

Background: SAP is an adaptor recruiting Fyn and is involved in immunity.
Results: SAP is critical for induction and progression of collagen-induced arthritis, an antibody-mediated autoimmune disease. Fyn-independent and Fyn-dependent effects of SAP in T cells mediate this function.
Conclusion: SAP promotes antibody-mediated autoimmunity by Fyn-independent and Fyn-dependent effects in T cells.
Significance: SAP is critical for antibody-mediated autoimmune diseases.
Keywords: Adaptor Proteins, Autoimmune Diseases, Immunology, SH2 Domains, Src
Abstract
Signaling lymphocytic activation molecule-associated protein (SAP) is an Src homology 2 domain-only adaptor involved in multiple immune cell functions. It has also been linked to immunodeficiencies and autoimmune diseases, such as systemic lupus erythematosus. Here, we examined the role and mechanism of action of SAP in autoimmunity using a mouse model of autoimmune arthritis, collagen-induced arthritis (CIA). We found that SAP was essential for development of CIA in response to collagen immunization. It was also required for production of collagen-specific antibodies, which play a key role in disease pathogenesis. These effects required SAP expression in T cells, not in B cells. In mice immunized with a high dose of collagen, the activity of SAP was nearly independent of its ability to bind the protein tyrosine kinase Fyn and correlated with the capacity of SAP to promote full differentiation of follicular T helper (TFH) cells. However, with a lower dose of collagen, the role of SAP was more dependent on Fyn binding, suggesting that additional mechanisms other than TFH cell differentiation were involved. Further studies suggested that this might be due to a role of the SAP-Fyn interaction in natural killer T cell development through the ability of SAP-Fyn to promote Vav-1 activation. We also found that removal of SAP expression during progression of CIA attenuated disease severity. However, it had no effect on disease when CIA was clinically established. Together, these results indicate that SAP plays an essential role in CIA because of Fyn-independent and Fyn-dependent effects on TFH cells and, possibly, other T cell types.
Introduction
Signaling lymphocytic activation molecule (SLAM)-associated3 protein (SAP, also known as SH2D1A) is a small intracellular adaptor molecule made up almost exclusively of an Src homology 2 (SH2) domain (1–4). It is expressed in T cells, natural killer (NK) cells, NK-T cells, and some B cells. SAP is mutated and inactivated in X-linked lymphoproliferative disease, a human immunodeficiency characterized by an uncontrolled immune response to Epstein-Barr virus infection. Previous analyses showed that SAP plays a pivotal role in several immune cell functions, including T helper 2 (TH2) cytokine production, T cell-dependent antibody production, follicular T helper (TFH) cell differentiation, NK-T cell development, CD8+ T cell-mediated cytotoxicity, and NK cell-mediated cytotoxicity (1–4).
The various biological activities attributed to SAP result from its ability to regulate the signals triggered by the SLAM family of receptors (1–4). This property is mediated through a dual signaling mechanism (5–8). On one hand, SAP couples SLAM family receptors to protein tyrosine phosphorylation signals because of its capacity to bind and activate the Src-related protein tyrosine kinase Fyn (9, 10). This ability is mediated by direct binding of SAP to Fyn via the arginine 78-based motif of SAP and the Src homology 3 domain of Fyn. On the other hand, SAP is able to prevent coupling of SLAM-related receptors to inhibitory effectors such as the Src homology 2 domain-containing phosphatases SHIP-1 and SHP-1 (6–8, 11). Most SAP functions are strongly dependent on the capacity to bind Fyn (9, 10, 12–15). However, this is not the case for T cell-dependent antibody production and TFH cell differentiation, which are reportedly Fyn-independent processes (15–17).
T cell-dependent B cell immunity leads to generation of high-affinity antibodies and B cell memory against protein antigens (18). When excessive, it can also be implicated in autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis (RA), and myasthenia gravis. Accumulating evidence indicates that T cell-dependent B cell responses are critically dependent on the ability of a subset of CD4+ T cells, the TFH cells, to initiate germinal center (GC) reaction (19–24). When contacted by antigen-specific TFH cells, GC B cells sharing the same antigen specificity undergo differentiation, isotype switching, and somatic hypermutation. SAP promotes initiation and progression of the GC reaction, at least in part by stabilizing conjugate formation between antigen-specific TFH cells and GC B cells (5, 16, 25, 26). This function necessitates the expression of SAP in T cells, not in B cells (15, 27). Although SAP is not needed for early TFH cell differentiation (also called “polarization”), it is required for completion of this process by enabling up-regulated expression of the TFH cell lineage commitment factor Bcl-6 (28).
SAP has been implicated in autoimmune diseases, in particular diseases associated with autoantibodies such as lupus (1–3). Constitutive SAP deficiency dramatically reduced the susceptibility of mice to pristane-induced lupus (29). This effect was associated with a pronounced reduction of autoantibody production. Likewise, a spontaneous germ line mutation in the SAP-encoding gene reduced the severity of lupus-associated glomerulonephritis and vasculitis in mice carrying a mutation of Fas (30). Lastly, lack of SAP attenuated enhanced GC formation, high levels of autoantibodies, and lupus-like renal pathology in mice carrying the Sanroque mutation (31). Transfer of TFH cells from Sanroque mice was sufficient to yield spontaneous GC formation in wild-type mice, suggesting that the autoimmunity in Sanroque mice was due, at least in part, to deregulated TFH cell functions. However, lack of SAP did not eliminate all signs of autoimmunity in these lupus models. This could be because other cell types, like TH1 cells or TH17 cells, were also involved in disease pathogenesis.
Critical questions remain to be addressed regarding the role and mechanism of action of SAP in antibody-mediated autoimmunity. First, whether SAP is critical for autoimmune conditions other than lupus requires evaluation. Second, the role of Fyn in the ability of SAP to mediate these processes needs to be clarified. Third, the relative contribution of SAP expression in TFH cells and, possibly, other T cell subsets should be examined. Fourth, the possibility that blockade of the SAP pathway could be used to delay or revert antibody-mediated autoimmune diseases in humans deserves to be assessed.
Here we addressed these issues using a broad range of approaches and genetically modified mouse models. Our data showed that SAP was absolutely required for induction of collagen-induced arthritis (CIA), a model of antibody-mediated autoimmunity. This effect required expression of SAP in T cells, not in B cells. The ability of SAP to bind Fyn was largely dispensable for the capacity to induce CIA and anti-collagen antibodies when a high dose of collagen was used for immunization. This correlated with a lack of requirement of the SAP-Fyn interaction for full differentiation of TFH cells. However, when a lower dose of collagen was utilized, the capacity of SAP to bind Fyn was critical for full disease induction and anti-collagen antibody production. This finding suggested that additional effects in TFH cells or in other T cell types might be involved in disease pathogenesis. Because the only other T cell defect observed in collagen-immunized mice lacking the SAP-Fyn interaction was reduced NK-T cell numbers, it is possible that this cell type was implicated. Such an effect possibly related to the ability of SAP-associated Fyn to promote activation of Vav-1, which is needed for NK-T cell development (32). Lastly, studies using an inducible SAP-deficient mouse showed that sustained SAP expression was needed for progression of CIA during the early phases of disease development. However, it was not required for maintenance of CIA when the disease was clinically established.
EXPERIMENTAL PROCEDURES
Mice
C57BL/6 and DBA/1J mice were from Harlan Sprague-Dawley (Montreal, Québec, Canada) and The Jackson Laboratory (Bar Harbor, ME), respectively. SAP-deficient (Sh2d1a−/), SAP R78A (Sh2d1aR78A/), conditional SAP-deficient (Sh2d1afl/), Fyn-deficient (Fyn−/−), Cd4-Cre, Mb1-Cre, and Ubc-CreERT2 mice were described previously (13, 27, 33–36). These animals were maintained in the C57BL/6 background. For all experiments, littermates were used as controls. Tamoxifen (TAM)-induced deletion of SAP was performed as detailed elsewhere (26). Animal experimentation was performed in agreement with the guidelines from the Canadian Council of Animal Care and was approved by the Animal Care Committee of the Clinical Research Institute of Montréal.
Collagen-induced Arthritis
To study CIA, mice were backcrossed for at least 12 generations to the DBA/1J background. To study the effect of tissue-specific or inducible deletion of SAP on CIA, Sh2d1afl/+ females in the DBA/1J background were crossed with Cre-expressing males in the C57BL/6 mice. F1 mice were used for experimentation. This was made possible by the observation that the F1 progeny of a cross between WT C57BL/6 mice and WT DBA/1J mice was susceptible to CIA. To trigger CIA, mice were injected intradermally at the base of the tail with 100 μg of bovine type II collagen (CII, catalog no. 20021, Chondrex, Redmond, WA) supplemented with complete Freund adjuvant (catalog no. 7001, Chondrex) and 200 μg of Mycobacterium tuberculosis in a final volume of 0.1 ml. In some experiments, mice received a second immunization 15 weeks after the first immunization. Arthritis development was monitored clinically twice a week and scored using a scale of 0–4 (37): 0, no erythema and swelling; 1, erythema and mild swelling confined to the mid-foot (tarsals) or ankle joint; 2, erythema and mild swelling extending from the ankle to mid-foot; 3, erythema and moderate swelling extending from the ankle to the metatarsal joints; and 4, erythema and severe swelling encompassing the ankle, foot, and digits. The clinical score was the sum of the scores for the four extremities. Mice were also bled at the indicated times after immunization to measure collagen-specific antibodies in serum using ELISA (see below).
Hapten Immunization
Mice were injected with equal volumes of hapten (at 20 μg/ml) and alum (catalog no. 77161, Thermo Scientific, Rockford, IL) in a final volume of 200 μl. The haptens were nitrophenyl (NP)(13)-ovalbumin (OVA) (catalog no. N-5051-100, Biosearch Technology) and phosphorylcholine (PC)(3)-OVA (catalog no. PC-5051-10, Biosearch Technology).
T Cell Responses and Intracellular Staining for Cytokines
Spleen cells were isolated 14 days after immunization with collagen and adjuvant. Cells were then reactivated for 4 h in vitro using phorbol myristate acetate (50 ng/ml) and ionomycin (100 ng/ml). Cytokine production in CD4+ T cells was examined by intracellular staining as outlined below.
ELISA
Antigen-specific antibody levels were measured by ELISA as described previously (27). To detect anti-collagen antibodies, ELISA-grade bovine CII (catalog no. 2012, Chondrex) or the CB11 fragment of bovine CII (catalog no. 9007, Chondrex) was used as coating antigen. To detect CII-specific IgE, 30 μl of protein A-Sepharose was added first to 300 μl of 1:100 diluted serum and incubated overnight to adsorb and eliminate IgG. The supernatants were then processed for ELISA. To detect hapten-specific Ig, NP(4)-BSA (catalog no. N-5050-10, Biosearch Technology) or PC(5)-BSA (catalog no. PC-1011-10, Biosearch Technology) was utilized as coating antigen. High-affinity collagen-specific antibodies were detected using the chaotropic agent NaSCN (1.0–2.0 M) as described (38).
Flow Cytometry
Anti-CD3 (mAb 145-2C11), anti-CD4 (mAb RM4.5), anti-PD-1 (mAb J43), anti-FoxP3 (mAb FJK-16s), anti-IL-17A (mAb eBio 17B7), anti-IFN-γ (mAb XMG1.2), anti-IL-4 (mAb BVD6–24G2), streptavidin-eFluor 450, and isotype-matched control antibodies were from eBiosciences (San Diego, CA). Anti-T and anti-B cell activation antigen (mAb GL-7), anti-CXCR5 (mAb 2G8), and anti-Bcl-6 (mAb K112-91) were from BD Biosciences. Phycoerythrin-conjugated mCD1d/PBS57 was obtained from the National Institutes of Health Tetramer Facility at Emory University (Atlanta, GA). Monoclonal antibody directed against SAP (mAb 1A9) was generated in our laboratory (27). Flow cytometry was performed using the CyAn ADP analyzer (Beckman Coulter, Mississauga, Ontario, Canada), followed by data analysis with FlowJo (Ashland, OR). Intracellular staining for cytokines or FoxP3 was performed using a BD Cytofix/Cytoperm Plus fixation/permeabilization kit (BD Biosciences) as outlined by the manufacturer. Bcl-6 expression was analyzed as outlined elsewhere (28).
Transfections, Immunoprecipitations, and Immunoblot Analyses
BI-141 is an antigen-specific mouse T cell line (39). Derivatives stably expressing full-length Ly108 with or without SAP or with mutant SAP R78A were generated by transfection using a cDNA encoding mouse Ly108. To study Vav-1 tyrosine phosphorylation induced by self-ligation of Ly108, BI-141 transfectants were seeded at the same concentration and cultured overnight. Then they were quickly collected, washed, and lysed with 1× TNE buffer (50 mm Tris (pH 8.0), 1% Nonidet P-40, and 2 mm EDTA) supplemented with protease and phosphatase inhibitors. Lysates were then processed for immunoprecipitation with anti-Vav-1 antibodies and immunoblot analyses as described elsewhere (40).
Statistical Analyses
Data were analyzed with Student's t test. p = 0.05 or p < 0.05 were viewed as statistically significant.
RESULTS
Critical Role of SAP in Collagen-induced Arthritis
CIA is induced by immunization of susceptible mouse strains with type II collagen. It recreates many of the clinical features of human RA, including some of its systemic inflammatory signs. It depends on production of anti-collagen antibodies, which lead to tissue damage. Recent evidence indicated that TFH cells and, to a lesser extent, TH17 cells are also critical for disease pathogenesis. To better understand the role of SAP in autoimmune disorders, the involvement of SAP in CIA was examined (Fig. 1) (37, 41). For this purpose, we first backcrossed a constitutive SAP-deficient mouse (Sh2d1a−/) for > 12 generations to the DBA/1J background, which is susceptible to CIA. Given that the SAP-encoding gene is X-linked, hemizygous SAP-deficient males, as well as WT control male littermates, were used for experimentation.
FIGURE 1.

SAP deficiency fully protects against collagen-induced arthritis. A–E, mice (males, 8- to 10 weeks old) in the DBA/1J background were immunized with CII in complete Freund adjuvant containing M. tuberculosis as detailed under “Experimental Procedures.” The incidence of RA and the mean clinical score over time were determined by clinical examination (A). The number (n) of mice studied is shown in brackets. Error bars represent mean ± S.E. of the clinical score. Clinical scores were as follows: 0, no erythema and swelling; 1, erythema and mild swelling confined to the mid-foot (tarsals) or ankle joint; 2, erythema and mild swelling extending from the ankle to mid-foot; 3, erythema and moderate swelling extending from the ankle to the metatarsal joints; and 4, erythema and severe swelling encompassing the ankle, foot, and digits. The clinical score was the sum of the scores for the four paws. Photographs of representative animals (RA score of 3 for the control mouse and 0 for the SAP-deficient mouse) are shown on the right. B, maximal clinical score for each paw observed during the experiment period. Levels of antibodies against CII (C), high-affinity antibodies against the collagen fragment CB11 (measured by addition of 1.5 m NaSCN during ELISA (D), and IgE antibodies against CII (E) were measured 105 days after immunization as detailed under “Experimental Procedures.” Individual mice are depicted by individual symbols, and the horizontal line shows mean values. p values are also shown. F–H, mice were immunized a second time (boost) with CII 105 days after the first immunization. RA incidence and mean clinical score (F) and maximal clinical score for each paw (G) were determined. H, levels of antibodies against CII were measured 70 days after the boost. CTL, control mice; SAP KO, SAP-deficient mice.
To trigger CIA, mice were immunized with a standard dose (100 μg) of bovine CII in complete Freund adjuvant containing M. tuberculosis. Disease development was then monitored clinically (Fig. 1A). Although 100% of WT mice developed CIA by ∼60 days, none of the SAP-deficient mice developed the disease. This was true whether disease incidence or mean clinical score was examined. It was also noted when the maximal clinical score of individual paws was analyzed (Fig. 1B). The lack of disease in SAP-deficient mice correlated with a marked reduction of the abundance of anti-collagen antibodies (Fig. 1, C and D, and data not shown). This was especially true for high-affinity IgG2a and IgG2b directed against a fragment of collagen, CB11 (Fig. 1D). Such antibodies were reduced by > 95% in SAP-deficient animals and are known to have a key role in disease pathogenesis (42). High-affinity antibodies were measured by adding the chaotropic agent NaSCN during ELISA. A pronounced diminution (by nearly 100%) in collagen-specific IgE was also seen (Fig. 1E). The resistance of SAP-deficient mice to CIA was also observed when mice were immunized a second time with collagen (Fig. 1, E–H). Hence, SAP was absolutely necessary for the pathogenesis of CIA. It was also critical for induction of anti-collagen antibodies.
Expression of SAP in T Cells, Not in B Cells, Is Required for CIA
To ascertain in which cell type(s) expression of SAP was needed to induce CIA, mice carrying a conditional (floxed) allele of the SAP-encoding gene (Sh2d1afl/) were also backcrossed to DBA/1J (27) (Fig. 2). The resulting mice were then bred with mice expressing the Cre recombinase under the control of the Cd4 promoter or the Mb1 promoter. These two promoters are efficient at deleting floxed alleles in T cells and B cells, respectively (33, 36). Note that the F1 progeny of a cross between WT C57BL/6 and WT DBA/1J was highly susceptible to CIA and produced anti-collagen antibodies of all Ig subsets (Fig. 2, A–D, and data not shown). Consequently, F1 males born from crosses between Sh2d1afl/ mice (in the DBA/1J background) and Cre-expressing mice (in the C57BL/6 background) were used for these experiments.
FIGURE 2.

SAP expression in T cells, not in B cells, is required for collagen-induced arthritis. A–D, F1 mice were generated from a cross between WT C57BL/6 and WT DBA/1J (B6.DBA, A) mice. Induction of RA (B and C) and anti-collagen (CII) antibodies (D) was monitored as detailed in Fig. 1. Antibody levels were measured 110 days after immunization. Individual mice are depicted by individual symbols, and the horizontal line shows mean values. p values are shown. E–H, mice expressing a conditional allele of the SAP-encoding gene (Sh2d1afl/, SAP cKO) were bred with mice expressing the Cre recombinase under the control of the CD4 promoter (Cd4-cre) (E). Induction of RA (F and G) and anti-CII antibodies (H) was monitored. Antibody levels were measured 28 days after immunization. I–L, Sh2d1afl/ mice were bred with mice expressing the Cre recombinase under the control of the MB1 promoter (Mb1-cre) (I). Induction of RA (J and K) and anti-CII antibodies (L) was monitored. Antibody levels were measured 28 days after immunization. CTL, control mice.
Breeding of Sh2d1afl/ mice with Cd4-Cre dramatically reduced the susceptibility to CIA (Fig. 2, E–G). In the experiment depicted, only one of five mice developed disease, and this disease was milder than that observed in control mice. The disease occurring in this mouse was presumably because of incomplete deletion of the floxed allele. Expression of Cd4-Cre also dramatically reduced production of anti-collagen antibodies (Fig. 2H). Efficient deletion of SAP in T cells was confirmed by intracellular staining with anti-SAP antibodies, as reported elsewhere (27) (data not shown). In comparison, breeding of Sh2d1afl/ mice with Mb1-Cre had no impact on disease development or production of antibodies against collagen (Fig. 2, I–L). These data implied that SAP expression in T cells, not in B cells, was required for development of CIA and production of anti-collagen antibodies.
The SAP-Fyn Interaction Is Not Critical for Induction of CIA at Standard Dose of Collagen
To understand the molecular mechanism by which SAP promoted CIA, disease susceptibility was also examined using a “knock-in” mouse strain in which SAP is expressed in normal amounts but cannot recruit Fyn (SAP arginine 78-to-alanine 78 (R78A) mouse) (13) (Fig. 3). The capacity of SAP to bind Fyn is critical for TH2 cytokine production and NK-T development but not TFH cell differentiation (9, 10, 12–17). SAP R78A mice were backcrossed to DBA/1J, and mice were immunized with the standard dose of collagen (100 μg). Strikingly, the SAP R78A mutation (Fig. 3, A and B) had no impact on disease incidence. 100% of SAP R78A mice developed CIA. However, these animals exhibited a small reduction of disease severity. SAP R78A mice also had no defect in the ability to develop anti-collagen IgG2a, although IgG2b was slightly reduced (by ∼40%) (Fig. 3C). A more pronounced reduction (by ∼80%) of anti-collagen IgE was seen (Fig. 3D).
FIGURE 3.
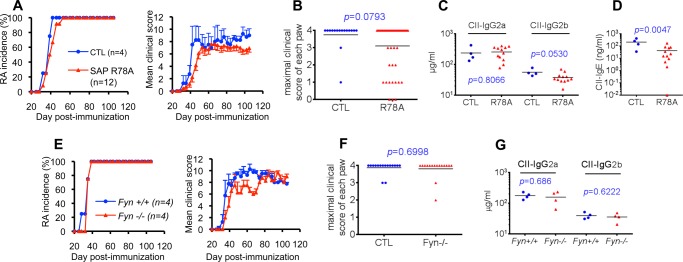
SAP-Fyn interaction is not required for CIA induction at the standard dose of collagen. A–D, SAP R78A mice. A, SAP R78A mice in DBA/1J background were immunized with 100 μg of CII. RA development was monitored as detailed in Fig. 1. B, the maximal clinical score for each paw is shown. CII-specific IgG2a and IgG2b (C) and IgE (D) were assayed 56 days after immunization. Individual mice are depicted by individual symbols, and the horizontal line shows mean values. p values are also shown. E–G, Fyn-deficient mice. Fyn-deficient mice in the DBA/1J background were immunized with 100 μg of CII. RA development (E) and maximal clinical score for each paw (F) were monitored. G, CII-specific IgG2a and IgG2b were measured 56 days after immunization. CTL, control mice.
Although the R78A mutation abolishes the ability of SAP to bind Fyn in in vitro binding studies (9, 10), it remains possible that a small amount of residual binding persists. Therefore, to ensure that lack of a pronounced effect of the SAP R78A mutation on CIA incidence was not due to residual Fyn-binding, similar experiments were conducted with Fyn-deficient mice (35) (Fig. 3, E–G). Like the SAP R78A mutation, lack of Fyn had little or no impact on CIA incidence or induction of anti-collagen antibodies. However, as was the case for the SAP R78A mutation, a small decrease in disease severity was seen.
These observations indicated that the SAP-Fyn interaction was largely dispensable for induction of CIA by the standard dose of collagen. However, it was required for maximal induction of some subsets of anti-collagen antibodies, in particular IgE.
The SAP-Fyn Association Is More Critical for CIA Induction at a Low Dose of Collagen
Even though the susceptibility to CIA was nearly preserved in SAP R78A mice, a small decrease in disease severity was observed. To address the possibility that the high dose of antigen (100 μg) used for immunization masked more pronounced differences, mice were also immunized with a lower dose of collagen. Thus, WT mice and SAP R78A mice were immunized with 2 μg of collagen and were monitored for disease development and production of anti-collagen antibodies as detailed above (Fig. 4). This antigen dose is likely closer to the amounts of endogenous antigens leading to autoimmunity in humans.
FIGURE 4.

SAP-Fyn interaction is critical for CIA induction at the low dose of collagen. SAP R78A mice in the DBA/1J background were immunized with 2 μg of CII. A, RA development was monitored as detailed in Fig. 1. B, maximal clinical score for each paw. CII-specific IgG2a (C), IgG2b (D), and IgE (E) were measured at the indicated times after immunization. Individual mice are depicted by individual symbols, and the horizontal line shows mean values. p values are also shown. CTL, control mice.
Under these conditions, fewer WT mice developed CIA (Fig. 4, A and B). Indeed, only ∼50% of WT animals developed disease in comparison to 100% when the standard dose of collagen was used (Fig. 1). In addition, in WT mice, onset of disease was slower, and disease intensity was lower (Fig. 4A). More importantly, when the lower dose of collagen was used, SAP R78A mice displayed a clearly diminished disease incidence (∼10%), in comparison to WT animals. Furthermore, although SAP R78A mice did not exhibit compromised production of anti-collagen IgG2a and IgG2b at day 28 after immunization, they displayed a significant reduction of these antibodies at days 56 (by ∼45–60%) and 105 (by ∼55–70%) (Fig. 4, C and D). A more pronounced decrease in anti-collagen IgE (by ∼80–90%) was also seen (Fig. 4E). These data implied that the SAP-Fyn interaction was more critical for induction of CIA and production of anti-collagen antibodies when a low dose of collagen was used for immunization.
The SAP-Fyn Interaction Is Dispensable for Full Differentiation of TFH Cells
Adoptive transfer studies using SAP-deficient T cells retrovirally transduced by a SAP R78A-encoding cDNA suggested that the SAP-Fyn interaction is not critical for TFH cell-dependent GC reaction and antibody production (15, 16). To examine whether this is also the case in SAP R78A mice, we quantitated TFH cells in spleens of WT, SAP-deficient, and SAP R78A mice immunized with collagen (Fig. 5). This was done 10 days after immunization at a time when the GC reaction was peaking. TFH cells were defined as CD4+CXCR5+PD-1+ cells (21, 43). There was no difference in the total number of TFH cells between any of these mice (Fig. 5A). There was also no significant difference in the number of “GC TFH cells,” which additionally express high levels of the activation marker GL-7 (17, 26) (Fig. 5B). In contrast, SAP-deficient mice exhibited a pronounced reduction (by ∼75%) of GC TFH cells expressing high levels of Bcl-6, the TFH cell lineage commitment factor (Fig. 5, C and D). These cells represent the most differentiated form of TFH cells and are significantly reduced in numbers in SAP-deficient animals (26, 28). However, the abundance of these cells was not compromised in SAP R78A mice. Similar results were obtained whether mice were immunized with the standard dose (Fig. 5, A–D) or the lower dose (E) of collagen.
FIGURE 5.

The SAP-Fyn interaction is not required for full TFH cell differentiation. A–D, mice were immunized with 100 μg of CII. Ten days later, splenocytes were isolated and analyzed by flow cytometry. A, numbers of TFH cells (defined as CD4+CXCR5+PD-1+ cells) in the spleen were determined. The gating strategy is shown on the left, whereas values for all mice are depicted by individual symbols on the right. The horizontal lines show mean values. p values are also shown. B, numbers of GC TFH cells (GL-7hiCD4+CXCR5+PD-1+ cells) were determined. The gating strategy is shown on the left, whereas numbers of GL-7lo and GL-7hi TFH cells for all mice are depicted on the right. C and D, Bcl-6 expression in GL-7lo and GL-7hi TFH cells was determined by intracellular staining. Histograms from representative mice are shown in C, and numbers of Bcl-6hiGL-7hi TFH cells for all mice are depicted in D. Bcl-6hi was defined as a mean fluorescence intensity of > 200. E, the experiment was performed as detailed in D, except that mice were immunized with 2 μg of CII, and analyses were performed 7 days after immunization. F and G, numbers of GC B cells (CD4−GL-7hiBcl-6hi cells) in spleens of mice immunized with 100 μg of CII were determined. The gating strategy is shown in F, whereas values for individual mice are depicted by individual symbols in G. SS, side scatter; Lin, linear; SPC, spleen cells; CTL, control mice; R78A, SAP R78A mice; KO, SAP-deficient mice.
We also examined the impact of the SAP R78A mutation on the generation of GC B cells (Fig. 5, F and G). GC B cells are CD19+ and correspond to the CD4-negative subpopulation of GL-7hiBcl-6hi cells (26). The number of GC B cells was markedly reduced (by ∼80%) in SAP-deficient mice in comparison to WT mice, as reported (15, 17, 26, 28). Nevertheless, the abundance of these cells was not diminished in SAP R78A mice.
These findings indicated that the SAP-Fyn association was not crucial for induction of full TFH cell differentiation and GC reaction following immunization with collagen. This likely explained the preserved susceptibility of SAP R78A mice, but not SAP-deficient mice, to CIA. Furthermore, it supported the idea that TFH cells played a critical role in CIA pathogenesis.
The SAP-Fyn Interaction Is Not Required for Maintenance or Reactivation of TFH Cell-dependent Antibody Responses
Although SAP R78A mice were prone to develop CIA, their disease susceptibility was clearly diminished compared with WT mice, particularly at the low dose of collagen. This observation raised the possibility that additional cellular mechanisms that depend on the SAP-Fyn interaction were also implicated in CIA pathogenesis. These mechanisms could implicate either TFH cells when these cells are fully differentiated or T cell types other than TFH cells.
In the case of TFH cells, the SAP-Fyn interaction could be needed for maintenance of TFH cell-dependent B cell responses after the initial induction phase is completed. Alternatively, it could be required for reactivation of TFH cell-dependent B cell responses by endogenous collagen released through tissue damage during disease progression. To address these possibilities, we switched to a hapten immunization protocol where T cell-dependent antibody responses and GC reaction are SAP-dependent (15, 26) (Fig. 6). SAP R78A mice and SAP-deficient mice were first immunized with a low dose (2 μg) of NP-OVA in the presence of alum (Fig. 6A). The ability of B cells to respond to the hapten, NP, is dependent on a TFH cell response against OVA. After 156 days, mice were immunized again with NP-OVA in addition to a second hapten, PC, also coupled to OVA. Production of antibodies against NP and PC was then examined. Production of anti-PC antibodies is facilitated by previous generation of OVA-specific memory TFH cells in response to primary immunization with NP-OVA.
FIGURE 6.

SAP-Fyn interaction is not required for maintenance or reactivation of TFH cell-dependent antibody responses. A, mice in the C57BL/6 background were immunized with a low dose (2 μg) of NP-OVA in the presence of alum. After 156 days, they were immunized again with low doses (2 μg each) of NP-OVA and PC-OVA. NP-specific IgM (B) and IgG1 (C) and high-affinity anti-NP IgG1 (measured by addition of 1.5 m NaSCN during ELISA, D) were measured at the indicated times after primary immunization with NP-OVA. Moreover, NP-specific IgG1 (E) and PC-specific IgG1 (F) were assayed 14 days (that is, on day 168) after the secondary immunization. Individual mice are depicted by individual symbols, and the horizontal line shows mean values. p values are shown. CTL, control mice; R78A, SAP R78A mice; KO, SAP-deficient mice.
As expected, SAP R78A mice displayed no significant reduction of production of anti-NP antibodies, either IgM or IgG1, after primary immunization with NP-OVA (Fig. 6, B and C). This was also the case for high-affinity IgG1 against NP, which was detected using NaSCN in ELISA (Fig. 6D). It was seen up to 154 days after primary immunization. In contrast, SAP-deficient mice exhibited a pronounced compromise in these responses (Fig. 6, B–D). More importantly, SAP R78A mice also exhibited no defect in the ability to increase levels of anti-NP antibodies or produce anti-PC antibodies after secondary immunization (Fig. 6, E and F). Once again, SAP-deficient mice had a severe reduction of these responses. These findings implied that the ability of SAP to interact with Fyn was not needed to maintain or reactivate TFH cell-dependent B cell responses to an antigen.
SAP-Fyn Interaction Is Not Needed for TH17 or TH2 Cytokine Production but Is Required for NK-T Cell Development
Another possible explanation for the reduced susceptibility of SAP R78A mice to CIA was that another T cell subset, in which the SAP-Fyn interaction plays a critical role, was implicated in disease pathogenesis. Along these lines, a recent report provided evidence that TH17 cells carry out an important function, albeit not as central as that of TFH cells, in CIA induction (44). Furthermore, previous studies showed that SAP expression and its ability to recruit Fyn are necessary for NK-T cell development and TH2 cell-mediated IL-4 production in vitro (1–4). These cell types can also affect the course of autoimmune diseases.
To ascertain whether SAP was needed for TH17 cell functions, SAP-deficient mice and SAP R78A mice in the DBA/1J background were immunized with collagen and adjuvant. After 14 days, splenic CD4+ T cells were restimulated with phorbol 12-myristate 13-acetate and ionomycin, and IL-17 production was detected by intracellular staining for IL-17A (Fig. 7A). There was no decrease in the number of IL-17A-producing CD4+ T cells in either SAP-deficient mice or SAP R78A mice in comparison to WT mice. There was also no reduction of the number of IFN-γ-producing cells in these mice (Fig. 7B). Likewise, the number of FoxP3-positive regulatory T cells, an immunosuppressive subset of CD4+ T cells, was not altered (Fig. 7C). In contrast, SAP-deficient mice and SAP R78A mice exhibited severe and partial decreases in NK-T cell numbers, respectively (Fig. 7D). This confirmed previous findings that SAP and the ability of SAP to bind Fyn are needed for NK-T cell development (12, 45–47).
FIGURE 7.
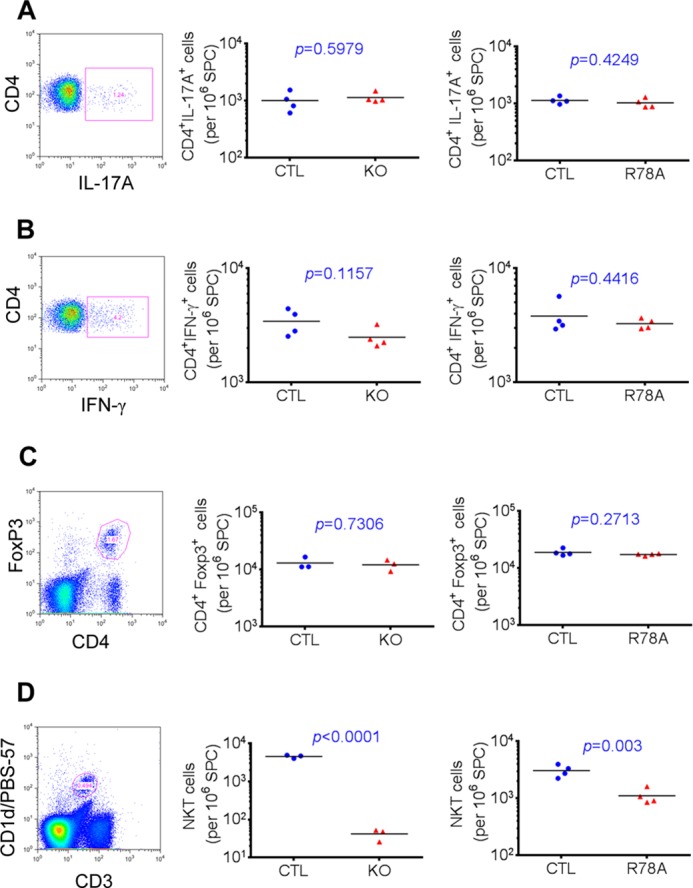
SAP is not needed for TH7 cytokine production. Mice in the DBA/1J background were immunized with 100 μg of collagen II. On day 14, splenocytes were isolated and reactivated at 37 °C for 4 h in the presence of Golgi Plug, phorbol myristate acetate, and ionomycin. Relative numbers of CD4+ T cells producing IL-17A (A) and IFN-γ (B) were determined by intracellular staining. Isolated splenocytes were also analyzed by flow cytometry for detection of regulatory T cells (C), which are CD4+FoxP3+, and NK-T cells (D), which are CD3+CD1d/PBS-57+. In all cases, the gating strategy is depicted on the left. On the right, values for individual mice are depicted by individual symbols, and the horizontal line shows mean values. p values are also shown. SPC, spleen cells; CTL, control mice; KO, SAP-deficient mice; R78A, SAP R78A mice.
We also tested whether the SAP-Fyn interaction was needed for the ability of T cells to produce the TH2 cytokine IL-4 in vivo (Fig. 8). To this end, mice were immunized with the low or high dose of collagen in the presence of adjuvant, and production of IL-4 was analyzed as detailed for Fig. 7. No defect in IL-4 production was observed when mice were immunized with the low dose of collagen (Fig. 8A). This was also the case for IL-17 and IFN-γ. Similar results were obtained when mice were injected with the high dose of collagen (Fig. 8B).
FIGURE 8.
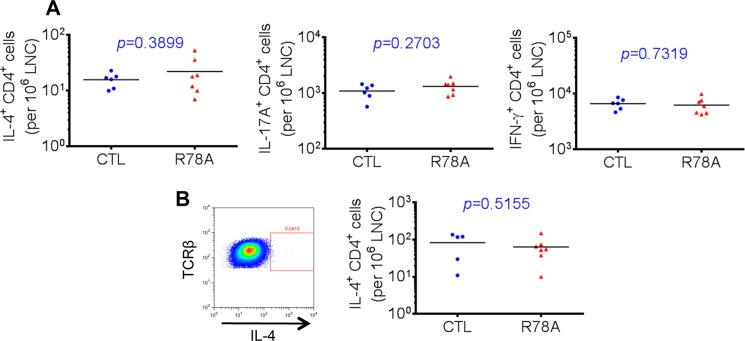
SAP-Fyn interaction is not needed for IL-4 production. Mice in the DBA/1J background were immunized with either a low dose (2 μg) or high dose (100 μg) of CII supplemented with 200 μg of M. tuberculosis in the hind footpads. On day 18, draining lymph node T cells were isolated and reactivated for 8 h at 37 °C in the presence of phorbol myristate acetate and ionomycin, followed by intracellular staining for cytokines. Golgi Stop was added during the last 6 h of stimulation. A, low dose of CII. Relative numbers of CD4+ T cells producing IL-4, IL-17A, and IFN-γ were determined by intracellular staining. B, high dose of CII. Relative numbers of CD4+ T cells producing IL-4 were determined by intracellular staining. Values for individual mice are depicted by individual symbols, and the horizontal line shows mean values. p values are also shown. CTL, control mice; R78A, SAP R78A mice.
Together, these findings revealed that the SAP-Fyn interaction was not needed for TH17 or TH2 cytokine production in response to collagen immunization. However, it was needed for NK-T cell development.
SAP-Fyn Interaction Promotes Tyrosine Phosphorylation of Vav-1 during Homotypic Cell-Cell Interactions
SAP and associated Fyn promote NK-T cell development by mediating activating signals that are triggered during homotypic T cell-T cell interactions in the thymus (12, 45–47). Ly108 appears to be the predominant SLAM family receptor triggering the function of SAP in this setting. Ly108 is a self-ligand.
To identify the downstream effector(s) of SAP-Fyn in this setting, we switched to a T cell line system (Fig. 9). This was necessary given that only a small fraction of thymocytes represents precursors of NK-T cells and that these cells cannot be isolated in sufficient numbers to allow signaling studies.
FIGURE 9.
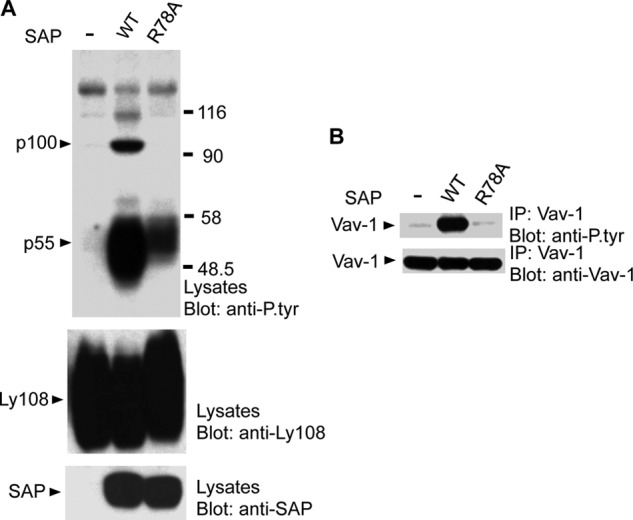
SAP-Fyn association is required for Vav-1 tyrosine phosphorylation during homotypic cell-cell interactions. BI-141 T cell transfectants expressing Ly108 in the absence of SAP or in the presence of WT SAP or SAP R78A were seeded overnight at the same confluency to allow homotypic cell-cell interactions and Ly108 self-ligation and then lysed. A, lysates were immunoblotted with antibodies against phosphotyrosine, Ly108, or SAP. The positions of the predominant substrates showing augmented tyrosine phosphorylation in cells expressing WT SAP are shown on the left. B, lysates were immunoprecipitated (IP) with antibodies against Vav-1 and probed by immunoblotting with antibodies against phosphotyrosine or Vav-1.
Hence, full-length Ly108 was expressed in the T cell line BI-141 in the absence of SAP or in the presence of either wild-type SAP or SAP R78A. Cells were then incubated at a fixed density overnight to enable homotypic cell-cell interactions and Ly108 self-ligation. Analyses of total cell lysates showed that wild-type SAP, but not SAP R78A, triggered a prominent increase in protein tyrosine phosphorylation (Fig. 9A). This increase implicated two predominant substrates: a p55, which represented Ly108 itself, and a p100.
The molecular mass of p100 was consistent with that of Vav-1, an exchange factor activated by tyrosine phosphorylation (48). To determine whether this was the case, Vav-1 was recovered by immunoprecipitation and probed by anti-phosphotyrosine immunoblotting (Fig. 9B). This experiment showed that tyrosine phosphorylation of Vav-1 was indeed markedly enhanced in cells expressing wild-type SAP but not SAP R78A. Because Vav-1 is required for NK-T cell development (32), these data suggested that the SAP-Fyn pathway was promoting NK-T cell development, at least in part, by activating Vav-1 during homotypic cell-cell interactions.
SAP Is Required for Progression, but Not for Maintenance, of CIA
It has been proposed that pharmacological blockade of the SAP pathway may be useful to treat ongoing antibody-mediated autoimmune diseases in humans (1–4). To test this idea, we ascertained the role of SAP in progression and maintenance of CIA after initiation of the disease (Figs. 10 and 11). For this purpose, conditional SAP-deficient mice (Sh2d1afl/) were bred with mice expressing a TAM-inducible Cre (UBC-Cre-ERT2) (34) (Fig. 10A). In the absence of TAM, this mutant Cre is cytosolic and inactive. However, in the presence of TAM, it translocates to the nucleus and triggers Cre-mediated deletion. Although SAP expression was normal in the absence of TAM treatment, it was reproducibly eliminated in > 90–95% of CD4+ T cells after a 5 day-treatment with TAM (Fig. 10, B and C, and data not shown). Ablation of SAP was stable for up to 100 days after TAM treatment (Fig. 10, B and C, and data not shown).
FIGURE 10.
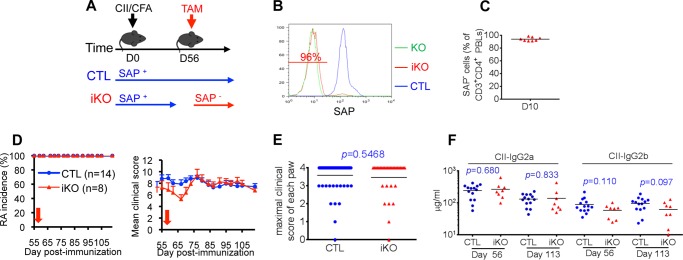
SAP is not required for maintenance of collagen-induced arthritis. A, mice expressing the conditional (“inducible”) allele of the SAP-encoding gene (Sh2d1afl/, SAP iKO) were bred with mice expressing the TAM-responsive Cre recombinase under the control of the ubiquitin C promoter (UBC-creERT2). They were then immunized with CII and complete Freund adjuvant (CFA). After 56 days, SAP was deleted by treatment of mice with TAM. B, expression of SAP in representative mice was examined by intracellular staining and flow cytometry using peripheral blood lymphocytes gated on CD3+CD4+ cells. Green line, SAP-deficient mouse; red line, SAP iKO mouse; blue line, control (CTL) mouse. The percentage of SAP-negative cells in the SAP iKO mouse is indicated. C, percentages of SAP-negative CD3+CD4+ T cells in multiple SAP iKO mice treated with TAM. Analyses were performed 10 days after TAM treatment. Induction of RA (D and E) and anti-CII antibodies (F) was monitored as detailed in Fig. 1. The time of TAM treatment is shown by a red arrow. Values for individual mice are depicted by individual symbols, and the horizontal line shows mean values. p values are also shown.
FIGURE 11.

SAP is required for progression of collagen-induced arthritis. A, this experiment was performed as detailed in Fig. 10, except that TAM treatment was initiated 29 days after immunization. B, anti-CII antibodies were measured 10 days (D10) after immunization of wild-type mice with CII when the clinical symptoms were not yet observed. C, high-affinity IgG2a antibodies against CII were measured on day 14 after CII injection using the chaotropic agent NaSCN. D, high-affinity IgG2a and IgG2b antibodies against CII were measured on the indicated days after CII injection. Induction of RA (E and F) and anti-CII antibodies (G) was monitored as detailed in Fig. 1. Time of TAM treatment is shown by a red arrow. Values for individual mice are depicted by individual symbols, and the horizontal line shows mean values. p values are also shown. H, the abundance of high-affinity anti-CII IgG2a was determined by performing ELISA in presence of progressively higher concentrations of NaSCN. CTL, control mice; iKO, inducible KO mice; CFA, complete Freund adjuvant.
The role of SAP in the various phases of CIA was examined by feeding mice with TAM at different times after collagen immunization. In the first group of mice, TAM was supplied 56 days after collagen injection when all mice had established clinical disease (Fig. 10, D and E). Under these conditions, there was no appreciable difference in disease incidence or severity between control mice, in which SAP was not deleted, and SAP-deficient mice. There was also no significant effect on the levels of anti-collagen antibodies (Fig. 10F).
In the second group of mice, TAM was given 29 days after collagen immunization (Fig. 11A). At that time, clinical manifestations of CIA had not yet developed or were minimal. However, anti-collagen antibodies were already detectable (Fig. 11, B–D). Indeed, anti-collagen IgM, IgG2a, or IgG2b were already detectable at day 10 (Fig. 11B). However, high-affinity antibodies were only detectable after day 28 (Fig. 11, C and D). Under these conditions, SAP deletion had a small impact on disease incidence (Fig. 11, E and F). However, disease severity was more significantly reduced, implying that CIA progression was compromised. Although there was no significant reduction of anti-collagen antibody levels in SAP-deleted mice, a subset of these animals exhibited lower levels of anti-collagen antibodies at day 106 (Fig. 11G). We also examined the impact of SAP deletion on the affinity of anti-collagen antibodies using progressively higher concentrations of NaSCN during the ELISA (Fig. 11H). Although there was a significant increase in the abundance of high-affinity antibodies between days 28 and 106 in control mice, this was not the case for SAP-deleted mice.
Thus, SAP was required for progression, but not for maintenance, of CIA. This requirement correlated with a role of SAP in the augmentation of high-affinity antibodies against collagen.
DISCUSSION
SAP plays a critical role in initiation and progression of antibody production as well as in generation of memory B cells and long-lived antibody-secreting cells in response to T cell-dependent protein antigens (1–4). It is also critical for development of autoimmunity in mouse models of lupus (29–31). These two activities were postulated to reflect the ability of SAP to promote full differentiation of TFH cells, which are required for GC reaction. However, SAP also carries out important functions in other T cell types, including TH2 cells and NK-T cells, which can also regulate B cell responses.
Here we examined further the role and mechanism of action of SAP in antibody-mediated autoimmunity. To this end, we studied CIA, a well established mouse model of T cell-dependent antibody-mediated autoimmunity. Our studies showed that constitutive SAP deficiency fully prevented the ability of DBA/1J mice to develop CIA. This was seen whether mice were immunized once or twice with collagen. This effect correlated with a marked defect in production of anti-collagen antibodies, including high-affinity IgG2a and IgG2b, which play a key role in CIA pathogenesis (37, 41). An even greater reduction of collagen-specific IgE was seen.
To identify the cell type in which SAP expression was required for CIA, we utilized a cell type-specific SAP-deficient mouse in which the Sh2d1a gene was deleted either in T cells or B cells. Removal of SAP in T cells, but not in B cells, compromised development of CIA and induction of anti-collagen antibodies. These findings were consistent with previous data obtained with hapten immunization protocols in which loss of SAP in T cells, but not in B cells, resulted in compromised antibody production and GC reaction (15, 27). Thus, the role of SAP in T cell-dependent B cell-mediated immunity and autoimmunity is consistently due to the function of SAP in T cells.
Subsequent studies with mice expressing a SAP mutant unable to recruit Fyn (SAP R78A mice) or with mice lacking Fyn showed that, at the dose of collagen typically used to induce CIA (100 μg), the role of SAP in CIA was largely Fyn-independent. Indeed, unlike SAP-deficient mice, all SAP R78A mice developed disease. This finding suggested that TFH cells, which require SAP but not the SAP-Fyn interaction for full differentiation (1–4), were critical to induce CIA. Other T cell subsets in which SAP plays a critical role, namely TH2 cells and NK-T cells, are much more dependent on the SAP-Fyn interaction for their function or development, respectively (1–4). This notion was further supported by the observation that SAP deficiency, but not the SAP R78A mutation, prevented the generation of Bcl-6hiGL-7hi TFH cells, the most differentiated subset of TFH cells.
Combined with earlier findings with mouse lupus models (29–31), these data support the idea that SAP has a central role in autoimmune diseases mediated by TFH cells and antibodies. Given that SAP is required for human TFH cell differentiation and that increased numbers of TFH cells and autoantibodies are seen in RA, systemic lupus erythematosus, thyroiditis, dermatomyositis, and Sjögren syndrome in humans (49–54), it is reasonable to propose that SAP also has a critical role in the pathogenesis of human autoimmune diseases. In mice, SAP may also have an important contribution in another recently described model of RA (55). This model was induced by transfer of primed OVA-specific CD4+ T cells and heat-aggregated OVA and was seemingly mediated by TFH cells. It should be pointed out, however, that some mouse RA models do not appear to be primarily caused by TFH cells. Models such as SKG and K/BxN seem largely mediated by other T cell subsets such as TH17 cells and, thus, could be much less dependent on SAP (56, 57).
Although induction of CIA at the standard dose of collagen was largely maintained in SAP R78A mice, a prominent defect in disease triggering was observed when a lower dose of collagen (2 μg) was utilized. In this situation, ∼50% of WT animals developed CIA, whereas only ∼10% of SAP R78A mice exhibited the disease. SAP R78A animals also showed reduced levels of anti-collagen IgG2a, IgG2b, and IgE in comparison to WT mice. The latter effects became more marked as the time interval after immunization increased. Thus, the ability of SAP to recruit Fyn was more critical for induction of CIA and anti-collagen antibodies when a lower dose of antigen was utilized. This dose of collagen is likely more akin to the amounts of self-antigens triggering autoimmune diseases in humans.
To explain the requirement of the SAP-Fyn interaction at the lower dose of collagen, several possibilities were considered. First, we found no indication that the association between SAP and Fyn was critical for full TFH cell differentiation, as measured by production of Bcl-6hiGL-7hi TFH cells and GC B cells, even at the low dose of antigen. Second, with sequential immunization by two distinct haptens coupled to the same antigen, there was also no evidence that the SAP-Fyn interaction was needed for maintenance or reactivation of antigen-specific TFH cells. Third, there was no indication that SAP was necessary for the function of TH17 or TH2 cells, which are also implicated in the autoimmunity (18, 44). Although it is possible that loss of the SAP-Fyn interaction interfered with the physiology of TFH cells, TH17 cells, or TH2 cells in an as yet unappreciated way, it is also plausible that the SAP R78A mutation affected the function of one or more other T cell types implicated in disease pathogenesis. Future studies in which SAP is selectively eliminated in individual T cells subsets such as TFH cells, TH17 cells, TH2 cells, or other T cells are needed to resolve adequately this issue. Unfortunately, the Cre transgenic mice available at this time do not allow such refined analyses.
One possibility is that the defect in NK-T cell development caused by loss of the SAP-Fyn interaction was responsible for the reduced CIA severity at the low dose of collagen. NK-T cells are activated by endogenous or exogenous lipid antigens (18). There are contradictory data regarding their involvement in CIA. Although initial reports suggested that NK-T cells have a pathogenic role in the disease (58), a more recent study in which the genetic background of mice was carefully controlled indicated that these cells have a protective role (59). Nonetheless, there is also published evidence that lipid-activated NK-T cells can provide help to protein antigen-specific B cells and augment B cell proliferation and antibody production (43). Moreover, activated NK-T cells were found to stimulate B cell proliferation outside GCs (60). Therefore, partial loss of NK-T cells in SAP R78A mice could result in compromised B cell responses outside GCs, thereby leading to reduced CIA induction. The impact of this defect could be more obvious when limiting doses of antigen are used for immunization.
Our data also suggest that the SAP-Fyn pathway promotes NK-T cell development, at least in part, by augmenting tyrosine phosphorylation and activation of Vav-1. In support of this, we found that self-engagement of the SLAM family receptor Ly108 in the context of homotypic cell-cell interactions, as occurs during NK-T cell development in the thymus, resulted in pronounced Vav-1 tyrosine phosphorylation. This effect was dependent on the ability of SAP to bind Fyn. Vav-1 is an exchange factor involved in Rac activation and plays a key role in NK-T cell development (32, 48). Hence, as was reported for NK cells (6), Vav-1 may be a key effector of the SAP-Fyn pathway in thymocyte precursors, leading to NK-T cell development.
Lastly, we used the CIA model to elucidate whether SAP was needed for progression and maintenance of autoimmune diseases mediated by antibodies. Using TAM-induced deletion of the SAP-encoding gene, it was observed that removal of SAP when CIA was clinically established (at day 56) had no effect on disease. However, when SAP was eliminated soon after collagen immunization (at day 29), at a time when anti-collagen antibodies were detectable but arthritis was clinically absent or minimal, there was attenuated disease progression. These data indicated that SAP was required not only for initiation but also for progression of CIA. Nonetheless, it had no appreciable role in disease maintenance. Such observations were consistent with our previous findings in hapten immunization experiments that SAP is required for initiation and progression, but not maintenance, of full TFH cell differentiation, T cell-dependent antibody production, and GC reaction (26). In all likelihood, when pathogenic long-lived antibody-secreting cells and memory B cells are established, TFH cells (and possibly other T cell subsets) are no longer critical for autoimmunity.
In summary, our data showed that SAP was critical for induction and progression, but not maintenance, of CIA. This function was the result of the action of SAP in T cells, not in B cells. Our findings also revealed that the ability of SAP to bind Fyn was largely dispensable for triggering of CIA when a standard dose of collagen was used. This correlated with a lack of requirement of the SAP-Fyn interaction for full TFH cell differentiation and GC reaction. However, the SAP-Fyn association was needed for maximal induction of CIA when a low dose of collagen was utilized. This influence could reflect a role of the SAP-Fyn association in as yet unappreciated aspects of TFH cell functions. Alternatively, it could result from the involvement of the SAP-Fyn interaction in other T cell types, such as NK-T cells. These findings also suggested the existence of qualitative, rather than purely quantitative, differences in the requirements for disease induction between the high and low doses of collagen.
Altogether, these findings further cemented the notion that the SAP pathway plays a pivotal role in the pathogenesis of antibody-mediated autoimmune conditions. Importantly, they also indicated that blockade of the SAP pathway may be a useful therapeutic modality against antibody-mediated autoimmunity during the early progression of autoimmunity. Indeed, in humans, the period of exposure to autoantibodies prior to onset of clinical manifestations, or between clinical episodes of autoimmune disease, may be quite long, perhaps months to years. Such patients may benefit from blockade of the SAP pathway.
Footnotes
- SLAM
- signaling lymphocytic activation molecule
- SAP
- signaling lymphocytic activation molecule-associated protein
- NK
- natural killer
- TFH
- follicular T helper
- RA
- rheumatoid arthritis
- GC
- germinal center
- CIA
- collagen-induced arthritis
- CII
- bovine type II collagen
- NP
- nitrophenyl.
REFERENCES
- 1. Schwartzberg P. L., Mueller K. L., Qi H., Cannons J. L. (2009) SLAM receptors and SAP influence lymphocyte interactions, development and function. Nat. Rev. Immunol. 9, 39–46 [DOI] [PubMed] [Google Scholar]
- 2. Veillette A., Dong Z., Latour S. (2007) Consequence of the SLAM-SAP signaling pathway in innate-like and conventional lymphocytes. Immunity 27, 698–710 [DOI] [PubMed] [Google Scholar]
- 3. Calpe S., Wang N., Romero X., Berger S. B., Lanyi A., Engel P., Terhorst C. (2008) The SLAM and SAP gene families control innate and adaptive immune responses. Adv. Immunol. 97, 177–250 [DOI] [PubMed] [Google Scholar]
- 4. Ma C. S., Nichols K. E., Tangye S. G. (2007) Regulation of cellular and humoral immune responses by the SLAM and SAP families of molecules. Annu. Rev. Immunol. 25, 337–379 [DOI] [PubMed] [Google Scholar]
- 5. Kageyama R., Cannons J. L., Zhao F., Yusuf I., Lao C., Locci M., Schwartzberg P. L., Crotty S. (2012) The receptor Ly108 functions as a SAP adaptor-dependent on-off switch for T cell help to B cells and NKT cell development. Immunity 36, 986–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dong Z., Davidson D., Pérez-Quintero L. A., Kurosaki T., Swat W., Veillette A. (2012) The adaptor SAP controls NK cell activation by regulating the enzymes Vav-1 and SHIP-1 and by enhancing conjugates with target cells. Immunity 36, 974–985 [DOI] [PubMed] [Google Scholar]
- 7. Dong Z., Cruz-Munoz M. E., Zhong M. C., Chen R., Latour S., Veillette A. (2009) Essential function for SAP family adaptors in the surveillance of hematopoietic cells by natural killer cells. Nat. Immunol. 10, 973–980 [DOI] [PubMed] [Google Scholar]
- 8. Zhao F., Cannons J. L., Dutta M., Griffiths G. M., Schwartzberg P. L. (2012) Positive and negative signaling through SLAM receptors regulate synapse organization and thresholds of cytolysis. Immunity 36, 1003–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Latour S., Roncagalli R., Chen R., Bakinowski M., Shi X., Schwartzberg P. L., Davidson D., Veillette A. (2003) Binding of SAP SH2 domain to FynT SH3 domain reveals a novel mechanism of receptor signalling in immune regulation. Nat. Cell Biol. 5, 149–154 [DOI] [PubMed] [Google Scholar]
- 10. Chan B., Lanyi A., Song H. K., Griesbach J., Simarro-Grande M., Poy F., Howie D., Sumegi J., Terhorst C., Eck M. J. (2003) SAP couples Fyn to SLAM immune receptors. Nat. Cell Biol. 5, 155–160 [DOI] [PubMed] [Google Scholar]
- 11. Sayos J., Wu C., Morra M., Wang N., Zhang X., Allen D., van Schaik S., Notarangelo L., Geha R., Roncarolo M. G., Oettgen H., De Vries J. E., Aversa G., Terhorst C. (1998) The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature 395, 462–469 [DOI] [PubMed] [Google Scholar]
- 12. Nunez-Cruz S., Yeo W. C., Rothman J., Ojha P., Bassiri H., Juntilla M., Davidson D., Veillette A., Koretzky G. A., Nichols K. E. (2008) Differential requirement for the SAP-Fyn interaction during NK T cell development and function. J. Immunol. 181, 2311–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davidson D., Shi X., Zhang S., Wang H., Nemer M., Ono N., Ohno S., Yanagi Y., Veillette A. (2004) Genetic evidence linking SAP, the X-linked lymphoproliferative gene product, to Src-related kinase FynT in T(H)2 cytokine regulation. Immunity 21, 707–717 [DOI] [PubMed] [Google Scholar]
- 14. Cannons J. L., Yu L. J., Hill B., Mijares L. A., Dombroski D., Nichols K. E., Antonellis A., Koretzky G. A., Gardner K., Schwartzberg P. L. (2004) SAP regulates T(H)2 differentiation and PKC-theta-mediated activation of NF-kappaB1. Immunity 21, 693–706 [DOI] [PubMed] [Google Scholar]
- 15. Cannons J. L., Yu L. J., Jankovic D., Crotty S., Horai R., Kirby M., Anderson S., Cheever A. W., Sher A., Schwartzberg P. L. (2006) SAP regulates T cell-mediated help for humoral immunity by a mechanism distinct from cytokine regulation. J. Exp. Med. 203, 1551–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qi H., Cannons J. L., Klauschen F., Schwartzberg P. L., Germain R. N. (2008) SAP-controlled T-B cell interactions underlie germinal centre formation. Nature 455, 764–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCausland M. M., Yusuf I., Tran H., Ono N., Yanagi Y., Crotty S. (2007) SAP regulation of follicular helper CD4 T cell development and humoral immunity is independent of SLAM and Fyn kinase. J. Immunol. 178, 817–828 [DOI] [PubMed] [Google Scholar]
- 18. Murphy K. (2012) Immunobiology, 8th Ed., Garland Science, New York [Google Scholar]
- 19. Yu D., Vinuesa C. G. (2010) The elusive identity of T follicular helper cells. Trends Immunol. 31, 377–383 [DOI] [PubMed] [Google Scholar]
- 20. Fazilleau N., Mark L., McHeyzer-Williams L. J., McHeyzer-Williams M. G. (2009) Follicular helper T cells. Lineage and location. Immunity 30, 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Crotty S. (2011) Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663 [DOI] [PubMed] [Google Scholar]
- 22. Vinuesa C. G., Cyster J. G. (2011) How T cells earn the follicular rite of passage. Immunity 35, 671–680 [DOI] [PubMed] [Google Scholar]
- 23. Tangye S. G., Deenick E. K., Palendira U., Ma C. S. (2012) T cell-B cell interactions in primary immunodeficiencies. Ann. N.Y. Acad. Sci. 1250, 1–13 [DOI] [PubMed] [Google Scholar]
- 24. Ma C. S., Deenick E. K., Batten M., Tangye S. G. (2012) The origins, function, and regulation of T follicular helper cells. J. Exp. Med. 209, 1241–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cannons J. L., Qi H., Lu K. T., Dutta M., Gomez-Rodriguez J., Cheng J., Wakeland E. K., Germain R. N., Schwartzberg P. L. (2010) Optimal germinal center responses require a multistage T cell:B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity 32, 253–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhong M. C., Veillette A. (2013) Critical role of SAP in progression and reactivation but not maintenance of T cell-dependent humoral immunity. Mol. Cell Biol. 33, 1223–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Veillette A., Zhang S., Shi X., Dong Z., Davidson D., Zhong M. C. (2008) SAP expression in T cells, not in B cells, is required for humoral immunity. Proc. Natl. Acad. Sci. U.S.A. 105, 1273–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Choi Y. S., Kageyama R., Eto D., Escobar T. C., Johnston R. J., Monticelli L., Lao C., Crotty S. (2011) ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34, 932–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hron J. D., Caplan L., Gerth A. J., Schwartzberg P. L., Peng S. L. (2004) SH2D1A regulates T-dependent humoral autoimmunity. J. Exp. Med. 200, 261–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Komori H., Furukawa H., Mori S., Ito M. R., Terada M., Zhang M. C., Ishii N., Sakuma N., Nose M., Ono M. (2006) A signal adaptor SLAM-associated protein regulates spontaneous autoimmunity and Fas-dependent lymphoproliferation in MRL-Faslpr lupus mice. J. Immunol. 176, 395–400 [DOI] [PubMed] [Google Scholar]
- 31. Linterman M. A., Rigby R. J., Wong R. K., Yu D., Brink R., Cannons J. L., Schwartzberg P. L., Cook M. C., Walters G. D., Vinuesa C. G. (2009) Follicular helper T cells are required for systemic autoimmunity. J. Exp. Med. 206, 561–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chan G., Hanke T., Fischer K. D. (2001) Vav-1 regulates NK T cell development and NK cell cytotoxicity. Eur. J. Immunol. 31, 2403–2410 [DOI] [PubMed] [Google Scholar]
- 33. Hobeika E., Thiemann S., Storch B., Jumaa H., Nielsen P. J., Pelanda R., Reth M. (2006) Testing gene function early in the B cell lineage in mb1-cre mice. Proc. Natl. Acad. Sci. U.S.A. 103, 13789–13794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ruzankina Y., Pinzon-Guzman C., Asare A., Ong T., Pontano L., Cotsarelis G., Zediak V. P., Velez M., Bhandoola A., Brown E. J. (2007) Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1, 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stein P. L., Lee H. M., Rich S., Soriano P. (1992) pp59fyn mutant mice display differential signaling in thymocytes and peripheral T cells. Cell 70, 741–750 [DOI] [PubMed] [Google Scholar]
- 36. Wolfer A., Bakker T., Wilson A., Nicolas M., Ioannidis V., Littman D. R., Lee P. P., Wilson C. B., Held W., MacDonald H. R., Radtke F. (2001) Inactivation of Notch 1 in immature thymocytes does not perturb CD4 or CD8T cell development. Nat. Immunol. 2, 235–241 [DOI] [PubMed] [Google Scholar]
- 37. Rosloniec E. F., Cremer M., Kang A. H., Myers L. K., Brand D. D. (2010) Animal models of IgA nephropathy. Curr. Protoc. Immunol. 15, 11–25 [Google Scholar]
- 38. Simmons M., Porter K. R., Hayes C. G., Vaughn D. W., Putnak R. (2006) Characterization of antibody responses to combinations of a dengue virus type 2 DNA vaccine and two dengue virus type 2 protein vaccines in rhesus macaques. J. Virol. 80, 9577–9585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhong M. C., Veillette A. (2008) Control of T lymphocyte signaling by Ly108, a signaling lymphocytic activation molecule family receptor implicated in autoimmunity. J. Biol. Chem. 283, 19255–19264 [DOI] [PubMed] [Google Scholar]
- 40. Chen R., Relouzat F., Roncagalli R., Aoukaty A., Tan R., Latour S., Veillette A. (2004) Molecular dissection of 2B4 signaling. Implications for signal transduction by SLAM-related receptors. Mol. Cell Biol. 24, 5144–5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brand D. D., Kang A. H., Rosloniec E. F. (2003) Immunopathogenesis of collagen arthritis. Springer Semin. Immunopathol. 25, 3–18 [DOI] [PubMed] [Google Scholar]
- 42. Myers L. K., Seyer J. M., Stuart J. M., Terato K., David C. S., Kang A. H. (1993) T cell epitopes of type II collagen that regulate murine collagen-induced arthritis. J. Immunol. 151, 500–505 [PubMed] [Google Scholar]
- 43. Galli G., Nuti S., Tavarini S., Galli-Stampino L., De Lalla C., Casorati G., Dellabona P., Abrignani S. (2003) CD1d-restricted help to B cells by human invariant natural killer T lymphocytes. J. Exp. Med. 197, 1051–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leavenworth J. W., Tang X., Kim H. J., Wang X., Cantor H. (2013) Amelioration of arthritis through mobilization of peptide-specific CD8+ regulatory T cells. J. Clin. Invest. 123, 1382–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nichols K. E., Hom J., Gong S. Y., Ganguly A., Ma C. S., Cannons J. L., Tangye S. G., Schwartzberg P. L., Koretzky G. A., Stein P. L. (2005) Regulation of NKT cell development by SAP, the protein defective in XLP. Nat. Med. 11, 340–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pasquier B., Yin L., Fondanèche M. C., Relouzat F., Bloch-Queyrat C., Lambert N., Fischer A., de Saint-Basile G., Latour S. (2005) Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J. Exp. Med. 201, 695–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chung B., Aoukaty A., Dutz J., Terhorst C., Tan R. (2005) Signaling lymphocytic activation molecule-associated protein controls NKT cell functions. J. Immunol. 174, 3153–3157 [DOI] [PubMed] [Google Scholar]
- 48. Bustelo X. R. (2001) Vav proteins, adaptors and cell signaling. Oncogene 20, 6372–6381 [DOI] [PubMed] [Google Scholar]
- 49. Ma C. S., Hare N. J., Nichols K. E., Dupré L., Andolfi G., Roncarolo M. G., Adelstein S., Hodgkin P. D., Tangye S. G. (2005) Impaired humoral immunity in X-linked lymphoproliferative disease is associated with defective IL-10 production by CD4+ T cells. J. Clin. Invest. 115, 1049–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Deenick E. K., Chan A., Ma C. S., Gatto D., Schwartzberg P. L., Brink R., Tangye S. G. (2010) Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity 33, 241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu K. T., Kanno Y., Cannons J. L., Handon R., Bible P., Elkahloun A. G., Anderson S. M., Wei L., Sun H., O'Shea J. J., Schwartzberg P. L. (2011) Functional and epigenetic studies reveal multistep differentiation and plasticity of in vitro-generated and in vivo-derived follicular T helper cells. Immunity 35, 622–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhu C., Ma J., Liu Y., Tong J., Tian J., Chen J., Tang X., Xu H., Lu L., Wang S. (2012) Increased frequency of follicular helper T cells in patients with autoimmune thyroid disease. J. Clin. Endocrinol. Metab. 97, 943–950 [DOI] [PubMed] [Google Scholar]
- 53. Simpson N., Gatenby P. A., Wilson A., Malik S., Fulcher D. A., Tangye S. G., Manku H., Vyse T. J., Roncador G., Huttley G. A., Goodnow C. C., Vinuesa C. G., Cook M. C. (2010) Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 62, 234–244 [DOI] [PubMed] [Google Scholar]
- 54. Morita R., Schmitt N., Bentebibel S. E., Ranganathan R., Bourdery L., Zurawski G., Foucat E., Dullaers M., Oh S., Sabzghabaei N., Lavecchio E. M., Punaro M., Pascual V., Banchereau J., Ueno H. (2011) Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 34, 108–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Platt A. M., Gibson V. B., Patakas A., Benson R. A., Nadler S. G., Brewer J. M., McInnes I. B., Garside P. (2010) Abatacept limits breach of self-tolerance in a murine model of arthritis via effects on the generation of T follicular helper cells. J. Immunol. 185, 1558–1567 [DOI] [PubMed] [Google Scholar]
- 56. Kouskoff V., Korganow A. S., Duchatelle V., Degott C., Benoist C., Mathis D. (1996) Organ-specific disease provoked by systemic autoimmunity. Cell 87, 811–822 [DOI] [PubMed] [Google Scholar]
- 57. Sakaguchi N., Takahashi T., Hata H., Nomura T., Tagami T., Yamazaki S., Sakihama T., Matsutani T., Negishi I., Nakatsuru S., Sakaguchi S. (2003) Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature 426, 454–460 [DOI] [PubMed] [Google Scholar]
- 58. Kim H. Y., Kim H. J., Min H. S., Kim S., Park W. S., Park S. H., Chung D. H. (2005) NKT cells promote antibody-induced joint inflammation by suppressing transforming growth factor β1 production. J. Exp. Med. 201, 41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Teige A., Bockermann R., Hasan M., Olofsson K. E., Liu Y., Issazadeh-Navikas S. (2010) CD1d-dependent NKT cells play a protective role in acute and chronic arthritis models by ameliorating antigen-specific Th1 responses. J. Immunol. 185, 345–356 [DOI] [PubMed] [Google Scholar]
- 60. Barral P., Eckl-Dorna J., Harwood N. E., De Santo C., Salio M., Illarionov P., Besra G. S., Cerundolo V., Batista F. D. (2008) B cell receptor-mediated uptake of CD1d-restricted antigen augments antibody responses by recruiting invariant NKT cell help in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 8345–8350 [DOI] [PMC free article] [PubMed] [Google Scholar]