

BRCA1 p220 participates in DNA damage responses. Dimitrov et al. find that miR-545 directly reduces p220 expression. miR-545 inhibition increased p220 expression, and aberrant p220-associated DNA damage responses and de novo DNA strand breaks accumulated. Strand breaks were a product of p220 overexpression and were also dependent on aberrant, overexpressed p220-driven recruitment of RAD51 to DNA damage sites. These results suggest that, like its loss, an excess of p220 function represents a threat to genome integrity.
Keywords: BRCA1, miR-545, DNA damage, cell cycle, G0/G1
Abstract
Endogenous BRCA1 p220 expression peaks in S and G2 when it is activated, and the protein participates in certain key DNA damage responses. In contrast, its expression is markedly reduced in G0/G1. While variations in transcription represent a significant part of p220 expression control, there is at least one other relevant process. We found that a microRNA, miR-545, that is expressed throughout the cell cycle down-modulates endogenous p220 mRNA and protein abundance directly in both G0/G1 and S/G2. When miR-545 function was inhibited by a specific antagomir, endogenous p220 expression increased in G0/G1, and aberrant p220-associated DNA damage responses and de novo DNA strand breaks accumulated. Analogous results were observed upon inhibition of miR-545 function in S/G2. Both sets of antagomir effects were mimicked by infecting cells with a p220 cDNA-encoding adenoviral vector. Thus, strand breaks were a product of p220 overexpression, and their prevention by miR-545 depends on its modulation of p220 expression. Breaks were also dependent on aberrant, overexpressed p220-driven recruitment of RAD51 to either spontaneously arising or mutagen-based DNA damage sites. Hence, when its level is not physiologically maintained, endogenous p220 aberrantly directs at least one DNA repair protein, RAD51, to damage sites, where their action contributes to the development of de novo DNA damage. Thus, like its loss, a surfeit of endogenous p220 function represents a threat to genome integrity.
BRCA1 is a high-penetrance, breast and ovarian cancer suppressor gene. Its p220 product performs its tumor suppression function. It is also a pivotal contributor to the maintenance of genome stability. The mechanisms underlying its organ-specific tumor-suppressive properties are poorly understood, although its role in genome stability control is a major factor (Li and Greenberg 2012; Roy et al. 2012; Silver and Livingston 2012).
BRCA1 is instrumental in the performance of several genome maintenance functions. Included are proper cell cycle checkpoint control, homologous recombination-mediated repair (HRR) of DNA double-strand breaks (DSBs), post-replicative repair of stalled replication forks (Moynahan et al. 1999; Greenberg et al. 2006; O'Donovan and Livingston 2010; Pathania et al. 2011; Schlacher et al. 2012; Tian et al. 2013), centrosome proliferation maintenance, proper spindle pole formation, and suppression of satellite RNA synthesis (Joukov et al. 2006; Pujana et al. 2007; Zhu et al. 2011). Some of these functions are also suspected of contributing to p220 tumor suppression activity.
Not surprisingly, BRCA1 mutant breast cancers exhibit gross chromosomal alterations and aneuploidy (Lakhani et al. 1998). Most of these tumors bear a strong morphological and molecular resemblance to sporadic basal-like sporadic breast cancer (BLC), in which BRCA1 mutations are absent (Perou et al. 2000; Foulkes et al. 2003; Sorlie et al. 2003; Turner et al. 2004). This suggests that defects in pathways in which BRCA1 functions, albeit not in BRCA1 itself, contribute to the development of this species of sporadic breast cancer (BLC). Indeed, unlike the case of sporadic ovarian cancer, somatic BRCA1 mutations are seldom identified in sporadic BLC (Futreal et al. 1994; Janatova et al. 2005; Zikan et al. 2007; Hennessy et al. 2010; Natrajan et al. 2012).
Against this backdrop, early reports identified a significant decrease of BRCA1 mRNA and protein expression in a subset of these tumors. For example, aberrant methylation of the BRCA1 promoter was detected in 11%–60% of sporadic breast tumors, correlating closely with low BRCA1 mRNA levels (Magdinier et al. 1998; Rice et al. 1998; Catteau et al. 1999; Esteller et al. 2000; Matros et al. 2005; Wei et al. 2005; Turner et al. 2007; Hsu et al. 2013). These findings suggest that epigenetic factors operate in the control of BRCA1 expression and that defects in this process predispose mammary epithelial cells to a deficiency in BRCA1-mediated genome integrity control and tumorigenesis (Thompson et al. 1995; Wilson et al. 1999; Reis-Filho and Tutt 2008; Santarosa and Maestro 2012).
p220 expression is cell cycle-dependent, being much lower in G0/G1 than in S/G2. At least part of the regulation that leads to these expressional differences is provided by major differences in BRCA1 transcription during the cell cycle (Gudas et al. 1996). One source of the regulation of BRCA1 expression is negative effects on its transcription by the Id4 and HMGA1 proteins (Beger et al. 2001; Baldassarre et al. 2003). However, the precise mechanisms by which Id4 and HMGA1 regulate BRCA1 expression are unknown.
The microRNA (miRNA) pathway represents a means of achieving post-transcriptional regulation of gene expression. miRNAs are small (22–24 nucleotides [nt]), endogenous noncoding RNAs expressed as independent genes or as mirtrons excised from an intron of a host gene. They bind in a sequence-dependent manner to the 3′ untranslated region (UTR) of target mRNAs and destabilize them and/or inhibit their translation (Bartel 2004; Guo et al. 2010). More than 50% of mammalian protein-coding genes are predicted to be regulated by miRNAs, and each miRNA can suppress the expression of multiple target mRNAs (Krek et al. 2005; Lewis et al. 2005; Lim et al. 2005; Bartel 2009; Friedman et al. 2009).
One or more miRNAs contribute to the physiological regulation of BRCA1 expression and, when abnormally expressed, the reduced levels of the protein detected in some cases of sporadic breast cancer. In support of this notion, two recent reports pointed to miR-146 family members (Garcia et al. 2011) and miR-182 (Moskwa et al. 2011) as normal suppressors of BRCA1 expression. Indeed, each compromised BRCA1 HR function when overexpressed. Currently, the physiological outcomes of such suppression are unknown.
However, an additional contribution to BRCA1 expression control is provided by a newly detected miRNA species, miR-545. We found that miR-545, a miRNA formerly unassociated with BRCA1 regulation, is expressed through much of the cell cycle and actively contributes to the control of endogenous BRCA1 p220 expression. It does so in both G0/G1 and S/G2, and its blockade results in both endogenous p220 overexpression and, surprisingly, de novo p220-dependant DNA damage. This system provides a new level of BRCA1 expression and DNA damage control.
Results
miR-545 suppresses endogenous BRCA1 p220 expression
To search for miRNAs that regulate BRCA1 expression, we examined a list of miRNAs that were predicted to target the 3′ UTR of BRCA1 mRNA in silico (TargetScan, release 4.0; http://www.targetscan.org). Indeed, when overexpressed, one of them, miR-545, significantly down-regulated expression of endogenous BRCA1 p220 (also known as p220) in immortalized primary mammary epithelial cells (IMECs). Transfection of these cells with a synthetic miR-545 mimic, but not with a mimic of the closely related miR-545* that differs by only one nucleotide in its seed sequence, led to a reduction of both BRCA1 mRNA and protein levels (Fig. 1A,B). No miR-545 target genes have been reported to date. Thus, its function has been unknown.
Figure 1.
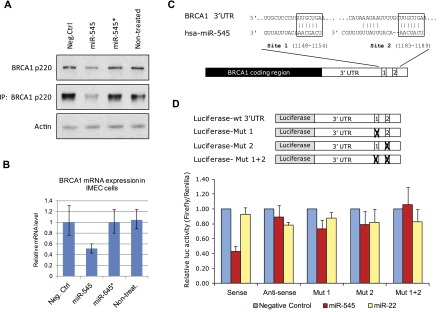
miR-545 suppresses expression of endogenous BRCA1 p220 by targeting two binding sites in the 3′ UTR of BRCA1 mRNA. (A) Western blot and immunoprecipitation (IP)-Western blot of BRCA1 p220 expression in human IMECs transfected with miR-545 mimic, miR-545* mimic, or a negative control reagent. Actin served as a loading control. (B) Expression of BRCA1 mRNA was quantified by quantitative RT–PCR (qRT–PCR) and normalized to expression levels of GAPDH mRNA and BRCA1 mRNA expression in negative control-transfected IMECs. Data represent the mean of assay results (n = 3). Error bars indicate the SD. (C,D) Two predicted miR-545 target sites between nucleotides 1148–1154 (site 1) and nucleotides 1183–1189 (site 2) of the BRCA1 3′ UTR were validated by dual-luciferase reporter (DLR) assay. The wild-type (wt) BRCA1 3′ UTR (in either the sense or antisense orientation) or 3′ UTR carrying a mutation in either or both putative miR-545 target sites was cloned downstream from a firefly luciferase reporter gene. Plasmids were cotransfected with a control, a Renilla luciferase expression vector plus and minus a miR-545 mimic, a miR-22 mimic, or a control reagent into 293T cells. Seventy-two hours later, firefly luciferase luminescence was quantified and normalized to the luminescence of the Renilla luciferase reporter, serving as an internal control. Data represent the mean of the assay results (n = 3). Error bars indicate the SD.
To determine whether these p220 expression phenomena are a result of direct targeting of BRCA1 mRNA, we assayed the effect of miR-545 on the expression of a BRCA1 3′ UTR-luciferase reporter gene. Two putative miR-545 target sites were identified at positions 1148–1154 and 1183–1189 of the BRCA1 3′ UTR (Fig. 1C). In one hypothesis, their close proximity, only 29 nt, represents an optimal interval for efficient cooperation by miRNA-containing RISC complexes in the down-regulation of p220 expression (Saetrom et al. 2007).
The wild-type BRCA1 3′ UTR (in either the sense or antisense orientation) and derivatives carrying a mutation in either or both putative miR-545 target sites were cloned downstream from a firefly luciferase reporter gene and transfected into HEK293 cells. Following cotransfection with either a miR-545 mimic, an unrelated miR-22 mimic, or a negative control reagent, specific suppression of the wild-type 3′ UTR-expressing reporter was achieved by miR-545 alone (Fig. 1D). Mutations in either of the predicted target sites partially reversed this effect, and mutations in both sites completely abolished it. Thus, miR-545 binds to two target sites in the BRCA1 3′ UTR and, in that setting, specifically suppresses the expression of the mRNA.
miR-545 modulates the efficiency of BRCA1-driven HRR
The well-established requirement for BRCA1 in HRR of DNA DSBs contributes to its role in the maintenance of genome integrity and in breast cancer suppression (Scully et al. 1997; Chen et al. 1999; Moynahan et al. 1999; Walsh and King 2007). Thus, we asked whether miR-545 affects HRR. Specifically, the effect of miR-545 in a standard HRR assay was studied in DR-U2OS cells, which carry a single, integrated HRR reporter (Fig. 2A; Pierce et al. 1999).
Figure 2.

Aberrant expression of miR-545 modulates BRCA1 p220 expression and HRR of DSB. (A) DR-U2OS cells carrying a single copy of a stably integrated direct repeat of inactivated GFP genes (DR-GFP) repair a DSB generated at a defined I-SceI site, yielding a GFP signal as reported (Pierce et al. 1999). Asynchronously growing DR-U2OS cells were transiently transfected with a miR-545 mimic, a miR-545 antagomir (α-miR-545), or negative control reagents (NC-A and NC-B, respectively). Twenty-four hours later, they were transfected with the I-SceI restriction enzyme. Following 48 h of incubation, the number of GFP-positive cells was quantified by flow cytometry. (B) Western blot showing the expression of BRCA1 p220 in DR-U2OS cells 72 h after transfection with a miR-545 mimic, a miR-545 antagomir (α-miR-545), or negative controls. Actin served as a loading control. (C) Histograms reveal the relative enrichment of GFP-positive cells in miR-545 mimic- and miR-545 antagomir-transfected cells compared with respective controls. Overexpression of miR-545 resulted in significant suppression of HR efficiency, and inhibition of miR-545 led to an increased rate of HR-directed DSB repair. Data represent the average of the relevant assays results (n ≥ 3). Error bars indicate the SEM. P-values were determined by a Student's t-test; (*) P < 0.05; (***) P < 0.001.
Asynchronous DR-U2OS cells were transfected with either a miR-545 mimic, a miR-545 antagomir, or respective negative controls. In comparison with control cells, miR-545 mimic-treated cells exhibited diminished levels of BRCA1 protein, mRNA (Fig. 2B; data not shown), and HRR function (Fig. 2C). On the other hand, inhibition of endogenous miR-545 by a miR-545 antagomir increased both BRCA1 expression and the amplitude of HRR (Fig. 2B,C).
To determine whether the increased HRR activity following miR-545 inhibition was a specific result of the up-regulation of BRCA1, DR-U2OS cells were depleted of BRCA1 p220, 53BP1, or both proteins simultaneously and transfected with either a miR-545 antagomir or a negative control (Supplemental Fig. 1). Acute depletion of RAP80, which resulted in an excessive, BRCA1-dependent increase in HRR (Coleman and Greenberg 2011; Hu et al. 2011), served as a positive control.
As expected, in the absence of p220, HRR activity in cells was significantly suppressed, and the simultaneous depletion of BRCA1 and 53BP1 partially restored this function (Bouwman et al. 2010; Bunting et al. 2010). However, inhibition of endogenous miR-545 function by the miR-545 antagomir failed to increase the amplitude of HRR in BRCA1- and 53BP1-codepleted cells. Thus, miR-545 inhibition of HRR-mediated DSB repair is BRCA1 depletion-dependent. Furthermore, miR-545 inhibition acted synergistically with RAP80 depletion and resulted in a significant, approximately threefold increase in HRR amplitude (Supplemental Fig. 1).
Therefore, when aberrantly expressed, miR-545 negatively affected both p220 synthesis and at least one p220-requiring function—HRR. Inhibition of endogenous miR-545 function resulted in the opposite outcome.
miR-545 controls the expression of endogenous p220 in G1 and S/G2
p220 exhibits a characteristic expression profile during the cell cycle (Chen et al. 1996; Ruffner and Verma 1997). Its expression is typically low during cycling G1 and lower still in quiescent cells. In contrast, it is significantly elevated at the G1/S transition and reaches peak levels in S and G2. To examine the cell cycle expression pattern of miR-545, T98G cells were synchronized in G0/G1 by serum starvation. After serum readdition, the cells progressed synchronously through the cell cycle, and the expression of miR-545 and BRCA1 p220 was analyzed.
As expected, p220 was expressed at very low levels in early to mid-G1, increased in late G1 (i.e., 12–16 h after release), and peaked in S/G2 (i.e., 24–28 h after serum addition) (Fig. 3A). p220 mRNA expression, which closely resembled that of p220 protein, was also low in early and mid-G1 (Fig. 3B). Endogenous miR-545 exhibited a similar expression pattern, with lower, but not absent, levels in early G1 and gradually increasing expression through S and G2, thereby mirroring both the nadir and the rise of p220 expression (Fig. 3C).
Figure 3.
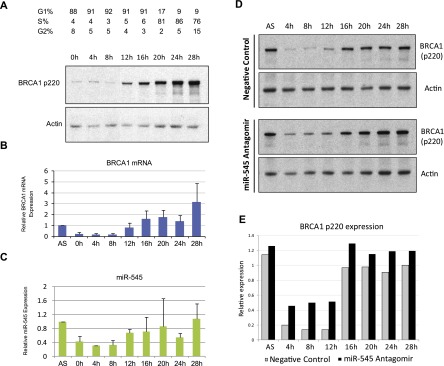
miR-545 modulates the endogenous expression of BRCA1 during the cell cycle. (A) T98G cells were synchronized in G0/G1 by serum starvation for 72 h. Upon release from the block by serum addition, the expression of BRCA1 p220 was examined at 4-h intervals by Western blot. (B) qRT–PCR analysis of BRCA1 mRNA expression at the same 4-h intervals in synchronously growing T98G cells. Relative expression was normalized to that of GAPDH mRNA and to BRCA1 mRNA levels in asynchronously growing (AS) T98G cells. Data represent the average of four independent experiments. Error bars indicate the SD. (C) qRT–PCR analysis of miR-545 in the same cells revealed a similar expression pattern, with relatively low, but not absent, expression in early and mid-G1 (0–8 h) and elevated levels in late G1 (12–16 h) and S/G2 (20–28 h). miR-545 expression was normalized to RNU6B expression and miR-545 level in asynchronous (AS) T98G cells. Data represent the average of four independent experiments. Error bars indicate the SD. (D) Western blots showing the expression of BRCA1 p220 during the cell cycle progression of synchronously proliferating T98G cells treated with either a control reagent (top panel) or miR-545 antagomir (α-miR-545) (bottom panel). (E) Quantitation of BRCA1 p220 expression by densitometry in the synchronously proliferating, miR-545 antagomir- or control-treated T98G cells shown in D. Actin served as a loading control in A and D.
These findings were confirmed in T24 bladder carcinoma cells, which were synchronized by contact inhibition. Upon replating these cells at lower density, they progressed synchronously, and BRCA1 p220 and miR-545 exhibited an analogous and seemingly coordinated expression pattern through the cell cycle (Supplemental Fig. 2A–D). Thus, similar kinetics of miR-545 and p220 expression were detected during the cell cycle in two different human cell lines.
Effective modulation of p220 abundance during the cell cycle might be vital for cell survival and proliferation. Its forced overexpression invariably leads to checkpoint activation and even cell death (Somasundaram et al. 1997; Harkin et al. 1999). In contrast, high-order depletion can result in proliferation arrest (Hakem et al. 1996; Deans et al. 2004).
The presence of miR-545 in G1, where there was detectable BRCA1 mRNA but low p220 abundance, suggested that endogenous miR-545 contributes to the physiologically low level of endogenous BRCA1 expression in G1. To test this possibility, we arrested T98G cells in G0/G1 by serum starvation and then exposed them to either a miR-545 antagomir or a negative control reagent. The cells were then released by serum refeeding, and p220 expression was analyzed at defined times thereafter (Fig. 3D). Inhibition of miR-545 function resulted in significant, twofold to threefold up-regulation of p220 in early G1 (i.e., 4–8 h after serum addition) (Fig. 3D,E; cf. Supplemental Fig. 3A,B). The p220 mRNA level was also significantly up-regulated at the same time points (Supplemental Fig. 3C). Likewise, miR-545 inhibition resulted in a consistent although less dramatic increase of p220 levels throughout S and G2 (i.e., 20–28 h after serum readdition) (Fig. 3D,E).
Increased expression of BRCA1 p220 in early G1 resulting from inhibition of miR-545 did not affect the cell cycle progression of these cells (Supplemental Fig. 3D). Thus, elevated BRCA1 p220 expression in G1 did not result in a relative enrichment of S-phase cells as a result of the culture progressing through G1 faster.
A similar effect of the miR-545 antagomir on p220 expression was observed in a different experimental setting. T24 cells, synchronized in G1 by addition of mimosine (Krude 1999), were treated with either a miR-545 antagomir or a negative control and analyzed for p220 expression (Supplemental Fig. 2E–G). Again, inhibition of miR-545 in G1 cells resulted in a significant increase of p220 and its mRNA.
These findings imply the existence of a physiological role for miR-545 in the control of p220 expression in both G0/G1 and S/G2.
Up-regulated p220 concentrates at sites of UV damage in G1 cells
The p220 DNA damage response (DDR) functions that operate in S/G2, like HRR, require the recruitment of p220 to readily detectable sites/foci of DNA damage. There, together with other repair factors, it forms distinct, multisubunit-containing supercomplexes (Greenberg et al. 2006; Kim et al. 2007; Liu et al. 2007; Sobhian et al. 2007; Wang et al. 2007; Chen et al. 2008).
DNA double-strand breaks (DSBs) in G1 are repaired predominantly through direct ligation of DNA ends, a product of the nonhomologous end-joining (NHEJ) pathway (Symington and Gautier 2011). This pathway choice in G1 is promoted by 53BP1- and RIF1-dependent suppression of DNA 5′ end resection that otherwise would be a key step in BRCA1-mediated HRR (Bouwman et al. 2010; Bunting et al. 2010; Bothmer et al. 2011; Chapman et al. 2013; Di Virgilio et al. 2013; Escribano-Diaz et al. 2013; Feng et al. 2013; Zimmermann et al. 2013).
Moreover, the dependence of HRR on the presence of a sister chromatid as a template precludes the appearance of HRR during G1, when there are much lower levels of p220 than in S/G2. This notwithstanding, there remains the question of whether low, endogenous p220 levels in G1 are required to prevent the protein from exercising aberrant DDR functions during that period. Thus, to determine whether abnormally high p220 expression in G1 triggers abnormal functional consequences, we asked whether the protein is aberrantly recruited to sites of acute DNA damage in miR-545 antagomir-expressing G1 cells.
Multiple, related cell lines were created. Each stably expressed a fluorescent indicator of passage through S/G2 (Sakaue-Sawano et al. 2008). G1 cells fail to express this reporter. Thus, using these cells, it was possible to score phenomena that occurred in G1 versus S/G2.
More specifically, T98G, U2OS, and IMECs (telomerase-IMECs) were stably transfected with a polypeptide composed of a Geminin degron fused to an Azami-Green fluorophore (mAG-hGEM) to form T98G-GEM, U2OS-GEM, and IMEC-GEM. In each, the reporter protein was selectively expressed in S/G2 nuclei. It did not compromise cell cycle progression (Sakaue-Sawano et al. 2008; see the Materials and Methods). After two rounds of fluorescence-activated cell sorting (FACS) selection, the majority of S/G2 and very few of the G1 cells from each cell line stably expressed the green fluorescent fusion protein (Supplemental Fig. 4). Homogeneous green, nuclear staining of mAG-hGEM-expressing S/G2 but not G1 cells allowed these populations to be distinguished from one another by fluorescent microscopy.
T98G-GEM cells, serum-starved and arrested in G0/G1, were transfected with a miR-545 antagomir or a negative control. This resulted in approximately twofold overexpression of p220 in the former (Supplemental Fig. 5A,B). After miR-545 transfection, these G0/G1 cells, still in serum-free medium, were irradiated with 10 Gy (irradiation [IR]), incubated for 1 h, and then analyzed for IR-induced foci (IRIF). γH2AX-containing foci were present in all nuclei.
While p220 was robustly recruited to these sites in S/G2, miR-545 antagomir-treated G1 cells failed to do so (Supplemental Fig. 5C). Identical experiments in asynchronously growing IMEC-GEM cells revealed similar results (Supplemental Fig. 5D–F).
In addition, we tested whether aberrantly overexpressed BRCA1 p220 in miR-545-inhibited IMEC-GEM cells was recruited to UV laser-induced DSB-containing lesions in G1. Asynchronous IMEC-GEM cells were transfected with either miR-545 antagomir or a negative control and depleted of RIF1 by specific siRNA or control-treated. Subnuclear, DSB-containing areas (stripes) were generated by 377-nm UV laser microirradiation, and, 1 h later, cells were fixed and analyzed for p220 localization by immunofluorescence (Supplemental Fig. 5G,H). γH2AX staining allowed depiction of UV laser-induced stripes. While p220 was readily detectable at stripes in S/G2 cells, it failed to localize there in miR-545-inhibited G1 cells. However, depletion of RIF1 resulted in the localization of p220 at stripes in miR-545 antagomir-treated but not in control-treated G1 IMECs, possibly reflecting the reappearance following RIF1 depletion of a key event in HRR; namely, DSB end resection (Chapman et al. 2013; Escribano-Diaz et al. 2013; Feng et al. 2013; Zimmermann et al. 2013).
Next, we asked whether abnormally up-regulated BRCA1 p220 is recruited to sites of UV damage in miR-545 antagomir-treated G1 cells. UV irradiation of cells through micropores of subnuclear size creates well-demarcated, intranuclear territories of UV damage that can be detected by anti-cyclobutane pyrimidine dimer (CPD) immunofluorescence (Katsumi et al. 2001). Again, serum-deprived, G0/G1-arrested T98G-GEM cells were transfected for 48 h with either a miR-545 antagomir or a negative control reagent. In the former culture, there was a significant increase in p220 (Fig. 4A). Cells were then irradiated through micropores with 30 J/m2 UV-C. Three hours later and still in serum-free medium, they were analyzed by immunofluorescence.
Figure 4.

Aberrantly expressed BRCA1 can localize at sites of UV damage in G1 cells. (A) Western blot showing BRCA1 p220 expression in G1-synchronized, UV-irradiated T98G-GEM cells transfected with miR-545 antagomir (α-miR-545) or control reagent in comparison with p220 expression in asynchronous (AS), untreated T98G-GEM cells. (B) Representative images of BRCA1 (red) localization at CPD-positive areas (green nuclear dots) in UV-irradiated, G1 (mAG-hGEM-negative) T98G-GEM cells transfected with either miR-545 antagomir or control reagent. Homogeneously green-stained nuclei represent mAG-hGEM-expressing S/G2 cells. (C) Quantitation of the average percentage of control- or miR-545 antagomir-transfected G1 T98G-GEM cells exhibiting BRCA1/CPD colocalization 3 h after UV irradiation. Error bars indicate the SEM (n = 3). (D) Representative images showing colocalization of BRCA1 (red) with CPD (green subnuclear areas) in miR-545 antagomir- or control-transfected G1 IMEC-GEM cells 3 h after UV irradiation. Homogeneously green-stained nuclei represent S/G2 cells. (E) Quantitation of the average percentage of miR-545 antagomir- or control-treated G1 IMEC-GEM cells exhibiting colocalization of BRCA1 with CPD 3 h after UV. Error bars indicate the SEM (n = 3). (F) Western blot of BRCA1 expression in serum-starved T98G-GEM cells transduced with either an adenoviral BRCA1 vector (Ad-BRCA1) or a control vector (Ad-0) or transfected with miR-545 antagomir or control reagent and UV-irradiated. (G) Representative images showing colocalization of BRCA1 (red) with CPD (green nuclear dots) in mAG-hGEM-negative, G1 T98G-GEM cells treated as described in F. (H) Quantitation of the average enrichment of BRCA1/CPD colocalization in G1 T98G-GEM cells treated as described in F. Error bars indicate the SEM (n = 3). (I) Representative images showing BRCA1 (red) and CPD (green nuclear dots) colocalization in UV-irradiated, G1 (mAG-hGEM-negative) T98G-GEM cells simultaneously transfected with miR-545 antagomir and control siRNA (siGL2) but not in miR-545 antagomir- or negative control-treated and siXPA-, siXPC- or siEXO1 oligonucleotide-treated cells. (J) Quantitation of the average enrichment of BRCA1/CPD colocalization in G1 T98G-GEM cells cotransfected with miR-545 antagomir or control reagent and XPA-, XPC-, EXO1-, or RPA-specific siRNAs or siGL2 control. Error bars indicate the SEM (n = 4). (*) P < 0.05; (**) P < 0.01 (Student's t-test). Bars, 10 μm.
Both antagomir- and control-treated cells exhibited recruitment of p220 to micropore sites of UV damage in S/G2 cells, as described previously (Pathania et al. 2011). Surprisingly, in G0/G1, where little p220 recruitment to micropore damage sites is observed (Pathania et al. 2011), miR-545 inhibition resulted in a major increase in the number of cells with overt p220/CPD colocalization (Fig. 4B,C). This cannot be attributed to an accumulation of late G1 cells that normally undergo an increase in p220 expression (Fig. 3A) because the antagomir failed to change the dynamics of cell cycle progression during these experiments (Supplemental Fig. 3D).
To determine whether these findings are more generally applicable, asynchronous U2OS-GEM, another cancer cell line, and IMEC-GEM cells derived from normal mammary epithelial cells were studied. Once again, p220 was readily detected at sites of UV-mediated DNA damage in S/G2 cells of both lines. Moreover, compared with control-treated IMEC-GEM and U2OS-GEM cells, miR-545 antagomir transfection resulted once again in a major increase of G1 cells exhibiting p220-CPD colocalization (Fig. 4D,E; Supplemental Fig. 6A–C).
To determine whether these phenomena depend on the irradiation dose, serum-starved, miR-545 antagomir- or control-transfected T98G-GEM cells were exposed to 5, 15, or 30 J/m2 UV-C. Relatively few control-transfected G1 cells concentrated p220 at CPD-containing UV lesions at any UV dose tested. In contrast, in the antagomir-transfected population, the number of such cells gradually increased with the dose. For example, after 30 J/m2, there was a major difference in the fraction of cells in the antagomir-treated, compared with the control-treated, population that revealed localization of p220 at these sites (Supplemental Fig. 7A,B). Furthermore, p220 recruitment to UV lesions in antagomir-treated G1 cells was a relatively late event, peaking at 3 h post-UV. In contrast, the protein robustly localized to the micropores in S/G2 cells even at 1 h and at all doses of irradiation (Supplemental Fig. 7C,D). These results suggest that elevated p220 expression in particular enables its G1 recruitment to UV damage sites and that this process is directed by a mechanism different from that which drives endogenous p220 to UV lesions in S phase (Pathania et al. 2011).
To test this hypothesis further, G0/G1 T98G-GEM cells were transfected with the miR-545 antagomir or a control reagent. Other aliquots were infected with a p220-encoding adenovirus or a control adenoviral vector. All cells were micropore UV-irradiated and, 3 h later, analyzed for recruitment of BRCA1 to UV lesions.
Ectopic p220 expression resulted in p220 overproduction and a major increase in p220-CPD-positive G1 cells (Fig. 4F–H). miR-545 antagomir-treated cells also exhibited enrichment of p220-CPD-positive G1 cells. However, simultaneous coexpression of the miR-545 antagomir and Adeno-p220 failed to significantly increase the number of p220-CPD-positive G1 cells when compared with cells expressing Adeno-p220 alone (Supplemental Fig. 7E,F). This implies that the UV damage sites were saturated after Adeno-p220 infection. Thus, an elevated abundance of p220 in G0/G1 is sufficient for its localization at UV damage sites in these cells.
Recruitment of aberrantly expressed BRCA1 p220 to UV-mediated DNA damage sites in G1 cells is dependent on nucleotide excision repair (NER) and DNA resection
Next, we asked whether localization of BRCA1 at UV lesions in G1 is dependent on the primary UV repair process; i.e., NER. Specifically, we depleted T98G-GEM and IMEC-GEM cells of either XPA or XPC using suitable siRNAs (Supplemental Fig. 8A). Cells were also transfected with a miR-545 antagomir or a control. They were then micropore UV-irradiated and analyzed for p220 localization.
As expected, in comparison with control cells, T98G-GEM cells exposed to a combination of control siRNA (siGL2) and the mir-545 antagomir exhibited a significant increase in G1 cells containing BRCA1 concentrated at UV lesions (Fig. 4I,J). XPA or XPC depletion strongly inhibited these effects (Fig. 4I,J). However, in S/G2 cells (intense green nucleus-containing cells), BRCA1 localized robustly at UV damage sites, and its recruitment was unaffected by XPA or XPC depletion. This is in agreement with its replication-dependent, but NER-independent, recruitment to these sites in S and G2 (Pathania et al. 2011). Thus, recruitment of p220 to UV lesions in G1 (and not in S/G2) cells is not entirely a nonspecific event. It depends on its abundance being sufficiently increased and a functioning NER pathway, the latter being a normal response to UV DNA damage.
NER is a rapid and robust repair process. However, when it fails to repair certain relatively resistant UV lesions, resection of the damaged DNA and processing of the surrounding chromatin take place. This results in checkpoint activation and engagement of alternative repair mechanisms (Giannattasio et al. 2010; Sertic et al. 2011). The delayed kinetics and the requirement for relatively high doses of irradiation for the localization of overexpressed, endogenous BRCA1 protein at UV lesions in G0/G1 cells suggested that recruitment in this instance was directed to those UV damage sites where NER was incomplete.
Thus, we asked whether p220 concentration at G1 UV lesions requires extensive DNA resection and generation of ssDNA at the sites of damage. Hence, we studied the effect of depleting G0/G1 cells of EXO1, an exonuclease that plays a role in UV-induced checkpoint activation in quiescent cells (Sertic et al. 2011).
G0/G1-synchronized T98G-GEM cells were treated with two different EXO1-specific siRNA species and miR-545 antagomir or a control reagent. Following 30 J/m2 UV-C exposure, p220 was detectable at UV damage sites in antagomir-treated G0/G1 cells, as expected. However, it failed to localize at micropore sites in EXO1-depleted, miR-545-inhibited cells (Fig. 4I,J). These results suggest strongly that BRCA1 recruitment requires DNA resection at UV lesion-containing DNA.
Moreover, depletion of RPA, which binds ssDNA and is instrumental in ATR and checkpoint activation, also abolished p220 localization to UV damage sites in G0/G1 cells (Fig. 4J; Supplemental Fig. 8B). Thus, excision-generated, RPA-coated ssDNA plays a crucial role in the G0/G1 BRCA1 localization process.
p220 can recruit RAD51 to sites of UV damage in G0/G1 cells
The recruitment of RAD51 to DSB in S/G2 is a critical step in the BRCA1 control of HRR and is dependent on an interaction of p220 with the PALB2–BRCA2–RAD51 complex (Greenberg et al. 2006; Xia et al. 2006; Sy et al. 2009). The aberrant localization of overexpressed, endogenous p220 at sites of UV damage in G0/G1 cells raised the question of whether RAD51 is also recruited to UV lesions in these cells.
Serum-starved T98G-GEM cells were transfected with either a miR-545 antagomir or a negative control. After exposure to 30 J/m2 UV-C, they were released into the cycle and analyzed for micropore RAD51 recruitment. As expected, RAD51 clearly localized to UV lesions in S/G2 cells (Pathania et al. 2011). Unexpectedly, it was also recruited to micropore sites in G1 cells (Fig. 5A). Although a smaller number of G1 cells exhibited RAD51 than p220 micropore recruitment, there was a significant increase in the miR-545 antagomir-treated, RAD51-positive population compared with the control (Fig. 5B). This observation was even more dramatic in IMEC-GEM and U2OS-GEM cells, where the number of RAD51-CPD-positive cells was ∼10-fold and approximately sevenfold higher than the control-treated population (Fig. 5D,E; Supplemental Fig. 6D,E). Moreover, RAD51 invariably colocalized with p220 in G1 cells (Fig. 5C,F).
Figure 5.

Overexpressed endogenous BRCA1 can recruit RAD51 to sites of UV damage in G1 cells. (A) Representative images showing the colocalization of RAD51 (red) with CPD (green nuclear dots) in serum-depleted, miR-545 antagomir (α-miR-545)- or negative control-transfected G1 (mAG-hGEM-negative) T98G-GEM cells 3 h after UV irradiation. Homogeneously green-stained nuclei represent mAG-hGEM-expressing S/G2 cells. (B) Quantitation of the average enrichment of G1 T98G-GEM cells exhibiting RAD51/CPD colocalization when treated as described in A. Error bars indicate the SEM (n = 3). (C) Representative images of colocalization of RAD51 (red) with BRCA1 (green) in serum-starved, miR-545 antagomir- or control-treated G1 T98G-GEM cells after UV. (D) Representative images showing colocalization of RAD51 (red) with CPD (green subnuclear dots) in UV-irradiated, G1 (mAG-hGEM-negative) IMEC-GEM cells after transfection with miR-545 antagomir or negative control reagent. (E) Quantitation of the average percentage of G1 IMEC-GEM cells treated as described in D that exhibit RAD51/CPD colocalization after UV irradiation. Error bars indicate the SEM (n = 3). (F) Representative images of colocalization of RAD51 (red) with BRCA1 (green) after UV irradiation of IMEC-GEM cells transfected with miR-545 antagomir or a negative control. (G) Representative images of colocalization of RAD51 (red) and CPD (green nuclear dots) in UV-irradiated, serum-starved T98G-GEM cells cotransfected with control siRNA (siGL2) and miR-545 antagomir compared with siBRCA1 oligonucleotide- and miR-545 antagomir- or negative control-transfected cells. (H) A histogram showing the average enrichment of G1 T98G-GEM cells exhibiting BRCA1/CPD (blue bars) and RAD51/CPD (red bars) colocalization when treated as described in G. Error bars indicate the SEM (n = 3). Bars, 10 μm.
To determine whether RAD51 recruitment in this setting was BRCA1-dependent, we depleted p220 from G0/G1 T98G-GEM cells using two different BRCA1-specific siRNA oligonucleotides in the presence of either a miR-545 antagomir or a negative control. Upon UV irradiation, BRCA1-depleted, antagomir-treated cells failed to localize RAD51 to sites of UV damage in both G1 and, as expected, S/G2 (Fig. 5G,H). We conclude that the accumulation of abnormally expressed, endogenous p220 at sites of UV damage in G1 cells drives corecruitment of RAD51, an abnormal event during this cell cycle interval.
Overexpressed endogenous BRCA1 localizes and recruits RAD51 to spontaneously arising 53BP1 nuclear bodies in G1 cells
In exponentially growing, undamaged cells, 53BP1 forms large nuclear bodies confined predominantly to G1. These endogenous structures originate in the preceding S and G2/M phase and contain unrepaired DNA DSBs that arose spontaneously. These include common fragile sites and costain with γH2AX, MDC1, and other markers of a DSB response (Harrigan et al. 2011; Lukas et al. 2011). We asked whether abnormally overexpressed, endogenous p220 in G1 cells localizes with 53BP1 at these sites.
Serum-starved T98G-GEM cells transfected with either a miR-545 antagomir or a negative control (cf. above) were analyzed by immunofluorescence for localization of the relevant proteins. Large 53BP1 foci appeared in a subset of G1 but not S/G2 cells (Fig. 6A). Indeed, we observed a marked increase in the number of cells containing p220 localized at 53BP1 nuclear bodies in miR-545-inhibited G1 cells compared with controls (Fig. 6A,B). Moreover, using γH2AX staining as another marker of these bodies, we observed a significant increase in the colocalization of RAD51 at these structures in the miR-545 antagomir-transfected cells, analogous to what happened in UV-treated cells (Fig. 6C,D). Similar phenomena were observed in G1 miR-545 antagomir-transfected IMEC-GEM cells (Supplemental Fig. 9A,B).
Figure 6.

Endogenous BRCA1 and RAD51 localize to 53BP1 nuclear bodies in G1 cells. Serum-starved T98G-GEM cells were transfected with either miR-545 antagomir or a negative control. Thirty-six hours later, they were fixed and analyzed by indirect immunofluorescence for the localization of the relevant proteins. (A) Representative images showing 53BP1 nuclear bodies (red) in G1, mAG-hGEM-negative T98G-GEM cells but not in homogeneously green, mAG-hGEM-positive S/G2 cells. BRCA1 (green foci) colocalized with 53BP1 nuclear bodies in miR-545 antagomir-treated (α-miR-545) but not in control-treated G1 cells. Bar, 10 μm. (B) Quantitation of the proportion of miR-545 antagomir- or control-treated G1 T98G-GEM cells exhibiting colocalization of BRCA1 with 53BP1. Data represent the mean of the assay results. Error bars indicate the SEM (n = 3). (C) Colocalization of RAD51 (red) with γH2AX (green), a marker of 53BP1 nuclear bodies, specifically in miR-545 antagomir-treated G1 cells (shown in the bottom panel). Bar, 10 μm. (D) Quantitation of the percentage of miR-545 antagomir- or control-transfected G1 cells exhibiting colocalization of RAD51 with γH2AX. Data represent the mean of the assay results. Error bars indicate the SEM (n = 3).
Thus, aberrantly overexpressed, endogenous p220 also concentrated at sites of DNA damage that were not exogenously generated. It did so during G1 in response to spontaneous DNA damage, possibly DSBs arising from replication stress incurred during a prior cell cycle interval (Lukas et al. 2011).
Accumulation of de novo DNA damage in miR-545-inhibited, endogenous BRCA1 p220-overproducing G1 and S/G2 cells
Thus, not only did overexpressed, endogenous p220 concentrate at sites of DNA damage in G0/G1, where it would not normally be detected, it also promotes at least one biochemical event; i.e., RAD51 recruitment to these sites at a time of the cell cycle when this does not normally occur. This raises the question of whether such unexpected p220 behavior in G1 is at least in part responsible for an abnormal DDR and accumulation of de novo DNA damage.
Since numerous 53BP1 foci contained endogenously overexpressed p220 (Supplemental Fig. 9C–E) and are suspected of harboring DNA breaks but lack sister chromatids (i.e., normal HRR donor substrates), we asked whether aberrant p220 overexpression and the resulting recruitment of RAD51 to DNA damage sites in G1 result in an abnormal outcome; e.g., de novo DNA damage. Comet assays were used to search for DNA strand breaks as a possible manifestation of such damage.
Asynchronous T98G mAG-hGEM cells were transfected with miR-545 antagomir or a control. Alternatively, they were infected with the p220-encoding adenovirus or a control adenovirus. All were then exposed to 30 J/m2 UV-C. G1 and S/G2 cells were separated by FACS and analyzed by comet assay (Fig. 7A,B). The assay was performed under both neutral and alkaline conditions.
Figure 7.

Formation of DNA strand breaks in BRCA1-overexpressing G1 and S/G2 cells. Exponentially growing T98G-GEM cells were transfected with miR-545 antagomir (α-miR-545) or a negative control or infected with a recombinant BRCA1-expressing adenoviral vector (Adeno-BRCA1) or control adenoviral vector (Adeno-Ctrl). Thirty-six hours later, one set of samples was irradiated with 30 J/m2 UV-C and incubated for an additional 3 h. G1 and S/G2 subpopulations were separated by FACS from both nonirradiated and UV-irradiated samples and analyzed for the presence of DNA breaks by single-cell gel electrophoresis (comet assay). (A,C) Western blot showing the expression of BRCA1 p220 in G1 and S/G2 populations of both UV-irradiated (A) and nonirradiated (C) T98G-GEM cells. Actin served as a loading control. (B,D) Quantitation of the average tail length of comets developed in miR-545 antagomir- or negative control-treated or Adeno-BRCA1- or control adenovirus-transduced T98G-GEM G1 and S/G2 cells. (E) Western blot showing the expression of BRCA1 p220 and RAD51 in T98G-GEM cells transfected with a miR-545 antagomir or a negative control and a RAD51-specific siRNA (siRAD51) or a negative control (siGL2) as indicated, incubated for 72 h, and separated by FACS into G1 and S/G2 populations. Lamin B1 served as a loading control. (F) Quantitation of the average tail length of comets developed in the miR-545 antagomir- or negative control-treated cells transfected either with RAD51-specific siRNA or control reagent (siGL2). Comet tails were detected and analyzed by CellProfiler software (Carpenter et al. 2006). Data represent the average tail length calculated from analysis of at least 100 cells per sample in three independent experiments. Error bars indicate the SEM. Statistical significance was determined by a Student's t-test; (***) P < 0.001; (NS) nonsignificant, P > 0.05.
In comparison with controls, there was a highly significant increase in average comet tail length in both miR-545 antagomir-treated and Adeno-p220-expressing G1 cells at alkaline pH (Fig. 7B). No such change occurred under neutral conditions (data not shown), implying that the tail length increase noted above was a product of ssDNA breakage.
The average comet tail length was also significantly increased in p220-overexpressing S/G2 cells, again after alkaline analysis (Fig. 7B). Thus, when UV-irradiated, overproduction of endogenous BRCA1 p220 in both G1 and S/G2 cells directly or indirectly induced DNA strand breaks.
To search for DNA breaks in the absence of exogenous DNA damage, we again assayed for comets. T98G-GEM cells were transfected with the miR-545 antagomir or a control reagent or were transduced with the Adeno-p220 vector or an adenovirus control, as noted above. Thirty-six hours later, G1 and S/G2 cells were isolated by FACS and analyzed for DNA breaks (Fig. 7C,D). Strikingly, in the alkaline assay, there was a highly significant increase in the average comet tail length in both miR-545 antagomir-treated and Adeno-BRCA1-expressing G1 cells compared with control cells (Fig. 7D). Again, no difference in the length of the comet tails was detected under neutral conditions (data not shown). The average comet tail length of BRCA1-overexpressing cells was significantly increased in S/G2 cells as well (Fig. 7D). Thus, overexpression of endogenous BRCA1 p220 leads to a significant increase in spontaneous DNA damage in both G0/G1 and S/G2 cells even in the absence of an exogenous mutagen.
The development of spontaneous DNA damage in cells that aberrantly overexpressed endogenous or exogenous p220 strongly suggested that this is a BRCA1-dependent phenomenon. To test this observation in another setting, T98G-GEM cells were depleted of BRCA1 p220 or both BRCA1 and 53BP1 using specific siRNAs. They were subsequently transfected with a miR-545 antagomir or a negative control. Seventy-two hours later, G1 and S/G2 cells were isolated by FACS and analyzed by alkaline comet assay for DNA break formation (Supplemental Fig. 10A,B).
Once again, compared with control cells, the average comet tail length was significantly increased in both G1 and S/G2 cells cotransfected with miR-545 antagomir and control siGL2 siRNA (Supplemental Fig. 10A,B). Depletion of BRCA1 alone resulted in increased de novo DNA damage, consistent with its well-established role in preservation of genome stability. However, simultaneous inhibition of miR-545 function and BRCA1 depletion did not elicit an increase in DNA break formation. Similarly, the inhibition of miR-545 in BRCA1- and 53BP1-codepleted cells failed to increase the average comet tail length. This points to a crucial role for p220 HRR function in the generation of de novo DNA damage in this setting.
Next, we asked whether inhibition of miR-545 in BRCA1 mutant cells results in a similar increase in the DNA breaks. HCC1937 is a human breast cancer cell line that carries a mutant BRCA1 allele (5382insC) and no wild-type allele. It produces a truncated protein and no wild-type p220 (Tomlinson et al. 1998). Isogenic HCC1937 cells reconstituted with either a wild-type BRCA1 cDNA or an empty vector (Scully et al. 1999) were transfected with miR-545 antagomir or a negative control. Seventy-two hours later, they were analyzed by alkaline comet assay. IMECs were treated identically and served as a control (Supplemental Fig. 10C,D).
miR-545 inhibition in IMECs resulted in a highly significant increase in the average comet tail length compared with controls (Supplemental Fig. 10D). Control-treated HCC1937 cells, not unexpectedly, exhibited a high basal level of comets that, surprisingly, decreased after miR-545 inhibition. Control-treated, wild-type BRCA1-reconstituted HCC1937 cells, however, exhibited short comets, and miR-545 antagomir in these cells led to no change in this parameter. This observation is consistent with the lack of a 3′ UTR sequence carrying miR-545 recognition sites in the BRCA1 cDNA clone used to reconstitute these cells. It thus reinforces the hypothesis that there is a specific, BRCA1-directed effect of miR-545 on genome stability.
These findings raise the question of whether inhibition of other miRNAs, such as miR-182, reported recently to suppress BRCA1 expression (Moskwa et al. 2011), elicits a similar effect on genome stability. Indeed, transfection of serum-starved, G0/G1-synchronized T98G-GEM cells with miR-182 antagomir resulted in aberrant overexpression of p220 in G1 and had an effect similar to that of miR-545 inhibition on de novo DNA damage generation (Supplemental Fig. 11).
Role of overexpressed BRCA1-dependent RAD51 recruitment in the generation of DNA strand breakage
Because BRCA1-dependent RAD51 recruitment was observed at DNA damage sites in G1 and S/G2 in both damaged and undamaged cells, we asked whether the coexistent DNA break formation was dependent on the BRCA1 p220 direction of aberrant recruitment of the RAD51 recombinase to these sites. Specifically, we compared the abundance of DNA breaks in miR-545-inhibited cells that were and were not RAD51-depleted.
Asynchronous T98G-GEM cells were transfected with either a miR-545 antagomir and a RAD51-specific siRNA or respective negative controls (Fig. 7E). Seventy-two hours later, G1 and S/G2 cells were separated by FACS and analyzed for DNA breaks by alkaline comet assay. In both miR-545-inhibited, p220-overproducing G1 and S/G2 cells, there was again a highly significant increase in the average comet tail length, while, after RAD51 depletion, it was completely abolished (Fig. 7F).
Thus, overexpressed, endogenous BRCA1 p220 directs the aberrant coconcentration of the RAD51 recombinase with it at DNA damage sites in G1 and S/G2. This results in the generation of de novo DNA breaks.
Discussion
The long-known, cell cycle-dependent regulation of p220 expression is the product of a complex regulatory process. The most prominent component is cell cycle-dependent BRCA1 transcription, which is considerably less active in G1 than in S/G2 (Gudas et al. 1996; Vaughn et al. 1996). Overlaid on it is a newly detected, post-transcriptional fine-tuning process directed by miR-545 that further modulates p220 mRNA and thereby p220 abundance.
miR-545 tuning operates through much of the cell cycle. In both G1 and S/G2, it translates into modest, physiological decreases in p220 abundance. Failure of this process resulted in modest but significant endogenous p220 overproduction. Surprisingly, even in undamaged cells, this translated into aberrant p220 DDR activity and de novo DNA damage. Thus, a relatively small increase in p220 abundance above normal translated into net DNA damage development. This alone implies that tight control of p220 abundance represents a physiologically important process.
This finding is supported and reinforced by examining other miRNAs that were recently reported to suppress p220 expression (Garcia et al. 2011; Moskwa et al. 2011). At least one of them, miR-182, when inhibited, resulted in a moderate up-regulation of the protein in G0/G1 that, like miR-545, translated into spontaneous DNA damage in unperturbed cells.
An insight into the molecular mechanism of the recruitment of aberrantly expressed p220 to DNA damage sites in G0/G1 emerges from an apparent paradox; namely, p220 was frequently detected at UV-induced lesions but much less so at sites of DSBs in these cells. BRCA1 p220 localization at UV lesions in G0/G1 was dependent on a functional NER pathway and extensive EXO1-mediated DNA resection at these sites.
In contrast, NHEJ-mediated repair of DSBs in G0/G1 does not itself involve the specific generation of ssDNA ends, and the surrounding chromatin bears little resemblance to DSB-induced chromatin modifications in S/G2 (Symington and Gautier 2011; Papamichos-Chronakis and Peterson 2012). Protection of the existing DNA ends by 53BP1 and RIF1 prevents the recruitment of the abnormally expressed p220 to DSB sites during this cell cycle interval (Zimmermann et al. 2013). Moreover, 53BP1 or RIF1 depletion restores end resection and facilitates p220 recruitment to sites of DNA DSBs in G0/G1 cells (Chapman et al. 2013; Escribano-Diaz et al. 2013; Feng et al. 2013).
Thus, given the similarity of these earlier findings from other groups to what was observed following p220 overexpression, we attribute the G0/G1 recruitment failure of overexpressed p220 to IRIF to a predictable lack of DSB end resection during this interval (Chapman et al. 2013; Escribano-Diaz et al. 2013; Feng et al. 2013). In contrast, p220 attraction to UV sites in G0/G1 took place in an environment in which EXO1-dependent end resection did occur (Giannattasio et al. 2010), possibly explaining the aforementioned paradox.
p220 also directed the RAD51 recombinase to DNA damage sites in G1. This is also an abnormal event, since, during G1, there is no physiological substrate available for HRR. Indeed, we found that readily detectable DNA strand breakage was a by-product of this p220 → RAD51-driven event.
The same cells modestly overexpressed p220 in S/G2. In these cells, the amplitude of ambient HRR activity also rose to abnormal levels, and p220-driven, RAD51-dependent DNA strand breakage was again detected. Thus, whenever endogenous p220 was overexpressed, it engaged in aberrant DDR activity. In this context, it directed at least one of its known partner proteins, RAD51, to colocalize with it, and their coparticipation contributed to the development of de novo DNA damage.
We and other groups have shown previously that yet another form of p220 tuning also occurs in S/G2. It is executed by the RAP80 complex and is directed at p220 functionality, per se, and not its expression (Coleman and Greenberg 2011; Hu et al. 2011). This process, too, is aimed at maintaining a physiological HRR response to DSB formation. Interrupting it triggers an outcome similar to that arising after blocking miR-545 function in S/G2; namely, excessive HRR amplitude and de novo DNA damage (Hu et al. 2011).
Thus, either a superabundance of p220 or failure to modulate its HRR function led to the same results—an excessive amplitude of p220 HRR function and net, new DNA damage. Hence, the miR-545 and RAP80 tuning systems provide seemingly complementary layers of p220 DNA repair function control.
The evidence for aberrant p220 DNA damage responsiveness after miR-545 blockade was readily apparent. Normally, p220 is uninvolved in DSB and photoproduct repair in G1 (Pathania et al. 2011). In fact, data presented here and elsewhere show that, when expressed at physiological levels, p220 avoids 53BP1-containing and UV damage sites during G1 (Pathania et al. 2011). However, this was not the case when the endogenous p220 level became abnormally elevated, as occurred after miR-545 inhibition or Adeno-BRCA1 infection. p220-driven recruitment of RAD51 to these sites was also readily detectable. It was either not detected or only weakly apparent in G1 cells that produce physiological quantities of BRCA1. Thus, these aberrant damage site protein localization phenomena represent qualitatively abnormal, p220-directed DDRs.
Cells that were not exposed to mutagen-induced DNA damage also responded abnormally to endogenous p220 overexpression in G1. The 53BP1-containing foci to which endogenous, overexpressed p220 was attracted during G1 in these cells likely contain DSBs that arose spontaneously in the former cell cycle (Harrigan et al. 2011; Lukas et al. 2011). One would have expected that any DSB repair during G1 would involve NHEJ, which is not a p220-dependent process.
Thus, the presence during G1 of two HRR-linked proteins at these sites—one, RAD51, dependent on the presence of the other, p220—again suggests that they are engaged in events that are not physiological. Conceivably, BRCA1 and RAD51 participate in illegitimate interhomolog or other interchromosomal recombination events at some of these 53BP1-containing structures in G1. These kinds of interchanges are a potential source of genomic disorder that, if sufficiently chronic and frequent, could prove to be carcinogenic.
The question of whether a breakdown in the miR-545/p220 tuning system is a cancer- promoting event is a valid one. This cannot be answered directly at present, given the paucity of relevant information. However, it seems reasonable to predict that chronic failure of the miR-545/p220 abundance-regulating system could, over time, result in chronic DNA damage. If not effectively repaired, this source of DNA damage could add to the risk of tumorigenesis at least in some cell types.
Materials and methods
Cell lines and culture conditions
T98G, T24, U2OS, DR-U2OS, and HEK293 cells were grown at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Gibco) in a 10% CO2-containing atmosphere. IMECs were obtained from Daniel Silver (Dana-Farber Cancer Institute) and maintained in complete MEGM medium (Lonza). The mAG-hGeminin(1/110)/pCSII-EF vector, which carries a fusion protein composed of the degron-containing fragment of human Geminin (amino acids 1–110) attached to the GFP mAG1, was obtained from Atsushi Miyawaki (RIKEN) (Sakaue-Sawano et al. 2008). T98G, U2OS, and IMECs were transduced with this recombinant vector, and, 48 h later, S/G2/M cells stably expressing the mAG-hGEM fusion protein were selected for green fluorescence by two subsequent rounds of FACS. These purified populations are referred to as T98G-GEM, U2OS-GEM, and IMEC-GEM.
Cell cycle synchronization
T98G and T98G-GEM cells were synchronized in G0/G1 by serum deprivation (Davis et al. 2001). Cells were plated at 1.5 × 104 cells per square centimeter in DMEM supplemented with 10% FBS (Gibco) and, 12 h later, washed once with PBS and incubated for an additional 72 h in DMEM containing 0.1% FBS. Cells were released from the proliferation block by readdition of 10% FBS-containing DMEM and allowed to progress synchronously through the cell cycle.
T24 cells were arrested in G0/G1 by contact inhibition after incubation for 72 h at confluency. Upon replating at lower density, the cells progressed synchronously through G1 and S/G2. Alternatively, T24 cells were arrested in late G1 by incubation in 0.5 mM mimosine-containing (Sigma-Aldrich) medium for 24 h (Krude 1999).
miRNA, siRNA reagents, and transfection
miRNA mimic reagents specific for miR-545 (PM12957) and miR-545* (PM12435) and negative controls were obtained from Ambion (Life Technologies). miR-545 antagomir (miRCURY LNA Power Inhibitor, no. 427261) and Negative Control B (no. 199021) were obtained from Exiqon. Transfections were performed using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer's instructions to achieve a final concentration of 100 nM for the mimic reagents or 10 nM for the antagomir reagent. Cells were analyzed 48 h after transfection.
The following siRNA reagents were used in this study: siBRCA1-1 (CAGCUACCCUUCCAUCAUA), siBRCA1-2 (GAAGGAGCUUUCAUCAUUC), siXPA (GCUACUGGCAUGGCUUU), siXPC (UGAAAUAUGAGGCCAUCUA), siRPA70 (AACACUCUAUCCUCUUUCAUGUU), siRAP80 (GUAUUGACUCGGAGACAAA), si53BP1 (GCCAGGUUCUAGAGGAUGA), siRIF1 (GCAUUGACUUCUCACCAUA), ON-TARGETplus RAD51 siRNA Smart Pool, and Luciferase GL2 Duplex control (all obtained from Dharmacon); and siEXO1-s17502 (GGCUAGGAAUGUGCAGACA) and siEXO1-s17503 (CUUUUGAACAGAUCGAUGA) (obtained from Ambion). All siRNA reagents were transfected into cells using Lipofectamine RNAiMAX (Life Technologies) in OptiMEM serum-free medium (Life Technologies). Cells were analyzed 72 h post-transfection.
Luciferase assay
Luciferase assays were performed using the Dual-Luciferase Reporter Assay system (Promega). The BRCA1 3′ UTR was PCR-amplified from genomic DNA and cloned in either the sense or antisense orientation into the pGL4 vector (Promega). Mutations in predicted miR-545 target sites at positions 1148–1154 and 1183–1189 were introduced with a QuickChange multisite-directed mutagenesis kit (Agilent Technologies). The wild-type or mutant reporters were cotransfetced into HEK293 cells with either miR-545 mimic, an unrelated miR-22 mimic, or a control reagent (all synthesized by Dharmacon) using Lipofectamine 2000 (Life Technologies). After 48 h, cells were processed according to the manufacturer's instructions (Promega), and luciferase activity was measured using a Veritas Microplate Luminometer (Turner Biosystems).
HR assay
HRR assay was performed in DR-U2OS containing a single, stably integrated copy of direct repeat GFP reporter as described previously (Xia et al. 2006). Exponentially growing DR-U2OS cells were transfected with either a miR-545 mimic, a miR-545 antagomir, or negative controls using RNAiMAX reagent (Life Technologies). After 24 h, cells were transfected with the pCBASce I-SceI-expressing vector using FuGene 6 reagent (Roche), and, 48 h later, GFP fluorescence was analyzed by FACS.
Quantitative RT–PCR (qRT–PCR) and TaqMan assays
Total RNA was extracted using TRIzol reagent according to the manufacturer's instructions (Life Technologies). Reverse transcription was carried out using SuperScript III (Life Technologies). Real-time PCR analysis of miRNA abundance was performed using specific TaqMan miRNA assays (no. 4380918 for has-miR-545 and no. 437381 for RNU6B, Applied Biosystems). PCR data were normalized to RNU6B expression levels and analyzed by the comparative CT method (Schmittgen et al. 2008).
BRCA1 mRNA expression was analyzed by qRT–PCR and normalized to GAPDH expression levels. The primers used were HBR16F (5′-GCAATGGAAGAAAGTGTGACG-3′), HBR22R (5′-GCCAAGGGTGAATGATGAAAG-3′), GAPDH-F (5′-GTGTTCCTACCCCCAATGTG-3′), and GAPDH-R (5′-GTCATTGAGCAATGCCAG-3′).
Western blotting and immunoprecipitation
Cells were lysed for 30 min on ice in NETN-420 lysis buffer (420 mM NaCl, 0.5 mM EDTA, 20 mM Tris-Cl at pH 8.0, 0.5% [v/v] Nonidet P-40 [NP-40]) supplemented with protease inhibitors (Roche). Protein concentration was measured using the Bio-Rad Protein Assay (Bio-Rad), and 10 μg of whole cell extract per sample was electrophoresed in an SDS–polyacrylamide gel. The separated proteins were transferred to a nitrocellulose membrane and blocked overnight at 4°C in blocking solution (5% non-fat dry milk [Bio-Rad] in phosphate-buffered saline/0.05% Tween 20 [PBS/Tween 20; Sigma-Aldrich]). Membranes were incubated with primary antibody in PBST for at least 1 h at room temperature, washed three time for 5 min each in PBS/Tween 20, incubated with HRP-conjugated secondary antibody in PBS/Tween 20 for 1 h at room temperature, and visualized using Western Lightning ECL reagent (PerkinElmer).
Immunoprecipitation was performed by incubation of the whole-cell lysates with rabbit polyclonal anti-BRCA1 antibody (Upstate-Millipore) or preimmune rabbit serum in nondenaturing NETN-150 buffer supplemented with protease inhibitors (Roche) for at least 1 h at 4°C and additional incubation for 3 h at 4°C following addition of protein A-coupled Sepharose beads. Then, Sepharose beads were extensively washed in NETN-150 buffer, boiled for 5 min at 95°C in Laemmli buffer (Bio-Rad), and centifugated, and the supernatants were subjected to SDS-PAGE.
The antibodies used for Western blotting include mouse monoclonal anti-BRCA1 (SD118, Millipore), mouse monoclonal anti-XPA (Clone 12F5, Thermo Scientific), rabbit polyclonal anti-XPC (H-300, Santa Cruz Biotechnology), rabbit polyclonal anti-EXO1 (Novus Biologicals), rabbit polyclonal anti-RAP80 (Bethyl Laboratories), and rabbit polyclonal anti-Actin (Sigma-Aldrich).
Indirect immunofluorescence and antibodies
Cells grown on glass coverslips were washed three times with PBS, fixed with 4% paraformaldehyde/PBS solution for 15 min at room temperature, washed twice with PBS, and permeabilized with PBS/0.05% Triton X-100/1% BSA (PBST-BSA) solution for 20 min at room temperature. Cells were then incubated with a primary antibody in PBST/BSA for 1 h at 37°C, washed twice with for 5 min in PBST, and incubated with rhodamine-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch) for 1 h at 37°C. Subsequent staining of UV-generated CPD lesions was performed as described previously (Pathania et al. 2011). Cells already stained for the primary protein of interest were fixed again as above for 15 min at room temperature, washed twice in PBST, exposed to 5 U of recombinant RNase-free DNase I for 25 min at 37°C, washed twice in PBST, incubated with anti-CPD antibody for 45 min at 37°C, washed again twice in PBST, and incubated with FITC-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch) for 1 h at 37°C. After the final wash in PBST, coverslips were mounted on slides with DAPI-containing mounting medium (Vectashield Laboratories) and kept at 4°C until analysis.
The following antibodies were used for indirect immunofluorescence: rabbit polyclonal anti-BRCA1 (Upstate Biotechnologies), mouse monoclonal anti-BRCA1 (D-9, Santa Cruz Biotechnology), mouse monoclonal anti-XPA (clone 12F5, Thermo Scientific), rabbit polyclonal anti-XPC (H-300, Santa Cruz Biotechnology), mouse monoclonal anti-RPA (Calbiochem), rabbit polyclonal anti-RAP80 (Bethyl Laboratories), rabbit polyclonal anti-RAD51 (Santa Cruz Biotechnology), mouse monoclonal anti-phospho-H2AX (Upstate-Millipore), rabbit polyclonal anti-53BP1 (Bethyl Laboratories), rabbit polyclonal anti-RIF1 (Bethyl Laboratories), and mouse monoclonal anti-CPD (Cosmo Bio).
UV and γ irradiation were performed as described previously (Pathania et al. 2011).
Comet assay
Alkaline and neutral comet assays were performed on isolated G1 and S/G2 populations of T98G-GEM cells using the Single-Cell Gel Electrophoresis Assay kit (Trevigen) according to the manufacturer's instructions. CellProfiler software (Carpenter et al. 2006) was used for analysis and quantitation of the results.
Adeno-BRCA1 vector
Ectopic expression of wild-type BRCA1 protein was performed by infecting cells with recombinant Adeno-BRCA1 or control adenoviral vector (Vector Biolabs) at a high multiplicity of infection (MOI = 200). Forty-eight hours later, cells were subjected to further analysis.
FACS was performed as described earlier (Pathania et al. 2011).
Laser-induced DNA stripes
Laser-generated DNA DSBs were generated using a P.A.L.M. MicroBeam laser microdissection system (Carl Zeiss MicroImaging, Inc.) as described previously (Sobhian et al. 2007; Hu et al. 2011). Cells grown on coverslips were incubated for 24 h in medium containing 10 μM BrdU (Sigma-Aldrich). Laser stripes were generated in ∼100 cells per coverslip with the above-noted laser (λ = 337 nm) using the 40× objective at 70% power. Cells were returned to a cell culture incubator at 37°C and fixed 90 min later following the indirect immunofluorescence protocol.
Statistical analysis
An unpaired, two-tailed Student's t-test was used to calculate the P-value of the data set comparisons throughout this study.
Acknowledgments
We thank Ronny Drapkin, Daniel Silver, Jean Feunteun, Todd Golub, and members of the Livingston laboratory for insightful discussions and suggestions. We are grateful to Serena Landini for technical advice and help. We thank Asako Sakaue-Sawano and Atsushi Miyawaki (RIKEN) for the mAG1-hGem-expressing and mKO2-hCdt1-expressing vectors, and Dipanjan Chowdhury for graciously sharing results prior to publication. This work was supported by funds from the Breast Cancer SPORE grant to Dana-Farber/Harvard Cancer Center (to S.D.D. and D.M.L.) and grants from the National Cancer Institute (5 P01CA80111), the Breast Cancer Research Foundation, and the Susan G. Komen Foundation for the Cure (SAC110022) (to D.M.L.). Generous support for this work was also provided by the National Institutes of Health Training grant (5T32CA00938-30) (to S.D.D.).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.225045.113.
References
- Baldassarre G, Battista S, Belletti B, Thakur S, Pentimalli F, Trapasso F, Fedele M, Pierantoni G, Croce CM, Fusco A 2003. Negative regulation of BRCA1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma. Mol Cell Biol 23: 2225–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP 2004. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116: 281–297 [DOI] [PubMed] [Google Scholar]
- Bartel DP 2009. MicroRNAs: Target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beger C, Pierce LN, Kruger M, Marcusson EG, Robbins JM, Welcsh P, Welch PJ, Welte K, King MC, Barber JR, et al. 2001. Identification of Id4 as a regulator of BRCA1 expression by using a ribozyme-library-based inverse genomics approach. Proc Natl Acad Sci 98: 130–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, et al. 2011. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell 42: 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 2010. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 17: 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 2010. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141: 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. 2006. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7: R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catteau A, Harris WH, Xu CF, Solomon E 1999. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: Correlation with disease characteristics. Oncogene 18: 1957–1965 [DOI] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ 2013. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 49: 858–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Farmer AA, Chen CF, Jones DC, Chen PL, Lee WH 1996. BRCA1 is a 220-kDa nuclear phosphoprotein that is expressed and phosphorylated in a cell cycle–dependent manner. Cancer Res 56: 3168–3172 [PubMed] [Google Scholar]
- Chen JJ, Silver D, Cantor S, Livingston DM, Scully R 1999. BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer Res 59: 1752s–1756s [PubMed] [Google Scholar]
- Chen L, Nievera CJ, Lee AY, Wu X 2008. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem 283: 7713–7720 [DOI] [PubMed] [Google Scholar]
- Coleman KA, Greenberg RA 2011. The BRCA1–RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J Biol Chem 286: 13669–13680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis PK, Ho A, Dowdy SF 2001. Biological methods for cell-cycle synchronization of mammalian cells. BioTechniques 30: 1322–1331 [DOI] [PubMed] [Google Scholar]
- Deans AJ, Simpson KJ, Trivett MK, Brown MA, McArthur GA 2004. Brca1 inactivation induces p27(Kip1)-dependent cell cycle arrest and delayed development in the mouse mammary gland. Oncogene 23: 6136–6145 [DOI] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. 2013. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339: 711–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. 2013. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 49: 872–883 [DOI] [PubMed] [Google Scholar]
- Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, et al. 2000. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst 92: 564–569 [DOI] [PubMed] [Google Scholar]
- Feng L, Fong KW, Wang J, Wang W, Chen J 2013. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem 288: 11135–11143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes WD, Stefansson IM, Chappuis PO, Begin LR, Goffin JR, Wong N, Trudel M, Akslen LA 2003. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J Natl Cancer Inst 95: 1482–1485 [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J, Miki Y, et al. 1994. BRCA1 mutations in primary breast and ovarian carcinomas. Science 266: 120–122 [DOI] [PubMed] [Google Scholar]
- Garcia AI, Buisson M, Bertrand P, Rimokh R, Rouleau E, Lopez BS, Lidereau R, Mikaelian I, Mazoyer S 2011. Down-regulation of BRCA1 expression by miR-146a and miR-146b-5p in triple negative sporadic breast cancers. EMBO Mol Med 3: 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Follonier C, Tourriere H, Puddu F, Lazzaro F, Pasero P, Lopes M, Plevani P, Muzi-Falconi M 2010. Exo1 competes with repair synthesis, converts NER intermediates to long ssDNA gaps, and promotes checkpoint activation. Mol Cell 40: 50–62 [DOI] [PubMed] [Google Scholar]
- Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM 2006. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev 20: 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudas JM, Li T, Nguyen H, Jensen D, Rauscher FJ III, Cowan KH 1996. Cell cycle regulation of BRCA1 messenger RNA in human breast epithelial cells. Cell Growth Differ 7: 717–723 [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, et al. 1996. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell 85: 1009–1023 [DOI] [PubMed] [Google Scholar]
- Harkin DP, Bean JM, Miklos D, Song YH, Truong VB, Englert C, Christians FC, Ellisen LW, Maheswaran S, Oliner JD, et al. 1999. Induction of GADD45 and JNK/SAPK-dependent apoptosis following inducible expression of BRCA1. Cell 97: 575–586 [DOI] [PubMed] [Google Scholar]
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP 2011. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol 193: 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD 2nd, Abkevich V, Potter J, Pruss D, Glenn P, et al. 2010. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol 28: 3570–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu NC, Huang YF, Yokoyama KK, Chu PY, Chen FM, Hou MF 2013. Methylation of BRCA1 promoter region is associated with unfavorable prognosis in women with early-stage breast cancer. PLoS ONE 8: e56256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM 2011. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev 25: 685–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janatova M, Zikan M, Dundr P, Matous B, Pohlreich P 2005. Novel somatic mutations in the BRCA1 gene in sporadic breast tumors. Hum Mutat 25: 319. [DOI] [PubMed] [Google Scholar]
- Joukov V, Groen AC, Prokhorova T, Gerson R, White E, Rodriguez A, Walter JC, Livingston DM 2006. The BRCA1/BARD1 heterodimer modulates Ran-dependent mitotic spindle assembly. Cell 127: 539–552 [DOI] [PubMed] [Google Scholar]
- Katsumi S, Kobayashi N, Imoto K, Nakagawa A, Yamashina Y, Muramatsu T, Shirai T, Miyagawa S, Sugiura S, Hanaoka F, et al. 2001. In situ visualization of ultraviolet-light-induced DNA damage repair in locally irradiated human fibroblasts. J Invest Dermatol 117: 1156–1161 [DOI] [PubMed] [Google Scholar]
- Kim H, Chen J, Yu X 2007. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 316: 1202–1205 [DOI] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. 2005. Combinatorial microRNA target predictions. Nat Genet 37: 495–500 [DOI] [PubMed] [Google Scholar]
- Krude T 1999. Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Exp Cell Res 247: 148–159 [DOI] [PubMed] [Google Scholar]
- Lakhani SR, Jacquemier J, Sloane JP, Gusterson BA, Anderson TJ, van de Vijver MJ, Farid LM, Venter D, Antoniou A, Storfer-Isser A, et al. 1998. Multifactorial analysis of differences between sporadic breast cancers and cancers involving BRCA1 and BRCA2 mutations. J Natl Cancer Inst 90: 1138–1145 [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20 [DOI] [PubMed] [Google Scholar]
- Li ML, Greenberg RA 2012. Links between genome integrity and BRCA1 tumor suppression. Trends Biochem Sci 37: 418–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM 2005. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433: 769–773 [DOI] [PubMed] [Google Scholar]
- Liu Z, Wu J, Yu X 2007. CCDC98 targets BRCA1 to DNA damage sites. Nat Struct Mol Biol 14: 716–720 [DOI] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, et al. 2011. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 13: 243–253 [DOI] [PubMed] [Google Scholar]
- Magdinier F, Ribieras S, Lenoir GM, Frappart L, Dante R 1998. Down-regulation of BRCA1 in human sporadic breast cancer; analysis of DNA methylation patterns of the putative promoter region. Oncogene 17: 3169–3176 [DOI] [PubMed] [Google Scholar]
- Matros E, Wang ZC, Lodeiro G, Miron A, Iglehart JD, Richardson AL 2005. BRCA1 promoter methylation in sporadic breast tumors: Relationship to gene expression profiles. Breast Cancer Res Treat 91: 179–186 [DOI] [PubMed] [Google Scholar]
- Moskwa P, Buffa FM, Pan Y, Panchakshari R, Gottipati P, Muschel RJ, Beech J, Kulshrestha R, Abdelmohsen K, Weinstock DM, et al. 2011. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell 41: 210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M 1999. Brca1 controls homology-directed DNA repair. Mol Cell 4: 511–518 [DOI] [PubMed] [Google Scholar]
- Natrajan R, Mackay A, Lambros MB, Weigelt B, Wilkerson PM, Manie E, Grigoriadis A, A'Hern R, van der Groep P, Kozarewa I, et al. 2012. A whole-genome massively parallel sequencing analysis of BRCA1 mutant oestrogen receptor-negative and -positive breast cancers. J Pathol 227: 29–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan PJ, Livingston DM 2010. BRCA1 and BRCA2: Breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 31: 961–967 [DOI] [PubMed] [Google Scholar]
- Papamichos-Chronakis M, Peterson CL 2012. Chromatin and the genome integrity network. Nat Rev Genet 14: 62–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania S, Nguyen J, Hill SJ, Scully R, Adelmant GO, Marto JA, Feunteun J, Livingston DM 2011. BRCA1 is required for postreplication repair after UV-induced DNA damage. Mol Cell 44: 235–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. 2000. Molecular portraits of human breast tumours. Nature 406: 747–752 [DOI] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M 1999. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 13: 2633–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujana MA, Han JD, Starita LM, Stevens KN, Tewari M, Ahn JS, Rennert G, Moreno V, Kirchhoff T, Gold B, et al. 2007. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet 39: 1338–1349 [DOI] [PubMed] [Google Scholar]
- Reis-Filho JS, Tutt AN 2008. Triple negative tumours: A critical review. Histopathology 52: 108–118 [DOI] [PubMed] [Google Scholar]
- Rice JC, Massey-Brown KS, Futscher BW 1998. Aberrant methylation of the BRCA1 CpG island promoter is associated with decreased BRCA1 mRNA in sporadic breast cancer cells. Oncogene 17: 1807–1812 [DOI] [PubMed] [Google Scholar]
- Roy R, Chun J, Powell SN 2012. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat Rev Cancer 12: 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffner H, Verma IM 1997. BRCA1 is a cell cycle-regulated nuclear phosphoprotein. Proc Natl Acad Sci 94: 7138–7143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saetrom P, Heale BS, Snove O Jr, Aagaard L, Alluin J, Rossi JJ 2007. Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res 35: 2333–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. 2008. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132: 487–498 [DOI] [PubMed] [Google Scholar]
- Santarosa M, Maestro R 2012. BRACking news on triple-negative/basal-like breast cancers: How BRCA1 deficiency may result in the development of a selective tumor subtype. Cancer Metastasis Rev 31: 131–142 [DOI] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51–BRCA1/2. Cancer Cell 22: 106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Lee EJ, Jiang J, Sarkar A, Yang L, Elton TS, Chen C 2008. Real-time PCR quantification of precursor and mature microRNA. Methods 44: 31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM 1997. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88: 265–275 [DOI] [PubMed] [Google Scholar]
- Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM 1999. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell 4: 1093–1099 [DOI] [PubMed] [Google Scholar]
- Sertic S, Pizzi S, Cloney R, Lehmann AR, Marini F, Plevani P, Muzi-Falconi M 2011. Human exonuclease 1 connects nucleotide excision repair (NER) processing with checkpoint activation in response to UV irradiation. Proc Natl Acad Sci 108: 13647–13652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver DP, Livingston DM 2012. Mechanisms of BRCA1 tumor suppression. Cancer Discov 2: 679–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA 2007. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316: 1198–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram K, Zhang H, Zeng YX, Houvras Y, Peng Y, Wu GS, Licht JD, Weber BL, El-Deiry WS 1997. Arrest of the cell cycle by the tumour-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/CiP1. Nature 389: 187–190 [DOI] [PubMed] [Google Scholar]
- Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et al. 2003. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci 100: 8418–8423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sy SM, Huen MS, Chen J 2009. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci 106: 7155–7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Gautier J 2011. Double-strand break end resection and repair pathway choice. Annu Rev Genet 45: 247–271 [DOI] [PubMed] [Google Scholar]
- Thompson ME, Jensen RA, Obermiller PS, Page DL, Holt JT 1995. Decreased expression of BRCA1 accelerates growth and is often present during sporadic breast cancer progression. Nat Genet 9: 444–450 [DOI] [PubMed] [Google Scholar]
- Tian F, Sharma S, Zou J, Lin SY, Wang B, Rezvani K, Wang H, Parvin JD, Ludwig T, Canman CE, et al. 2013. BRCA1 promotes the ubiquitination of PCNA and recruitment of translesion polymerases in response to replication blockade. Proc Natl Acad Sci 110: 13558–13563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson GE, Chen TT, Stastny VA, Virmani AK, Spillman MA, Tonk V, Blum JL, Schneider NR, Wistuba II, Shay JW, et al. 1998. Characterization of a breast cancer cell line derived from a germ-line BRCA1 mutation carrier. Cancer Res 58: 3237–3242 [PubMed] [Google Scholar]
- Turner N, Tutt A, Ashworth A 2004. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer 4: 814–819 [DOI] [PubMed] [Google Scholar]
- Turner NC, Reis-Filho JS, Russell AM, Springall RJ, Ryder K, Steele D, Savage K, Gillett CE, Schmitt FC, Ashworth A, et al. 2007. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene 26: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Vaughn JP, Davis PL, Jarboe MD, Huper G, Evans AC, Wiseman RW, Berchuck A, Iglehart JD, Futreal PA, Marks JR 1996. BRCA1 expression is induced before DNA synthesis in both normal and tumor-derived breast cells. Cell Growth Differ 7: 711–715 [PubMed] [Google Scholar]
- Walsh T, King MC 2007. Ten genes for inherited breast cancer. Cancer Cell 11: 103–105 [DOI] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ 2007. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316: 1194–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Grushko TA, Dignam J, Hagos F, Nanda R, Sveen L, Xu J, Fackenthal J, Tretiakova M, Das S, et al. 2005. BRCA1 promoter methylation in sporadic breast cancer is associated with reduced BRCA1 copy number and chromosome 17 aneusomy. Cancer Res 65: 10692–10699 [DOI] [PubMed] [Google Scholar]
- Wilson CA, Ramos L, Villasenor MR, Anders KH, Press MF, Clarke K, Karlan B, Chen JJ, Scully R, Livingston D, et al. 1999. Localization of human BRCA1 and its loss in high-grade, non-inherited breast carcinomas. Nat Genet 21: 236–240 [DOI] [PubMed] [Google Scholar]
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM 2006. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 22: 719–729 [DOI] [PubMed] [Google Scholar]
- Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM 2011. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 477: 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikan M, Janatova M, Pavlista D, Pohlreich P 2007. High frequency of BRCA1/2 and p53 somatic inactivation in sporadic ovarian cancer. J Genet 86: 169–171 [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T 2013. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339: 700–704 [DOI] [PMC free article] [PubMed] [Google Scholar]