

Background: HisJ is a bilobal periplasmic binding protein that mediates basic amino acid transport.
Results: The apo-HisJ solution NMR structure was solved. The individual lobes of HisJ were isolated, and domain 1 binds histidine.
Conclusion: Histidine binding at domain 1 initiates large conformational changes in HisJ.
Significance: Intact hinge regions that bridge the two lobes of HisJ are critical for function.
Keywords: Amino Acid Transport, Isothermal Titration Calorimetry, Ligand-binding Protein, NMR, Protein Domains, Protein Dynamics, Protein Structure
Abstract
Escherichia coli HisJ is a type II periplasmic binding protein that functions to reversibly capture histidine and transfer it to its cognate inner membrane ABC permease. Here, we used NMR spectroscopy to determine the structure of apo-HisJ (26.5 kDa) in solution. HisJ is a bilobal protein in which domain 1 (D1) is made up of two noncontiguous subdomains, and domain 2 (D2) is expressed as the inner domain. To better understand the roles of D1 and D2, we have isolated and characterized each domain separately. Structurally, D1 closely resembles its homologous domain in apo- and holo-HisJ, whereas D2 is more similar to the holo-form. NMR relaxation experiments reveal that HisJ becomes more ordered upon ligand binding, whereas isolated D2 experiences a significant reduction in slower (millisecond to microsecond) motions compared with the homologous domain in apo-HisJ. NMR titrations reveal that D1 is able to bind histidine in a similar manner as full-length HisJ, albeit with lower affinity. Unexpectedly, isolated D1 and D2 do not interact with each other in the presence or absence of histidine, which indicates the importance of intact interdomain-connecting elements (i.e. hinge regions) for HisJ functioning. Our results shed light on the binding mechanism of type II periplasmic binding proteins where ligand is initially bound by D1, and D2 plays a supporting role in this dynamic process.
Introduction
Periplasmic binding proteins (PBPs)2 were originally identified as a class of proteins located near the surface of Gram-negative bacteria, which are released by mild osmotic shock and are able to bind a variety of small molecules (1, 2). PBPs are directly involved in the acquisition of nutrients such as phosphate, sulfate, some carbohydrates, and amino acids. Uptake of amino acids is important for bacteria as they contribute to the cytoplasmic amino acid pool (without the need for de novo biosynthesis) and are incorporated during protein translation. In Gram-negative bacteria basic amino acids can be transported across the outer membrane through general or specific porins such as OmpF or LamB. In Escherichia coli, once histidine gains access to the periplasm it can be transported into the cytoplasm via low and high affinity uptake systems. The low affinity system is known as the general aromatic transport system, and it is only able to transport histidine at concentrations of >0.1 mm through its cytoplasmic membrane permease, AroP (3). The high affinity histidine system is composed of the histidine-binding protein (HisJ), a PBP, and a permease (HisQMP2). HisQM is a heterodimer that forms the integral membrane portion of the permease, and the HisP homodimer is the cognate cytoplasmic ATP-binding subunit (4). HisJ is able to bind histidine with high affinity (Kd ∼30 nm) (5, 6) and deliver it to HisQMP2 from where it is then translocated into the cytoplasm in an ATP-dependent manner. A second PBP, the lysine-, arginine-, and ornithine-binding protein (LAOBP), which shares 70% sequence identity with HisJ, is also able to serve HisQMP2 and binds histidine with an ∼16-fold lower affinity than HisJ (7).
PBPs are distinguished based on their ligand specificity and structural features such as the number of interdomain cross-links (i.e. hinge regions) (8–13). In types I, II, and III PBPs, the two structural domains are connected by three, two, and one hinge regions, respectively. By-products of having multiple hinge regions are the structural domains that are folded discontinuously. For example, in the type II PBP HisJ, domain 1 (D1) is formed by two subdomains where one (D1A) is folded first, then domain 2 (D2), and finally the second subdomain (D1B) of D1 (Fig. 1A). The apo-forms of type II PBPs (e.g. LAOBP- and glutamine-binding protein) (14–16) have been crystallized in an open conformation wherein there are few interdomain contacts except for regions adjacent to the hinge regions. For these PBPs, ligand binding occurs in the interdomain cleft region and is accompanied by significant closing and twisting motions that bring the domains together (14, 17). Although no structure for apo-HisJ has previously been reported, it is expected to adopt an open conformation similar to the related LAOBP protein (18–22). Two crystal structures for HisJ complexed with histidine have been solved for the proteins from E. coli (PDB 1HSL) (23) and Salmonella typhimurium (PDB 1HPB) (24) whose sequences are essentially identical (98.3%), except for four residues that are not directly involved in function. Holo-HisJ adopts a closed conformation similar to that of LAOBP bound to various ligands (14).
FIGURE 1.
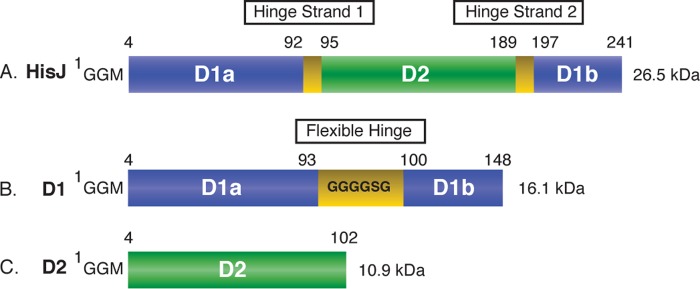
Schematic diagram of the protein constructs used in this study. The numbering for the HisJ (A), D1 (B), and D2 (C) constructs is indicated and includes the N-terminal GGM extension that is a remnant of the purification procedure. D1, D2, and hinge strand residues are colored blue, green, and yellow, respectively.
In this work, we solved the solution structure for apo-HisJ in an open conformation and also confirmed that the structure of the holoprotein in aqueous solution closely resembles that in the crystal structures. We then examined the type and strength of interactions that mediate domain-domain interactions in the HisJ protein using NMR as our primary investigative tool. Our approach was to dissect this protein by expressing each domain individually and to perform protein interaction studies with them in the absence of interdomain connectivity. Our results support the idea that the individual domains of HisJ retain the same global fold as in the WT protein; however, there are apparent functional differences related to the absence of an intact hinge region. The two domains of HisJ were unable to interact in the absence of ligand; moreover, in the presence of histidine only D1 was found to bind histidine. These results strongly support the idea that ligand binding is initiated by the D1 domain of HisJ and that the intact linker region is required for transmission of the “closing” signal to the D2 region.
EXPERIMENTAL PROCEDURES
Cloning of E. coli HisJ and Construct Generation
The mature HisJ protein (residues 1–238), without periplasmic signal sequence, was amplified from the E. coli K12 genome through PCR with oligonucleotides containing 5′-NdeI and 3′-XhoI (New England Biolabs) restriction sites and cloned into the pET15-MHL vector. Domain 1 (D1, residues 1–93 and 193–238) and domain 2 (D2, residues 94–192) constructs of HisJ are described in Fig. 1. For cloning of D1, subdomain A (residues 1–93) was amplified with oligonucleotides containing 5′-NdeI and 3′-KpnI restriction sites, whereas subdomain B (residues 193–238) was amplified with oligonucleotides containing 5′-KpnI and 3′-XhoI restriction sites. Subdomains A and B were digested with KpnI and ligated using T4 ligase (Fermentas). The newly created D1 construct was then amplified by PCR and cloned into the pET15-MHL vector using the 5′-NdeI and 3′-XhoI restriction sites. A flexible linker (Gly-Gly-Gly-Gly-Ser-Gly) was inserted between subdomains A and B of D1 to prevent inadvertently constraining either subdomain in a non-native conformation (Fig. 1). The D2 construct was amplified by PCR and cloned into the pET15-MHL vector using the 5′-NdeI and 3′-XhoI restriction sites.
The pET15-MHL vector was kindly donated by the Structural Genomics Consortium (University of Toronto) and attaches a hexa-histidine tag and a tobacco etch virus (TEV) protease cleavage site to the N terminus of the expressed protein. Cleavage of the expressed protein with TEV protease results in the addition of Gly-Gly-Met to the N terminus. Constructs were verified through nucleotide sequencing at the University of Calgary Genetic Analysis Laboratory. Of note, the HisJ residue numbering in this study includes the additional three amino acids at the N terminus of the protein constructs and thus differs from the native HisJ sequence (e.g. Ala-4 of this paper is equivalent to Ala-1 of the native HisJ sequence).
Sample Preparation
For overexpression of HisJ, D1, and D2, E. coli BL21 (DE3) cells with the vector of interest were grown in 1 liter of Luria-Bertani (LB) in the presence of 100 μg/ml ampicillin. For NMR experiments, isotope-labeled HisJ, D1, and D2 were prepared using the general isotope-labeling strategy outlined by Tugarinov et al. (25) and Huang and Vogel (26). Briefly, E. coli BL21 cells (DE3) were grown in 1 liter of D2O M9 medium containing 3 g/liter d-[U-1H,13C]glucose (Cambridge Isotope Laboratories) as the sole carbon source and 1 g/liter 15NH4Cl (Cambridge Isotope Laboratories) as the nitrogen source. Depending on the experiment and the protein, H2O, M9 media and unlabeled d-glucose and NH4Cl were used in place of their labeled counterparts. For HisJ and D2, cells were grown to an absorbance at 600 nm (A600) of ∼0.8 at 37 °C and induced with 0.8 mm isopropyl β-d-1-thiogalactopyranoside for 3 h. For expression of D1, cells were grown to an A600 of ∼0.5 at 37 °C and induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside at room temperature for ∼16 h. Cells were then harvested and resuspended in 20 mm Tris, 0.1 m NaCl, and 20 mm imidazole (pH 8) and lysed by a French pressure cell. The cell lysate was centrifuged at 18,500 × g for 45 min at 4 °C. Nickel affinity column chromatography was performed using chelating Sepharose (GE Healthcare), where bound proteins were washed with 20 mm Tris, 0.1 m NaCl, and 20 mm imidazole (pH 8), and proteins were eluted with 20 mm Tris, 0.1 m NaCl, and 250 mm imidazole (pH 8). SDS-PAGE was used to confirm protein homogeneity. The purified proteins were then buffer exchanged into 20 mm Tris, 0.1 m NaCl, 0.5 mm EDTA, 3 mm l-glutathione (reduced; Sigma), and 0.3 mm (−)-glutathione (oxidized; Sigma (pH 8)) and subjected to TEV protease digestion (∼ 4 h at 30 °C) to remove the N-terminal histidine tag. The solution was then buffer exchanged into 20 mm Tris, 0.1 m NaCl (pH 8) and applied to a chelating Sepharose column where the cleaved histidine tag and TEV protease are bound, and histidine tag free HisJ, D1, and D2 are found in the flow-through. The proteins were subsequently dialyzed into 5 mm Tris (pH 8) for anion-exchange chromatography using a DEAE-Sepharose Resource Q column (GE Healthcare) and eluted with a linear gradient of 0.1 m NaCl in 5 mm Tris (pH 8). This final anion-exchange chromatography step was used to further purify the proteins and remove any bound histidine. The protein concentrations were determined through a theoretical extinction coefficient (ϵ280) calculated using the ExPASy ProtParam program. For D1, Bradford assays (27) were used to confirm the protein concentration determined by ϵ280.
In general, protein samples were prepared in 50 mm Na3PO4 (pH 7). For NMR experiments, 0.5 mm 2,2-dimethyl-2-silapentane-5-sulfonate was added for peak referencing as well as 10% v/v D2O. Holo-HisJ NMR samples were prepared by adding 5 mm histidine. NMR samples for backbone assignments of HisJ and D1 contained ∼1 mm U-13C,15N-1H/2H-labeled protein, although for D2 ∼1 mm 13C,15N-labeled protein was utilized. Methyl assignments for apo-HisJ were acquired on the same U-13C,15N-1H/2H-labeled sample as the backbone assignments according to Mulder and co-workers (28). 13C,15N-Labeled apo-HisJ (∼1 mm) was used in the acquisition of NOESY NMR experiments. NMR spin relaxation experiments were performed on ∼1 mm U-15N-1H/2H-HisJ and -D1 and ∼1 mm 15N-labeled D2. Residual dipolar coupling (RDC) experiments were performed using the same labeling scheme as the NMR spin relaxation experiment samples except that ∼0.5 mm protein was used, and 150 mm NaCl was added to the buffer. RDC experiments for holo-HisJ were performed with 50 mm NaCl to prevent disruption of histidine binding (6). NMR titration experiments were performed with ∼0.3 mm 15N-D1 or -D2 and with 100 mm NaCl. Of note, unless otherwise specified, histidine refers to l-histidine.
ITC Experiments
All ITC experiments were performed using a Microcal VP-ITC microcalorimeter (GE Healthcare). All samples were prepared in a buffer containing 20 mm HEPES (pH 7.0) and 100 mm NaCl. The syringe was filled with 0.5 mm histidine that was injected into the ITC cell containing 20 μm protein (D1, D2, or HisJ) solution at 30 °C. The heat of dilution was measured separately by injecting titrant into the ITC buffer and was shown to be negligible. After subtraction of the heat of dilution, the data were fit to a one-site binding model (MicroCal Origin version 7.0 software) to obtain the stoichiometry (N), enthalpy (ΔH), and association constant (Ka), which are converted to Kd values using the relationship shown in Equation 1,
and where the reported values are the average and S.D. of three independent titrations.
NMR Experiments
All NMR experiments for the backbone chemical shift assignments, titrations, and relaxation experiments were performed at 298 K on Bruker Avance 500- or 700-MHz NMR spectrometers equipped with triple resonance inverse cryoprobes with a single axis z-gradient. Sequential assignments of HN, N, C′, Cα, and Cβ resonances of apo- and holo-HisJ, D1, and D2 were achieved using two-dimensional 1H,15N-HSQC and three-dimensional HNCACB, HN(CO)CACB, HN(CA)CO, and HNCO experiments. For apo- and holo-HisJ and D1 proteins, TROSY versions of these experiments were used (29, 30). For apo-HisJ, methyl group assignments were acquired using a three-dimensional C-TOCSY-CHD2 experiment (28). NMR chemical shift assignments for apo-, holo-, D1-, and D2-HisJ have been deposited in the Biological Magnetic Resonance Data Bank (BioMagResBank) under the accession codes 19242, 19245, 19243, and 19244, respectively.
NMR spin relaxation experiments were conducted at a static magnetic field strength of 500 MHz on [U-15N]1H/2H apo- and holo-HisJ and 15N-D1 and -D2 samples. 15N R1 values were performed using sensitivity-enhanced inversion-recovery pulse sequences (31) with T delays of 14, 70, 210, 420, 560 (2×), 700 (2×), 882, and 1060 ms. 15N R2 values were acquired using sensitivity-enhanced Carr-Purcell-Meiboom-Gill (CPMG)-type T2 experiments with pulsed field gradients (31), with T delays of 8 (2×), 16 (2×), 40, 48, 64 (2×), 72, 80, and 96 ms. {1H} 15N NOE experiments were recorded with and without a 1H presaturation period of 5 s and an interscan delay of 5 s. Relaxation dispersion experiments were performed with TROSY-based CPMG experiments at 50.684 MHz for the 15N frequency (32, 33). CPMG spectra were acquired at frequencies (νCPMG) of 40 and 1000 Hz. The NMR relaxation dispersion (R2 eff) was calculated as shown in Equation 2,
A mixing time of 120 ms was used in the three-dimensional 15N-edited NOESY-TROSY (34) and constant time 13C-edited NOESY-HSQC experiment acquired on a 13C,15N-apo-HisJ sample. NOESY experiments were collected using a nonuniform sampling procedure (30%) where processing was performed by MDDGUI 1.0 software (Arrowsmith Laboratory, University Health Network, University of Toronto), which requires MddNMR 2.0 (35). The 13C-edited NOESY-HMQC was acquired in 99.9% D2O.
1DNH RDC were measured on partially aligned U-15N-1H/2H apo- and holo-HisJ and 15N-D1 and -D2 samples. The alignment media used included Pf1 phage (Asla Lab) at concentrations of 10 mg/ml for apo- and holo-HisJ and 12 mg/ml for D1 and D2 (36). 1DNH RDCs were measured using an in-phase/anti-phase 1H,15N-HSQC experiment with 2048 × 512 complex points (37). The PALES (38) software was used to fit the measured HN-N RDCs to those back-calculated from the crystal structure of the holo-(histidine) HisJ complex (PDB 1HSL) and models of D1 and D2 built from the apo- and holo-HisJ structures. Of note, the deposited holo-HisJ structure (1HSL) contains four point mutations (N5K, S22A, E107A, and S184A) that were corrected to match the HisJ sequence from E. coli K12 (GenBankTM accession number BAA16155.1) using MODELLER (39) prior to analysis. Only well structured regions of D1, D2, and HisJ were chosen for analysis based on {1H} 15N NOE values >0.65 from NMR spin relaxation experiments (see below).
CSPs upon ligand binding were examined on a per residue basis using a combined chemical shift difference, where applicable, according to Mulder et al. (40) for backbone atoms, where Ri denotes the scaling factor of nucleus i. The scaling factors used were RN = 6.4, RC′ = 3.0, RCα, and RCβ = 3.2 as shown in Equation 3.
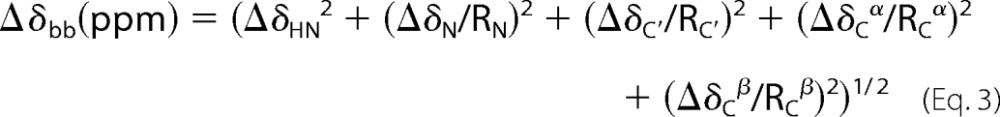 |
All data were processed with the NMRPipe (41) software package and analyzed using the program Sparky (42). Protein backbone dihedral angle data were calculated using the program TALOS+ (43), and all chemical shifts were referenced to 2,2-dimethyl-2-silapentane-5-sulfonate (44).
NMR Analysis
For NMR spin relaxation experiments, the values of R1 and R2 were obtained by fitting the extracted peak intensities to a mono-exponential decay curve. Peak intensities were extracted using the relaxation module in Sparky (42), and data fitting was performed by Curvefit version 1.4 (A. G. Palmer, Columbia University) using the script “sparky2rate.” Uncertainties in the peak heights were determined from duplicate measurements, and uncertainties in R1 and R2 were estimated from Jackknife simulations (45). {1H} 15N NOE values were determined from the experiments with and without irradiation. Data points with errors larger than 25% were excluded from the analysis. Relaxation data used in the rotational diffusion analysis were subject to filtering as described by Bax and co-workers (46) The rotational correlation time for the global tumbling (τm) of each residue was estimated from R2/R1 ratio using the program R2R1_tm (A. G. Palmer, Columbia University), and the overall rotational correlation times (τc) were determined by averaging the per residue τm values.
Apo-HisJ Structure Calculation
An initial template apo-HisJ structure was generated by threading the HisJ sequence onto the apo-LAOBP structure (PDB 2LAO) using Swiss-Model (47). For the structure calculation, deuterium isotope-corrected backbone chemical shifts (48, 49) were used to determine the position of secondary structure elements and dihedral backbone angle (Φ,Ψ) (43) values utilizing the programs chemical shift index (CSI) (50) and TALOS+ (43), respectively. Of note, TALOS+ restraints were used for all residues except proline. Secondary structure elements were implemented in structure calculations using artificial hydrogen bond restraints according to Tugarinov et al. (51). RDC (52, 53) and distance restraints were also used as input for the structure calculations. Distance restraints were binned according to Tugarinov et al. (51), where the majority of HN-HN NOEs were bound to 1.8–5.0 Å, except for residues in helices (1.8–4.0 Å). CH3-CH3 and CH3-HN NOEs were binned to 1.8–8.0 and 1.8–6.0 Å, respectively, except for intraresidue HN-CH3 NOEs (1.8–4.0 Å). Apo-HisJ structures were calculated with Xplor-NIH software (54), using torsion angle simulated annealing according to a two-stage, low temperature protocol (55–57). In stage 1, the starting model undergoes Powell energy minimization, 10 ps of torsion angle dynamics (at 200 K), and 4 ps of simulated annealing at each step wherein the temperature was incrementally decreased from 200 to 20 K at a 10 K step size. During simulated annealing, the force constants were increased except the backbone torsion angle restraints for ϕ and ψ, which are fixed at 300 kcal mol−1 rad−2. 200 structures were generated in stage 1, and the lowest energy structure was selected as the starting model for the next stage. Stage 2 involves 10 ps of torsion angle dynamics at 20 K, which is followed by 6 ps of simulated annealing at each temperature step from 20 to 1 K in 1 K increments. In stage 2, the backbone dihedral angle force constant was ramped down from 300 to 50 kcal mol−1 rad−2, and the backbone angle restraints were derived from the lowest energy structure from stage 1. 100 structures were calculated in stage 2, and an ensemble of the 30 lowest energy structures was generated and validated using the program Protein Structure Validation Suite (58). The atomic coordinates for the 30 lowest energy structure ensemble of apo-HisJ has been deposited in the PDB under the accession code 2M8C. All molecular graphics were created using PyMOL (59).
RESULTS
NMR Characterization
The domains of HisJ were cloned, expressed, and characterized in order to assess their global folds and to examine their ability to interact with each other in the presence or absence of ligand. 1H-15N (TROSY) HSQC spectra of all protein constructs used in this study show well folded structures (Fig. 2). From the backbone experiments, 99.7% of all possible 1HN, 15N, 13Cα, 13Cβ, and 13C′ resonance assignments for both apo- and holo-HisJ (complexed with histidine) were assigned. In both proteins, the 1HN, 15N, and 13Cβ resonance assignments of Ser-25 could not be determined. The overall completeness of the backbone resonance assignment for apo-HisJ represents a slight improvement from a previously published NMR assignment (60). In particular, apo-HisJ backbone amide resonances for Ile-59, Ser-72–Gln-81, Gln-146, and Lys-228 could be resolved through the use of perdeuteration and TROSY (29). The backbone resonance assignments for the D1 and D2 constructs were also assigned to near-completeness at 99.4 and 97.8%, respectively. For D1, the missing assignments belonged to residues Ser-25 and Glu-78 (1HN and 15N). For D2, the missing assignments included Asn-12 and Thr-34 (1HN and 15N) and Pro-17 13Cβ.
FIGURE 2.

1H,15N (TROSY) HSQC spectra of protein constructs from this study. A, 1H,15N-HSQC NMR spectra of 2H,15N-apo-HisJ (red), 15N-D1 (blue), and 15N-D2 (green) protein constructs. Residues from the inserted flexible linker region of D1 are labeled. B, 1H,15N TROSY-HSQC spectra of 2H,15N-apo-HisJ (red) and 2H,15N-holo-HisJ (black).
Using a two-stage low temperature simulated annealing protocol, the apo-HisJ NMR structure was determined in an open conformation where the two lobes of the protein are spread apart (Fig. 3A; Table 1). Similar to other PBPs, each lobe of HisJ displays a mixed α/β-fold (Rossmann-like) characterized by a central β-sheet (61). In contrast, the holo-HisJ crystal structure (PDB 1HSL; Fig. 3B) displays a closed conformation with the two lobes making extensive interdomain contacts. Comparison of the apo- and holo-HisJ forms using combined backbone CSP analysis reveals several regions with large values (Fig. 3C) that can be attributed to the conformational transition that occurs upon ligand binding as well as modifications to the chemical environment of ligand-interacting residues.
FIGURE 3.
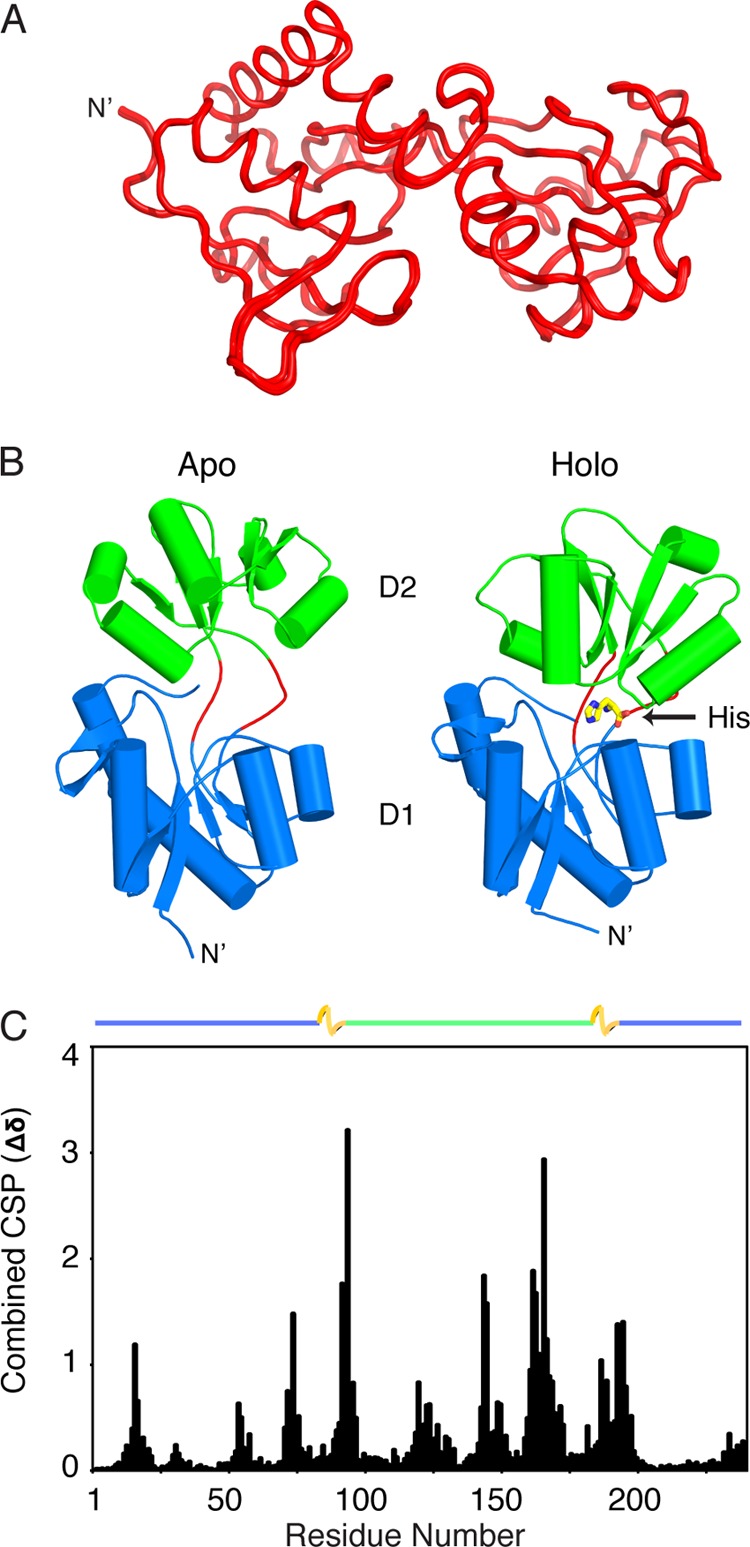
NMR structure of apo-HisJ. A, 30 lowest energy structures of apo-HisJ (red) overlaid (see Table 1 for structural statistics). B, side-by-side comparison of the lowest energy NMR structure of apo-HisJ and the crystal structure of holo-HisJ (PDB 1HSL). The first (D1) and second domain (D2) of each structure is colored blue and green, respectively. Holo-HisJ adopts a closed conformation in comparison with apo-HisJ, which results in histidine (yellow) being engulfed. Of note, hinge regions are colored red. C, combined NMR CSP for apo-HisJ and holo-HisJ (complexed with histidine). The domain organization of HisJ is noted above the figure where D1 subdomains (blue), D2 (green), and hinge regions (yellow) are indicated.
TABLE 1.
Experimental restraints and structural statistics for the 30 lowest total energy structures of apo-HisJ
r.m.s.d. means root mean square deviation.
| No. of experimental restraints | |
| Upper distance restraints from NOE | |
| Total | 1370 |
| Short/medium range | 917 |
| Long range | 453 |
| 1DNH RDCs | 185 |
| Dihedral angle restraints (TALOS+) | 407 |
| Hydrogen bonding restraints | 74 |
| Average r.m.s.d. values from experimental data | |
| Distance restraint violation | 0.065 Å |
| Dihedral angle restraint violation | 0.217° |
| RDC restraint violation | 0.104 Hz |
| Average r.m.s.d. values from idealized covalent geometry | |
| Bonds | 0.003 Å |
| Angles | 0.434° |
| Impropers | 0.444° |
| PROCHECK Ramachandran analysis for the folded regions | |
| Residues in most favored regions | 84.8% |
| Residues in additional allowed regions | 13.5% |
| Residues in generously allowed regions | 1.7% |
| Residues in disallowed regions | 0% |
| Coordinate precision of folded regions | |
| Backbone | 0.13 ± 0.06 Å |
| All heavy atoms | 0.18 ± 0.06 Å |
CSPs indicate that the chemical environments of the individual HN-N chemical shifts from D1 and D2 compare favorably with those of apo-HisJ (Fig. 4A). For the D1 protein, significant CSPs for residues 93 and 100–104 were observed. These residues are located adjacent to the artificial D1 linker and are part of the two interdomain linkers (residues 92–95 and 189–197) in the full-length HisJ constructs. For D2, residues at the N′ and C′ termini experience significant (>1 S.D.) CSPs, as well as residues 73, 76–77, and 79–83 that correspond to residues 163, 166–167, and 169–173, respectively, in the full-length protein (Fig. 4A). Despite these differences, the majority of the domain chemical shifts compare favorably with those from apo-HisJ. Similarly, analysis of calculated backbone dihedral angles (Φ and Ψ) reveals large changes at regions adjacent to the flexible D1 linker and termini regions of D2 compared with apo-HisJ (Fig. 4B).
FIGURE 4.
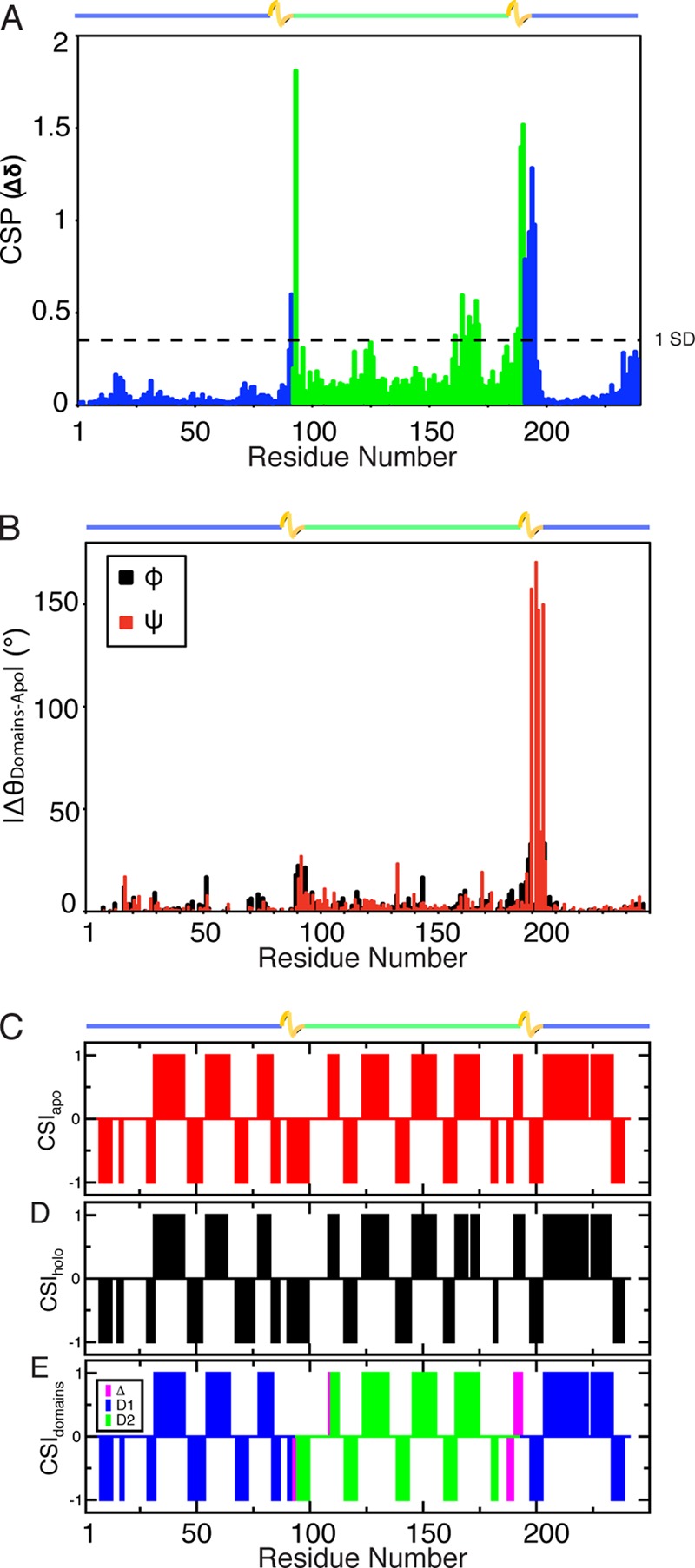
Structural similarity of the isolated domain constructs and their equivalent domains in apo-HisJ. A, HN-N CSP of D1 (blue) and D2 (green) compared with apo-HisJ. The horizontal line indicates CSPs greater than 1.0 S.D. from the mean. B, comparison of the backbone dihedral angles of D1 and D2 versus apo-HisJ that were calculated using the program TALOS+. C and D, CSI secondary structure prediction results for apo- and holo-HisJ, respectively. E, CSI results for D1 (blue) and D2 (green). Residues where differences in secondary structure (compared with apo-HisJ) are detected are colored purple. CSI results are interpreted as +1 for α-helical propensity, −1 for β-strand prediction, and 0 for random coil. Of note, the inserted flexible linker of D1 was excluded in the comparison. The domain organization of HisJ is noted above the figures.
Secondary structure distribution as determined by the CSI (50) data reveals that both apo- and holo-HisJ have similar composition and distribution of secondary structure elements (Fig. 4, C and D). Furthermore, secondary structure element distribution of the solution holo-HisJ protein compares favorably to that of the E. coli holo (histidine)-HisJ crystal structure (PDB 1HSL, data not shown). Comparison of CSI predictions for D1 and D2 versus the full-length apo-HisJ protein reveals an almost perfect level of conservation except for residues immediately adjacent to the D1 flexible linker and at the N′ and C′ termini of D2 (Fig. 4E).
HN-N RDCs were used to assess the folding of D1 and D2 and examine the orientations of these domains in holo-HisJ. HN-N RDCs provide information on the average orientation of interatomic HN-N vectors relative to the molecular alignment tensor (52). The RDC is used to define the HN-N bond vector with respect to the external magnetic field and can be used to assess domain orientation (62). The high degree of correlation between the measured HN-N RDCs for apo-HisJ and those back-calculated from the initial starting model (see “Experimental Procedures”; Table 2; Fig. 5A) suggested that apo-HisJ adopts an open conformation, which was later confirmed with our NMR structure. Holo-HisJ HN-N RDCs have a high degree of agreement with the closed conformation holo-HisJ crystal structure (PDB 1HSL; Fig. 5B), whereas D1 HN-N RDCs have similar levels of correlation with domains from either the apo-open or holo-closed HisJ structures (Table 2; Fig. 5C). Interestingly, D2 HN-N RDCs have a higher correlation with the equivalent domain in the holo-HisJ crystal structure than the apo-HisJ NMR structure (Fig. 5D). These RDC results indicate that the D1 and D2 HN-N bond vectors closely resemble those found in the apo-open and holo-closed HisJ structures; however, D2 shows greater differences with respect to the apo-form of which Ser-97 (full-length Ser-187) has the largest variation (Fig. 5E).
TABLE 2.
RDC analysis of HN-N bond vectors from apo- and holo-HisJ and D1 and D2
Experimental values are compared with those back-calculated from the structures of apo- and holo-HisJ. A high R value and a low Q value indicate close agreement.
| Protein | No. of residues | Structure | R | Q |
|---|---|---|---|---|
| % | ||||
| Apo-HisJ | 184 | Apo-open (starting model) | 0.95 | 0.30 |
| 184 | Holo-closed (PDB 1HSL) | 0.78 | 0.63 | |
| Holo-HisJ | 202 | Apo-open (NMR) | 0.80 | 0.60 |
| 202 | Holo-closed (PDB 1HSL) | 0.94 | 0.34 | |
| D1 | 115 | D1 model (apo-NMR) | 0.94 | 0.34 |
| 115 | D1 model (holo-crystal) | 0.95 | 0.30 | |
| D2 | 82 | D2 model (apo-NMR) | 0.83 | 0.56 |
| 82 | D2 model (holo-crystal) | 0.94 | 0.36 |
FIGURE 5.

Correlation between isolated D1 and D2 and their wild-type equivalents in apo- and holo-HisJ as revealed by RDC analysis. A, high R and low Q values (see Table 2) were obtained from fitting apo-HisJ RDCs and the starting model used in the structure calculations (see “Experimental Procedures”). B, similarly, holo-HisJ RDCs were fit to the holo-HisJ crystal structure (PDB 1HSL). C, D1 could be fit to either the lowest energy apo-HisJ NMR structure and the holo-HisJ structure. D, isolated D2 shows a better correlation with its domain in holo-HisJ than with the apo-HisJ NMR structure. E, absolute value difference plot in measured and back-calculated RDCs between isolated D2 and the same domain in either the apo- or holo-HisJ structure is plotted as a function of D2 residue number, and outliers are indicated by the horizontal line.
NMR Spin Relaxation
NMR relaxation experiments (R1, R2, and {1H} 15N NOE) can provide information on protein tumbling and dynamics on the picosecond to nanosecond time scale. Apo- and holo-HisJ are rigid proteins with highly flexible terminal regions (residues 1–8 and 241; Fig. 6A). Compared with the holo-form, apo-HisJ has increased flexibility at Gly-145 and residues 196 and 197 from the 2nd interdomain linker. Furthermore, residues 93 and 95 of apo-HisJ have broadened peak line widths compared with their holo-form counterparts suggesting an increase in conformational averaging perhaps due to increased dynamics. D1 has increased mobility at the N′- and C′-terminal ends as well as the Gly linker region (residues 94–99) but is otherwise rigid (Fig. 6B). Similar to apo-HisJ, D2 behaves as a globular rigid protein except for residue Gly-55 (corresponding to full-length Gly-145) and the N′- and C′-terminal residues (Fig. 6C). Rotational diffusion analysis of the R1 and R2 data allowed for calculations of average rotational correlation times (τc) from the τm values of each residue. Apo- and holo-HisJ (26.5 kDa) have similar correlation times of 13.83 ± 1.12 and 13.79 ± 1.23 ns (Fig. 6D), respectively, despite the difference in conformation. The D1 and D2 domains within the apo- and holo-HisJ proteins have similar τc values suggesting that the domains tumble dependently in solution. Isolated D1 and D2 have correlation times of 8.59 ± 0.36 and 5.33 ± 0.31 ns, respectively, indicating that these proteins tumble as monomers in solution (Fig. 6, E and F). For D1, measurement of similar τc values for D1a and D1b subdomains indicate that they tumble dependently and provide further evidence to suggest that D1 is folded similarly to the D1 domain within HisJ.
FIGURE 6.

NMR spin relaxation properties of the HisJ constructs used in this study. {1H} 15N NOE spin relaxation parameters and rotational correlation time for the global tumbling (τm) of each residue for apo- (red) and holo- (black) HisJ (A and D) and isolated D1 (B and E) and D2 (C and F). Of note, residues in the inserted flexible linker in D1 (B) are colored in light blue. The domain organization of HisJ is noted above the plots in A and D.
CPMG relaxation dispersion experiments indicate areas that are involved in slow chemical exchange (millisecond to microsecond). Relaxation dispersion data indicate that apo-HisJ experiences slow conformational motions, although holo-HisJ does not (Fig. 7A). From the D1 domain of apo-HisJ, Ala-18 undergoes elevated (R2 eff 40 Hz/1000 Hz = 2.0) slow motions and residues 98, 197, and 198 located proximal to the hinge regions also experience significant dispersion (R2 eff 40 Hz/1000 Hz >1.3). Residues 121, 125, 151, 152, 161, 163, 171, and 174 from the D2 domain of apo-HisJ also show significant slow motions. The majority of the residues experiencing chemical exchange occurs in the D2 region of apo-HisJ, although nearly all of the D1 R2 eff ratios remain unperturbed. Isolated D1 experiences no significant chemical exchange (Fig. 7B), whereas residues in the latter portion of D2 show elevated R2 eff values (>1.25), including C-terminal Lys-102 (R2 eff = 1.53; Fig. 7C). Overall, isolated D2 residues (8, 31, 35, 61, 71, 73, and 81) experience a significant reduction in slow motions compared with the equivalent residues in apo-HisJ.
FIGURE 7.
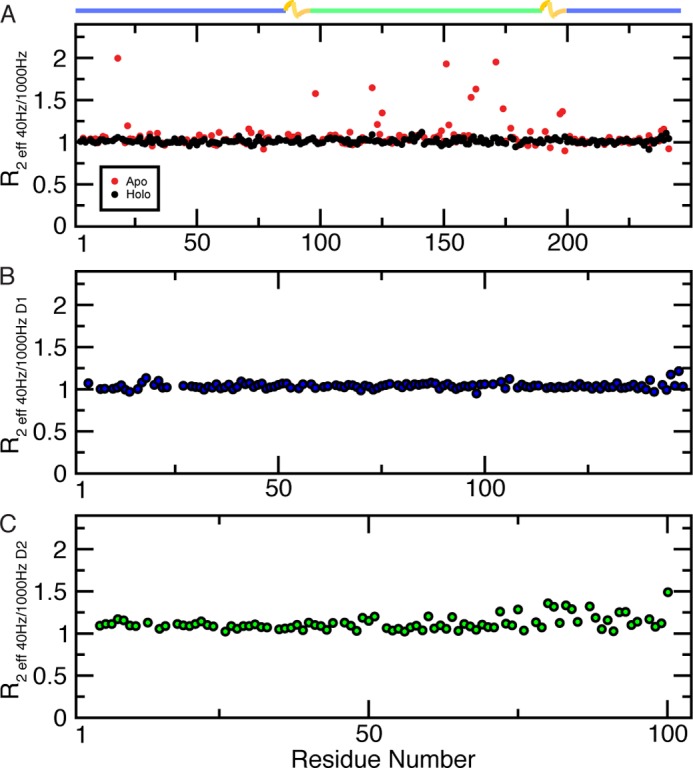
Slow motion (millisecond to microsecond) dynamics from backbone CPMG experiments. A, apo- and holo-HisJ are depicted in red and black, respectively. The isolated D1 (blue) (B) and D2 (green) (C) constructs are also shown. The R2 eff ratio was calculated from experiments acquired at νCPMG of 40 and 1000 Hz and are plotted per residue.
Ligand Binding
Previous studies have established that HisJ binds histidine with a higher affinity than other basic amino acids (5, 6, 21, 63, 64). ITC and NMR were used to examine the binding of histidine to apo-HisJ, D1, and D2. Apo-HisJ exhibited an exothermic interaction with histidine with a binding constant (Kd) of 114 ± 16 nm using ITC (Fig. 8). The thermodynamic parameters for histidine binding to HisJ are ΔH = −61.2 ± 0.126 kJ/mol and TΔS = −23 ± 0.412 kJ/mol, which indicate that it is driven by a favorable enthalpy term. Binding constants and thermodynamics of histidine binding to D1 and D2 could not be determined as the molecules did not interact, the interaction was too weak to be detected, or the reaction did not produce a discernable heat signal (data not shown). NMR titrations were performed to examine CSP changes in the HSQC spectra of D1 and D2 upon addition of histidine. Based on the apo-HisJ NMR titration with histidine (Fig. 3C), it was expected that many CSPs would be observed in the experiments with the isolated domains. In the D1 titration, a small number of signals shift in a fast exchange manner (on the NMR time scale) upon histidine addition, which indicates weak binding (Kd Ser-73 ∼80 μm; Fig. 9, A and B). In the isolated D2 NMR titration, no significant CSPs were observed suggesting that D2 is not able to bind histidine (Fig. 9C), which is consistent with the ITC results. From the E. coli HisJ crystal structure (PDB 1HSL), both D1 and D2 residues make direct contact with the histidine ligand (Table 3). The majority of these residues experience significant CSPs when apo-HisJ is titrated with histidine; however, in the individual D1 titration, significant CSPs (>1 S.D.) for only Tyr-17, Ala-18, Ser-73, and Ser-75 were detected.
FIGURE 8.
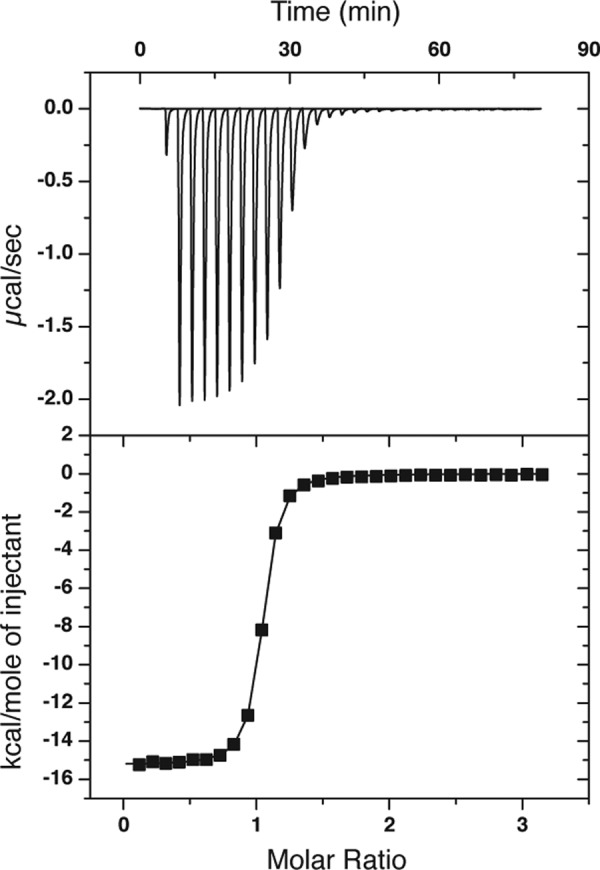
Binding of histidine to HisJ by ITC. A representative binding isotherm obtained by injecting histidine into apo-HisJ is shown.
FIGURE 9.
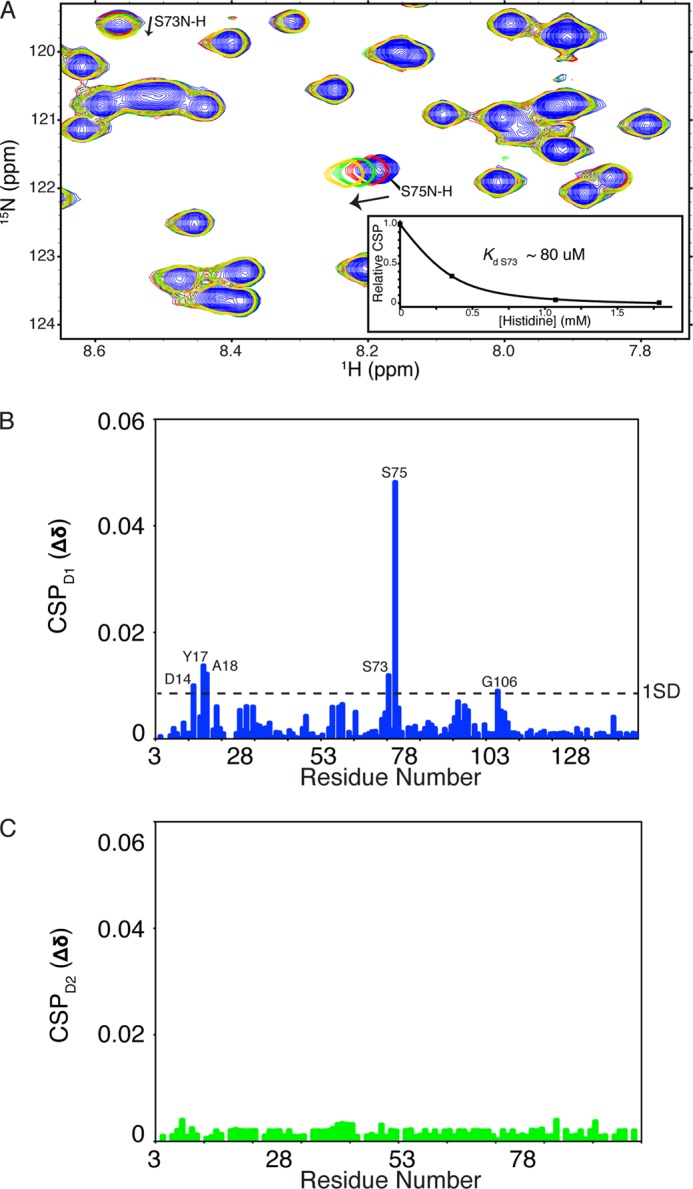
NMR titrations of histidine binding to 15N-D1 (A and B) and 15N-D2 (C). A, region of the 15N-D1 HSQC experiment. The majority of the peaks do not change position during the experiment; however, Ser-73 and Ser-75 shift steadily upon each addition (see arrow). Inset in A is the plot of Ser-73 peak position relative to histidine concentration and the Kd value as determined by the NMRViewJ titration analysis module (One Moon Scientific).
TABLE 3.
Analysis of interdomain and interhinge strand contacts
Contact surface area was calculated using contact map analysis software (92).
| Interaction partners | Residue (D1) | Residue (D2) | Contact surface area |
|---|---|---|---|
| Å2 | |||
| Apo-HisJ | |||
| D1–D2 | 19 | 170 | 39.6 |
| 20 | 166 | 33.8 | |
| 234 | 166 | 24.2 | |
| 236 | 165 | 34.5 | |
| 236 | 166 | 28.9 | |
| Hinge 1–hinge 2 | 194 | 93 | 33.5 |
| Holo-HisJ | |||
| D1–D2 | 16 | 146 | 32.7 |
| 16 | 147 | 23.2 | |
| 17 | 167 | 42.9 | |
| 17 | 164 | 31.8 | |
| 17 | 146 | 28.9 | |
| 18 | 171 | 25.4 | |
| 19 | 170 | 27.9 | |
| 55 | 123 | 24.4 | |
| 56 | 121 | 36.9 | |
| 56 | 144 | 35.4 | |
| 234 | 166 | 32.3 | |
| 236 | 166 | 23.1 | |
| 236 | 170 | 27.3 | |
| Hinge 1–Hinge 2 | 194 | 95 | 32.6 |
| D1/D2–histidine | 17 | 48.1 | |
| 55 | 18.1 | ||
| 72 | 21.7 | ||
| 73 | 32 | ||
| 75 | 30.6 | ||
| 80 | 31.6 | ||
| 120 | 19.6 | ||
| 123 | 21 | ||
| 124 | 42.8 | ||
| 125 | 21 | ||
| 146 (H2O-mediated) | 0.6 | ||
| 164 | 26.8 | ||
Interdomain Interactions
Analysis of the lowest energy apo-HisJ NMR structure and holo-HisJ crystal structure (PDB 1HSL) reveals that the number of residues making significant (>1 S.D.) contact area between the two protein lobes increases from 5 to 13, which results in an ∼2.4-fold increase in interdomain contact surface area (Table 3). Of these, important contributions are made from D1 residues 16–18 and 55 and 56, which contact D2 residues 121–123, 144–147, and 164–171 to help stabilize the closed holo-form.
NMR titrations were performed with D1 and D2 to examine the ability of the isolated domains to interact with each other. 15N-D1 was titrated with unlabeled D2, and changes in peak position and intensity were monitored; however, no significant changes were observed (data not shown). Moreover, 15N-D2 was titrated with unlabeled D1, and no significant changes were noted except for Asp-100, which lies at the C terminus of the D2 construct (data not shown). The addition of histidine to an equimolar mixture of 15N-D1 and 15N-D2 did not result in any significant chemical shifts (data not shown) apart from those observed from the 15N-D1-histidine titration (Fig. 9, A and B). Similar to the D1-histidine ITC titration (data not shown), ITC experiments performed on a 1:1 molar ratio of D1/D2 with histidine did not produce a detectable heat change (data not shown).
DISCUSSION
Dynamics of Ligand Binding
Our solution structure of apo-HisJ shows that it adopts an open conformation in the absence of ligand and that the ordering of dynamic residues in the hinge regions contributes to HisJ transitioning to a closed form in the presence of histidine (Fig. 3B). This large scale conformational change is reminiscent of the “Venus flytrap” mechanism of ligand binding used by other PBPs (65), such as LAOBP and maltose-binding protein (MBP). Although the apo-HisJ solution structure shows an average open conformation, we also present CPMG dynamics data that support a minor apo-conformation(s) based on an increase in chemical exchange for D2 (Fig. 7A). This result is corroborated by HisJ MD simulations that show that apo-HisJ is able to sample a closed conformation (66). Previous studies with the type I MBP revealed a minor (5%) apo-closed conformation that was identified and solved using RDC analysis in combination with paramagnetic relaxation enhancement (67). This minor MBP conformation has also been sampled by MD simulations (68), although other efforts have used a combined NMR and CS-Rosetta approach to identify these “invisible” protein states (69). In contrast, a minor apo-conformation was not detectable for the type II glutamine-binding protein (70), indicating that not all structurally related PBPs undergo an NMR identifiable conformational transition in the absence of ligand.
For HisJ, binding of ligand results in the formation of a stable closed conformation that is characterized by an ordering of dynamic residues on both fast and slow NMR time scales. In comparison, the apo-form of the type II glutamine-binding protein has two rigid hinge strands that are stabilized by inter-hinge strand hydrogen bonds and whose residues are directly involved in ligand binding (70, 71). Tjandra and co-workers (70, 72) have suggested that variation in hinge flexibility among PBPs is related to ligand binding specificity and mediates the open-closed conformational transitions. Mutational studies of the interdomain hinge regions of the type I ribose-binding protein and glucose/galactose-binding protein have also been shown to alter hinge region dynamics and concomitantly the intrinsic binding affinity of these proteins for their ligands (73). Our results support an intimate role between hinge region dynamics and ligand binding in PBPs that is not dependent on contact between the hinge and ligand.
Multifunctional Roles of D1 and D2
In this study, the main polypeptide chain break points for our isolation of the individual D1 and D2 domains of HisJ were chosen to coincide with the hinge regions. RDC, CSI, CSP, dihedral angle backbone analysis, and NMR spin relaxation data indicate that these two isolated domains are folded similarly compared with their homologous domains in apo-HisJ, except for terminal regions and residues adjacent to the inserted flexible linker in D1. The other main structural differences originated in D2. CSP, RDC, and NMR relaxation dispersion data indicate two loop regions in D2 (residues 30–35 near α-helix 2 and 72–75 near α-helix 4) are significantly affected in the absence of intact interdomain linkers (Fig. 10, A and B).
FIGURE 10.
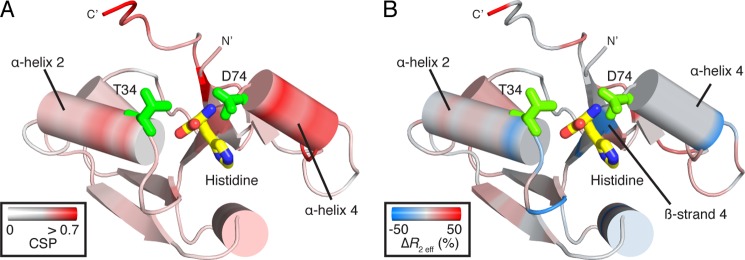
Mapping of CSPs (A) and changes in R2 eff chemical exchange (B) onto the D2 structure from holo-HisJ. A, CSPs arise from differences in isolated D2 and the same domain in apo-HisJ. Significant changes (indicated in red) are localized near α-helix 2 and 4, and key ligand-binding residues (green) Thr-34 (full-length Thr-124) and Asp-74 (full-length Asp-164) are indicated. B, relative change in R2 eff (D2-apo) (%) where loss and gain in chemical exchange of D2 compared with apo-HisJ are indicated in blue and red, respectively. Significant decreases in R2 eff are found near residue Thr-34 and adjacent to Asp-74 on β-strand 4.
From the holo-HisJ crystal structure (PDB 1HSL), it is observed that the majority (∼60%) of the direct contact surface area with histidine occurs through residues in the D1 domain. This has led to the idea that D1 and D2 may have different roles in PBP ligand binding (14), where D1 serves as the initial ligand acceptor. To date, experimental evidence to support such a model in PBPs is limited to the structure of the type I E. coli leucine, isoleucine, and valine-binding protein where leucine was soaked into the crystal and bound to D1 (PDB 2LIV) (74) and theoretical targeted MD simulations of the same protein (75). MD simulations of LAOBP also indicated that an initial D1-ligand contact step was required for an open-to-closed conformational change to occur (76). In this study, we provide direct experimental evidence that isolated D1 of HisJ, but not D2, is able to bind histidine in a similar manner as the full-length HisJ. D1 residues Tyr-17, Ser-73, and Ser-75 show significant CSPs upon histidine titration. These residues along with Leu-55 and Ser-72, which also have high CSP values, comprise the histidine binding pocket of D1 (Fig. 9B). These affected residues are identical to those perturbed by addition of histidine to the full-length HisJ protein (Fig. 3C) and those that participate in histidine coordination in the in the holo-HisJ crystal structure. The ability of D1 to bind histidine supports a model where this domain is involved in the initial ligand binding step of HisJ and related proteins (e.g. LAOBP) (14, 24). Furthermore, the apparent reduced affinity of D1 for histidine is in agreement with the notion that a D2 interaction is required to further increase the affinity of HisJ for its ligand.
The inability of isolated D2 to interact with histidine indicates that it plays a supporting role in ligand binding. Previous studies have proposed that PBP-ligand binding occurs by an induced-fit mechanism wherein domain closure is stimulated by a ligand binding event (14, 21, 24, 70, 77, 78). In the case of HisJ, we suggest that histidine binding at D1 triggers a closing event that rapidly brings D2 in close proximity with D1. In the absence of interdomain connectivity, D2 is unable to make contact with the ligand; D1 and D2 are not able to interact; and a conformational change leading to a closed holo-form is not communicated.
The changes in chemical environment and structure of D2 loop regions (residues 30–35 and 72–75) in the absence of interdomain connectivity directly affects residues involved in both ligand binding and domain-domain contacts (Fig. 10A). Similarly, a loss in R2 chemical exchange for residues 31, 35, 71, and 73 in isolated D2 agrees with this notion (Fig. 10B). Previous studies have shown that mutations to Thr-34 (full-length T124A) and Asp-74 (D164A/E165K) reduce HisJ binding affinity for histidine by >150-fold (24, 64, 79). Moreover, a T124A HisJ mutant was unable to form a stable, closed-liganded conformation (64). Mutations to other residues in the same D2 loop regions have also been shown to disrupt histidine binding and the ability to adopt a closed conformation (64). In particular, our analysis reveals that loop residues 72–75 and the proceeding α-helix 4 of D2 are extensively involved in making interdomain contacts in both apo- and holo-HisJ (Table 3). This indicates that in the absence of intact hinge regions, chemical environmental changes in D2 affect the ability of this domain to interact with ligand and D1. In this regard, mutation of S95F from the 1st hinge strand has been previously shown to affect the ability of HisJ to bind histidine (Kd >5 μm) and adopt a closed conformation (24).
Currently, the body of information regarding the interaction of PBPs and their inner membrane ABC transporters is growing (80, 81). Studies involving MBP and its ABC transporter MalFGK2 have established that a large extracellular loop (MalF-P2) is responsible for mediating interactions with MBP and inducing a conformational change that leads to release of its ligand (82–87). Previous work by Ames and co-workers (88–90) has established that HisJ docks onto HisQMP2, and mutational studies have shown that regions on D1 and D2 have a role in forming a stable complex with the permease and in signaling active transport of the ligand. HisQMP2 is not predicted to have a large periplasmic signaling loop (91); thus, it remains to be established what the molecular mechanism is that induces ligand release from PBPs with transporters that lack large extracellular loops. Our study with the individual lobes of HisJ suggests that a change in PBP conformation that separates the two lobes would significantly weaken the association with the ligand and could facilitate ligand release. A ligand-dependent transport rate for HisQMP2 has also been observed, but the biochemical nature of this regulation has not been elucidated (21). Future solution NMR studies of HisJ bound to other basic amino acids (i.e. arginine, ornithine, and lysine) may show differences that are not apparent from the crystal structures of holo-LAOBP (PDB 1LAH, 1LAG, 1LAF, and 1LST). Moreover, it is unclear if both lobes of HisJ are required to stimulate transport, which could be studied using the isolated domain constructs described in this work.
This HisJ study is the first to structurally and functionally characterize the individual lobes of a PBP. We have also attempted to perform similar studies for PBPs that have a single hinge region (ferric enterobactin-binding protein) and three hinge regions (MBP) but have not yet been successful.3
Acknowledgments
We thank the late Dr. Deane McIntyre for maintenance of the NMR spectrometers in Calgary, Canada. We also thank Dr. Hiroaki Ishida for helpful discussions.
This work was supported in part by an operating grant from the Canadian Institutes of Health Research (Novel Alternatives to Antibiotics Program).
The atomic coordinates and structure factors (code 2M8C) have been deposited in the Protein Data Bank (http://wwpdb.org/).
B. C. H. Chu and H. J. Vogel, unpublished results.
- PBP
- periplasmic binding protein
- CSI
- Chemical Shift Index
- CSP
- chemical shift perturbation
- D1
- domain 1
- D2
- domain 2
- HSQC
- heteronuclear single quantum coherence
- LAOBP
- lysine, arginine, ornithine binding protein
- MBP
- maltose binding protein
- MD
- molecular dynamics
- PDB
- Protein Data Bank
- RDC
- residual dipolar coupling
- TEV
- tobacco etch virus
- CPMG
- Carr-Purcell-Meiboom-Gill
- ITC
- Isothermal titration calorimetry.
REFERENCES
- 1. Ames G. F., Lever J. (1970) Components of histidine transport: histidine-binding proteins and HisP protein. Proc. Natl. Acad. Sci. U.S.A. 66, 1096–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nossal N. G., Heppel L. A. (1966) The release of enzymes by osmotic shock from Escherichia coli in exponential phase. J. Biol. Chem. 241, 3055–3062 [PubMed] [Google Scholar]
- 3. Brown K. D. (1970) Formation of aromatic amino acid pools in Escherichia coli K-12. J. Bacteriol. 104, 177–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Doige C. A., Ames G. F. (1993) ATP-dependent transport systems in bacteria and humans: relevance to cystic fibrosis and multidrug resistance. Annu. Rev. Microbiol. 47, 291–319 [DOI] [PubMed] [Google Scholar]
- 5. Ames G. F., Lever J. E. (1972) The histidine-binding protein J is a component of histidine transport. Identification of its structural gene, hisJ. J. Biol. Chem. 247, 4309–4316 [PubMed] [Google Scholar]
- 6. Lever J. E. (1972) Quantitative assay of the binding of small molecules to protein: comparison of dialysis and membrane filter assays. Anal. Biochem. 50, 73–83 [DOI] [PubMed] [Google Scholar]
- 7. Nikaido K., Ames G. F. (1992) Purification and characterization of the periplasmic lysine-, arginine-, ornithine-binding protein (LAO) from Salmonella typhimurium. J. Biol. Chem. 267, 20706–20712 [PubMed] [Google Scholar]
- 8. Lee Y. H., Deka R. K., Norgard M. V., Radolf J. D., Hasemann C. A. (1999) Treponema pallidum TroA is a periplasmic zinc-binding protein with a helical backbone. Nat. Struct. Biol. 6, 628–633 [DOI] [PubMed] [Google Scholar]
- 9. Quiocho F. A., Ledvina P. S. (1996) Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes. Mol. Microbiol. 20, 17–25 [DOI] [PubMed] [Google Scholar]
- 10. Chu B. C., Vogel H. J. (2011) A structural and functional analysis of type III periplasmic and substrate binding proteins: their role in bacterial siderophore and heme transport. Biol. Chem. 392, 39–52 [DOI] [PubMed] [Google Scholar]
- 11. Claverys J. P. (2001) A new family of high-affinity ABC manganese and zinc permeases. Res. Microbiol. 152, 231–243 [DOI] [PubMed] [Google Scholar]
- 12. Tam R., Saier M. H., Jr. (1993) Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 57, 320–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krewulak K. D., Peacock R. S., Vogel H. J. (2004) in Periplasmic Binding Proteins Involved in Bacterial Iron Uptake (Crosa J. H., Mey A. R., Payne S. M., eds) pp. 113–129, American Society for Microbiology, Washington, D. C [Google Scholar]
- 14. Oh B. H., Pandit J., Kang C. H., Nikaido K., Gokcen S., Ames G. F., Kim S. H. (1993) Three-dimensional structures of the periplasmic lysine/arginine/ornithine-binding protein with and without a ligand. J. Biol. Chem. 268, 11348–11355 [PubMed] [Google Scholar]
- 15. Hsiao C. D., Sun Y. J., Rose J., Wang B. C. (1996) The crystal structure of glutamine-binding protein from Escherichia coli. J. Mol. Biol. 262, 225–242 [DOI] [PubMed] [Google Scholar]
- 16. Kang C. H., Shin W. C., Yamagata Y., Gokcen S., Ames G. F., Kim S. H. (1991) Crystal structure of the lysine-, arginine-, ornithine-binding protein (LAO) from Salmonella typhimurium at 2.7-Å resolution. J. Biol. Chem. 266, 23893–23899 [PubMed] [Google Scholar]
- 17. Sun Y. J., Rose J., Wang B. C., Hsiao C. D. (1998) The structure of glutamine-binding protein complexed with glutamine at 1.94 Å resolution: comparisons with other amino acid-binding proteins. J. Mol. Biol. 278, 219–229 [DOI] [PubMed] [Google Scholar]
- 18. Cedel T. E., Cottam P. F., Meadows M. D., Ho C. (1984) A high-resolution 1H-NMR investigation of the histidine-binding protein J of Salmonella typhimurium. Substrate-induced conformational changes. Biophys. Chem. 19, 279–287 [DOI] [PubMed] [Google Scholar]
- 19. Post J. F., Cottam P. F., Simplaceanu V., Ho C. (1984) Fluorine-19 nuclear magnetic resonance study of 5-fluorotryptophan-labeled histidine-binding protein J of Salmonella typhimurium. J. Mol. Biol. 179, 729–743 [DOI] [PubMed] [Google Scholar]
- 20. Zukin R. S., Klos M. F., Hirsch R. E. (1986) Conformational dynamics of two histidine-binding proteins of Salmonella typhimurium. Biophys. J. 49, 1229–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wolf A., Lee K. C., Kirsch J. F., Ames G. F. (1996) Ligand-dependent conformational plasticity of the periplasmic histidine-binding protein HisJ. Involvement in transport specificity. J. Biol. Chem. 271, 21243–21250 [DOI] [PubMed] [Google Scholar]
- 22. Wolf A., Shaw E. W., Nikaido K., Ames G. F. (1994) The histidine-binding protein undergoes conformational changes in the absence of ligand as analyzed with conformation-specific monoclonal antibodies. J. Biol. Chem. 269, 23051–23058 [PubMed] [Google Scholar]
- 23. Yao N., Trakhanov S., Quiocho F. A. (1994) Refined 1.89-Å structure of the histidine-binding protein complexed with histidine and its relationship with many other active transport/chemosensory proteins. Biochemistry 33, 4769–4779 [DOI] [PubMed] [Google Scholar]
- 24. Oh B. H., Kang C. H., De Bondt H., Kim S. H., Nikaido K., Joshi A. K., Ames G. F. (1994) The bacterial periplasmic histidine-binding protein. Structure/function analysis of the ligand-binding site and comparison with related proteins. J. Biol. Chem. 269, 4135–4143 [PubMed] [Google Scholar]
- 25. Tugarinov V., Kanelis V., Kay L. E. (2006) Isotope labeling strategies for the study of high-molecular-weight proteins by solution NMR spectroscopy. Nat. Protoc. 1, 749–754 [DOI] [PubMed] [Google Scholar]
- 26. Huang H., Vogel H. J. (2013) Purification and stable isotope-labeling of calcium-and integrin-binding protein 1 for structural and functional NMR studies. Methods Mol. Biol. 963, 99–113 [DOI] [PubMed] [Google Scholar]
- 27. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 28. Otten R., Chu B., Krewulak K. D., Vogel H. J., Mulder F. A. (2010) Comprehensive and cost-effective NMR spectroscopy of methyl groups in large proteins. J. Am. Chem. Soc. 132, 2952–2960 [DOI] [PubMed] [Google Scholar]
- 29. Eletsky A., Kienhöfer A., Pervushin K. (2001) TROSY NMR with partially deuterated proteins. J. Biomol. NMR 20, 177–180 [DOI] [PubMed] [Google Scholar]
- 30. Salzmann M., Wider G., Pervushin K., Senn H., Wuthrich K. (1999) TROSY-type triple resonance experiments for sequential NMR assignments of large proteins. J. Am. Chem. Soc. 121, 844–848 [Google Scholar]
- 31. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., Kay L. E. (1994) Backbone dynamics of a free and a phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 [DOI] [PubMed] [Google Scholar]
- 32. Loria J. P., Rance M., Palmer A. G., 3rd (1999) A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J. Biomol. NMR 15, 151–155 [DOI] [PubMed] [Google Scholar]
- 33. Mulder F. A., Mittermaier A., Hon B., Dahlquist F. W., Kay L. E. (2001) Studying excited states of proteins by NMR spectroscopy. Nat. Struct. Biol. 8, 932–935 [DOI] [PubMed] [Google Scholar]
- 34. Zhu G., Kong X. M., Sze K. H. (1999) Gradient and sensitivity enhancement of 2D TROSY with water flip-back, 3D NOESY-TROSY and TOCSY-TROSY experiments. J. Biomol. NMR 13, 77–81 [DOI] [PubMed] [Google Scholar]
- 35. Orekhov V. Y., Ibraghimov I., Billeter M. (2003) Optimizing resolution in multidimensional NMR by three-way decomposition. J. Biomol. NMR 27, 165–173 [DOI] [PubMed] [Google Scholar]
- 36. Hansen M. R., Mueller L., Pardi A. (1998) Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat. Struct. Biol. 5, 1065–1074 [DOI] [PubMed] [Google Scholar]
- 37. Ottiger M., Delaglio F., Bax A. (1998) Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 131, 373–378 [DOI] [PubMed] [Google Scholar]
- 38. Zweckstetter M., Bax A. (2000) Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J. Am. Chem. Soc. 122, 3791–3792 [Google Scholar]
- 39. Eswar N., John B., Mirkovic N., Fiser A., Ilyin V. A., Pieper U., Stuart A. C., Marti-Renom M. A., Madhusudhan M. S., Yerkovich B., Sali A. (2003) Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 31, 3375–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mulder F. A., Schipper D., Bott R., Boelens R. (1999) Altered flexibility in the substrate-binding site of related native and engineered high-alkaline Bacillus subtilisins. J. Mol. Biol. 292, 111–123 [DOI] [PubMed] [Google Scholar]
- 41. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 42. Goddard T. D., Kneller D. G. (2003) Sparky 3, Version 3.113, University of California, San Francisco [Google Scholar]
- 43. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wishart D. S., Bigam C. G., Yao J., Abildgaard F., Dyson H. J., Oldfield E., Markley J. L., Sykes B. D. (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6, 135–140 [DOI] [PubMed] [Google Scholar]
- 45. Mosteller F., Tukey J. W. (1977) Data Analysis and Regression: A Second Course in Statistics, pp. 133–163, Addison-Wesley, Reading, MA [Google Scholar]
- 46. Barbato G., Ikura M., Kay L. E., Pastor R. W., Bax A. (1992) Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry 31, 5269–5278 [DOI] [PubMed] [Google Scholar]
- 47. Guex N., Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 48. Gardner K. H., Rosen M. K., Kay L. E. (1997) Global folds of highly deuterated methyl-protonated proteins by multidimensional NMR. Biochemistry 36, 1389–1401 [DOI] [PubMed] [Google Scholar]
- 49. Venters R. A., Farmer B. T., 2nd, Fierke C. A., Spicer L. D. (1996) Characterizing the use of perdeuteration in NMR studies of large proteins. J. Mol. Biol. 264, 1101–1116 [DOI] [PubMed] [Google Scholar]
- 50. Wishart D. S., Sykes B. D. (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4, 171–180 [DOI] [PubMed] [Google Scholar]
- 51. Tugarinov V., Choy W. Y., Orekhov V. Y., Kay L. E. (2005) Solution NMR-derived global fold of a monomeric 82-kDa enzyme. Proc. Natl. Acad. Sci. U.S.A. 102, 622–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tjandra N., Omichinski J. G., Gronenborn A. M., Clore G. M., Bax A. (1997) Use of dipolar 1H-15N and 1H-13C couplings in the structure determination of magnetically oriented macromolecules in solution. Nat. Struct. Biol. 4, 732–738 [DOI] [PubMed] [Google Scholar]
- 53. Tolman J. R., Flanagan J. M., Kennedy M. A., Prestegard J. H. (1995) Nuclear magnetic dipole interactions in field-oriented proteins: information for structure determination in solution. Proc. Natl. Acad. Sci. U.S.A. 92, 9279–9283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schwieters C. D., Kuszewski J. J., Tjandra N., Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 55. Chou J. J., Li S., Bax A. (2000) Study of conformational rearrangement and refinement of structural homology models by the use of heteronuclear dipolar couplings. J. Biomol. NMR 18, 217–227 [DOI] [PubMed] [Google Scholar]
- 56. Gifford J. L., Ishida H., Vogel H. J. (2011) Fast methionine-based solution structure determination of calcium-calmodulin complexes. J. Biomol. NMR 50, 71–81 [DOI] [PubMed] [Google Scholar]
- 57. Huang H., Vogel H. J. (2012) Structural basis for the activation of platelet integrin αIIbβ3 by calcium- and integrin-binding protein 1. J. Am. Chem. Soc. 134, 3864–3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bhattacharya A., Tejero R., Montelione G. T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795 [DOI] [PubMed] [Google Scholar]
- 59. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 60. Igarashi S., Osawa M., Ozawa S., Shimada I. (2010) Backbone resonance assignments for the ligand binding subunit of the histidine permease complex (HisJ) from Escherichia coli, under histidine-bound and unbound states. Biomol. NMR Assign. 4, 17–20 [DOI] [PubMed] [Google Scholar]
- 61. Fukami-Kobayashi K., Tateno Y., Nishikawa K. (1999) Domain dislocation: a change of core structure in periplasmic binding proteins in their evolutionary history. J. Mol. Biol. 286, 279–290 [DOI] [PubMed] [Google Scholar]
- 62. Tjandra N., Bax A. (1997) Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 278, 1111–1114 [DOI] [PubMed] [Google Scholar]
- 63. Miller D. M., 3rd, Olson J. S., Pflugrath J. W., Quiocho F. A. (1983) Rates of ligand binding to periplasmic proteins involved in bacterial transport and chemotaxis. J. Biol. Chem. 258, 13665–13672 [PubMed] [Google Scholar]
- 64. Wolf A., Shaw E. W., Oh B. H., De Bondt H., Joshi A. K., Ames G. F. (1995) Structure/function analysis of the periplasmic histidine-binding protein. Mutations decreasing ligand binding alter the properties of the conformational change and of the closed form. J. Biol. Chem. 270, 16097–16106 [DOI] [PubMed] [Google Scholar]
- 65. Felder C. B., Graul R. C., Lee A. Y., Merkle H. P., Sadee W. (1999) The Venus flytrap of periplasmic binding proteins: an ancient protein module present in multiple drug receptors. AAPS PharmSci. 1, E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chu B. C., Chan D. I., DeWolf T., Periole X., Vogel H. J. (2013) Molecular dynamics simulations reveal that Apo-HisJ can sample a closed conformation. Proteins 10.1002/prot.24396 [DOI] [PubMed] [Google Scholar]
- 67. Tang C., Schwieters C. D., Clore G. M. (2007) Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature 449, 1078–1082 [DOI] [PubMed] [Google Scholar]
- 68. Bucher D., Grant B. J., Markwick P. R., McCammon J. A. (2011) Accessing a hidden conformation of the maltose binding protein using accelerated molecular dynamics. PLoS Comput. Biol. 7, e1002034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Korzhnev D. M., Religa T. L., Banachewicz W., Fersht A. R., Kay L. E. (2010) A transient and low-populated protein-folding intermediate at atomic resolution. Science 329, 1312–1316 [DOI] [PubMed] [Google Scholar]
- 70. Bermejo G. A., Strub M. P., Ho C., Tjandra N. (2010) Ligand-free open-closed transitions of periplasmic binding proteins: the case of glutamine-binding protein. Biochemistry 49, 1893–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bermejo G. A., Strub M. P., Ho C., Tjandra N. (2009) Determination of the solution-bound conformation of an amino acid binding protein by NMR paramagnetic relaxation enhancement: use of a single flexible paramagnetic probe with improved estimation of its sampling space. J. Am. Chem. Soc. 131, 9532–9537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pistolesi S., Tjandra N., Bermejo G. A. (2011) Solution NMR studies of periplasmic binding proteins and their interaction partners. Biomol. Concepts 2, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ortega G., Castaño D., Diercks T., Millet O. (2012) Carbohydrate affinity for the glucose-galactose binding protein is regulated by allosteric domain motions. J. Am. Chem. Soc. 134, 19869–19876 [DOI] [PubMed] [Google Scholar]
- 74. Sack J. S., Saper M. A., Quiocho F. A. (1989) Periplasmic binding protein structure and function. Refined x-ray structures of the leucine/isoleucine/valine-binding protein and its complex with leucine. J. Mol. Biol. 206, 171–191 [DOI] [PubMed] [Google Scholar]
- 75. Trakhanov S., Vyas N. K., Luecke H., Kristensen D. M., Ma J., Quiocho F. A. (2005) Ligand-free and -bound structures of the binding protein (LivJ) of the Escherichia coli ABC leucine/isoleucine/valine transport system: trajectory and dynamics of the interdomain rotation and ligand specificity. Biochemistry 44, 6597–6608 [DOI] [PubMed] [Google Scholar]
- 76. Silva D. A., Domínguez-Ramírez L., Rojo-Domínguez A., Sosa-Peinado A. (2011) Conformational dynamics of l-lysine, l-arginine, l-ornithine binding protein reveals ligand-dependent plasticity. Proteins 79, 2097–2108 [DOI] [PubMed] [Google Scholar]
- 77. Ames G. F. (1986) Bacterial periplasmic transport systems: structure, mechanism, and evolution. Annu. Rev. Biochem. 55, 397–425 [DOI] [PubMed] [Google Scholar]
- 78. Stockner T., Vogel H. J., Tieleman D. P. (2005) A salt-bridge motif involved in ligand binding and large-scale domain motions of the maltose-binding protein. Biophys. J. 89, 3362–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Prossnitz E. (1991) Determination of a region of the HisJ binding protein involved in the recognition of the membrane complex of the histidine transport system of Salmonella typhimurium. J. Biol. Chem. 266, 9673–9677 [PubMed] [Google Scholar]
- 80. Schneider E., Eckey V., Weidlich D., Wiesemann N., Vahedi-Faridi A., Thaben P., Saenger W. (2012) Receptor-transporter interactions of canonical ATP-binding cassette import systems in prokaryotes. Eur. J. Cell Biol. 91, 311–317 [DOI] [PubMed] [Google Scholar]
- 81. Locher K. P. (2009) Structure and mechanism of ATP-binding cassette transporters. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Böhm S., Licht A., Wuttge S., Schneider E., Bordignon E. (2013) Conformational plasticity of the type I maltose ABC importer. Proc. Natl. Acad. Sci. U.S.A. 110, 5492–5497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Daus M. L., Grote M., Schneider E. (2009) The MalF P2 loop of the ATP-binding cassette transporter MalFGK2 from Escherichia coli and Salmonella enterica serovar typhimurium interacts with maltose binding protein (MalE) throughout the catalytic cycle. J. Bacteriol. 191, 754–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jacso T., Grote M., Daus M. L., Schmieder P., Keller S., Schneider E., Reif B. (2009) Periplasmic loop P2 of the MalF subunit of the maltose ATP binding cassette transporter is sufficient to bind the maltose binding protein MalE. Biochemistry 48, 2216–2225 [DOI] [PubMed] [Google Scholar]
- 85. Jacso T., Schneider E., Rupp B., Reif B. (2012) Substrate transport activation is mediated through second periplasmic loop of transmembrane protein MalF in maltose transport complex of Escherichia coli. J. Biol. Chem. 287, 17040–17049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Oldham M. L., Chen J. (2011) Snapshots of the maltose transporter during ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 108, 15152–15156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Oldham M. L., Chen J. (2011) Crystal structure of the maltose transporter in a pretranslocation intermediate state. Science 332, 1202–1205 [DOI] [PubMed] [Google Scholar]
- 88. Ames G. F., Spurich E. N. (1976) Protein-protein interaction in transport: periplasmic histidine-binding protein J interacts with P protein. Proc. Natl. Acad. Sci. U.S.A. 73, 1877–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Prossnitz E., Nikaido K., Ulbrich S. J., Ames G. F. (1988) Formaldehyde and photoactivatable cross-linking of the periplasmic binding protein to a membrane component of the histidine transport system of Salmonella typhimurium. J. Biol. Chem. 263, 17917–17920 [PubMed] [Google Scholar]
- 90. Liu C. E., Liu P. Q., Wolf A., Lin E., Ames G. F. (1999) Both lobes of the soluble receptor of the periplasmic histidine permease, an ABC transporter (traffic ATPase), interact with the membrane-bound complex. Effect of different ligands and consequences for the mechanism of action. J. Biol. Chem. 274, 739–747 [DOI] [PubMed] [Google Scholar]
- 91. Eckey V., Weidlich D., Landmesser H., Bergmann U., Schneider E. (2010) The second extracellular loop of pore-forming subunits of ATP-binding cassette transporters for basic amino acids plays a crucial role in interaction with the cognate solute binding protein(s). J. Bacteriol. 192, 2150–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sobolev V., Eyal E., Gerzon S., Potapov V., Babor M., Prilusky J., Edelman M. (2005) SPACE: a suite of tools for protein structure prediction and analysis based on complementarity and environment. Nucleic Acids Res. 33, W39–W43 [DOI] [PMC free article] [PubMed] [Google Scholar]