

Abstract
Both Duchenne and golden retriever muscular dystrophy (GRMD) are caused by dystrophin deficiency. The Duchenne muscular dystrophy sartorius muscle and orthologous GRMD cranial sartorius (CS) are relatively spared/hypertrophied. We completed hierarchical clustering studies to define molecular mechanisms contributing to this differential involvement and their role in the GRMD phenotype. GRMD dogs with larger CS muscles had more severe deficits, suggesting that selective hypertrophy could be detrimental. Serial biopsies from the hypertrophied CS and other atrophied muscles were studied in a subset of these dogs. Myostatin showed an age-dependent decrease and an inverse correlation with the degree of GRMD CS hypertrophy. Regulators of myostatin at the protein (AKT1) and miRNA (miR-539 and miR-208b targeting myostatin mRNA) levels were altered in GRMD CS, consistent with down-regulation of myostatin signaling, CS hypertrophy, and functional rescue of this muscle. mRNA and proteomic profiling was used to identify additional candidate genes associated with CS hypertrophy. The top-ranked network included α-dystroglycan and like-acetylglucosaminyltransferase. Proteomics demonstrated increases in myotrophin and spectrin that could promote hypertrophy and cytoskeletal stability, respectively. Our results suggest that multiple pathways, including decreased myostatin and up-regulated miRNAs, α-dystroglycan/like-acetylglucosaminyltransferase, spectrin, and myotrophin, contribute to hypertrophy and functional sparing of the CS. These data also underscore the muscle-specific responses to dystrophin deficiency and the potential deleterious effects of differential muscle involvement.
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder caused by mutations in the dystrophin gene and occurs in approximately 1 in 3500 live male births.1 DMD boys show signs of skeletal muscle weakness, evidenced by a delay in walking until approximately 18 months and loss of ambulation by the teenage years. Necrotic muscle ultimately fails to regenerate and is replaced with fibrous connective tissue and fat. Molecular and cellular mechanisms underlying gradual muscle deterioration are poorly understood.
Animal models of DMD include the mdx mouse and golden retriever muscular dystrophy (GRMD) dog.2,3 Despite sharing the same fundamental genetic and biochemical lesions, remarkable phenotypic variation occurs among dystrophin-deficient individuals and muscles. Mdx mice have a relatively mild phenotype,4 whereas affected dogs have clinical and pathological features consistent with those of DMD.5 Even among DMD patients, who all lack dystrophin except for rare revertant fibers, symptoms can vary markedly.6 Dogs with GRMD also demonstrate pronounced phenotypic variation, as some dogs lose the ability to walk within the first 6 months of life, whereas others remain ambulatory to 10 years of age or older.7–9
In GRMD neonatal dogs, flexor muscles such as the sartorius are generally more severely involved than extensors, potentially due to their role in crawling.10,11 Early dystrophic histopathological changes seen in these diseased muscles include myofiber necrosis evidenced by hyaline fibers, mineralization, edema, and inflammation, with associated regeneration.10 Presumably, as dogs subsequently begin to walk, weight-bearing extensor muscles such as the vastus lateralis (VL) are more predisposed to injury and display these same acute dystrophic changes. With regard to individual muscle variation in DMD, extensors that undergo eccentric contraction (eg, quadriceps femoris) are particularly vulnerable to early weakness and wasting.12 On the other hand, the extraocular muscles are largely spared.13
In DMD patients, most muscles atrophy over time, but some, such as the gastrocnemius, undergo gross enlargement.14 On the basis of early histological studies of dystrophic muscle biopsies, this calf hypertrophy was initially attributed to deposition of fat and fibrotic tissue and was termed pseudohypertrophy.15 However, in a series of 350 neuromuscular patients, including 9 with Becker muscular dystrophy, quantitative ultrasound demonstrated that calf hypertrophy was most often due to an actual increase in contractile tissue.16 Mdx mice17 and dystrophin-deficient cats18 also have muscle hypertrophy in the absence of significant fat and connective tissue infiltration. The sartorius muscle is particularly intriguing in both DMD and GRMD. Humans have a single muscle, whereas dogs have cranial and caudal bellies. Serving principally as a hip flexor, the sartorius extends from the pelvis to the proximal tibia in people. Both heads of the canine sartorius also arise from the pelvis, but they insert at different sites (caudal, proximal tibia; cranial, distal femur). The cranial sartorius (CS) muscle of neonatal GRMD dogs sustains extensive necrosis19 and then regenerates, often undergoing dramatic true hypertrophy.9,20 In DMD patients, the sartorius muscle is relatively spared and may hypertrophy late in the disease process.21,22
Studies showing variable phenotypes among dystrophin-deficient species, individuals, and muscles suggest that factors other than dystrophin deficiency, so-called secondary effects, are involved in the disease process.23 Determining the molecular underpinnings of the variable clinical and histopathological response to dystrophin deficiency should provide insight into disease pathogenesis and an opportunity to identify potential targets for therapy. Phenotypic–molecular correlations are inherently limited in DMD patients due to unavoidable restrictions of muscle sampling. Animal studies are potentially more powerful because multiple muscles can be sampled at different ages, thus allowing clearer distinction of factors contributing to disease progression. We chose to use the GRMD model of DMD for this study because of the availability of archived biopsy samples of multiple muscles from affected dogs at two ages and corresponding systematic functional data that could be correlated with mRNA and protein expression findings.
Hierarchical clustering of several phenotypic markers, including CS muscle size, tibiotarsal joint angle,7 and flexor and extensor torque,8 was first performed in a group of GRMD and normal dogs. Consistent with our prior studies,9 severely affected dogs tended to have larger CS muscles. To achieve a better understanding of the molecular signals that drive muscle hypertrophy, we extended a prior, largely pathological study of differential muscle involvement in the GRMD model.19 Proteins that are well known to influence muscle size [myostatin (MSTN)]24,25 or potentially compensate for dystrophin deficiency [utrophin (UTRN)]26 were assessed in a subset of the dogs evaluated by hierarchical clustering. MSTN showed an age-dependent decrease and an inverse correlation with the degree of CS hypertrophy. Regulators of MSTN at the protein (AKT1) and miRNA (miR-539 and miR-208b targeting myostatin mRNA) level were altered, consistent with down-regulation of MSTN signaling, CS hypertrophy, and functional rescue of this muscle. The growth factor myotrophin (MTPN) was increased in the CS. These studies were augmented by analysis of mRNA, miRNA, and proteomic profiles from several GRMD muscles at two different ages to elucidate additional hypertrophic pathways. Although UTRN was also uniformly increased in GRMD muscles, there was no association with CS size. Other membrane-associated proteins, including α-dystroglycan, like-acetylglucosaminyltransferase (LARGE), and β-spectrin, were increased in the GRMD CS, consistent with a role in membrane stabilization. These results indicate that several muscle proteins may act together to stabilize myofibers and promote muscle growth. Our findings also further substantiate that differential muscle involvement can exaggerate the GRMD phenotype. This suggests that care should be taken with treatments targeting specific pathways, such as MSTN, that could selectively exaggerate muscle hypertrophy.
Materials and Methods
Overall Experimental Approach
Two different data sets were collected. Functional information (Figure 1) was analyzed on a cohort of GRMD (n = 49) and normal (n = 4) dogs at 6 months of age, creating a heat map that draws associations among a set of functional endpoints. From the dogs included in the heat map, we selected eight GRMD dogs with variable phenotypes, as well as the four normal dogs, for further analysis. Histopathological lesions and myofiber diameter, molecular signatures [mRNA, microarray profiling, and quantitative real-time PCR (qPCR)], and protein expression patterns (proteomic profiling, Western blot analysis, and immunofluorescence microscopy) were assessed in archived samples from three different muscles at both 4 to 9 weeks and 6 months of age. (A flowchart of the experimental design is shown in Supplemental Figure S1.) Four dogs used for mRNA profiling and five other age-matched phenotyped dogs, but not mRNA profiled, were studied at the miRNA level. An additional six GRMD dogs from the heat map were analyzed by light microscopy for myofiber measurement at 4 to 9 weeks and by qPCR at 6 months of age to further confirm the MSTN mRNA findings.
Figure 1.

A schematic diagram of the canine pelvic limb that demonstrates functional outcome measures in golden retriever muscular dystrophy. The cranial sartorius (CS; line of diamonds) muscle is a hip flexor; the long digital extensor (dashed line) muscle is a tarsal joint flexor; the VL (dotted line) muscle is a stifle extensor. As CS circumference and tarsal joint flexor strength increase and tibiotarsal joint angle and extensor torque decrease, overall phenotype becomes more severe (see Figure 2). This study focused on the endpoint of CS circumference (hypertrophy).
Animals
All dogs were used and cared for according to principles outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Dogs were housed at the Kornegay laboratory either at the University of Missouri–Columbia or University of North Carolina at Chapel Hill. GRMD dogs were identified at 1 day of age based on dramatic elevation of serum creatine kinase (CK) levels.5 Genotype was confirmed by PCR when CK results were ambiguous. Characteristic clinical signs subsequently developed.
Functional Outcome Measures
The phenotypic variables assessed in these dogs included CS muscle circumference, tibiotarsal joint angle, and tibiotarsal joint tetanic extensor and flexor isometric torque (Figure 1 and Supplemental Table S1). The major flexors of the tibiotarsal joint are the cranial tibialis, peroneus longus, and long digital extensor (LDE). The LDE, as the name indicates, also serves as a digital extensor. In our previous studies, we found the LDE to be significantly atrophied in GRMD compared to normal dogs. Accordingly, it has been used as a comparator for the hypertrophied CS. However, for the sake of drawing inferences between tibiotarsal joint flexion and the molecular analysis in this study, either the cranial tibialis or peroneus longus might have been a better choice.
Methods for measuring tibiotarsal joint angle and torque have been described.7,8 To measure CS circumference, the muscle was isolated at the time of surgical biopsy. Nylon suture was placed around the muscle at approximately the midsection and tightened to snugly encircle the muscle belly. The two ends of the suture were secured with a pair of hemostats and then cut on the muscle side of the hemostat. The length in mm was divided by body weight in kg (mm/kg). An average of two measurements was recorded.20
Muscle Biopsies
Biopsy samples from the CS, LDE, and VL muscles were collected surgically, snap frozen in liquid nitrogen–cooled SUVA34A (a Freon analog; DuPont Fluorochemicals, Wilmington, DE), and subsequently archived at −80°C until analysis.
RNA Extraction
Total RNA was extracted from approximately 30 mg of flash-frozen muscle using TRIzol (Invitrogen, Carlsbad, CA). The extraction procedure was performed as recommended by the manufacturer. Total RNA was stored at −80°C until analysis.
mRNA Expression Profiling
Genome-wide mRNA analysis was performed on RNA from the CS, LDE, and VL biopsy samples using microarrays (Affymetrix Canine 2.0 GeneChip; Affymetrix, Santa Clara, CA); 72 chips were used for the analysis [12 dogs (N1 to N4 and D6, D11, D20, D21, D38, D40, D43, and D49) (Supplemental Table S1), two time points, three muscles]. Single-stranded cDNA, followed by double-stranded cDNA, and finally biotinylated complimentary (c)RNA were generated in a cDNA/cRNA reaction.27 Biotinylated cRNA was fragmented into 50- to 200-bp segments and subsequently hybridized to the oligonucleotides in the Canine 2.0 GeneChip. Hybridization was performed with an Affymetrix GeneChip 640 Hybridization Oven for 16 hours at 45°C (3200 × g). The fluorescence of hybridized biotinylated cRNA was washed and stained using the Affymetrix GeneChip 450 Fluidics Station. Probe-set fluorescence was detected and averaged to determine the relative abundance of the original mRNA by an Affymetrix GeneChip 3000 Scanner. We focused on the CS mRNA profiles for this study.
miRNA Expression Profiling
Genome-wide miRNA analysis was performed on total RNA from the CS and VL samples using microarrays (Affymetrix miRNA 3.0 GeneChip); 12 chips were used for the analysis (12 canine samples from a total of 9 dogs: 3 GRMD CS, 3 normal CS, 3 GRMD VL, and 3 normal VL muscles; at one time point 7.5 to 8 months). Four of these dogs were the same that were used for mRNA profiling above (N2, N4, D21, and D38), whereas five were different (two normal dogs, Charles and Baker; and three GRMD, Caroline, Flash, and Hagatha), not mRNA profiled or included in the heat map. The FlashTag Biotin HSR RNA Labeling Kit protocol was performed as recommended by the manufacturer (Affymetrix). Briefly, total RNA was poly-A tailed and then ligated with biotin HSR. RNA labeling was confirmed with an enzyme-linked oligosorbent quality control assay. Hybridization was performed with an Affymetrix GeneChip 640 Hybridization Oven for 16 hours at 48°C (3200 × g). Hybridized biotinylated miRNA was washed and stained using the Affymetrix GeneChip 450 Fluidics Station. Probe-set fluorescence was detected and averaged to determine the relative abundance of the original mRNA by an Affymetrix GeneChip 3000 Scanner.
Bioinformatics
The relative probe-set hybridization signal of the mRNA profiles was interpreted by PLIER algorithm and visualized using MATLAB hierarchical clustering (MathWorks, Natick, MA). Normalization of miRNA profiles was performed by Partek (St. Louis, MO). Ingenuity Pathway Analysis (IPA) version 16542223 (Ingenuity Systems, Redwood City, CA) was used to determine significant molecular networks within CS muscle mRNA profiles.
cDNA Synthesis
Generation of cDNA was performed using an Applied Biosystems’ High Capacity cDNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA). Samples were heated to 25°C for 10 minutes followed by 37°C for 2 hours on a heating plate. qPCR reaction parameters using Applied Biosystems Universal PCR TaqMan Master Mix (Life Technologies) were as follows: stage 1: 50°C for 2 minutes; stage 2: 95°C for 10 minutes; stage 3 repeated for 40 to 45 cycles: 95°C for 15 seconds, followed by 60°C for 1 minute.
qPCR
Triplicate reactions were performed with an Applied Biosystems 7900 HT Fast Real-Time PCR system (Life Technologies) for the eight arrayed GRMD dogs. MSTN was assessed in an additional six GRMD dogs (D14, D23, D28, D36, Athena, and Sprite). The endogenous control, hypoxanthine guanine phosphoribosyltransferase 1 (HPRT1), was selected due to its consistent value in all expression profiles. Probes for DAG1, LARGE, MSTN, and HPRT1 were selected from Applied Biosystems (Life Technologies) and were used as sense/antisense primers (DAG1 assay ID: Cf03023355_m1; LARGE assay ID: Cf02642045_m1; HPRT1 assay ID: Cf02626258_m1; and MSTN assay ID: Cf02704228_m1). All quantitative real-time (q)PCR results were normalized to control samples.
Immunofluorescence and Light Microscopy
Samples from the CS, LDE, and VL muscles of the eight GRMD and four normal dogs were evaluated at 6 months for immunofluorescence of target proteins and histopathological changes. Due to the small size of the muscle biopsy at 4 to 9 weeks, five of eight GRMD samples for the CS, LDE, and VL were completely used during the RNA extraction procedure, leaving samples from only three GRMD dogs for evaluation. Frozen sections of skeletal muscle were thawed on Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA) using a Leica Microsystems Cryostat (Leica, Bannockburn, IL) at −25°C. Immunostaining and H&E staining were completed on 6- to 8-μm and 10-μm sections, respectively. Minimal Feret’s diameter (ImageJ version 1.46r; NIH, Bethesda, MD) method was used to measure myofiber diameter in H&E sections.28 The entire field was analyzed, and 200 to 1000 myofibers per sample were measured. The myofiber membrane was outlined and minimal Feret’s diameter calculated with ImageJ.
α-Dystroglycan mouse monoclonal IgM antibody (IIH6) was from the Kevin P. Campbell laboratory (Iowa City, IA). UTRN mouse monoclonal IgG antibody (MANCHO3 clone 8A4) was purchased from Developmental Studies Hybridoma Bank (Iowa City, IA). β-Sarcoglycan mouse monoclonal IgG antibody (B-SARC; Novocastra) was purchased from Leica Biosystems (Buffalo Grove, IL). α-, δ-, and γ-Sarcoglycan were probed with similar Novocastra antibodies, but no detectable cross-reaction was visible in canine muscle. β-Spectrin 1 protein (SPTBN1; non-erythrocytic) mouse monoclonal IgG antibody (B-SPEC; Novocastra) was from Leica Biosystems. ARP52859_P050 was used to probe MTPN (Aviva Systems Biology, San Diego, CA). No detectable immunoreaction occurred when dog muscle was probed with α-spectrin antibodies from Leica Biosystems. Secondary antibodies were Cy3 conjugates from Jackson ImmunoResearch Laboratories (West Grove, PA). Primary and secondary antibodies were diluted according to the manufacturer’s recommendations. Sections were incubated with blocking buffer (10% horse serum and 1× PBS) for 1 hour. Sectioned samples were then incubated with the primary antibody for 16 to 24 hours and with the secondary antibody for 1 hour. Cryosections were washed with blocking buffer three times for 5 minutes each before and after applying primary and secondary antibodies. Samples were analyzed in triplicate with an Axiovert 200 M (immunofluorescence) microscope (Carl Zeiss Microscopy, Jena, Germany) or an Olympus light microscope (light microscopy) (Olympus, Tokyo, Japan).
Western Blot Analysis
Muscle proteins were isolated in radioimmunoprecipitation assay buffer, proteinase inhibitor (Bio-Rad, Hercules, CA), and phosphatase inhibitor (Roche Diagnostics, Mannheim, Germany) and quantified with DC protein assay (Bio-Rad) at 650 nm. Proteins (10 μg or 50 μg) were dissolved in 4× lithium dodecyl sulfate running buffer (Invitrogen) and 10× reducing agent (Invitrogen) and loaded into an SDS NU-PAGE gel (4% to 12% Bis-Tris or 3% to 8% Tris acetate gel) (Invitrogen) and subsequently separated via electrophoresis at 160 V for 1 hour. Proteins were transferred to a polyvinylidene difluoride membrane by a semidry transfer method (Bio-Rad Trans-Blot SD Semi-Dry Electrophoretic Transfer Cell) and incubated in blocking buffer (5% milk in 1× Tris-buffered saline–Tween) for 16 to 24 hours at 4°C. The membrane was subsequently incubated with primary antibody (dilution: 1:1000) [IIH6 for α-dystroglycan; MANCHO3 clone 8A4 for UTRN; ARP52859_P050 for MTPN; 587F11 for phosphorylated-AKT1 mouse monoclonal IgG antibody (587F11; Cell Signaling Technology, Danvers, MA)] for 18 to 24 hours at 4°C and horseradish peroxidase–conjugated secondary antibody (dilution 1:5000) for 1 hour at room temperature. The B-SARC and B-SPEC antibodies from Leica Biosystems used for immunofluorescence did not produce a detectable signal for immunoblots in dog samples. Membranes were washed three times for 5 minutes each with 1× Tris-buffered saline–Tween after primary and secondary antibody incubation. The membranes were incubated with ECL Plus (GE Healthcare, Cleveland OH) for 1 minute, then subsequently exposed to a radiographic film and developed. Protein gels were stained with Bio-Safe Coomassie Blue (Bio-Rad) for 10 minutes and washed with water overnight at 4°C. Blot and gel samples were quantified using a GS-800 Calibrated Densitometer (Bio-Rad) with Quantity One software version 4.5.2 (Bio-Rad). Membrane bands were normalized to myosin heavy chain from the SDS NU-PAGE gel.
Proteomic Profiling
Label-free quantitative mass spectrometry was performed in CS samples from three each GRMD and normal dogs at age 4 to 9 weeks and 6 months. These included four dogs (N1, N3, N4, and D40) from those previously mRNA arrayed and two that were not arrayed (D23 and D36). CS muscles (50 10-μm sections) were cut on the cryostat and placed in a microcentrifuge tube at −80°C. Protein was isolated and quantified, as detailed in Western Blot Analysis. Protein (50 μm) was dissolved and run electrophoretically on a 4% to 12% Bis-Tris gel (Invitrogen), and gels were stained for 1 hour. Individual protein bands were sliced and prepared for in-gel digestion.29 Resulting peptides from each band were analyzed using NanoLC (Eksigent, Dublin, CA) connected to an LTQ Orbitrap XL instrument (ThermoFisher Scientific, Waltham, MA) using a label-free method.30 Proteins were identified using the SEQUEST algorithm in the BioWorks Browser version 3.3.1 SP1 (ThermoFisher Scientific) against the National Center for Biotechnology Information genome biology canine RNA database. The SEQUEST search files were uploaded into ProteoIQ software version 2.3.07 (Nusep, Bogart, GA) for parsing and spectral counting of two or more independent peptides mapping to the same parent protein. Proteins were retained for further analyses if both peptides XCorr >2.2, peptide probability >0.98, and protein probability >0.95. Total spectra counts for each group were used to estimate relative abundance. Proteins of interest were identified and molecular functions were determined based on data in Online Mendelian Inheritance of Man (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/omim, last accessed March 6, 2012).
Statistical Analysis
Prism software version 5.04 (GraphPad Software, La Jolla, CA) was used for statistical testing. A correlation coefficient was used to correlate the mRNA and protein profiles with CS circumference at 6 months of age. Spearman correlation was used to test significance between expression and phenotype values. A nonparametric t-test was used to test significance between GRMD and normal dog expression values. Paired t-test was used to test significance of GRMD samples between one another.
Results
Associations of Phenotypic Measures in GRMD Dogs at 6 Months
The values for four phenotypic measures are provided in Supplemental Table S1. Measures were visualized using hierarchical clustering (heat map), where the color green indicates a relatively lower value for each variable and red a higher value (Figure 2). Normal dogs had larger tibiotarsal joint angles, tetanic extensor and flexor torques, and smaller CS circumferences. The 49 GRMD dogs showed marked variability in each measure. Mildly affected dogs had proportionally larger tibiotarsal joint angles and tetanic extensor torques and smaller CS circumferences and tetanic flexor torques. The opposite pattern was seen in severely affected dogs (Figure 2).
Figure 2.

Heat map of unsupervised hierarchical clustering showed that increased cranial sartorius (CS) circumference, increased flexor, and decreased extensor tetanic torque, and decreased tibiotarsal joint angle were associated with a more severe phenotype. More severely affected dogs had proportionally larger CS circumferences (hypertrophy), stronger tetanic flexor torques, reduced tibiotarsal joint angles (contractures), and weaker tetanic extensor torques at 6 months. Mildly affected dogs had a reverse pattern of involvement. The eight golden retriever muscular dystrophy (D) and four normal (N) dogs selected for histopathological and molecular analyses are identified with black areas: golden retriever muscular dystrophy dogs (left to right): D6, D11, D20, D21, D38, D40, D43, and D49; normal (W/T) dogs (left to right): N1, N2, N3, and N4. Red, increased; green, decreased.
Myofiber Hypertrophy Is Seen in the GRMD CS Muscle at 6 Months
Histopathological changes in eight GRMD dogs with mild-to-severe phenotypes were contrasted with one another and four age-matched normal controls (see Figure 2 for dogs selected). Three muscles (CS, VL, and LDE) were assessed in three of the eight GRMD dogs at 4 to 9 weeks and in all eight at 6 months of age. Muscles in the four normal dogs were assessed at both ages. Muscles from age-matched controls were histologically normal (Figure 3, A, C, and E). All GRMD muscles had lesions of necrotizing myopathy. These changes varied depending on the age and muscle. At 4 to 9 weeks, each of the GRMD muscles had multifocal clusters of necrotic myofibers, mononuclear cell inflammation, expansion of the perimysial and endomysial spaces, and groups of small, basophilic regenerating myofibers. Changes were generally more pronounced in the CS compared to the VL or LDE at this younger age. By 6 months, lesions of active necrosis had largely resolved in the CS (Figure 3B), whereas the VL and LDE still showed small group myofiber necrosis and regeneration (Figure 3, D and F). The minimal Feret’s diameter (means ± SD) of GRMD CS myofibers at 4 to 9 weeks (14.7 ± 5.1 μm) and 6 months (61.1 ± 16.7 μm) was larger relative to those from age-matched normal littermates (9.3 ± 3 μm at 4 to 9 weeks; P < 0.05; and 36.7 ± 6.2 μm at 6 months; P < 0.001), as well as the GRMD LDE (10 ± 3.0 μm at 4 to 9 weeks; P = 0.06; and 41.4 ± 18.3 μm at 6 months; P < 0.01) and VL (7.6 ± 2.0 μm at 4 to 9 weeks; P < 0.05; and 39.7 ± 13.5 μm at 6 months; P < 0.01) muscles (Figure 4, A and B). These data indicate that GRMD CS hypertrophy begins as early as age 4 to 9 weeks when dogs are relatively mildly affected.
Figure 3.

Histopathological features of golden retriever muscular dystrophy (GRMD) and normal dogs at 6 months of age, showing relative myofiber hypertrophy and less pronounced acute dystrophic changes in the cranial sartorius (CS). Cranial sartorius muscle of a normal dog (N3; A) and a GRMD dog (D40; B). Note that the myofibers are larger compared to normal and other GRMD muscles. Endomysial connective tissue is increased, and there are increased numbers of central nuclei. Patchy increased cellularity associated with inflammation is present. Long digital extensor (LDE) muscle of a normal dog (N4) (C) and a GRMD dog (D11) (D) illustrating myofiber size variation, inflammatory cell infiltration, and increased endomysial connective tissue. Vastus lateralis (VL) muscle of a normal dog (N4) (E) and a GRMD dog (D43) (F). F: The arrows indicate hyaline fibers and associated connective tissue, inflammatory cells, and small regenerating basophilic myofibers. Original magnification, ×100 (A–F). Scale bars: 50 μm (A–F).
Figure 4.

Myofiber hypertrophy in golden retriever muscular dystrophy (GRMD) cranial sartorius (CS) and myostatin (MSTN) expression is inversely correlated with bodyweight-corrected CS circumference (mm/kg). A: Myofiber diameter is increased in GRMD (D) CS at 4 to 9 weeks compared to normal (N) and GRMD vastus lateralis (VL). GRMD CS tend to be greater than long digital extensor (LDE; P = 0.06). GRMD LDE and VL are not significantly different from respective normal samples. B: Myofiber diameter is increased in GRMD CS at 6 months compared to normal, GRMD LDE, and GRMD VL. GRMD LDE and VL are not significantly different from respective normal samples. C:MSTN qPCR expression revealed decreased expression in GRMD muscle, with the greatest decrease in the hypertrophied CS at 6 months (6M; n = 8 arrayed dogs). D:MSTN mRNA was inversely correlated with CS size at 6 months in the CS of GRMD dogs of the discovery data set (r = −0.73; r2 = 0.53; P < 0.05; n = 8 arrayed dogs). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
MSTN Is Associated with CS Muscle Hypertrophy
Because MSTN expression is strongly correlated with muscle size,24,25 we hypothesized that levels would be decreased in the hypertrophied CS muscle. The CS and VL, but not the LDE, showed strong age-related decreased MSTN mRNA expression. MSTN expression was most profoundly decreased in the hypertrophied CS at 6 months of age (average fold change = −25; P < 0.05; n = 8) (Figure 4C), at which time there was also an inverse correlation between qPCR levels and CS muscle circumference (r = −0.73; r2 = 0.53; P < 0.05; n = 8) (Figure 4D). We confirmed the size-related decrease in MSTN (average fold change = −6.5 compared to normal) in the CS and VL muscles compared to normal controls with qPCR in six additional GRMD dogs, four of which were included in the heat map (Athena, Sprite, D14, D23, D28, and D36; not shown).
miRNAs that Modulate MSTN Are Up-Regulated in the Hypertrophied CS
We were intrigued by this differential expression of MSTN in GRMD CS compared to the other dystrophic muscles and sought to define possible upstream modulators of MSTN. MSTN has a highly conserved 3′–untranslated region with multiple miRNA binding sites. We hypothesized that altered regulation of miRNAs could account for the observed differential expression of MSTN in GRMD muscle. Using Affymetrix GeneChip 3.0 miRNA profiles, we found increased target miRNAs predicted to regulate MSTN in the hypertrophied GRMD [miR-539 (fold change = +3; P < 0.05) and miR-208b (fold change = +2; P < 0.05)]. Moreover, GRMD CS showed greater increases in miR-539 and miR-208b compared to normal CS and VL and GRMD VL samples (Figure 5, A and B). These data suggest that differential regulation of upstream miRNAs targeting MSTN contributes to CS muscle hypertrophy.
Figure 5.

Regulators of myostatin are increased in the hypertrophied golden retriever muscular dystrophy (GRMD) cranial sartorius (CS) muscle. A: cfa-miR-208b_st was increased in GRMD (D) CS compared to normal (N) CS (fold change = +2). B: cfa-miR-539_st was increased in GRMD CS compared to normal CS (fold change = +3). C: AKT1 phosphorylation at serine 473 was increased in GRMD and positively correlated with CS circumference at 6 months (r = +0.78; r2 = 0.6; P < 0.05; n = 8). miRNA profiling was used to detect target miRNAs. Western blot and densitometry was used to detect AKT1 phosphorylation status. Numbers to the left of the membranes and gels indicate molecular weight (kDa). MHC, myosin heavy chain. ∗P < 0.05
AKT1 Phosphorylation Is Associated with Increased Muscle Size in the Hypertrophied CS in GRMD Dogs at 6 Months
MSTN has been shown to regulate muscle growth, at least in part, through regulation of AKT1. Inhibition of MSTN leads to an increase in myotube diameter and Akt phosphorylation.31 To query differential AKT1 phosphorylation status as an additional regulator of MSTN, we defined the phosphorylation status of AKT1 in the different muscles in the dogs by Western blot. AKT1 phosphorylation (normalized to myosin heavy chain) was increased in the hypertrophied CS muscle (Figure 5C) and positively correlated with CS circumference at 6 months (r = +0.78; r2 = 0.6; P < 0.05; n = 7) (Figure 5C). AKT1 phosphorylation remained unchanged between normal and GRMD LDE samples (data not shown), suggesting that the MSTN/AKT1 pathway changes were specific to the hypertrophied CS muscles.
Thus, two regulators of MSTN, miRNAs and AKT1 phosphorylation status, are consistent with active down-regulation of MSTN in the CS. The resulting hypertrophy illustrates a muscle-specific response to dystrophin deficiency.
UTRN Increases in GRMD Muscles But Is Not Specific to the CS and Does Not Correlate with CS Size
We hypothesized that the autosomal homologue of dystrophin, UTRN, would be increased at the myofiber membrane of GRMD CS and that its expression would correlate with CS hypertrophy. Immunofluorescent UTRN expression was faint with microscopy, but more pronounced in all three GRMD versus normal muscles at both time points (not shown). On Western blots, UTRN was also increased in each of the three GRMD muscles (CS, 190%; LDE, 167%; and VL, 197% of normal levels) at 6 months (Figure 6). However, UTRN protein expression was not significantly correlated with CS size in GRMD dogs (data not shown).
Figure 6.

Western blot shows increased utrophin (UTRN) expression in golden retriever muscular dystrophy (GRMD) cranial sartorius (CS) muscle at 6 months. In GRMD (D) CS, UTRN was expressed at 190% of the level of normal dogs (N) at 6 months. Levels in the long digital extensor and vastus lateralis were at 167% and 197%, respectively, of age-matched normal samples (not shown). Numbers to the left of the membranes and gels indicate molecular weight (kDa). MHC, myosin heavy chain.
Novel Molecular Networks Are Quantitatively Associated with Variable CS Muscle Hypertrophy
We sought to find novel molecular signatures associated with muscle hypertrophy in response to dystrophin deficiency. mRNA microarray profiles were performed on the CS, LDE, and VL muscles from eight GRMD dogs and four normal dogs from the heat map at both 4 to 9 weeks and 6 months. Using quantitative trait analyses, we queried for mRNAs that were strongly correlated with the degree of CS hypertrophy at 6 months, using a rigorous threshold for correlation coefficients (r ≥ ±0.94; r2 ≥ 0.88) and stringent P values associated with those r values (P ≤ 0.001). Two hundred fifty transcripts met these correlation coefficient and P value criteria, and were studied further (Supplemental Table S2).
The 250 transcripts that were associated with CS hypertrophy were entered into IPA software to determine the most significant molecular networks associated with muscle hypertrophy. The top-ranked pathway included genes coding for two dystrophin-associated mRNAs, namely DAG1 (highest correlation with CS hypertrophy; P = 0.00009; r = 0.98) and LARGE enzyme responsible for O-linked glycosylation of DAG1 (fourth lowest correlated transcript; P = 0.001; r = 0.94) (Figure 7).32 We have previously shown that levels of the protein product of DAG1, α-dystroglycan, in the GRMD peroneus longus muscle correlate with contraction tension and CK levels, suggesting that it could play a role in stabilizing the myofiber membrane.33 We chose to focus on validation of these two transcripts due to our previous results and their close relationship with the dystrophin–glycoprotein complex. A brief description of the other transcripts in the highest-ranked IPA network from Figure 7 can be found in Supplemental Table S3.
Figure 7.

DAG1 and LARGE are in the top-ranked network associated with cranial sartorius hypertrophy. Transcripts associated with cranial sartorius size (n = 250) were entered into ingenuity pathway analysis (IPA), and ranked networks were generated. We focused on DAG1 and LARGE (black arrows) in the top-ranked network (shown) due to their association with the dystrophin-glycoprotein complex.
DAG1 and LARGE mRNAs Are Up-Regulated in all Dystrophic Muscles
To determine whether increased DAG1 and LARGE mRNAs were specific to the hypertrophied CS, we queried the probe sets for these candidates in all 72 microarrays and also performed qPCR to validate the microarray findings. This analysis showed that the DAG1 and LARGE mRNAs were most up-regulated in CS at both 4 to 9 weeks and 6 months of age (Table 1). DAG1 and LARGE qPCR expression values correlated directly with CS circumference at 6 months (Supplemental Figure S2), validating the microarray data. Also similar to the microarray data, qPCR analysis of all 72 GRMD muscles for DAG1 and LARGE showed up-regulation compared to normal, but reached significance only for DAG1 mRNA in GRMD CS at 6 months (P ≤ 0.05) and the VL at 4 to 9 weeks (P ≤ 0.05) (Table 1).
Table 1.
Array and qPCR Values for DAG1 and LARGE in GRMD and Normal Muscle
| Transcript | Muscle | Normal array |
GRMD array |
GRMD qPCR |
|||
|---|---|---|---|---|---|---|---|
| 4–9 weeks | 6 months | 4–9 weeks | 6 months | 4–9 weeks | 6 months | ||
| DAG1 | CS | 439 ± 51 (3) | 220 ± 28 (3)∗ | 664 ± 93 (8) | 562 ± 51 (8)∗ | +1.3 ± 0.21 (8) | +1.6 ± 0.11 (8)∗ |
| DAG1 | LDE | 522 ± 59 (4) | 370 ± 29 (4) | 459 ± 43 (8)†‡ | 418 ± 42 (8)†‡ | +1.27 ± 0.1 (8) | +1.27 ± 0.18 (8) |
| DAG1 | VL | 548 ± 84 (4) | 355 ± 51 (4) | 643 ± 40 (8)‡ | 417 ± 37 (8)‡ | +1.7 ± 0.28 (8) | +1.8 ± 0.47 (8) |
| LARGE | CS | 146 ± 19 (4) | 120 ± 11 (4) | 121 ± 6 (8)†‡ | 198 ± 20 (8)†‡ | +1.4 ± 0.22 (8) | +1.4 ± 0.15 (8) |
| LARGE | LDE | 72 ± 10 (4) | 107 ± 4 (4) | 87 ± 8 (8)†‡ | 100 ± 3 (8)†‡ | +1 ± 0.09 (8)‡ | +1.29 ± 0.25 (8)‡ |
| LARGE | VL | 98 ± 9 (4) | 109 ± 11 (4)∗ | 105 ± 6 (8) | 131 ± 12 (8)∗ | +1.4 ± 0.09 (8)∗§ | +1.7 ± 0.36 (8) |
Array values are reported as expression units. qPCR values are reported as fold change relative to normal. All values are means ± SEM (n).
P < 0.05 versus age/muscle-matched normal.
P < 0.05 versus age (4 to 9 weeks versus 6 months) within the same disease (normal or GRMD) and muscle (CS, LDE, or VL) group.
P < 0.05 versus muscle (CS versus LDE or VL) within the same genotype (normal or GRMD) and age (4 to 9 weeks or 6 months) group.
P < 0.05 versus muscle (LDE versus VL) within same genotype (normal or GRMD) and age (4 to 9 weeks or 6 months) group.
Glycosylated α-Dystroglycan Protein Decreases in GRMD Muscles Compared to Normal But Proportionally Greater in the GRMD CS
Glycosylated α-dystroglycan protein was markedly reduced compared to normal but present on immunostaining in all GRMD muscles at both ages. α-Dystroglycan expression was most pronounced in the CS at 6 months (Figure 8B), where immunoblot levels were at 33% of normal (not shown). The degree of expression did not correlate with the degree of CS hypertrophy, although this analysis was highly subjective. Despite multiple attempts, we could not produce interpretable immunoblots for glycosylated α-dystroglycan protein in the LDE and VL muscles. By comparison, β-sarcoglycan protein immunostaining in myofibers was uniform in normal dogs (Figure 8D) but reduced and patchy among the three GRMD muscles, regardless of age (Figure 8, E and F).
Figure 8.

Immunofluorescence microscopy of α-dystroglycan (DAG1), β-sarcoglycan (SGCB), and β-spectrin (SPTBN1) reveals variable expression in golden retriever muscular dystrophy (GRMD) muscles at 6 months. A: In normal muscle, DAG1 is expressed at the membrane in every myofiber observed. B and C: DAG1 is variably expressed at the membrane in GRMD cranial sartorius (CS; B), long digital extensor (LDE), and vastus lateralis (VL) muscles (C), but appears to have greater intensity in the CS. D: SGCB is expressed at the membrane of every myofiber in normal dogs. E and F: In GRMD dogs, SGCB is expressed at the membrane of myofibers in the CS (E), LDE, and VL (F), but less than normal samples. No differences in intensity are seen among the three GRMD muscles. G: SPTBN1 is expressed at the myofiber membrane in all normal muscles. H and I: The intensity of expression in the GRMD CS (H) is greater than normal and other affected muscles (I). J: Myotrophin (MTPN) is expressed in the perimembranous region of myofibers in normal muscle. K: GRMD CS muscle shows perimembranous staining. L: GRMD VL samples at 6 months showed slightly increased perimembranous myofiber MTPN. Original magnification, ×100 (A–L). Scale bars: 50 μm (A–L).
Spectrin Increases in Hypertrophied CS Muscles of GRMD Dogs
Both α- and β-spectrin spectral counts were increased on proteomic profiling (mass spectrometry) in GRMD CS compared to normal at 4 to 9 weeks and 6 months (Table 2). By immunostaining, β-spectrin was uniformly expressed at the myofiber membrane in both normal (Figure 8G) and GRMD muscles, with greater intensity in the CS (Figure 8H) compared to other GRMD muscles (Figure 8I). By contrast, laminin-α2 protein expression, as detected by proteomic profiling, was reduced at 4 to 9 weeks and absent at 6 months (Table 2). However, immunostaining of laminin-α2 in normal and GRMD muscle samples showed strong and similar staining, suggesting that the reductions seen by proteomics were due to lack of sensitivity of this technique (data not shown).
Table 2.
Total Spectral Counts for Spectrin, Myotrophin, and Laminin-α2 from Proteomic Profiling Results
| Genotype | Age | SPTAN1 | SPTBN1 | MTPN | LAMA2 |
|---|---|---|---|---|---|
| Normal | 4–9 weeks | 43 | 7 | 7 | 54 |
| 6 months | 36 | 1 | 5 | 3 | |
| GRMD | 4–9 weeks | 65 | 19 | 12 | 12 |
| 6 months | 65 | 3 | 15 | 0 |
Sum of spectral counts for each group is shown. There are n = 3 profiles for normal and GRMD at 4 to 9 weeks and at 6 months (12 total profiles).
LAMA2, laminin-α2; MTPN, myotrophin; SPTAN1, α-spectrin; SPTBN1, β-spectrin.
Myotrophin Increases in Hypertrophied CS But Not in Other Muscles
In assessing proteomic profiling data for other growth factors that could contribute to CS hypertrophy, we were struck by increased total spectral counts for MTPN at both 4 to 9 weeks and 6 months (Table 2). Levels of MTPN were also increased on immunoblot analysis at 4 to 9 weeks and 6 months in the CS GRMD samples profiled with both proteomics (Figure 9A) and mRNA microarrays (Figure 9B). No immunoblot signal for MTPN was seen in normal or GRMD LDE or VL muscles (Figure 9B). There was faint perimembranous, but no cytoplasmic, myofiber staining for MTPN, in all three muscles of both normal and GRMD dogs (Figure 8, J and L). Additional perimembranous staining of MTPN was also seen in small GRMD myofibers at 4 to 9 weeks and hypertrophied fibers at 6 months, consistent with a unique expression pattern (Figure 8K).
Figure 9.
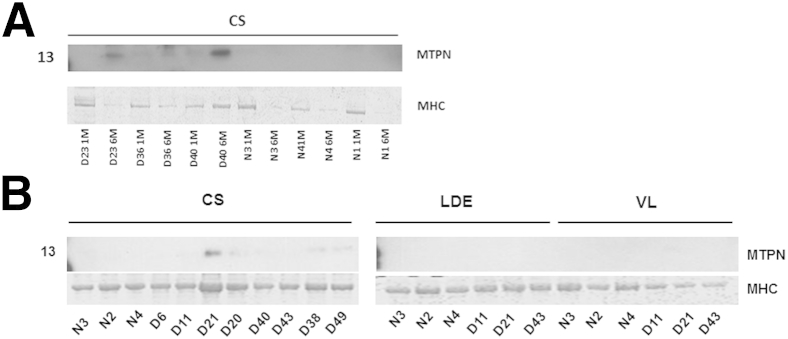
Myotrophin (MTPN) is increased on immunoblots of the cranial sartorius (CS) at 4 to 9 weeks and 6 months in golden retriever muscular dystrophy (GRMD) dogs. A: MTPN is increased on immunoblots of GRMD (D) CS muscles assessed by proteomics. B: MTPN is increased in GRMD CS muscles on immunoblots of arrayed dogs, but not expressed in normal (N) samples or GRMD long digital extensor (LDE) or vastus lateralis (VL) muscles. Numbers to the left of the membranes and gels indicate molecular weight (kDa). MHC, myosin heavy chain.
Discussion
Mechanisms responsible for relative sparing or hypertrophy of dystrophin-deficient muscles are poorly defined. The GRMD CS muscle, which undergoes early necrosis followed by remarkable hypertrophy,9,19 provides a platform on which factors contributing to hypertrophy can be explored. Studies reported here tested the hypothesis that specific age-related molecular signatures support growth and hypertrophy of this muscle. To identify a cohort of GRMD dogs to be included in molecular studies, unsupervised hierarchical clustering of functional outcome measures was first completed in 49 6-month-old GRMD dogs. Consistent with our earlier studies,19,34,35 there was dramatic phenotypic variation. Mildly affected dogs had proportionally smaller CS muscles, lower tetanic flexor torque, and larger tibiotarsal joint angles and tetanic extensor torque. The opposite pattern was seen in severely affected dogs. Variably affected GRMD dogs from the larger cohort were assessed at the mRNA and protein level to identify molecular fingerprints that could explain differential muscle involvement, specifically CS hypertrophy.
In keeping with previous studies,9,19,20 histopathological analysis of GRMD muscles showed lesions of necrotizing myopathy that varied depending on the age and muscle. Changes were generally more pronounced in the CS compared to the VL or LDE at the younger (4 to 9 week) age. There was expansion of the perimysial and endomysial spaces in all muscles at both time points. Lesions of active necrosis had largely resolved in the CS at 6 months, whereas the VL and LDE still showed small group myofiber necrosis and regeneration. We had previously shown that the mean lesser (Feret’s) diameter of CS myofibers is normal at 4 weeks19 but increased at 6 months.9 This work was extended by measurement of myofiber diameter in all three muscles at 4 to 9 weeks and 6 months in the dogs in this study. CS myofiber diameter was increased at both ages, suggesting that molecular players are involved early in muscle hypertrophy.
We were particularly interested in factors that were either involved in regulation of muscle mass or could potentially serve as dystrophin surrogates to stabilize the myofiber membrane. Studies initially focused on MSTN because of its well-established role as a potent negative growth regulator of skeletal muscle mass.24,25 As reported previously in DMD36 and the mdx mouse,37 we hypothesized that MSTN would be decreased in GRMD muscle proportional to the degree of hypertrophy. This hypothesis was supported by results of qPCR, because MSTN levels in GRMD muscles, and especially the hypertrophied CS at 6 months, were lower than normal. Moreover, MSTN levels correlated inversely with CS circumference at 6 months, in keeping with an active role in muscle hypertrophy. Although our prior studies and the myofiber measurements made here suggest that the CS undergoes true hypertrophy, connective tissue is increased at 6 months and even more so at later ages.9 Previous studies have shown that MSTN can increase myofibroblast proliferation and inhibition of MSTN decreases fibrosis in dystrophic muscle.38 Interestingly, although GRMD MSTN mRNA decreased proportionally to normal levels in the CS and VL at the two ages studied, levels in the LDE were stable, perhaps contributing to the severe wasting that has been documented in this muscle.9
To determine the molecular pathways mediating the differential expression of MSTN, we performed studies of miRNAs targeting MSTN mRNA 3′–untranslated region and AKT1 phosphorylation status. miRNA profiling of two miRNAs, miR-208b and miR-539, predicted to down-regulate MSTN, showed two to three times higher levels in GRMD CS relative to normal CS and VL or GRMD VL, consistent with a role of these miRNAs in MSTN down-regulation and associated CS hypertrophy. The serine threonine kinase AKT1 was also assessed. It has previously been shown that MSTN knockout leads to increased phosphorylation at AKT serine 473 and myotube hypertrophy.31 Therefore, we hypothesized that AKT1 could mediate hypertrophy in CS muscle. AKT1 phosphorylation on Western blot analysis correlated with CS muscle size in GRMD dogs at 6 months, suggesting a relationship between AKT1 and muscle hypertrophy. By contrast, a previous study showed that AKT1 and AKT2 knockout mice, in which MSTN was blocked with an activin receptor IIB fusion protein, still had increased muscle growth and function.39 These results suggested that AKT1 is not essential for activin receptor IIB–mediated inhibition of muscle size and function.39 On the contrary, our data showed a strong correlation between AKT phosphorylation and GRMD CS muscle size. Consistent with these results, AKT1 phosphorylation status in the LDE muscle, which atrophies to one-half normal size in GRMD, did not differ in GRMD and normal samples. Our data suggest that AKT1 and MSTN are important myogenic factors, along with others, responsible for muscle-specific responses to dystrophin deficiency, such as CS hypertrophy.
Without the dystrophin–glycoprotein complex, the myofiber membrane is subject to tears, allowing entry of calcium, activation of proteases, and subsequent cell necrosis. Dystrophin homologs and other proteins could potentially stabilize the myofiber membrane in the absence of dystrophin. Although dystrophin was predictably uniformly absent on immunoblotting and immunofluorescence microscopy in all GRMD samples at both ages, the autosomal protein, UTRN, was increased. This was consistent with other studies showing that overexpression of UTRN ameliorated the dystrophic process in mdx mice40 and GRMD dogs.41 A lack of UTRN up-regulation was also offered as a potential explanation for the more severe cardiac phenotype in GRMD versus the mdx mouse.42 However, because UTRN was increased in all GRMD muscles studied here, and CS levels did not correlate with size at 6 months, it did not appear to play a specific role supporting hypertrophy.
To determine novel molecular pathways that could drive muscle hypertrophy or stabilize the muscle cell membrane, mRNA was analyzed in the GRMD CS muscle. We hypothesized that transcripts correlating with CS hypertrophy (r > 0.9, P < 0.001, n = 250 transcripts) could be involved in muscle hypertrophy. The top-ranked network in the IPA that correlated with CS muscle hypertrophy included two genes that encode DAG1 and its glycosyltransferase, LARGE, of particular interest because of their close relationship with dystrophin and associated membrane proteins. The DAG1 mRNA codes for a single propeptide that is subsequently cleaved to form α and β protein subunits. α-Dystroglycan is extracellular and binds to merosin α-2 laminin in the basement membrane, whereas β-dystroglycan is a transmembrane protein and binds to dystrophin. In the absence of dystrophin, the dystroglycans and other proteins of the dystrophin–glycoprotein complex should be decreased by up to 80% to 90% in dystrophic muscle.43,44 Indeed, we have previously shown that α-dystroglycan protein levels are reduced by approximately 75% in the GRMD peroneus longus muscle.33 However, this same study suggested that α-dystroglycan expression could stabilize the dystrophic membrane in that levels correlated with both contraction tension and CK values. Consistent with this finding, levels of DAG1 and LARGE mRNA both correlated with CS hypertrophy at 6 months of age, leading us to hypothesize that the protein product, α-dystroglycan, would be increased as well. Thereby, just as apparently occurs with the GRMD peroneus longus,33 α-dystroglycan could substitute for dystrophin, playing a role hypothesized for UTRN and other membrane-associated proteins, such as spectrin.45 To confirm this hypothesis, it was critical to demonstrate that α-dystroglycan had actually been translated from the mRNA; alternatively, the array data could just reflect a positive feedback loop whereby mRNA levels were increased subsequent to loss of the respective protein as the membrane underwent necrosis. Immunofluorescence microscopy and immunoblotting revealed α-dystroglycan expression in GRMD muscles, with 6-month levels being higher in the CS muscle at 33% normal. Unlike our mRNA findings, levels did not appear to correlate with CS circumference.
Another study found that overexpression of GalNAc transferase increased glycosylated α-dystroglycan at the myofiber membrane and lessened dystrophic lesions in mdx mice.46 In keeping with LARGE playing an active role in muscle hypertrophy, mRNA levels in the GRMD CS at 6 months were higher than those for normal CS or other GRMD muscles at this same age. Although these data suggest that LARGE may facilitate CS hypertrophy, further studies are needed to determine whether LARGE actually enhances glycosylation of α-dystroglycan in the GRMD CS muscle. A separate paper found that increased α- and β-dystroglycan in transgenic mdx mice did not ameliorate dystrophic lesions.47 But these mice also had less UTRN expression, leading the authors to suggest the transgenic dystroglycan isoform differed from the one binding UTRN at the myofiber membrane.47 Along these same lines, dystroglycan typically colocalizes with the sarcoglycan complex at the myofiber membrane, and its expression often parallels that of sarcoglycan. Similar to what occurs with α-dystroglycan, we found decreased but persisting expression of β-sarcoglycan in all GRMD versus normal muscles. Interestingly, mdx mice expressing sarcoglycans, including β-sarcoglycan, lived longer and had an improved phenotype compared to the double-mutant sarcoglycan/mdx mouse.48 Therefore, persisting expression of β-sarcoglycan in the GRMD dogs of this study could provide an analogous protective function.
Proteomic profiling via mass spectrometry was performed to identify other novel growth factors and dystrophin-associated proteins that could contribute to CS hypertrophy. Although the analysis was hindered by the lack of a mature protein database for dogs, we followed up on target proteins of interest. We chose to focus on MTPN, a 12-kDa growth factor that is expressed in small, regenerating myofibers of DMD,49 mdx skeletal muscle,49 and GRMD VL muscle.50 In the profiled dogs, MTPN was up-regulated in the CS at 4 to 9 weeks and even more so at 6 months. The specificity of such a response was not supported by another study showing increased MTPN on proteomic profiles of the VL muscle from 4-month-old GRMD dogs.50 This may be due to the increased sensitivity of mass spectrometry of VL samples performed in the previous study50 compared to Western blot of VL samples in our study.
On a similar note, we wanted to assess other proteins that could help support the cytoskeleton and membrane, and queried members of the dystrophin–glycoprotein complex in the proteomic profiles. Similar to the immunoblot and immunofluorescence studies, dystrophin was absent from GRMD dogs (not shown). Laminin-α2 protein expression might logically have been expected in the hypertrophied CS muscle, but levels were reduced at 4 to 9 weeks and absent at 6 months, consistent with disruption of the overall complex. Our failure to detect laminin may have occurred because of limitations of the LTQ Orbitrap XL instrument to detect low peptide levels. This was supported by the detection of laminin expression in the basal lamina of normal and GRMD samples by immunostaining (not shown). In addition, α-dystroglycan and laminin-α2 showed reductions in GRMD muscles by mass spectrometry, but these were likely due to lack of sensitivity of proteomic profiling for large, highly glycosylated proteins.51 We found increased levels of the α and β chains of spectrin in GRMD CS at 6 months and even more so at 4 to 9 weeks compared to normal. The higher levels at 4 to 9 weeks may have occurred because of the predominance of smaller myofibers at this age, with an associated increased membrane surface area. Immunostaining for β-spectrin was also identified in GRMD CS at 6 months, seemingly with greater intensity than the other dystrophic muscles, consistent with a role in membrane stabilization necessary for growth. Although these findings are intriguing, another recent study did not find differential spectrin expression in human dystrophic muscle and recommended its use as an internal control for dystrophin immunofluorescence.52 The differences in β-spectrin expression may have been due to the amount of hypertrophy seen in the CS in this study compared to the typical atrophy seen in DMD samples.52
The differential involvement of tibiotarsal joint flexors and extensors, as reflected by their force values, was discussed in a prior paper.8 Although this method does not directly assess function of the CS or VL muscles, flexor and extensor muscles tend to show similar patterns of disease. This is well represented by the analogous changes seen in α-dystroglycan levels of the GRMD peroneus longus33 and those of the CS reported here. Findings from the LDE must be interpreted in the context of its dual function as a tarsal (ankle) flexor and digital extensor. Despite an increase in flexor torque in GRMD dogs at 6 months, the LDE undergoes atrophy/wasting.9 Thus, changes in the LDE likely do not reflect the molecular basis for increased tarsal flexor force. We also saw differential expression patterns of the targeted genes/proteins (MSTN, α-dystroglycan, spectrin, and MTPN) between the CS and LDE muscles. Taken together, our results suggest that multiple factors contribute to CS hypertrophy in GRMD dogs. Perhaps, most importantly, the potent negative growth regulator MSTN was down-regulated and inversely correlated with CS circumference at 6 months of age. Moreover, increases of the growth factor, MTPN, were specific to the hypertrophied CS, perhaps providing another pathway for hypertrophy. We were also intrigued by dystrophin surrogates that could stabilize the muscle cell membrane. Although DAG1 and LARGE mRNAs and α-dystroglycan, β-sarcoglycan, and UTRN proteins were all proportionally increased in the GRMD CS, they were also variably expressed in the LDE and VL samples, even though these muscles typically atrophy. Early necrosis in the CS versus the more delayed onset of lesions in the VL and LDE may lead to different patterns of disease. We believe that the convergence of factors driving both differentiation and regeneration in muscles that undergo early necrosis can lead to a disordered response and selective hypertrophy. Coincident or subsequent up-regulation of additional dystrophin-associated proteins, including β-dystroglycan, sarcospan, syntrophins, and other sarcoglycans, could further stabilize myofiber membranes in dystrophin-deficient individuals and should be evaluated in GRMD.
mRNA, miRNA, and proteomic data were evaluated at 4 to 9 weeks and 6 months of age to take advantage of available functional data for correlation at 6 months. Due to a small number of samples left for histopathological/morphometric and molecular analyses, we could not fully correlate 4- to 9-week data with functional measures at 6 months. Given that necrosis occurs in the GRMD CS muscle within the first few days of life, it might be necessary to evaluate molecular data at even earlier time points to gain a full understanding of factors that drive muscle hypertrophy. This work should ideally be expanded to determine whether these proteins could be manipulated for therapeutic benefit. As an example, delivery of antisense oligonucleotides to inhibit the respective mRNA at a young age, with subsequent histopathological, morphometric, and molecular analysis of the CS muscle at 6 months, might elucidate underlying mechanisms. In addition, adeno-associated virus vector delivery of respective genes to the CS muscle of young GRMD dogs should be considered.
Acknowledgments
We thank Janet Bogan, Daniel Bogan, and Jennifer Dow for care of the dogs and data collection, Ramya Marathi for her technical assistance with proteomic profiling techniques, and Amanda Field and Mamta Giri for technical assistance with the miRNA profiling and analysis.
Footnotes
Supported by a neurology research fellowship from the American College of Veterinary Internal Medicine, a National Institute of Arthritis and Musculoskeletal and Skin Diseases Pediatric Extramural Loan Repayment Program award, and F32 National Research Service Award grant 1F32AR060703-01 (P.P.N.), a pilot grant from the NICHD National Center for Medical Rehabilitation Research (5R24HD050846 to E.P.H.), NINDS grant 1U24NS059696-01A1, the Muscular Dystrophy Association (E.P.H.), the Co-operative Program in Translational Research: Proposal for Establishment of the National Center for Canine Models of Duchenne Muscular Dystrophy, and a Muscular Dystrophy Association Infrastructure grant to the Translational Research Advisory Committee (J.N.K.).
Current address of P.M., Department of Human Genetics, Graduate School of Public Health, University of Pittsburg, Pittsburg, PA; of S.J.S., Veterinary Emergency and Specialty Center of Santa Fe, Santa Fe, NM.; of J.N.K., Department of Veterinary Integrative Biosciences, College of Veterinary Medicine, Texas A&M University, College Station, TX.
Supplemental Data
A flowchart showing the overall experimental approach for this study. Functional outcome testing was performed on a group of GRMD and normal dogs, followed by analysis of histopathological lesions, mRNA expression (microarray profiling and qPCR), and protein [proteomic profiling, immunofluorescence microscopy (IF), and Western blotting] expression. 6M, 6 months.
DAG1 and LARGE array and qPCR values correlated with golden retriever muscular dystrophy (GRMD) bodyweight-corrected cranial sartorius (CS) circumference (mm/kg) at 6 months. Note the strong correlation with CS size and LARGE (r2 = 0.86; r = 0.93; P = 0.0019) expression and, to a lesser extent, that of DAG1 (r2 = 0.56; r = 0.75; P = 0.05).
References
- 1.Hoffman E.P., Brown R.H., Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Sicinski P., Geng Y., Ryder-Cook A.S., Barnard E.A., Darlison M.G., Barnard P.J. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 3.Kornegay J.N., Tuler S.M., Miller D.M., Levesque D.C. Muscular dystrophy in a litter of golden retriever dogs. Muscle Nerve. 1988;11:1056–1064. doi: 10.1002/mus.880111008. [DOI] [PubMed] [Google Scholar]
- 4.Pastoret C., Sebille A. Mdx mice show progressive weakness and muscle deterioration with age. J Neurol Sci. 1995;129:97–105. doi: 10.1016/0022-510x(94)00276-t. [DOI] [PubMed] [Google Scholar]
- 5.Valentine B.A., Cooper B.J., de Lahunta A., O’Quinn R., Blue J.T. Canine X-linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: clinical studies. J Neurol Sci. 1988;88:69–81. doi: 10.1016/0022-510x(88)90206-7. [DOI] [PubMed] [Google Scholar]
- 6.Arechavala-Gomeza V., Kinali M., Feng L., Guglieri M., Edge G., Main M., Hunt D., Lehovsky J., Straub V., Bushby K., Sewry C.A., Morgan J.E., Muntoni F. Revertant fibres and dystrophin traces in Duchenne muscular dystrophy: implication for clinical trials. Neuromuscul Disord. 2010;20:295–301. doi: 10.1016/j.nmd.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Kornegay J.N., Sharp N.J., Schueler R.O., Betts C.W. Tarsal joint contracture in dogs with golden retriever muscular dystrophy. Lab Anim Sci. 1994;44:331–333. [PubMed] [Google Scholar]
- 8.Kornegay J.N., Bogan D.J., Bogan J.R., Childers M.K., Cundiff D.D., Petroski G.F., Schueler R.O. Contraction force generated by tarsal joint flexion and extension in dogs with golden retriever muscular dystrophy. J Neurol Sci. 1999;166:115–121. doi: 10.1016/s0022-510x(99)00118-5. [DOI] [PubMed] [Google Scholar]
- 9.Kornegay J.N., Cundiff D.D., Bogan D.J., Bogan J.R., Okamura C.S. The cranial sartorius muscle undergoes true hypertrophy in dogs with golden retriever muscular dystrophy. Neuromuscul Disord. 2003;13:493–500. doi: 10.1016/s0960-8966(03)00025-7. [DOI] [PubMed] [Google Scholar]
- 10.Valentine B.A., Cooper B.J. Canine X-linked muscular dystrophy: selective involvement of muscles in neonatal dogs. Neuromuscul Disord. 1991;1:31–38. doi: 10.1016/0960-8966(91)90040-y. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen F., Cherel Y., Guigand L., Goubault-Leroux I., Wyers M. Muscle lesions associated with dystrophin deficiency in neonatal golden retriever puppies. J Comp Path. 2002;126:100–108. doi: 10.1053/jcpa.2001.0526. [DOI] [PubMed] [Google Scholar]
- 12.Edwards R.H., Newham D.J., Jones D.A., Chapman S.J. Role of mechanical damage in the pathogenesis of proximal myopathy in man. Lancet. 1984;1:548–552. doi: 10.1016/s0140-6736(84)90941-3. [DOI] [PubMed] [Google Scholar]
- 13.Karpati G., Carpenter S. Small-caliber skeletal muscle fibers do not suffer deleterious consequences of dystrophic gene expression. Am J Med Genet. 1986;25(4):653–658. doi: 10.1002/ajmg.1320250407. [DOI] [PubMed] [Google Scholar]
- 14.Cros D., Harnden P., Pellissier J.F., Serratrice G. Muscle hypertrophy in Duchenne muscular dystrophy. A pathological and morphometric study. J Neurol. 1989;236:43–47. doi: 10.1007/BF00314217. [DOI] [PubMed] [Google Scholar]
- 15.Gowers W.R. Clinical lectures on pseudohypertrophic muscular paralysis. Lancet. 1879;2 1, 37, 73,113. [Google Scholar]
- 16.Reimers C.D., Schlotter B., Eicke B.M., Witt T.N. Calf enlargement in neuromuscular diseases: a quantitative ultrasound study in 350 patients and review of the literature. J Neurol Sci. 1996;143:46–56. doi: 10.1016/s0022-510x(96)00037-8. [DOI] [PubMed] [Google Scholar]
- 17.Coulton G.R., Curtin N.A., Morgan J.E., Partridge T.A. The mdx mouse skeletal muscle myopathy: II. Contractile properties. Neuropathol Appl Neurobiol. 1988;14:299–314. doi: 10.1111/j.1365-2990.1988.tb00890.x. [DOI] [PubMed] [Google Scholar]
- 18.Gaschen F.P., Hoffman E.P., Gorospe J.R., Uhl E.W., Senior D.F., Cardinet G.H., 3rd, Pearce L.K. Dystrophin deficiency causes lethal muscle hypertrophy in cats. J Neurol Sci. 1992;110:149–159. doi: 10.1016/0022-510x(92)90022-d. [DOI] [PubMed] [Google Scholar]
- 19.Childers M.K., Okamura C.S., Bogan D.J., Bogan J.R., Sullivan M.J., Kornegay J.N. Myofiber injury and regeneration in a canine homologue of Duchenne muscular dystrophy. Am J Phys Med Rehabil. 2001;80:175–181. doi: 10.1097/00002060-200103000-00004. [DOI] [PubMed] [Google Scholar]
- 20.Liu J.M., Okamura C.S., Bogan D.J., Bogan J.R., Childers M.K., Kornegay J.N. Effects of prednisone in canine muscular dystrophy. Muscle Nerve. 2004;30:767–773. doi: 10.1002/mus.20154. [DOI] [PubMed] [Google Scholar]
- 21.Liu G.C., Jong Y.J., Chiang C.H., Jaw T.S. Duchenne muscular dystrophy: MR grading system with functional correlation. Radiology. 1993;186:475–480. doi: 10.1148/radiology.186.2.8421754. [DOI] [PubMed] [Google Scholar]
- 22.Scopinaro F., Manni C., Miccheli A., Massa R., De Vincentis G., Schillaci O., Ierardi M., Danieli R., Banci M., Lorio F. Muscular uptake of Tc-99m MIBI and TI-201 in Duchenne muscular dystrophy. Clin Nucl Med. 1996;21:792–796. doi: 10.1097/00003072-199610000-00009. [DOI] [PubMed] [Google Scholar]
- 23.Hoffman E.P., Gorospe R.M. The animal models of Duchenne muscular dystrophy: windows on the pathophysiological consequences of dystrophin deficiency. Curr Top Membr. 1991;38:113–155. [Google Scholar]
- 24.McPherron A.C., Lawler A.M., Lee S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 25.Lee S.J. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol. 2004;20:61–86. doi: 10.1146/annurev.cellbio.20.012103.135836. [DOI] [PubMed] [Google Scholar]
- 26.Matsumura K., Ervasti J.M., Ohlendieck K., Kahl S.D., Campbell K.P. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 27.Tumor Analysis Best Practices Working Group Expression profiling—best practices for data generation and interpretation in clinical trials. Nat Rev Genet. 2004;5:229–237. doi: 10.1038/nrg1297. [DOI] [PubMed] [Google Scholar]
- 28.Briguet A., Courdier-Fruh I., Foster M., Meier T., Magyar J.P. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul Disord. 2004;14:675–682. doi: 10.1016/j.nmd.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Jensen O.N., Wilm M., Shevchenko A., Mann M. Sample preparation methods for mass spectrometric peptide mapping directly from 2-DE gels. Methods Mol Biol. 1999;112:513–530. doi: 10.1385/1-59259-584-7:513. [DOI] [PubMed] [Google Scholar]
- 30.Saratsis A.M., Yadavilli S., Magge S., Rood B.R., Perez J., Hill D.A., Hwang E., Kilburn L., Packer R.J., Nazarian J. Insights into pediatric diffuse intrinsic pontine glioma through proteomic analysis of cerebrospinal fluid. Neuro Oncol. 2012;14:547–560. doi: 10.1093/neuonc/nos067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morissette M.R., Cook S.A., Buranasombati C., Rosenberg M.A., Rosenzweig A. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am J Physiol Cell Physiol. 2009;297:C1124–C1132. doi: 10.1152/ajpcell.00043.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hara Y., Kanagawa M., Kunz S., Yoshida-Moriguchi T., Satz J.S., Kobayashi Y.M., Zhu Z., Burden S.J., Oldstone M.B., Campbell K.P. Like-acetylglucosaminyltransferase (LARGE)-dependent modification of dystroglycan at Thr-317/319 is required for laminin binding and arenavirus infection. Proc Natl Acad Sci U S A. 2011;108:17426–17431. doi: 10.1073/pnas.1114836108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ervasti J.M., Roberds S.L., Anderson R.D., Sharp N.J., Kornegay J.N., Campbell K.P. Alpha-dystroglycan deficiency correlates with elevated serum creatine kinase and decreased muscle contraction tension in golden retriever muscular dystrophy. FEBS Lett. 1994;350:173–176. doi: 10.1016/0014-5793(94)00748-9. [DOI] [PubMed] [Google Scholar]
- 34.Kornegay J.N., Childers M.K., Bogan D.J., Bogan J.R., Nghiem P., Wang J., Fan Z., Howard J.F., Jr., Schatzberg S.J., Dow J.L., Grange R.W., Styner M.A., Hoffman E.P., Wagner K.R. The paradox of muscle hypertrophy in muscular dystrophy. Phys Med Rehabil Clin N Am. 2012;23:149–172. doi: 10.1016/j.pmr.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kornegay J.N., Bogan J.R., Bogan D.J., Childers M.K., Grange R.W. Golden retriever muscular dystrophy (GRMD): developing and maintaining a colony and physiological functional measurements. Methods Mol Biol. 2011;709:105–123. doi: 10.1007/978-1-61737-982-6_7. [DOI] [PubMed] [Google Scholar]
- 36.Chen Y.-W., Nagaraju K., Bakay M., McIntyre O., Rawat R., Shi R., Hoffman E.P. Early onset of inflammation and later involvement of TGFβ in Duchenne muscular dystrophy. Neurology. 2005;65:826–834. doi: 10.1212/01.wnl.0000173836.09176.c4. [DOI] [PubMed] [Google Scholar]
- 37.Tseng B.S., Zhao P., Pattison J.S., Tseng B.S., Zhao P., Pattison J.S., Gordon S.E., Granchelli J.A., Madsen R.W., Folk L.C., Hoffman E.P., Booth F.W. Regenerated mdx mouse skeletal muscle shows differential mRNA expression. J Appl Physiol. 2002;93:537–545. doi: 10.1152/japplphysiol.00202.2002. [DOI] [PubMed] [Google Scholar]
- 38.Bo Li Z., Zhang J., Wagner K.R. Inhibition of myostatin reverses muscle fibrosis through apoptosis. J Cell Sci. 2012;125(Pt 17):3957–3965. doi: 10.1242/jcs.090365. [DOI] [PubMed] [Google Scholar]
- 39.Goncalves M.D., Pistilli E.E., Balduzzi A., Birnbaum M.J., Lachey J., Khurana T.S., Ahima R.S. Akt deficiency attenuates muscle size and function but not the response to ActRIIB inhibition. PLoS One. 2010;5:e12707. doi: 10.1371/journal.pone.0012707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tinsley J., Deconinck N., Fisher R., Kahn D., Phelps S., Gillis J.M., Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998;4:1441–1444. doi: 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- 41.Cerletti M., Negri T., Cozzi F., Colpo R., Andreetta F., Croci D., Davies K.E., Cornelio F., Pozza O., Karpati G., Gilbert R., Mora M. Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Ther. 2003;10:750–757. doi: 10.1038/sj.gt.3301941. [DOI] [PubMed] [Google Scholar]
- 42.Townsend D., Turner I., Yasuda S., Martindale J., Davis J., Shillingford M., Kornegay J.N., Metzger J.M. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest. 2010;120:1140–1150. doi: 10.1172/JCI41329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ervasti J.M., Ohlendieck K., Kahl S.D., Gaver M.G., Campbell K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 44.Ohlendieck K., Campbell K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol. 1991;15:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lai Y., Thomas G.D., Yue Y., Yang H.T., Li D., Long C., Judge L., Bostick B., Chamberlain J.S., Terjung R.L., Duan D. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119:624–635. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen H.H., Jayasinha V., Xia B., Hoyte K., Martin P.T. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci U S A. 2002;99:5616–5621. doi: 10.1073/pnas.082613599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoyte K., Jayasinha V., Xia B., Martin P.T. Transgenic overexpression of dystroglycan does not inhibit muscular dystrophy in mdx mice. Am J Pathol. 2004;164:711–718. doi: 10.1016/S0002-9440(10)63158-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li D., Long C., Yue Y., Duan D. Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum Mol Genet. 2009;18:1209–1220. doi: 10.1093/hmg/ddp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Furukawa Y., Hashimoto N., Yamakuni T., Ishida Y., Kato C., Ogashiwa M., Kobayashi M., Kobayashi T., Nonaka I., Mizusawa H., Song S.Y. Down-regulation of an ankyrin repeat-containing protein, V-1, during skeletal muscle differentiation and its re-expression in the regenerative process of muscular dystrophy. Neuromuscul Disord. 2003;13:32–41. doi: 10.1016/s0960-8966(02)00185-2. [DOI] [PubMed] [Google Scholar]
- 50.Guevel L., Lavoie J.R., Perez-Iratxeta C., Rouger K., Dubreil L., Feron M., Talon S., Brand M., Megeney L.A. Quantitative proteomic analysis of dystrophic dog muscle. J Proteome Res. 2011;10:2465–2478. doi: 10.1021/pr2001385. [DOI] [PubMed] [Google Scholar]
- 51.Stalnaker S.H., Aoki K., Lim J.M., Porterfield M., Liu M., Satz J.S., Buskirk S., Xiong Y., Zhang P., Campbell K.P., Hu H., Live D., Tiemeyer M., Wells L. Glycomic analyses of mouse models of congenital muscular dystrophy. J Biol Chem. 2011;286:21180–21190. doi: 10.1074/jbc.M110.203281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor L.E., Kaminoh Y.J., Rodesch C.K., Flanigan K.M. Quantification of dystrophin immunofluorescence in dystrophinopathy muscle specimens. Neuropathol Appl Neurobiol. 2012;38:591–601. doi: 10.1111/j.1365-2990.2012.01250.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A flowchart showing the overall experimental approach for this study. Functional outcome testing was performed on a group of GRMD and normal dogs, followed by analysis of histopathological lesions, mRNA expression (microarray profiling and qPCR), and protein [proteomic profiling, immunofluorescence microscopy (IF), and Western blotting] expression. 6M, 6 months.
DAG1 and LARGE array and qPCR values correlated with golden retriever muscular dystrophy (GRMD) bodyweight-corrected cranial sartorius (CS) circumference (mm/kg) at 6 months. Note the strong correlation with CS size and LARGE (r2 = 0.86; r = 0.93; P = 0.0019) expression and, to a lesser extent, that of DAG1 (r2 = 0.56; r = 0.75; P = 0.05).