

Abstract
Primitive neuroectodermal tumor (PNET) is a broad term that includes a wide array of lesions with varying differentiating potential affecting both the central and peripheral nervous system. Peripheral PNET (pPNET), a variant of PNET, is known to arise in many places throughout the body; involvement of the mandible is however rare. A review of the scientific literature describes only seven reports of pPNET to be arising from the mandible. Given their insidious clinical symptoms, variable locations and rarity, the accurate diagnosis of peripheral PNETs poses a challenge for clinicians. PNETs exhibits characteristic immunophenotypical and genetic features that distinguish them from other small round cell tumors. A multidisciplinary approach is necessary to manage patients affected by PNET. There is however no consensus about the best therapeutic strategy. We recently got to treat a young patient with mandibular PNET; the clinical course as well as the histopathology with immunohistochemistry correlation of this rare entity is discussed.
Keywords: Mandibular tumor, primitive neuroectodermal tumor, round cell tumors
INTRODUCTION
Primitive neuroectodermal tumor (PNET) is a broad term that includes a wide array of lesions with varying differentiating potential affecting both the central and peripheral nervous system.[1,2] It is a predominately a neural, nonepithelial neoplasm similar to Ewing sarcoma. Peripheral PNET (pPNET), a variant of PNET, is known to arise in many places throughout the body; involvement of the mandible is however rare. A review of the scientific literature describes only seven reports of pPNET to be arising from the mandible.[3,4,5,6,7] This report describes the course and management of a pPNET located in the mandibular ramus in a 22-year-old female patient. The clinical presentation as well as the histopathology with immunohistochemistry (IHC) correlation of this rare entity is discussed.
CASE REPORT
A 22-year-old female presented with a complaint of insidious onset painless swelling in the left side of the face which had increased in size over 4 months. Clinical examination revealed a firm, well-circumscribed swelling measuring 7 × 7 cm in the left parotid region with numbness in the chin region [Figure 1]. The angle of the mandible was widened by the tumor resulting in a left-sided intraoral bulge. Otorhinolaryngological findings were otherwise normal; there was no evidence of cervical lymphadenopathy or any systemic symptoms.
Figure 1.
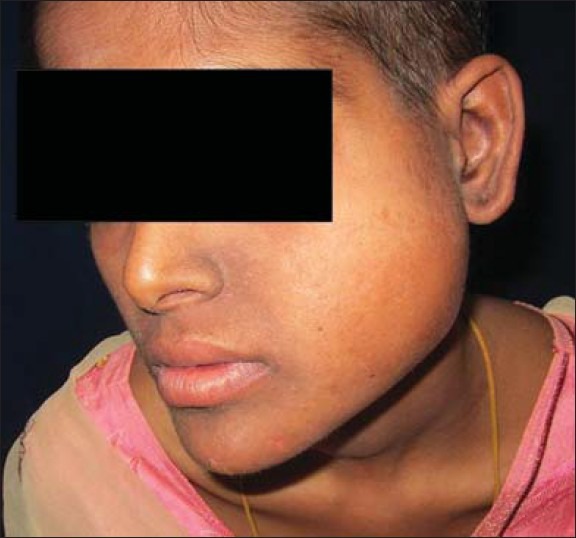
Clinical photograph at presentation
Computed tomography (CT) revealed a heterodense mass lesion measuring 8 × 7 cm at its greatest dimension causing bony destruction of the ascending mandibular ramus and with extension into the skull base. The tumor caused effacement of the fat and muscle planes of the infratemporal fossa and the masticator spaces. It was seen pushing the parotid space laterally and the parapharyngeal space medially. The left temporomandibular joint was obliterated by the tumor [Figures 2a–c]. A bone scintigram showed an increased abnormal activity in the left side of mandible corresponding to the tumor [Figure 2d]. A bone marrow aspiration and a bone marrow biopsy were within normal limits. An extraoral trucut biopsy from the tumor on histopathologic examination revealed a diffused collection of small malignant round to oval cells with hyperchromatic nuclei, inconspicuous nucleoli, with vacuolated to pale cytoplasm and high mitotic activity, and scattered blood vessels with peritheliomatous arrangements of the tumor cells. The tumor cells on immunohistochemistry were positive for vimentin, CD99, Bcl2, chromogranin, and S-100. It was negative for keratin, LCA, CD3, CD 20, CD56, and Alk 1. Fifty percentage of the tumor cells showed strong nuclear positivity to Ki-67. The histopathology along with immunohistochemistry correlation favored a diagnosis of PNET of the mandible [Figures 3a–d].
Figure 2.
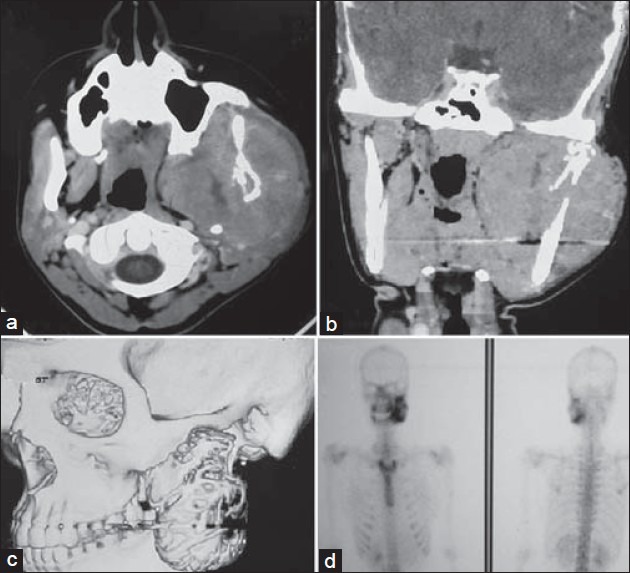
CT scan: (a) Axial section, (b) Coronal section, and (c) Reconstructed image showing a heterodense mass lesion measuring 8 × 7 cm at its greatest dimension, causing bony destruction of the ascending mandibular ramus, with extension into the skull base. The tumor caused effacement of the fat and muscle planes of the infratemporal fossa and the masticator space, (d) A bone scintigram showed an increased abnormal activity in the left side of the mandible corresponding to the tumor
Figure 3.
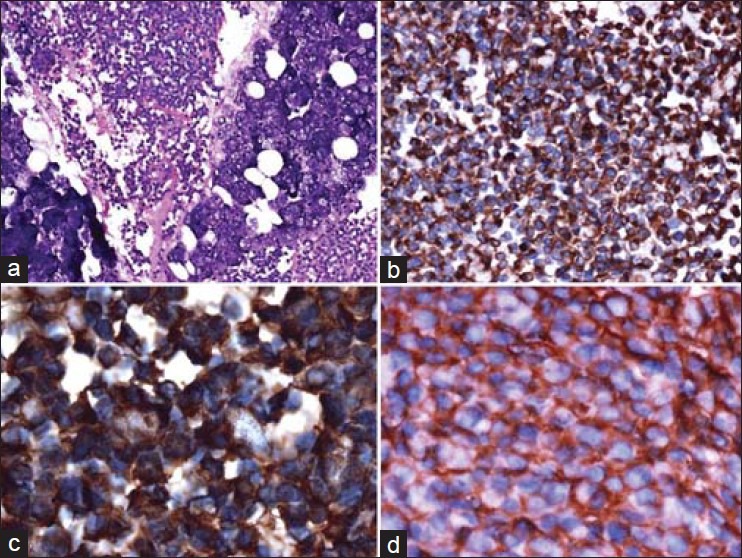
(a) A diffuse collection of small malignant round to oval cells with hyperchromatic nuclei, inconspicuous nucleoli, with vacuolated to pale cytoplasm and high mitotic activity, and scattered blood vessels with peritheliomatous arrangements of the tumor cells (H and E, ×20), (b) Tumor cells showing immunopositivity to vimentin (IHC, ×20), (c) Tumor cells showing immunopositivity to S-100 (IHC, ×100), (d) Tumor cells showing immunopositivity to CD-99 (IHC, ×40)
The patient was planned for a multimodality treatment. She initially received four cycles of vincristine, adriamycin, cyclophosphamide alternating with ifosfamide and etoposide (VAC/IE) based chemotherapy. In view of the extensive soft tissue involvement, she was initially considered for external beam radiotherapy as a part of her local therapy. A static response was noted after 54 Grey (Gy) of radiotherapy, which was confirmed on a subsequent CT of the mandible which showed a necrotic residue with patchy contrast uptake. She was subsequently taken for a definitive surgery after the sixth cycle of chemotherapy. En bloc excision of the tumor along with a posterior segmental mandibulectomy from the level of left first molar to left condyle and left temporomandibular joint was done [Figures 4a–d]. An ‘L’ shaped reconstruction plate was anchored to the zygoma and the remnant mandible to bridge the bony defect, soft tissue cover was achieved by an anterolateral thigh free flap. [Figures 5a and b] The postoperative histopathology revealed no viable tumor and a 100% tumor necrosis. She further went on to complete her further courses of chemotherapy and is presently disease free.
Figure 4.
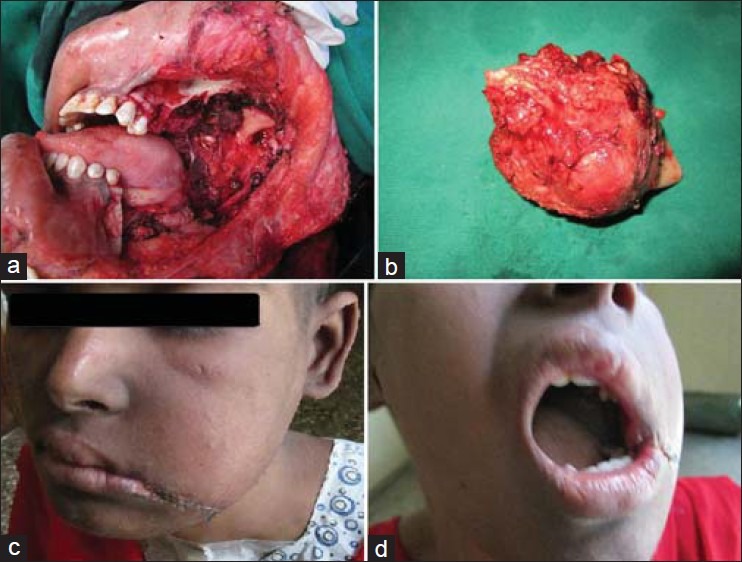
(a) Intraoperative photograph following en bloc resection of the tumor, (b) Clinical photograph of the operated specimen, (c) Postoperative extraoral clinical photograph, (d) Postoperative intraoral clinical photograph
Figure 5.
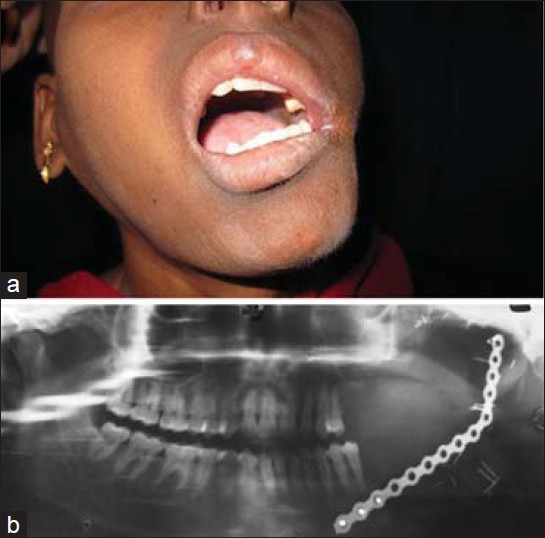
(a) Postoperative intraoral clinical photograph after 10 months showing the soft tissue reconstruction by anterolateral thigh free flap, (b) Postoperative orthopantomogram showing the bony defect following en bloc removal of tumor involving the posterior segmental mandibulectomy from the level of the left first molar to the left condyle and left temporomandibular joint. An ‘L’ shaped reconstruction plate was anchored to the zygoma and the remnant mandible to bridge the bony defect
DISCUSSION
PNET is predominately neural, nonepithelial neoplasm which is included in the differential diagnosis of small round cell tumors. Previously, it was thought that these tumors exclusively localized to the central nervous system (CNS). However, with the recognition of PNET outside of the central nervous system, the current understanding of the origin of the PNET has changed. PNET are now classified into two types, based on their location in the body: Peripheral PNET (pPNET) and CNS PNET (cPNET). pPNET is commonly known to arise in the thoracopulmonary region, retroperitoneum and kidneys. pPNET rarely arises from the mandible, there are only seven reports in the scientific literature. PNET is a malignancy which usually peaks in adolescents and young adults with no gender bias, although some studies have shown a male preponderance. PNETs exhibits characteristic immunophenotypical and genetic features that distinguish them from other small round cell tumors. The pPNET tumors were initially included within the extra-skeletal Ewing tumors, with which they share microscopic and genetic alterations but differ in extent of neuronal differentiation. These tumors share the chromosomal translocation, t(11:22)(q24:q12).[6] This genetic anomaly leads to the creation of a fusion protein consisting of EWS and FLI-1 gene products.[8] Many authors consider pPNET and extraskeletal Ewing's sarcoma to represent extremes of the same group of tumors, PNET tends to represent the somewhat more differentiated end of tumor spectrum. Proper discrimination between the two can at times be difficult, and hence discrimination criteria have been recommended to aid in diagnosis of the pPNET.[6] Features of the pPNET include: Well-defined histological evidence with rosette formation, immunoreactivity to two or more neural markers, and/or electron micrographic evidence of neural differentiation and neurosecretory granules with microtubules and intermediate filaments.
A multidisciplinary approach is necessary to manage patients affected by PNET. There is however no consensus about the best therapeutic strategy. Generally, PNET and extraosseous Ewing Sarcoma share the same treatment protocols.[9] Treatment options include chemotherapy and surgical excision with or without radiation; some authors consider chemotherapy with or without radiotherapy as the first line of treatment. PNETs are tumors which evolve rapidly with poor prognosis. The advent of multiagent chemotherapy has markedly improved the prognosis from a dismal 5 to 15% to an approximately 75% 5-year survival. The amount of chemotherapy-induced necrosis is an important prognostic finding. These patients should be followed-up for life to rule out recurrence, metastasis, and treatment-related malignancies. In conclusion, given their rarity and insidious clinical symptoms, the accurate diagnosis of mandibular PNETs poses unique clinical challenges; knowledge of this tumor can help clinicians precisely manage these patients.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Mills SE. Neuroectodermal neoplasms of the head and neck with emphasis on neuroendocrine carcinoma. Mod Pathol. 2002;15:264–78. doi: 10.1038/modpathol.3880522. [DOI] [PubMed] [Google Scholar]
- 2.Nikitakis NG, Salama AR, O’Malley BW, Jr, Ord RA, Papadimitriou JC. Malignant peripheral primitive neuroectodermal tumor-peripheral neuroepithelioma of the head and neck: A clinicopathologic study of five cases and review of the literature. Head Neck. 2003;25:488–98. doi: 10.1002/hed.10260. [DOI] [PubMed] [Google Scholar]
- 3.Ozer E, Kanlikama M, Karakurum G, Sirikci A, Erkilic S, Aydin A. Primitive neuroectodermal tumour of the mandible. Int J Pediatr Otorhinolaryngol. 2002;65:257–61. doi: 10.1016/s0165-5876(02)00183-0. [DOI] [PubMed] [Google Scholar]
- 4.Sundine MJ, Bumpous JM. Primitive neuroectodermal tumor of the mandible: Report of a rare case. Ear Nose Throat J. 2003;82:211–4. [PubMed] [Google Scholar]
- 5.Alrawi SJ, Tan D, Sullivan M, Winston J, Loree T, Hicks W, et al. Peripheral primitive neuroectodermal tumor of the mandible with cytogenetic and molecular biology aberrations. J Oral Maxillofac Surg. 2005;63:1216–21. doi: 10.1016/j.joms.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 6.Votta TJ, Fantuzzo JJ, Boyd BC. Peripheral primitive neuroectodermal tumor associated with the anterior mandible: A case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:592–7. doi: 10.1016/j.tripleo.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 7.Kanaya H, Hirabayashi H, Tanigaito Y, Baba K. Ewing's sarcoma/primitive neuroectodermal tumour of the mandible: Report of a rare case and review of the literature. J Otolaryngol. 2007;36:E15–8. doi: 10.2310/7070.2006.0150. [DOI] [PubMed] [Google Scholar]
- 8.O’sullivan MJ, Perlman EJ, Furman J, Humphrey PA, Dehner LP, Pfeifer JD. Visceral primitive peripheral neuroectodermal tumors: A clinicopathologic and molecular study. Hum Pathol. 2001;32:1109–15. doi: 10.1053/hupa.2001.28247. [DOI] [PubMed] [Google Scholar]
- 9.Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewings sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]