2004; Pluquet et al. 2005), both of which decrease p53 turnover. To test this, we performed pulse-chase analysis with 35S metabolic labeling and found that upon knockdown of GSK3β, p53 protein stability was markedly increased (Supplemental Fig. S7A). As a result, decreased p53 turnover would then compensate for decreased p53 mRNA translation due to GSK3β deficiency. Thus, we measured the level of newly synthesized p53 protein by 35S metabolic labeling. We showed that the level of de novo synthesized p53 protein was significantly decreased upon knockdown of GSK3β in HCT116 cells (Fig. 7A,B, right panels). These results suggest that GSK3β regulates p53 mRNA translation through Ser195 phosphorylation of RNPC1.
Figure 7.

The PI3K–Akt pathway regulates p53 expression via GSK3-mediated phosphorylation of RNPC1. (A) Knockdown of GSK3β attenuates RNPC1 phosphorylation at Ser195 and consequently inhibits p53 mRNA translation. (Left panels) HCT116 cells were transduced with lentivirus particles expressing a control shRNA or an shRNA against GSK3β for 72 h followed by treatment with 250 ng/mL doxorubicin for 16 h. Cell lysates were collected and subjected to Western blot analysis with antibodies against RNPC1, GSK3β, and actin. (Right panel) Cells treated as described in the left panel were 35S-labeled for 20 min followed by immunoprecipitation with anti-p53. (Right panel) The immunocomplexes were resolved by SDS-PAGE, and p53 was visualized by autoradiography. The relative level of p53 was measured by densitometry, and the relative fold change is shown below each pair. (B) The experiment was performed as in A except that 500 nM camptothecin was used. (C) Inhibition of Akt kinase leads to activation of GSK3β kinase accompanied by increased levels of p-RNPC1 and p53 expression. The levels of p-AKT, p-GSK3β, GSK3β, RNPC1, p53, and actin were measured in HCT116 and HCT116(p53−/−) cells mock-treated or treated with 0–2 μM MK2206 for 2 h. (D) The effect of the Akt inhibitor MK2206 on p53 expression is RNPC1-dependent. The levels of p-GSK3β, RNPC1, p53, and actin were measured in HCT116 and MCF7 cells with or without RNPC1 knockdown for 3 d followed by treatment with 0–2 μM MK2206 for 2 h. (E) Ser195 phosphorylation converts RNPC1 from a repressor to an activator of p53 mRNA translation. RNPC1 interacts with eIF4E on p53 mRNA and inhibits p53 mRNA translation by sequestering eIF4E from interacting with p53 mRNA. Ser195 phosphorylation by GSK3 blocks the interaction of RNPC1 with eIF4E, which abrogates RNPC1 to inhibit p53 mRNA translation. Additionally, Ser195 phosphorylation enhances the interaction of RNPC1 with eIF4G on p53 mRNA and then recruits eIF4E to promote p53 mRNA translation.
It is well known that Akt kinase phosphorylates GSK3β at Ser9 and then inhibits GSK3β activity (Cross et al. 1995). Thus, we examined whether the effect of GSK3β on RNPC1-mediated p53 mRNA translation is modulated by the Akt kinase inhibitor (MK2206) and PI3K inhibitor (wortmannin). PI3K is an activator of Akt kinase, and inhibition of PI3K weakens Akt kinase activity and subsequently increases GSK3β activity (Cross et al. 1995). We showed that upon treatment with the Akt inhibitor MK2206, the level of p-Akt and GSK3β S9 phosphorylation was decreased in both p53-proficient and p53-deficient HCT116 cells (Fig. 7C). We also showed that upon treatment with MK2206, the level of p-RNPC1 and p53 was increased in a dose-dependent manner (Fig. 7C). Moreover, we showed that upon knockdown of RNPC1, p53 expression was increased (Fig. 7D, lanes 4,10), consistent with our previous study (Zhang et al. 2011). However, the Akt kinase inhibitor MK2206 was unable to further increase p53 expression (Fig. 7D, cf. lanes 4 and 5,6 and lanes 10 and 11,12), suggesting that the increased expression of p53 upon treatment with MK2206 was RNPC1-dependent. Similarly, we found that upon treatment with the PI3K inhibitor wortmannin, GSK3β S9 phosphorylation was decreased, which led to increased RNPC1 phosphorylation and increased p53 expression (Supplemental Fig. S7B).
Discussion
Translational regulation of p53 is a major mechanism by which p53 activity is controlled (Zhang and Chen 2008). Previously, we showed that RNPC1 interacts with eIF4E and the p53 5′ and 3′ UTRs (Zhang et al. 2011). The interaction of RNPC1 with eIF4E on p53 mRNA specifically sequesters eIF4E from interacting with p53 mRNA, leading to repression of p53 mRNA translation (Fig. 7E; Zhang et al. 2011). Here, we showed that p-RNPC1 is still capable of interacting with the p53 5′ UTR, but Ser195 phosphorylation alters the conformation of RNPC1 protein (Fig. 6H) and then weakens its interaction with eIF4E on p53 mRNA, making eIF4E free to bind to p53 mRNA. This is likely responsible for Ser195 phosphorylation abrogating the negative effect of RNPC1 on p53 mRNA translation. However, we also found that p-RNPC1 and S195D actually stimulate p53 mRNA translation by enhancing the binding of eIF4E to p53 mRNA. How do we explain this? It is well known that eIF4G is a scaffold protein that interacts with eIF4E and other components of the translation machinery and then stabilizes the eIF4F complex (Haghighat and Sonenberg 1997; von der Haar et al. 2000). We showed that p-RNPC1 has a high affinity toward eIF4G and that the interaction of S195D with the p53 3′ UTR is required for promoting p53 mRNA translation. In addition, we showed that the domain in eIF4G for binding to RNPC1 (Supplemental Fig. S6B,C) is separate from the domain for binding to eIF4E (Haghighat et al. 1995). Furthermore, the domain in RNPC1 for binding eIF4E (Supplemental Fig. S5A–D) is also separate from the domain for binding to eIF4G (Supplemental Fig. S6D,E). Consistent with these observations, RNPC1(Δ189–204), which lacks residues 189–204 and is capable of interacting with eIF4G but not eIF4E, increases p53 mRNA translation via the p53 3′ UTR (Fig. 6I–K). Thus, we hypothesize that upon binding to p53 mRNA, p-RNPC1 preferentially interacts with eIF4G, which then recruits eIF4E for the assembly of the eIF4F complex to increase p53 translation (Fig. 7E). Our model is consistent with several other pathways that regulate mRNA translation via phosphorylation. One is 4E-BP, which interacts with and then inhibits eIF4E activity. Interestingly, hyperphosphorylation prevents 4E-BP from binding to eIF4E, thus abrogating the inhibitory effect of 4E-BP on eIF4E (Heesom and Denton 1999). Another example is Maskin, which associates with eIF4E and inhibits the assembly of the eIF4F complex. Phosphorylation prevents Maskin from interacting with eIF4E, which then allows for translational activation of cytoplasmic polyadenylation element-containing mRNAs (Barnard et al. 2005; Cao et al. 2006). Nevertheless, modulation of RNPC1 activity by phosphorylation is unique; that is, Ser195 phosphorylation not only abrogates the inhibitory activity of RNPC1 toward p53 mRNA translation via eIF4E, but also enhances the interaction of RNPC1 with eIF4G and then promotes p53 mRNA translation. As a result, phosphorylation of RNPC1 at Ser195 converts RNPC1 from a repressor to an activator of p53 mRNA translation (Fig. 7E). Together, our data suggest that the p53–RNPC1 loop can be modulated by phosphorylation and possibly other mechanisms to increase or decrease p53 activity for cancer therapy.
GSK3β is shown to possess two opposing activities toward p53. For example, GSK3β was found to increases p53 activity through p53–GSK3β complex formation in the nucleus (Watcharasit et al. 2002, 2003) and through phosphorylation of TIP60 and TIP60-mediated acetylation of p53 (Charvet et al. 2011). In contrast, GSK3β was found to phosphorylate MDM2, which then promotes p53 degradation (Kulikov et al. 2005). In addition, GSK3β can directly phosphorylate p53, and phosphorylated p53 at Ser315 and Ser376 is recognized as a suitable Mdm2 substrate for degradation (Qu et al. 2004; Pluquet et al. 2005). Indeed, we found that GSK3β possesses two opposing activities toward p53. We showed that knockdown of GSK3β increased p53 protein stability (Supplemental Fig. S7A) but decreased p53 mRNA translation via RNPC1 (Fig. 7A,B). As a result, the overall level of p53 protein was not significantly altered in cells upon knockdown of GSK3β (Fig. 7A,B). We note that knockdown of GSK3β has a mild effect on S195 phosphorylation under a nonstress condition (Fig. 2C; Supplemental Fig. S2D). Thus, DNA damage makes GSK3β competent to phosphorylate RNPC1 (Fig. 2E; Supplemental Fig. S2E), consistent with a previous report that GSK3β is activated by endoplasmic reticulum (ER) stress to phosphorylate p53 (Qu et al. 2004) and by DNA damage to phosphorylate TIP60 (Charvet et al. 2011). Importantly, when the signal from the PI3K–Akt pathway is blocked, which leads to activation of GSK3β kinase, p53 expression is increased in a RNPC1-dependent manner (Fig. 7C,D; Supplemental Fig. S7B), suggesting that the opposing function of GSK3β to inhibit p53 expression via phosphorylation of Mdm2/p53 is suppressed in the absence of the PI3K–Akt signaling. Indeed, the PI3K–Akt pathway is found to activate Mdm2, which subsequently promotes p53 degradation (Gottlieb et al. 2002). Altogether, further understanding of how GSK3β regulates RNPC1 phosphorylation and p53 activity under both basal and stress conditions is warranted, especially considering that inhibitors of GSK3, which are currently being explored as a therapeutic agent for neurological diseases, may increase the potential of tumor incidence (Luo 2009; Sutherland 2011).
Materials and methods
Reagents
The list of supplies is provided in the Supplemental Material.
Plasmids
The cloning strategy and primers used are listed in the Supplemental Material.
Cell culture and cell line generation
RKO, MCF7, HCT116, p21-null HCT116, and p53-null HCT116 cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Hyclone) as previously described (Zhang and Chen 2007). p53−/−; RNPC1−/− double-knockout MEFs were generated as described previously (Zhang et al. 2011) and cultured in DMEM supplemented with 10% fetal bovine serum, 55 μM β-mercaptoethanol, and 1× MEM nonessential amino acid solution (Cellgro). The cell lines that inducibly express RNPC1 or HA-tagged RNPC1 were generated and cultured as previously reported (Shu et al. 2006; Zhang et al. 2010). The stable cell lines that inducibly express HA-tagged S195D or S195A were generated based on the Tet-on-inducible system as previously described (Harms and Chen 2007). Briefly, a pcDNA4 vector containing HA-tagged S195D or S195A was transfected into RKO, MCF7, HCT116, p21-null HCT116, and p53-null HCT116 cells that also express a tetracycline repressor (pcDNA6). The RNPC1-expressing cells were selected with zeocin and confirmed by Western blot analysis. To induce expression of HA-tagged S195D or S195A, 0.5 μg/mL doxycycline, a tetracycline analog, was added to the medium for various times. HCT116 cell lines that inducibly expressed untagged S195D or S195A were similarly generated and used as described above. The cell lines that can inducibly express an shRNA against RNPC1 were generated and cultured as previously reported (Zhang et al. 2010).
Western blot analysis and immunoprecipitation
Western blot analysis was performed as previously described (Zhang and Chen 2007). Cell lysates suspended in 2× SDS sample buffer were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and probed with the indicated antibodies. The immunoreactive bands were visualized by enhanced chemiluminescence (Pierce) and quantified by densitometry with the software LabWorks (UVP). The immunoprecipitation assay was performed as previously described (Zhang and Chen 2007). Briefly, cells were lysed in 0.2% Triton lysis buffer (25 mM Tris at pH 7.4, 25 mM NaCl, 0.2% Triton X-100) supplemented with 100 μg/mL proteinase inhibitor cocktail followed by incubation with 1 μg of antibody or control IgG. The immunocomplexes were brought down by protein A/G beads and subjected to Western blot analysis.
35S metabolic labeling
The metabolic labeling was performed as described (Bonifacino 2001). Briefly, cells were preincubated in methionine-free DMEM for 1 h and then labeled with 100 μCi/mL 35S-methionine (PerkinElmer) for 10 min. The incorporation of 35S-methionine into newly synthesized proteins was measured by TCA precipitation. 35S-labeled lysates (1 × 107 counts per minute [cpm]) were immunoprecipitated with 1.0 μg of anti-p53. The immunocomplexes were resolved in SDS-PAGE gels and then subjected to autoradiography.
RNA isolation and RT–PCR analysis
The methods and primers used are listed in the Supplemental Material.
RNA-ChIP
RNA-ChIP was performed as described (Peritz et al. 2006). Briefly, cells (2 × 107) were uninduced or induced to express wild-type RNPC1, S195D, or S195A for 24 h. Cell extracts were prepared with immunoprecipitation buffer (100 mM KCl, 5 mM MgCl2, 10 mM HEPES, 1 mM DTT, 0.5% NP-40) and then incubated with 2 μg of anti-HA, anti-RNPC1, or an isotype control IgG overnight at 4°C. The RNA–protein immunocomplexes were brought down by protein A/G beads followed by RT–PCR.
RNAi
Scrambled siRNAs (5′-GCAGUGUCUCCACGUACUAdTdT-3′) and siRNAs against GSK3β (5′-CTGCATTTATCGTTAACCTAAdTdT-3′) were purchased from Dharmacon. For siRNA transfection, siLentFect lipid reagent (Bio-Rad) was used according to the user's manual. For lentiviral production, lentivirus vectors (pLKO.1-puro) expressing shRNA of interest were purchased from Sigma. The sequences were 5′-CGCTGAGTACTTCGAAATGTC-3′ for control luciferase shRNA and 5′-CCGATTGCGTTATTTCTTCTA-3′ for GSK3β shRNA. Ten micrograms of a lentivirus vector expressing shRNA along with packaging plasmids, 5 μg of pRSV-REV, 5 μg of pMDL g/p RRE, and 5 μg of VSVG were cotransfected into 1 × 107 293T cells by the ExpressFect transfection system (Denville Scientific) according to the user's manual. After 48 h, the supernatant containing shRNA-expressing lentivirus was filtered and concentrated by ultracentrifugation (28,000 rpm for 1 h at 4°C). The concentrated lentiviral particles were then transduced into cells followed by puromycin selection (1 μg/mL) for 72 h.
In vitro kinase assay
GST-tagged RNPC1, RNPC1b, and S195A were expressed in bacteria BL21 and purified with glutathione sepharose beads. One microgram of each purified protein was incubated in the presence or absence of 20 U of GSK3β (New England Biolabs) in a reaction buffer (20 mM Tris-HCl, 10 mM MgCl2, 5 mM DTT, 200 mM ATP, 10 μCi [r-32P] ATP) for 30 min at 30°C. The reaction mixtures were subjected to SDS-PAGE. Gels were transformed to a nitrocellulose membrane and then autoradiographed for 24 h. After exposure to film, the blot was probed with anti-GST antibody to reveal the positions and quantities of RNPC1 proteins.
V8 protease sensitivity assay
Extracts were collected from HCT116 cells induced to express HA-tagged wild-type RNPC1, S195A, or S195D for 24 h and then immunoprecipitated with antibody against RNPC1. The immunocomplexes were digested with 1 μg of V8 protease (endoproteinase Glu-C) in 50 mM ammonium bicarbonate (pH 7.8) for 1 h at 37°C. The reactions were stopped by the addition of 2 mM PMSF and immediately subjected to SDS-PAGE.
Acknowledgments
We thank Dr. Jim Woodgett (University of Toronto, Canada) for sharing GSK3β−/− MEFs. This work is supported in part by NIH grants CA076069, CA102188, and CA121137. M.Z., J.Z., Xiangling Chen, and S.-J.C. did the experiments and/or analyzed the data; Xinbin Chen supervised the project and analyzed the data; and M.Z., J.Z., and Xinbin Chen wrote the manuscript. All authors read and commented on the draft version of the manuscript and approved the final version.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.221739.113.
References
- Barnard DC, Cao Q, Richter JD 2005. Differential phosphorylation controls Maskin association with eukaryotic translation initiation factor 4E and localization on the mitotic apparatus. Mol Cell Biol 25: 7605–7615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi RM, Nebreda AR 2003. Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem J 372: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS 2001. Metabolic labeling with amino acids. Curr Protoc Protein Sci 17: 3.7.1–3.7.10 [DOI] [PubMed] [Google Scholar]
- Cao Q, Kim JH, Richter JD 2006. CDK1 and calcineurin regulate Maskin association with eIF4E and translational control of cell cycle progression. Nat Struct Mol Biol 13: 1128–1134 [DOI] [PubMed] [Google Scholar]
- Charvet C, Wissler M, Brauns-Schubert P, Wang SJ, Tang Y, Sigloch FC, Mellert H, Brandenburg M, Lindner SE, Breit B, et al. 2011. Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol Cell 42: 584–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kastan MB 2010. 5′–3′-UTR interactions regulate p53 mRNA translation and provide a target for modulating p53 induction after DNA damage. Genes Dev 24: 2146–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder T, et al. 2006. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 10: 529–541 [DOI] [PubMed] [Google Scholar]
- Cho SJ, Zhang J, Chen X 2010. RNPC1 modulates the RNA-binding activity of, and cooperate with, HuR to regulate p21 mRNA stability. Nucleic Acids Res 38: 2256–2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Frame S 2001. The renaissance of GSK3. Nat Rev Mol Cell Biol 2: 769–776 [DOI] [PubMed] [Google Scholar]
- Cross DAE, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA 1995. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785–789 [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR 2003. GSK-3: Tricks of the trade for a multi-tasking kinase. J Cell Sci 116: 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldstein O, Ben-Hamo R, Bashari D, Efroni S, Ginsberg D 2012. RBM38 is a direct transcriptional target of E2F1 that limits E2F1-induced proliferation. Mol Cancer Res 10: 1169–1177 [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P 2001. GSK3 takes centre stage more than 20 years after its discovery. Biochem J 359: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajjar M, Candeias MM, Malbert-Colas L, Mazars A, Fujita J, Olivares-Illana V, Fahraeus R 2012. The p53 mRNA–Mdm2 interaction controls Mdm2 nuclear trafficking and is required for p53 activation following DNA damage. Cancer Cell 21: 25–35 [DOI] [PubMed] [Google Scholar]
- Gottlieb TM, Leal JF, Seger R, Taya Y, Oren M 2002. Cross-talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 21: 1299–1303 [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS 2001. The multifaceted roles of glycogen synthase kinase 3Î2 in cellular signaling. Prog Neurobiol 65: 391–426 [DOI] [PubMed] [Google Scholar]
- Haghighat A, Sonenberg N 1997. eIF4G dramatically enhances the binding of eIF4E to the mRNA 5′-cap structure. J Biol Chem 272: 21677–21680 [DOI] [PubMed] [Google Scholar]
- Haghighat A, Mader S, Pause A, Sonenberg N 1995. Repression of cap-dependent translation by 4E-binding protein 1: Competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J 14: 5701–5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms KL, Chen XB 2007. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53–DNA binding activity. Cancer Res 67: 3145–3152 [DOI] [PubMed] [Google Scholar]
- Harms K, Nozell S, Chen X 2004. The common and distinct target genes of the p53 family transcription factors. Cell Mol Life Sci 61: 822–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesom KJ, Denton RM 1999. Dissociation of the eukaryotic initiation factor-4E/4E-BP1 complex involves phosphorylation of 4E-BP1 by an mTOR-associated kinase. FEBS Lett 457: 489–493 [DOI] [PubMed] [Google Scholar]
- Hentze MW 1997. Translation—elF4G: A multipurpose ribosome adapter? Science 275: 500–501 [DOI] [PubMed] [Google Scholar]
- Jenssen TK, Kuo WP, Stokke T, Hovig E 2002. Associations between gene expressions in breast cancer and patient survival. Hum Genet 111: 411–420 [DOI] [PubMed] [Google Scholar]
- Jonsson G, Staaf J, Olsson E, Heidenblad M, Vallon-Christersson J, Osoegawa K, de Jong P, Oredsson S, Ringner M, Hoglund M, et al. 2007. High-resolution genomic profiles of breast cancer cell lines assessed by tiling BAC array comparative genomic hybridization. Genes Chromosomes Cancer 46: 543–558 [DOI] [PubMed] [Google Scholar]
- Kapasi P, Chaudhuri S, Vyas K, Baus D, Komar AA, Fox PL, Merrick WC, Mazumder B 2007. L13a blocks 48S assembly: Role of a general initiation factor in mRNA-specific translational control. Mol Cell 25: 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse J-P, Gu W 2009. Modes of p53 regulation. Cell 137: 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulikov R, Boehme KA, Blattner C 2005. Glycogen synthase kinase 3-dependent phosphorylation of Mdm2 regulates p53 abundance. Mol Cell Biol 25: 7170–7180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveille N, Elkon R, Davalos V, Manoharan V, Hollingworth D, Vrielink JO, le Sage C, Melo CA, Horlings HM, Wesseling J et al. 2011. Selective inhibition of microRNA accessibility by RBM38 is required for p53 activity. Nat Commun 2: 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Oren M 2009. The first 30 years of p53: Growing ever more complex. Nat Rev Cancer 9: 749–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J 2009. Glycogen synthase kinase 3β (GSK3β) in tumorigenesis and cancer chemotherapy. Cancer Lett 273: 194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazan-Mamczarz K, Galban S, Lopez de Silanes I, Martindale JL, Atasoy U, Keene JD, Gorospe M 2003. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci 100: 8354–8359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peritz T, Zeng F, Kannanayakal TJ, Kilk K, Eiriksdottir E, Langel U, Eberwine J 2006. Immunoprecipitation of mRNA–protein complexes. Nat Protoc 1: 577–580 [DOI] [PubMed] [Google Scholar]
- Pluquet O, Qu LK, Baltzis D, Koromilas AE 2005. Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3β. Mol Cell Biol 25: 9392–9405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L, Huang S, Baltzis D, Rivas-Estilla AM, Pluquet O, Hatzoglou M, Koumenis C, Taya Y, Yoshimura A, Koromilas AE 2004. Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3β. Genes Dev 18: 261–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajyaguru P, She M, Parker R 2012. Scd6 targets eIF4G to repress translation: RGG motif proteins as a class of eIF4G-binding proteins. Mol Cell 45: 244–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley T, Sontag E, Chen P, Levine A 2008. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9: 402–412 [DOI] [PubMed] [Google Scholar]
- Shu L, Yan W, Chen X 2006. RNPC1, an RNA-binding protein and a target of the p53 family, is required for maintaining the stability of the basal and stress-induced p21 transcript. Genes Dev 20: 2961–2972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland C 2011. What are the bona fide GSK3 substrates? Int J Alzheimers Dis 2011: 505607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi M, Absalon MJ, McLure KG, Kastan MB 2005. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and Nucleolin. Cell 123: 49–63 [DOI] [PubMed] [Google Scholar]
- von der Haar T, Ball PD, McCarthy JEG 2000. Stabilization of eukaryotic initiation factor 4E binding to the mRNA 5′-cap by domains of eIF4G. J Biol Chem 275: 30551–30555 [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C 2009. Blinded by the light: The growing complexity of p53. Cell 137: 413–431 [DOI] [PubMed] [Google Scholar]
- Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, Johnson GV, Jope RS 2002. Direct, activating interaction between glycogen synthase kinase-3β and p53 after DNA damage. Proc Natl Acad Sci 99: 7951–7955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watcharasit P, Bijur GN, Song L, Zhu JH, Chen XB, Jope RS 2003. Glycogen synthase kinase-3 β (GSK3β) binds to and promotes the actions of p53. J Biol Chem 278: 48872–48879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Pan W 2010. GSK3: A multifaceted kinase in Wnt signaling. Trends Biochem Sci 35: 161–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu E, Zhang J, Chen X 2013. MDM2 expression is repressed by the RNA-binding protein RNPC1 via mRNA stability. Oncogene 32: 2169–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Zhang J, Zhang Y, Jung YS, Chen X 2012. p73 expression is regulated by RNPC1, a target of the p53 family, via mRNA stability. Mol Cell Biol 32: 2336–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen X 2007. ΔNp73 modulates nerve growth factor-mediated neuronal differentiation through repression of TrkA. Mol Cell Biol 27: 3868–3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen X 2008. Posttranscriptional regulation of p53 and its targets by RNA-binding proteins. Curr Mol Med 8: 845–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Jun Cho S, Chen X 2010. RNPC1, an RNA-binding protein and a target of the p53 family, regulates p63 expression through mRNA stability. Proc Natl Acad Sci 107: 9614–9619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Cho SJ, Shu L, Yan W, Guerrero T, Kent M, Skorupski K, Chen H, Chen X 2011. Translational repression of p53 by RNPC1, a p53 target overexpressed in lymphomas. Genes Dev 25: 1528–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]