

Background: RAD54 performs branch migration and stimulation of RAD51 DNA strand exchange.
Results: Streptonigrin inhibits branch migration through specific binding to RAD54 but spares stimulation of RAD51-mediated DNA strand exchange.
Conclusion: RAD54 ATPase activity is relatively less important in the stimulation of DNA pairing than in branch migration.
Significance: Small-molecule inhibitors are instrumental to study the mechanisms of RAD54 activities.
Keywords: ATPases, DNA Binding Protein, DNA Repair, Homologous Recombination, Small Molecules
Abstract
RAD54, an important homologous recombination protein, is a member of the SWI2/SNF2 family of ATPase-dependent DNA translocases. In vitro, RAD54 stimulates RAD51-mediated DNA strand exchange and promotes branch migration of Holliday junctions. It is thought that an ATPase-dependent DNA translocation is required for both of these RAD54 activities. Here we identified, by high-throughput screening, a specific RAD54 inhibitor, streptonigrin (SN), and used it to investigate the mechanisms of RAD54 activities. We found that SN specifically targets the RAD54 ATPase, but not DNA binding, through direct interaction with RAD54 and generation of reactive oxygen species. Consistent with the dependence of branch migration (BM) on the ATPase-dependent DNA translocation of RAD54, SN inhibited RAD54 BM. Surprisingly, the ability of RAD54 to stimulate RAD51 DNA strand exchange was not significantly affected by SN, indicating a relatively smaller role of RAD54 DNA translocation in this process. Thus, the use of SN enabled us to identify important differences in the effect of the RAD54 ATPase and DNA translocation on two major activities of RAD54, BM of Holliday junctions and stimulation of DNA pairing.
Introduction
Homologous recombination (HR)2 is important for the repair of most harmful types of DNA damage, including DNA double strand breaks and interstrand cross-links, and for chromosome segregation in meiosis (1, 2). During double strand break repair, the DNA ends are nucleolitically resected first to create 3′-ssDNA overhangs. RAD51 binds to the resected ssDNA, creating a contiguous nucleoprotein filament (3). The RAD51-ssDNA filament searches for a homologous dsDNA to use it as a template for double strand break repair. The filament invades the dsDNA by performing strand exchange between homologous DNA molecules, which results in formation of a joint molecule also known as a D loop. In joint molecules, a DNA polymerase extends the invading ssDNA strand. The cross structure that forms at the point of DNA strand exchange is known as a Holliday junction (4, 5). The Holliday junction can move along the DNA axis in a process known as branch migration (BM), which involves the stepwise breakage of DNA base pairs on the two opposite arms followed by the subsequent formation of matching opposing base pairs on the two remaining arms. BM may lead to either dissociation or extension of joint molecules, depending on the specific HR pathway.
There are several auxiliary proteins that stimulate RAD51. One of the most important among them is RAD54, a member of the SNF2/SWI2 family of ATPase-dependent DNA translocases that is conserved in all eukaryotes (6). In Saccharomyces cerevisiae, RAD54 mutants are among the three most ionizing radiation-sensitive mutants along with RAD51 and RAD52 mutants (7). In mice, RAD54 knockouts are sensitive to ionizing radiation during early development and hypersensitive to the DNA cross-linking agent mitomycin C at every stage of development (8). Furthermore, mutations in the RAD54 gene lead to genetic instability and cancer (9, 10). RAD54 physically interacts with RAD51 (11–13) and stimulates its DNA strand exchange activity in vitro (14–16). In addition, RAD54 can bind specifically to Holliday junctions, form oligomeric complexes, and promote their BM by translocating along the DNA in an ATPase-dependent manner (17, 18). In vivo, BM of Holliday junctions is thought to occur in the late stage of HR during postsynapsis when heteroduplex extension or dissociation is required for completion of HR (19–21). In addition to BM, the DNA translocation activity of RAD54 is implicated in disruption or remodeling of DNA complexes with proteins, e.g. histones or RAD51 (22–24).
Despite extensive studies, the exact mechanism of stimulation of RAD51 DNA strand exchange by RAD54 remains unknown, and the role of the RAD54 ATPase activity in this stimulation is controversial. It has been shown that RAD54 forms a co-complex with RAD51-ssDNA filaments, stabilizing the filament in a manner that is independent of ATP hydrolysis by RAD54 (22, 25). However, RAD54 mutants defective in ATP hydrolysis fail to stimulate RAD51 DNA strand exchange, indicating that additional downstream mechanisms are important for the stimulation (14, 16, 26). It has been suggested that, during the search for homology, binding of dsDNA by RAD54 and its ATPase-dependent translocation along the RAD51-ssDNA filament may stimulate DNA strand exchange by either providing rapid delivery of the incoming dsDNA for the homology sampling by RAD51 or by locally disrupting the dsDNA base pairs, making them accessible for the homology search by the RAD51-ssDNA filament (14, 26, 27). Although RAD54 lacks canonical DNA helicase activity, it may cause disruption of base pairs because of transient positive and negative supercoils that form in DNA as a byproduct of DNA translocation (27–29). However, although these hypothetical mechanisms are appealing, they lack solid evidence for the role of ATPase-dependent dsDNA translocation by RAD54 in stimulation of RAD51 DNA pairing activity. Moreover, the inability of the RAD54 ATPase-defective mutants could be attributed to their excessively stable complexes with dsDNA that disrupt the search for homology by RAD51 rather than to their deficiency in DNA translocation. In addition, several other proteins that stimulate DNA strand exchange of RAD51 either do not have an ATPase-dependent DNA translocation ability, like HOP2-MND1 (30, 31) and RAD51AP1 (32, 33), or do not require it for RAD51 stimulation, like BLM (34). These data indicate that DNA translocation may not be an essential attribute of RAD51-stimulatory proteins.
To understand whether the ATPase-dependent dsDNA translocation by RAD54 is similarly important for stimulation of DNA strand exchange and for BM of Holliday junctions, we employed a specific small-molecule inhibitor that selectively disrupts RAD54 ATPase activity and analyzed its effect on RAD54 BM and stimulation of DNA strand exchange activity of RAD51. In contrast to the effect of mutations, the inhibitory effect of small-molecule inhibitors can be gradually modulated in a concentration- and time-dependent manner.
Using high-throughput screening of a library of 2000 compounds, we identified streptonigrin (SN) as a specific inhibitor of RAD54 BM activity3. SN is an aminoquinone compound that was first isolated from Streptomyces flocculus (35). SN was found to have antitumor activity on a broad range of cancers, with the highest efficacy against malignant lymphomas, squamous cell carcinoma of the cervix, breast cancer, malignant melanoma, and head/neck cancers (36). It is proposed that the antitumor activity of SN can be attributed to its ability to cause DNA damage by generating reactive oxygen species (ROS) through cycles of reduction and auto-oxidation of the quinone group. In addition, in vitro SN has an ability to inhibit topoisomerase II by trapping it in a “cleavable complex” with DNA, which may lead to formation of DNA double strand breaks in vivo (37).
We studied the mechanism of inhibition of RAD54 BM by SN. Our results demonstrated that SN binds specifically to RAD54 and inhibits its ATPase activity by generating ROS. At the same time, SN caused only a slight inhibition of DNA binding by RAD54. Furthermore, we found that SN differentially affected two RAD54 key activities: BM of Holliday junctions and stimulation of RAD51 DNA strand exchange. Although SN inhibited BM with approximately the same efficiency as the ATPase, the RAD54 ability to stimulate RAD51-mediated DNA strand exchange was not significantly affected by SN. Thus, our data indicate that RAD54 ATPase activity and ATPase-dependent dsDNA translocation play a more important role in BM than in stimulation of DNA strand exchange promoted by RAD51.
EXPERIMENTAL PROCEDURES
Chemicals, Proteins, and DNA
SN and lapachol were purchased from Sigma-Aldrich. The toxoflavin analog was a gift from the Broad Institute Probe Development Center. Escherichia coli RuvAB protein was a gift from Dr. Michael Cox. Human RAD51 and RAD54 were purified as described (16, 38). GST-RAD54 was treated with thrombin (GE Healthcare) that was added as a powder to GST-RAD54 in buffer containing 20 mm Tris HCl (pH 7.5), 400 mm KCl, 2.5 mm DTT, and 30% glycerol at a final concentration of 58 units/mg of GST-RAD54 for 5 h at 4 °C on ice. Tagless protein was then fractionated by a Superdex-200 column (60 ml) equilibrated with 20 mm KH2PO4 (pH) 7.5, 0.5 mm EDTA, 10% glycerol, 10 mm 2-mercaptoethanol, and 500 mm KCl. RAD54 fractions were collected and dialyzed overnight with storage buffer containing 1 mm DTT. The oligonucleotides (supplemental Table S1) were purchased from IDT Inc. in a desalted form. Oligonucleotides and pUC19 supercoiled dsDNA substrates were prepared as described (39, 40). The DNA concentrations are expressed as moles of nucleotides or, when indicated, as moles of molecules.
Measuring the Effect of SN on BM Activity of RAD54 and RuvAB
RAD54 (100 nm) was incubated with SN in the indicated concentrations or DMSO (2%) for 5 min at 30 °C in buffer containing 25 mm Tris acetate (pH 7.5), 100 μg/ml BSA, 3 mm magnesium acetate, 2 mm DTT (unless indicated otherwise), 2 mm ATP, and the ATP-regenerating system (30 units/ml creatine phosphokinase and 20 mm creatine phosphate). When indicated, SN was substituted with lapachol or toxoflavin analog. When needed, CuSO4 was added to the reaction mixture at indicated concentrations. BM was initiated by addition of 32P-labeled synthetic PX junction (partial X-junction) (oligos 71/169/170/171) (33 nm, molecules) and was carried out for 5 min. Aliquots (10 μl) were withdrawn, and the DNA products were deproteinized by treatment with stop buffer (6.2 μl of 1.9% SDS, 20% glycerol, 0.03% bromphenol blue, and 3.6 mg/ml proteinase K) for 5 min at 37 °C and analyzed by electrophoresis in 10% polyacrylamide gels (17:1) in Tris borate-EDTA buffer (90 mm Tris borate (pH 8.3) and 1 mm EDTA). The gels were dried on DE81 chromatography paper (Whatman) and quantified using a Storm 840 PhosphorImager (GE Healthcare).
RuvA (140 nm) and RuvB (360 nm) were incubated with SN (50 μm) or DMSO (2%) for 5 min at 37 °C in reaction buffer containing 25 mm Tris acetate (pH 7.5), 100 μg/ml BSA, 10 mm magnesium acetate, 2 mm DTT, 2 mm ATP, and the ATP-regenerating system (10 units/ml creatine phosphokinase and 10 mm creatine phosphate). BM was initiated by addition of 32P-labeled synthetic PX junction (71/169/170/171) (33 nm, molecules) at 37 °C. Aliquots (10 μl) were withdrawn, and DNA products were deproteinized and analyzed as described above for DNA products of RAD54-promoted BM.
Measuring the Effect of SN on RAD51 DNA Strand Exchange
1 μmRAD51 was incubated with ssDNA oligonucleotide (oligo 1, 63-mer) (3 μm) in buffer containing 33 mm Hepes (pH 7.0), 100 μg/ml BSA, 5 mm CaCl2, 1 mm DTT, and 2 mm ATP for 15 min at 37 °C. Then SN was added at the indicated concentrations, and incubation continued for another 30 min. DNA strand exchange was initiated by addition of 32P-labeled dsDNA oligonucleotide (oligos 45/55, 31-mers) (6 μm) for 6 min. The DNA products were deproteinized and analyzed as described above for DNA products of RAD54 BM.
Measurement of SN Binding to RAD54 by Surface Plasmon Resonance
Analyses were performed on a ProteOn XPR36 surface plasmon resonance protein interaction array system (Bio-Rad). High-density GLH sensor chips were preconditioned with two 10 s pulses of 50 mm NaOH, 100 mm HCl, and 0.5% SDS. Then the system was equilibrated with PBS-T buffer (20 mm Na-phosphate, 150 mm NaCl, and 0.005% polysorbate 20 (pH 7.4)). Individual ligand flow channels were activated for 5 min at 25 °C with a mixture of 1-ethyl-3-(3-dimethylaminopropyl carbodiimide hydrochloride) (0.2 m) and sulfo-N-hydroxysuccinimide (0.05 m). Immediately after surface activation, either RAD51 or RAD54 was injected across ligand flow channels for 5 min at a flow rate of 30 μl min−1. Excess active ester groups on the sensor surface were capped by a 5-min injection of 1 m ethanolamine HCl (pH 8.5). This resulted in the coupling of RAD51 and RAD54 to 19,000 response units (an arbitrary unit that corresponds to 1 pg/mm2), respectively. The S.D. in the immobilization level from the six spots within each channel was less than 4%. 2-fold dilutions of SN in 5% DMSO, prepared in running buffer containing 25 mm Tris acetate (pH 7.5), 3 mm magnesium acetate, 1 mm ATP, and 1 mm DTT, were injected over the control and RAD54 surfaces at a flow rate of 100 μl min−1 for a 2-min association phase, followed by a 15-min dissociation phase at 25 °C using the “one-shot” functionality of the ProteOn (41). The effect of DTT on the interaction of SN with RAD54 was assessed in a similar manner as reported above but using buffer without DTT. Specific regeneration of the surfaces between injections was not needed because of rapid dissociation kinetics of the interaction. Data were analyzed using the ProteOn Manager software version 3.0 (Bio-Rad). The responses of a buffer injection and responses from the reference flow cell to which RAD51 was immobilized were subtracted to account for nonspecific binding. Experimental data were fitted globally to a simple 1:1 binding model. The interaction parameters generated from six data sets were used to define the association (ka) and dissociation (kd) rate constants. The equilibrium dissociation constant (KD) was calculated from the ratio of the rate constants (KD) (41).
ATP Hydrolysis Assay
The hydrolysis of ATP by RAD54 was monitored continuously by a spectrophotometer as described previously (42). The oxidation of NADH, coupled to ADP phosphorylation, resulted in a decrease in absorbance at 340 nm, measured by a Hewlett-Packard 8453 diode array spectrophotometer using UV light-visible ChemStation software. The rate of ATP hydrolysis was calculated from the rate of change in absorbance using the following formula: rate of A340 decrease (s) × 9880 = rate of ATP hydrolysis (μm/min). RAD54 (20 nm) was incubated with SN at the indicated concentrations for 5 min at 30 °C in buffer containing 25 mm Tris acetate (pH 7.5), 5 mm magnesium acetate, 2 mm DTT, 2 mm ATP, 3 mm phosphoenolpyruvate, pyruvate kinase (20 units/ml), lactate dehydrogenase (20 units/ml), and NADH (200 μg/ml). ATP hydrolysis was initiated by the addition of PX junction (71/169/170/171) (1 nm, molecules).
Measuring the Effect of SN on the RAD54 ATPase under D Loop Formation Conditions
To form the filament, 800 nm RAD51 was incubated with2.4 μm ssDNA (oligo 90, 90-mer) in reaction buffer containing 20 mm Tris HCl (pH 7.5), 1.5 mm MgCl2, 0.5 mm CaCl2, 1 mm DTT, 2 mm ATP, 3 mm phosphoenolpyruvate, 20 units/ml pyruvate kinase, 20 units/ml lactate dehydrogenase, and 200 μg/ml NADH for 15 min at 30 °C. 120 nm RAD54 was preincubated with SN at the indicated concentrations for 5 min at 30 °C in dilution buffer containing 20 mm Tris HCl (pH 7.5), 100 μg/ml BSA, 2 mm DTT, 100 mm KCl, and 10% glycerol and then added to the reaction mixture containing RAD51-ssDNA filaments or the reaction buffer alone in a control. 62 μm supercoiled pUC19 dsDNA was added to initiate ATP hydrolysis. The rate of ATP hydrolysis was measured using a spectrophotometer as described above.
RAD54 DNA Binding Assay
100 nm RAD54 was incubated with SN in the indicated concentrations or DMSO (2%) for 5 min at 30 °C in binding buffer containing 25 mm Tris acetate (pH 7.5), 5 mm magnesium acetate, 100 μg/ml BSA, 2 mm DTT, 2 mm ATP, and 10% glycerol. After 5 min, immobile 32P-labeled PX junction (oligos 175/174/176/181) (33 nm, molecules) was added, followed by a 5-min incubation at 30 °C. DNA-protein complexes were resolved by electrophoresis in 6% nondenaturing polyacrylamide gels (29:1) in 0.25× Tris borate-EDTA buffer (22.5 mm Tris borate (pH 8.3) and 0.5 mm EDTA). The gels were dried on DE81 chromatography paper (Whatman) and quantified using a Storm 840 PhosphorImager (GE Healthcare).
Triple-helix Displacement Assay
The triple-helix assay was a modification of a procedure described previously (43, 44). SspI-linearized pMJ5 (44) (50 nm, molecules) and 32P-labeled triplex-forming oligonucleotide (TFO) (oligo TFO, 22-mer) (100 nm, molecules) were mixed in buffer containing 25 mm MES (pH 5.5), and 10 mm MgCl2 and incubated for 15 min at 57 °C, followed by overnight cooling at room temperature. 50 μm SN or 2% DMSO was incubated for 5 min at 30 °C in reaction buffer containing 35 mm Tris HCl (pH 7.2), 3 mm MgCl2, 100 μg/ml BSA, 1 mm DTT, 50 mm KCl, 3 mm ATP, and the ATP-regenerating system (30 units/ml creatine phosphokinase and 15 mm creatine phosphate). Then, 100 nm RAD54 was added to the reaction buffer and incubated for 5 min at 30 °C followed by addition of the triple-helix substrate (pMJ5 + TFO) (100 nm, molecules) to start translocation. 10-μl aliquots were withdrawn and quenched with 5 μl of stop buffer containing 3% SDS, 250 mm MOPS (pH 5.5), 15% glycerol, and 0.02% bromphenol blue, and DNA products were analyzed in 1.2% agarose gels (40 mm Tris acetate (pH 5.5), 5 mm sodium acetate, and 1 mm MgCl2) at 3.5 V/cm for 2 h at 4 °C. The gels were dried on DE81 chromatography paper (Whatman) and quantified using a Storm 840 PhosphorImager (GE Healthcare).
Measuring RAD54 Degradation by Electrophoresis on SDS-PAGE
700 ng/10 μl or 6.3 μm RAD54 was incubated for the indicated times at 37 °C with 50 μm SN and 25 μm CuSO4 in reaction buffer containing 25 mm Tris acetate (pH 7.5), 3 mm magnesium acetate, 2 mm DTT, and 2 mm ATP. The reactions were quenched with 5 μl of 4× Laemmli buffer (40% glycerol, 0.6 m β-mercaptoethanol, 8% SDS, 0.02% bromphenol blue, and 224 mm Tris HCl (pH 6.8)) and incubated for 5 min at 55 °C. The protein products were resolved by electrophoresis in a 10% SDS-PAGE gel and visualized using Coomassie Blue or Western blot analysis. In the latter case, protein products were transferred from SDS-PAGE gels to PVDF membranes and blotted with the following antibodies: RAD54 c-terminal (C-15) (catalog no. sc-34517, Santa Cruz Biotechnology), donkey anti-goat IgG-HRP (catalog no. sc-2056, Santa Cruz Biotechnology), and RAD54 N-terminal (D-18) (catalog no. sc-5849, Santa Cruz Biotechnology).
D Loop Formation
800 nmRAD51 was incubated with 2.4 μm 32P-labeled ssDNA (oligo 90, 90-mer) in reaction buffer containing 20 mm Tris HCl (pH 7.5), 1.5 mm MgCl2, 0.5 mm CaCl2, 100 μg/ml BSA, 1 mm DTT, 2 mm ATP, and the ATP-regenerating system (30 units/ml creatine phosphokinase and 20 mm creatine phosphate). The RAD51 nucleoprotein filaments were allowed to form for 10 min at 30 °C. RAD54 (120 nm or indicated otherwise) and SN (50 μm or indicated otherwise) or 2% DMSO were added to the filaments for 5 min at 30 °C. 62 μm supercoiled pUC19 DNA was added to initiate D loop formation. The DNA products were deproteinized by treatment with stop buffer (0.7% SDS, 7.7% glycerol, 0.01% bromphenol blue, 1.4 mg/ml proteinase K, and 10 mm EDTA) for 5 min at 37 °C and analyzed by electrophoresis in 1% agarose in 1× TAE buffer (40 mm Tris acetate (pH 8.0) and 1 mm EDTA). The gels were dried on DE81 chromatography paper (Whatman) and quantified using a Storm 840 PhosphorImager (GE Healthcare).
In Fig. 12, 1 μm RAD51 was incubated with 3 μm 32P-labeled ssDNA (oligo 90, 90-mer) in reaction buffer containing 25 mm Tris acetate (pH 7.5), 0.5 mm CaCl2, 100 μg/ml BSA, 2 mm DTT, 1 mm ATP, and the ATP-regenerating system (30 units/ml creatine phosphokinase and 20 mm creatine phosphate) for 15 min at 37 °C (34). 100 or 200 nm RAD54 or RAD54 K189R was added, followed by addition of 50 μm supercoiled pUC19 DNA to initiate D loop formation. The DNA products were deproteinized and analyzed as described above.
FIGURE 12.
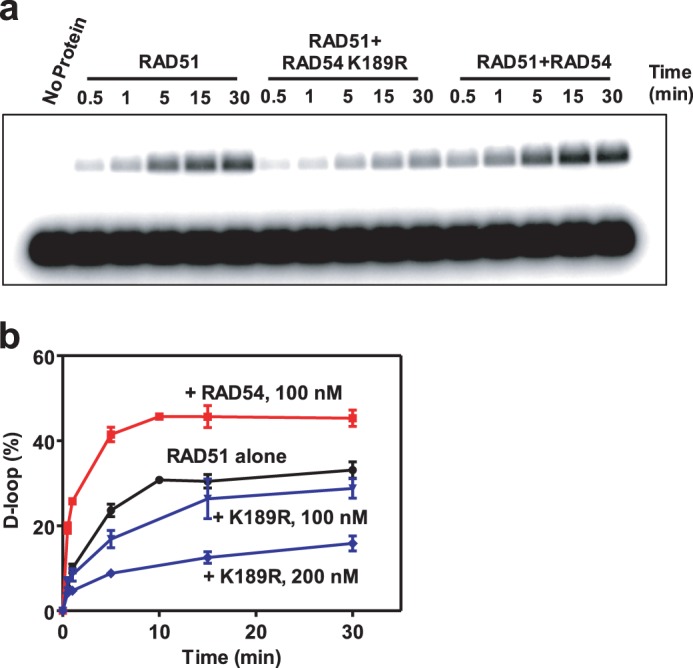
The RAD54-K189R ATPase-deficient mutant inhibits RAD51-promoted D loop formation. a, D loop formation promoted by 1 μm RAD51 alone or in the presence of 200 nm RAD54 or 200 nm RAD54 K189R between 32P-labeled ssDNA (oligo 90, 90-mer) (3 μm, nts) and pUC19 dsDNA (50 μm, nts) was analyzed by electrophoresis in a 1% agarose gel. b, the kinetics of RAD51-promoted D loop formation in the presence of 100 nm RAD54 and 100 nm and 200 nm RAD54 K189R presented as a graph. Error bars represent the S.E.
DNA Coaggregation Assay
This assay measures the homology-independent interaction of the RAD51-ssDNA filaments with dsDNA in the presence of RAD54. 800 nm RAD51 was incubated with 2.4 μm nts 32P-labeled ssDNA (oligo 71, 90-mer) in reaction buffer containing 20 mm Tris HCl (pH 7.5), 1.5 mm MgCl2,0.5 mm CaCl2, 100 μg/ml BSA, 1 mm DTT, 2 mm ATP, and the ATP-regenerating system (30 units/ml creatine phosphokinase and 15 mm creatine phosphate). The RAD51 nucleoprotein filaments were allowed to form for 10 min at 30 °C. 200 nm RAD54 and the indicated concentrations of SN or 2% DMSO were added to the filaments and incubated for 5 min at 30 °C. Coaggregation was initiated by 62 μm supercoiled pUC19 DNA. Coaggregates were collected by centrifugation at 14,000 × g for 5 min at room temperature. The supernatant was collected, and the pellet was redissolved in the same volume of reaction buffer (10 μl). Both the pellet and supernatant were treated with 6.2 μl of stop buffer containing 1.9% SDS, 20% glycerol, 0.03% bromphenol blue, and 3.6 mg/ml proteinase K and incubated for 5 min at 37 °C. The DNA products were analyzed by electrophoresis in 1% agarose-TAE (40 mm Tris acetate (pH 8.0) and 1 mm EDTA) gels. The gels were stained with 2 μg/ml ethidium bromide and visualized under UV light using an AlphaImager 3400 (Alpha Innotech). The yield of coaggregates was determined using a known quantity of supercoiled pUC19 DNA as a reference.
Measuring the Effect of the K189R Mutation on Dissociation of RAD54 from dsDNA
16.5 nm RAD54 or RAD54-K189R ATPase-deficient mutant was incubated in the presence of 2% DMSO or 50 μm SN in binding buffer containing 25 mm Tris acetate (pH 7.5), 6 mm magnesium acetate, 100 μg/ml BSA, 2 mm DTT, 4 mm ATP, and 10% glycerol for 5 min at 30 °C. To form protein-DNA complexes, an equimolar mixture of two 32P-labeled dsDNA fragments (1313 bp and 1373 bp) (1 μm, bp total) produced by cleavage of pUC19 with AflIII and ScaI were added to the reactions at 30 °C. After 10 min, identical unlabeled competitor dsDNA (74 μm, bp) fragments were added for the indicated periods of time, followed by fixation with glutaraldehyde (0.25%) for 15 min. DNA-protein complexes were analyzed by electrophoresis in 1.5% agarose gels in 1× TAE buffer (40 mm Tris acetate (pH 8.0) and 1 mm EDTA). The gels were dried on DE81 chromatography paper (Whatman) and quantified using a Storm 840 PhosphorImager (GE Healthcare).
RESULTS
SN Specifically Inhibits RAD54 BM Activity
Using high-throughput screening of a 2000-compound library, we identified an antitumor antibiotic, SN (Fig. 1a), as a potential inhibitor of RAD54 BM3. To validate this result, we performed a BM assay using 32P-labeled partial Holliday junction (PX) substrates (oligos 169/71/170/171) (supplemental Table S1) that contain three dsDNA arms and one ssDNA arm. We showed previously that RAD54 has the highest affinity for PX junctions (18). In these PX junctions, spontaneous BM was reduced by incorporating a single heterologous bp on one of the four DNA arms that creates one mismatch in each of the two branch migration products (Fig. 1b). The products of BM were analyzed by electrophoresis in 10% polyacrylamide gels. SN inhibited RAD54 BM in a dose-dependent manner with an IC50 of 16.5 μm (Fig. 1, c and d).
FIGURE 1.

SN inhibits RAD54 BM. a, the structure of SN. b, schematic of BM of PX junctions (oligo 169/71/170/171, supplemental Table S1). The asterisk indicates the 32P label at the DNA 5′ end. c, the effect of 0–50 μm SN on 100 nm RAD54 BM of PX junctions (33 nm, molecules). The DNA products were run on a 10% polyacrylamide gel. M denotes the marker (oligos 169/171) corresponding to the BM product. d, the data from c presented as a graph. Error bars represent mean ± S.E.
We then tested whether SN shows specificity in inhibition of RAD54. We tested the effect of SN on the prokaryotic enzyme RuvAB that promotes BM of Holliday junctions similar to RAD54 (4, 45, 46) but does not share structural homology with it. 50 μm SN had no significant effect on RuvAB BM but strongly inhibited RAD54 BM (Fig. 2, a and b). We further tested the specificity of SN inhibition of RAD54 by using another structurally unrelated protein, human RAD51, that possesses DNA strand exchange activity. In our assay, RAD51 formed a filament on a 63-mer ssDNA (oligo 1) that then promoted DNA strand exchange with a homologous 32P-labeled dsDNA (oligo 45/55, 31-mers) (Fig. 2c, top panel). No inhibition of RAD51 DNA strand exchange was observed at SN concentrations as high as 500 μm (Fig. 2, c and d). Thus, our results defined SN as a specific inhibitor of the BM activity of RAD54.
FIGURE 2.

SN inhibition is specific to RAD54. a, the effect of 50 μm SN on 140 and 360 nm RuvAB and 100 nm RAD54. BM of PX junctions (33 nm, molecules) was analyzed by electrophoresis in 10% polyacrylamide gels. M denotes markers corresponding to the product of helicase (middle band) and BM (lowest band) which were produced by annealing of oligos 169/71 and 169/171, respectively. b, the data from a presented as a graph. c, the effect of SN on RAD51 DNA strand exchange. The experimental schematic is shown in the top panel. 1 μm RAD51 was incubated with 3 μm ssDNA (oligo 1, 63-mer). Then, 0–500 μm SN was added, and DNA strand exchange was initiated by adding 6 μm dsDNA (oligos 45/55, 31-mers). The DNA products were analyzed by electrophoresis in 10% polyacrylamide gels. The asterisk indicates the 32P-label at the DNA 5′ end. d, the data from c presented as a graph. Error bars represent the S.E.
SN Binds Directly to RAD54
Because SN shows specificity in inhibiting RAD54 BM, we suggested that it may exert its effect through direct binding to RAD54. To test this hypothesis, we performed surface plasmon resonance analyses. Briefly, RAD54 and RAD51 were covalently immobilized to the surface of a sensor chip, and a concentration series of SN was injected over these surfaces. The response from the RAD51 sensor chip used as a reference was subtracted from the response from the RAD54 surface. In addition, the response from a buffer-only injection was subtracted. We found that SN interacts specifically and robustly with RAD54 with a 1:1 stoichiometry and an equilibrium dissociation constant (KD) of 9.1 ± 3.79 μm (Fig. 3a). The kinetics of this interaction are characterized by a relatively slow association rate (ka = 2.03 (± 0.8) × 103 m/s) coupled with rapid dissociation (kd = 1.92 (± 1.3) × 10−2s−1). Because the quinone group of SN can exist in both an oxidized and reduced form, we tested whether the binding of SN to RAD54 was mapped to either of these forms by testing the interaction in the absence of reducing agent. As can be seen in Fig. 3b, removal of the reducing agent DTT completely abolished the interaction of SN with RAD54, suggesting that the reduced form of SN is the active form.
FIGURE 3.
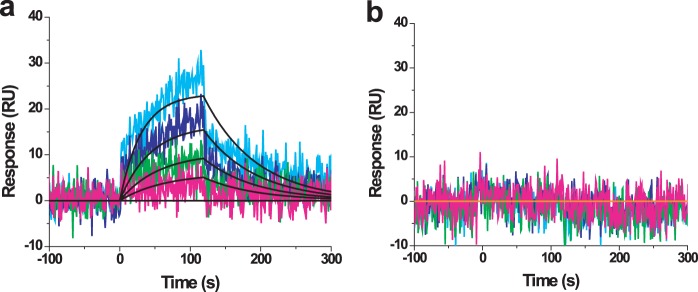
SN binds directly to RAD54 in the presence of DTT (a) but not in its absence (b). Colored lines represent actual binding data at SN concentrations of 18.8, 9.38, 4.69, 2.34, and 0 μm. Black lines show the fit to a simple 1:1 Langmuir isotherm. SN-RAD54 ka = 2.03 (± 0.8) × 103 m−1 s−1, kd = 1.92 (± 1.3) × 10−2 s−1, and KD = 9.1 ± 3.79 μm. Error bars represent the S.D. RU, response unit.
SN inhibits ATP Hydrolysis by RAD54 and DNA Translocation but Shows Only Slight Inhibition of DNA Binding
To determine the mechanism of RAD54 BM inhibition by SN, we analyzed the effect of SN on basic RAD54 activities required for BM of the Holliday junction, including ATP hydrolysis, DNA binding, and DNA translocation. We found that SN inhibited the RAD54 ATPase on PX substrates (oligos 169/71/170/171) in a dose-dependent manner with an IC50 of 14.4 μm (Fig. 4a), similar to the IC50 in BM (16.5 μm) (Fig. 1d).
FIGURE 4.
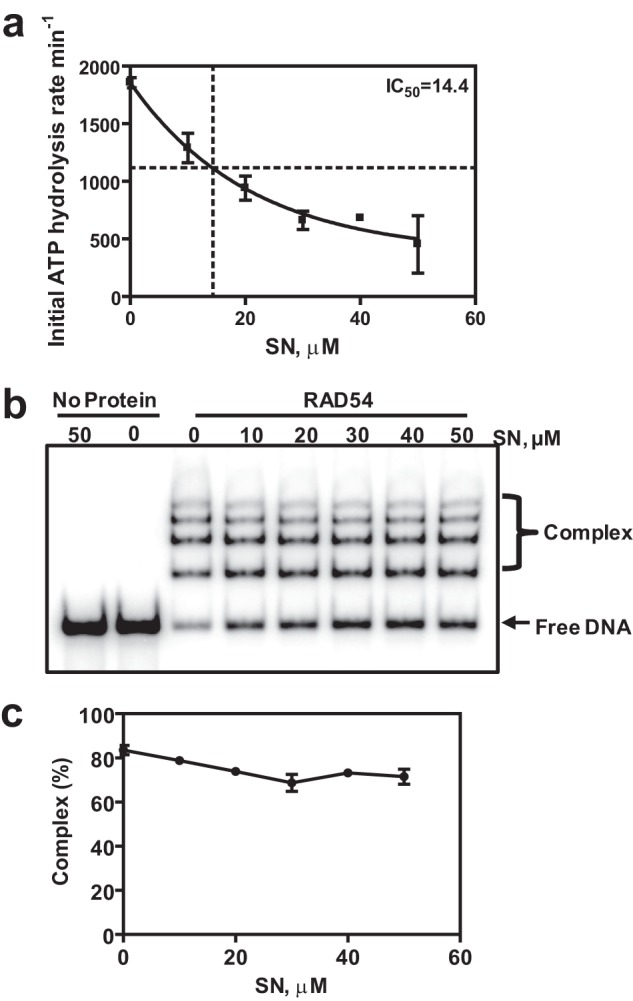
SN inhibits RAD54 ATPase but does not significantly disrupt RAD54-DNA complexes. a, the effect of 0–50 μm SN on 20 nm RAD54 ATPase in the presence of PX junctions (oligos 169/71/170/171) (1 nm, molecules). Hydrolysis of ATP was monitored using a spectrophotometric assay. b, the effect of SN (0–50 μm) on the formation of 100 nm RAD54 complexes with PX junctions (33 nm, molecules) was analyzed using electrophoresis in a 4% polyacrylamide gel. c, the data from b presented as a graph. Error bars represent the S.E.
Because RAD54 is a DNA-dependent ATPase, we wished to explore whether SN inhibited ATP hydrolysis by disrupting DNA binding by RAD54. To assess this possibility, we used an electrophoretic mobility shift assay in which RAD54 was incubated with PX DNA (oligos 169/71/170/171) in the presence or absence of SN. In the absence of SN, RAD54, as expected, formed high-order nucleoprotein complexes with the PX DNA. Addition of 50 μm SN caused only ∼15% inhibition of RAD54 nucleoprotein complex formation (Fig. 4, b and c).
We then wished to explore whether SN inhibited the ability of RAD54 to translocate along DNA, which requires the concerted actions of DNA binding and ATP hydrolysis. To test this, we used a triple-helix displacement assay (43, 44) (Fig. 5a). Briefly, a pyrimidine-rich TFO (oligo TFO, 22-mer) was annealed to a purine-rich target duplex sequence located in pMJ5 plasmid DNA (44) under acidic conditions. The resulting triple-helix structure contains T-AT and C+-GC triplets formed by Hoogsteen hydrogen bonding (47). The triple helix is stable at physiological pH. However, when the TFO is displaced by RAD54, it will be unable to rebind plasmid DNA. Under BM conditions, we found that 50 μm SN showed a more than 2-fold inhibition of TFO displacement by RAD54 (Fig. 5, b and c). Thus, the mechanism of inhibition of RAD54 BM by SN involves a reduction in ATPase activity and DNA translocation but only minimal disruption of RAD54 interactions with DNA.
FIGURE 5.

SN inhibits triple-helix disruption by RAD54. a, experimental schematic. The asterisk represents the 32P label on the TFO (oligo TFO, 22-mer). b, the effect of 50 μm SN on 100 nm RAD54 triple-helix (10 nm, molecules) disruption was analyzed by electrophoresis in 1.2% agarose gels at 4 °C. c, the data from the gel presented as a graph. Error bars represent the S.E.
Both Specific Binding and ROS Generation Are Required for RAD54 Inhibition by SN
In the presence of reducing agents, SN can generate ROS through cycles of reduction/auto-oxidation of the quinone group. ROS may cause protein damage and inactivation (48). Here we wished to assess the role of ROS generated by SN in inhibition of RAD54 BM activity.
First, we tested whether any quinone-containing compounds known to produce ROS would inhibit the RAD54 BM activity nonspecifically. We found that, under tested conditions, lapachol and a toxoflavin analog (49, 50), in contrast to SN, did not significantly inhibit RAD54 BM, indicating that specific binding of SN is required for RAD54 inhibition (Fig. 6, a–c).
FIGURE 6.

Inhibition of RAD54 requires both specific binding of SN and generation of ROS. a, the structures of lapachol and the toxoflavin analog compared with SN. The quinone groups are shown in blue boxes. b, the effect of 20 μm quinone-containing compounds shown in a on 100 nm RAD54 BM. c, the effect of increasing concentrations of 0–50 μm SN or lapachol on 100 nm RAD54 BM. d, the effect of 0–10 mm DTT concentration on 100 nm RAD54 inhibition by 50 μm SN. For each tested DTT concentration, the percentage of inhibition was determined relatively to the rate of BM in the absence of SN. e, 0.050 mg/ml catalase partially alleviates 50 μm SN inhibition of 100 nm RAD54 BM (30 units/ml creatine phosphokinase and 19 mm creatine phosphate). Error bars represent the S.E.
Next, we tested whether quenching of ROS will alleviate RAD54 inhibition. When RAD54 was incubated in the presence of 50 μm SN over a range of DTT concentrations (0–10 mm), we found that, in the absence of DTT, the inhibitory effect was relatively small (∼40%) (Fig. 6d). This result was consistent with poor binding of SN to RAD54 observed in the absence of reducing agent (Fig. 3b) or poor ROS generation, which requires reducing agent. This inhibitory effect increases sharply with the increase in DTT concentration, reaching a maximum (∼100%) inhibition at 0.5 mm DTT, consistent with both stimulation of SN binding to RAD54 and stimulation of ROS generation. However, a further increase in DTT concentration alleviated inhibition of RAD54 BM by SN. This effect is consistent with the quenching of ROS by DTT through donating electrons to the radicals and indicates an important role of ROS in the inhibition of RAD54 BM activity.
Among ROS, the hydroxyl radical is thought to have an especially harmful effect on biological macromolecules. Hydroxyl radicals are produced in the reaction between hydrogen peroxide and superoxide radicals that are generated during SN reduction/auto-oxidation. To further test the role of ROS, and hydroxyl radicals particularly, in RAD54 inhibition, we used the enzyme catalase that converts hydrogen peroxide to molecular oxygen and water, thereby reducing the production of hydroxyl radicals. We found that catalase partially alleviates the inhibition of RAD54 BM by SN (50 μm) (Fig. 6e).
It has been shown previously that the rate of SN reduction increases significantly when SN forms a complex with Cu2+ (51, 52). If ROS generated during SN reduction/auto-oxidation plays a role in RAD54 inactivation, one could expect that Cu2+ may enhance the inhibitory effect of SN. To test this proposal, we examined the effect of Cu2+ on RAD54 inhibition by SN (10 μm). Indeed, we found that Cu2+ increases the inhibitory effect of SN. At 6 μm of Cu2+, the increase in inhibition was ∼2-fold (Fig. 7).
FIGURE 7.
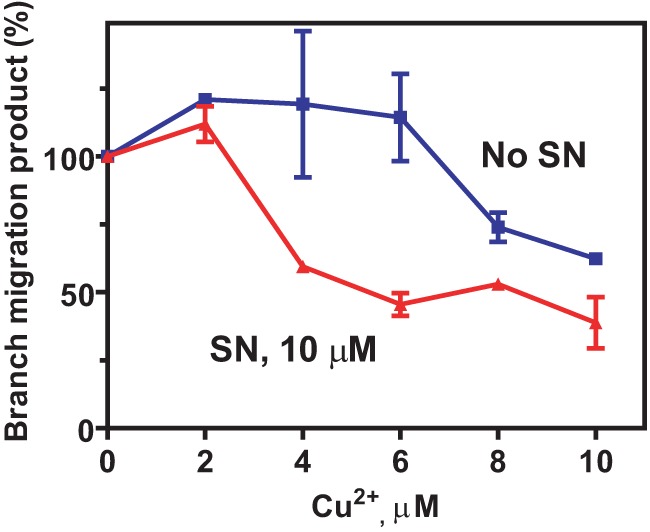
Cu+ enhances inhibition of RAD54 BM by SN. The effect of 0–10 μm Cu2+ concentration on inhibition of 100 nm RAD54 BM by 10 μm SN. The reactions were carried out for 5 min. Error bars represent the S.E.
Taken together, these results show that SN inhibits RAD54 BM through a mechanism that involves specific binding to RAD54 and generation of ROS through cycles of reduction/auto-oxidation of the SN quinone group. Importantly, all tested ROS-generating compounds did not significantly inhibit RAD54 BM or RAD51 DNA strand exchange through nonspecific interactions.
Mapping of the Fragments Induced in RAD54 by SN
It is known from previous studies that ROS cause protein fragmentation and aggregation (48). Indeed, treatment of the RAD54 or RAD54 ΔN67 truncated mutant lacking the first 67 N-terminal amino acid residues with 50 μm SN for 30 min at 37 °C showed aggregation visualized by SDS-PAGE and Coomassie staining (Fig. 8a). Addition of 25 μm Cu2+ to the reaction mixture containing SN increases RAD54 aggregation in a time-dependent manner (supplemental Fig. S1a) and, with an extended incubation (> 20 min), causes fragmentation of RAD54 (Fig. 8, a and b, and supplemental Fig. S1a). Incubation of RAD54 with 25 μm or 50 μm Cu2+ alone did not significantly increase RAD54 aggregation or fragmentation (supplemental Fig. S1, a and b). The effect was specific for RAD54 because SN-Cu2+ had no noticeable effect on RAD51 (supplemental Fig. S1b). We then wished to identify specific RAD54 fragments produced during protein treatment with SN-Cu2+ using Western blotting with specific antibodies raised against the N-terminal (D-18, catalog no. 5849, Santa Cruz Biotechnology) and the C-terminal regions (C-15, catalog no. 34517, Santa Cruz Biotechnology) of RAD54. Previously, a similar “ROS target mapping approach” was used successfully to map the binding site of a native metal in E. coli primase (53). First, using a RAD54 ΔN67 truncated protein, we confirmed the specificity of the antibodies (Fig. 8b). Then, using the N-terminal-specific antibodies, we were able to visualize a major protein fragment of ∼36 kDa (Fig. 8b, left panel). With the C-terminal-specific antibodies, we identified a fragment of ∼48 kDa. This fragment was found in both RAD54 and ΔN67 RAD54 truncated protein, indicating its C-terminal localization (Fig. 8b, right panel). Formation of two major fragments migrating at these sizes was consistent with a fragmentation site located within the conserved helicase domain III that is involved in coordination of the γ-phosphate group of ATP (54, 55) (Fig. 8c). Thus, the location of this cleavage site in RAD54 is consistent with the inhibition of the ATPase activity by SN observed in this study.
FIGURE 8.
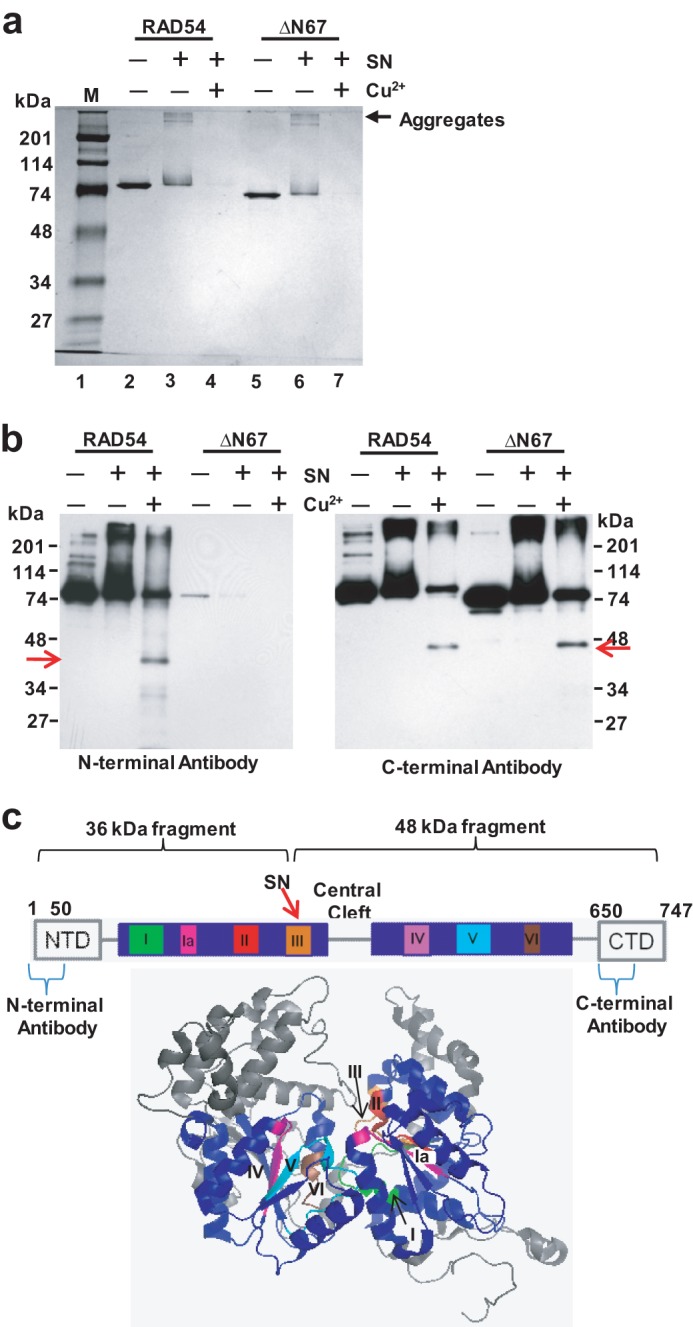
Mapping of the fragments induced in RAD54 by SN in the presence of Cu2+. a, fragmentation and aggregation of 700 ng RAD54 and 700 ng RAD54 ΔN67 by 50 μm SN and 25 μm Cu2+. The reactions were carried out for 30 min at 37 °C, and the products were visualized by electrophoresis on a 10% SDS-polyacrylamide gel after staining with Coomassie Blue. M denotes Bio-Rad prestained SDS-PAGE standards broad range. b, specific RAD54 fragments (arrows) were identified by Western blotting using N-terminal- or C-terminal-specific RAD54 antibodies. c, schematic of RAD54 highlighting the seven conserved helicase motifs and the predicted binding site of SN. Shown below is the x-ray crystal structure of zebrafish RAD54 (PDB code 1Z3I). The seven conserved helicase motifs are highlighted in various colors in the two motor domains labeled in blue.
SN Does Not Significantly Affect Stimulation of DNA Pairing by RAD54
The results described above demonstrate that SN efficiently inhibits RAD54 BM. Here, we wished to test whether SN also inhibits the ability of RAD54 to stimulate DNA pairing promoted by RAD51, another important activity of RAD54.
To measure the effect of SN on RAD51 DNA pairing in the presence or absence of 120 nm RAD54, we used the D loop assay (Fig. 9a). First, the RAD51-ssDNA filament was formed, and then RAD54 and SN, at the indicated concentrations, were added. The DNA pairing was initiated by adding supercoiled pUC19 dsDNA to the reaction mixture, and the DNA products (D loops) were analyzed by electrophoresis in 1% agarose gels (Fig. 9b). We found that SN did not significantly inhibit the ability of RAD54 to stimulate RAD51 in D loop formation, with only a 1.3-fold decrease in the yield of D loops at 50 μm SN (Fig. 9c).
FIGURE 9.
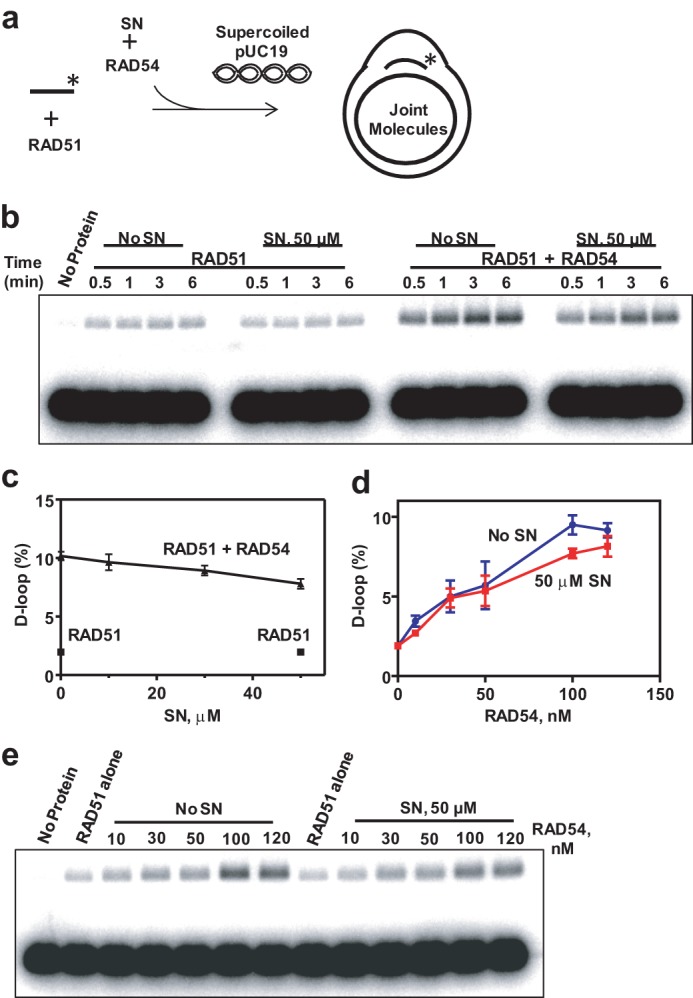
SN does not significantly inhibit RAD54-dependent stimulation of DNA strand exchange promoted by RAD51. a, schematic of D loop formation. The asterisk indicates the 32P label at the DNA 5′ end. b, the effect of 50 μm SN on D loop formation promoted by 800 nm RAD51 in the presence of 120 nm RAD54 between 32P-labeled ssDNA (oligo 90, 90-mer) (2.4 μm, nts) and pUC19 dsDNA (62 μm, nts) was analyzed by electrophoresis in a 1% agarose gel. c, the effect of the SN concentration on D loop formation presented as a graph. The reactions were carried out for 3 min. d and e, the effect of 50 μm SN on D loop formation promoted by 800 nm RAD51 in the presence of indicated RAD54 concentrations between 32P-labeled ssDNA (oligo 90, 90-mer) (2.4 μm, nts) and pUC19 dsDNA (62 μm, nts). The reactions were carried out for 5 min. D loops were analyzed by electrophoresis in a 1% agarose gel (e). Graphical data representation is shown in d.
We then examined whether SN would have a greater inhibitory effect on D loop formation at lower RAD54 concentrations. However, we found that D loop formation was not significantly inhibited by SN at RAD54 concentrations as low as 10 nm RAD54 (Fig. 9, d and e).
In contrast, under D loop conditions, SN reduced the RAD54 ATPase ∼9-fold in the presence of the RAD51-ssDNA filament (Fig. 10a). We then wished to explore whether this inhibition of the RAD54 ATPase activity by SN is due to inhibition of the ability of RAD54 to bind dsDNA in the presence of the RAD51-ssDNA filament. To assess this possibility, we used a DNA coaggregation assay in which RAD54, along with the indicated concentrations of SN, was incubated with RAD51-ssDNA nucleoprotein filaments (Fig. 10b). In the presence of SN, no significant inhibition of DNA coaggregation mediated by RAD54 was observed (Fig. 10c). This result is in agreement with the results of our previous electrophoretic mobility shift experiments (Fig. 4, b and c), indicating that SN does not have a large effect on DNA binding by RAD54 alone or in the presence of the RAD51-ssDNA filament.
FIGURE 10.
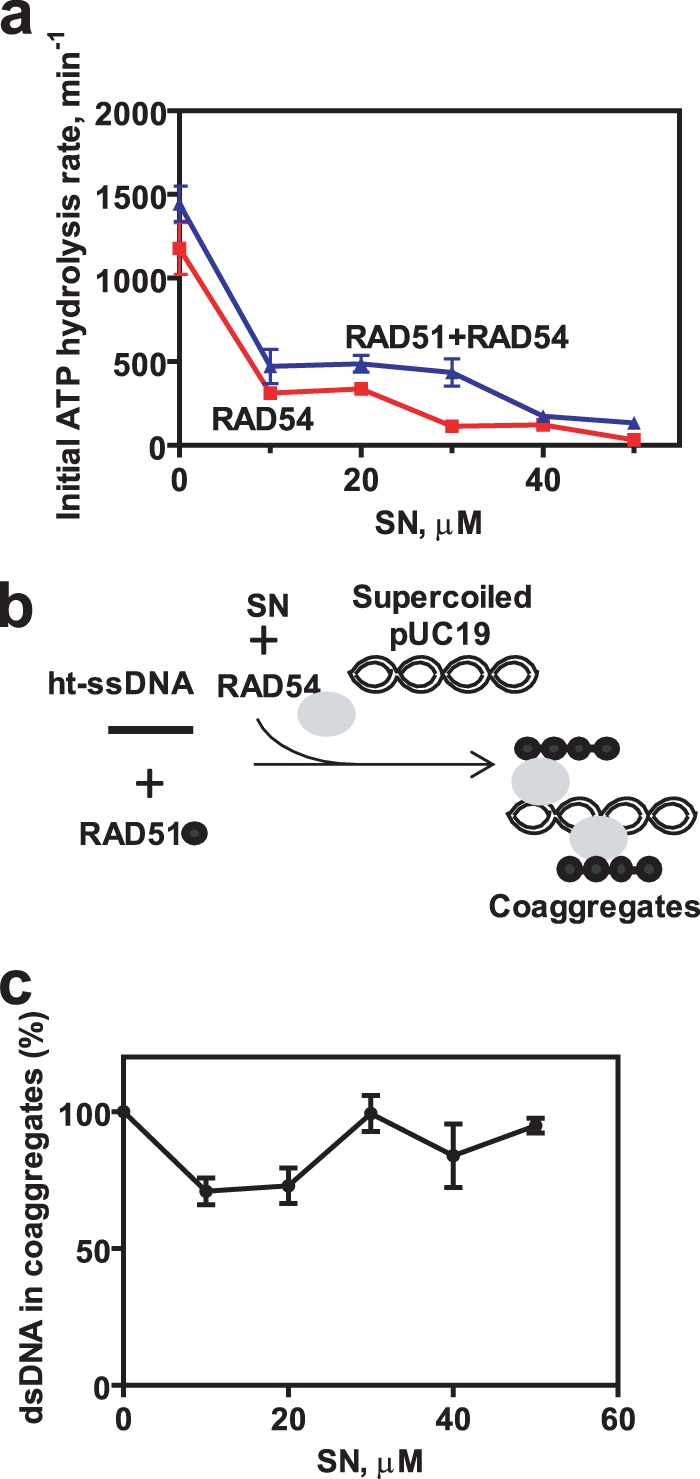
SN inhibits RAD54 ATPase but not coaggregation with dsDNA under DNA strand exchange conditions. a, the effect of SN concentration on RAD54 ATPase under conditions of D loop formation in the presence or absence of RAD51-ssDNA filaments. b, schematic of the DNA coaggregation assay. ht-ssDNA denotes heterologous ssDNA. c, 800 nm RAD51 was incubated with 32P-labeled ssDNA (oligo 71, 90-mer) (2.4 μm, nts) to form nucleoprotein filaments. 200 nm RAD54 and the indicated concentrations of SN or 2% DMSO were added to the filaments. Coaggregation was initiated by adding pUC19 DNA (62 μm, nts). Coaggregates were analyzed by electrophoresis in 1% agarose-TAE gels, and the data are represented as a graph. Error bars represent the S.E.
The K189R Mutation More Strongly Inhibits RAD54 Dissociation from dsDNA Than SN
A relatively small inhibition by SN observed in the D loop assay indicates a minor role of the RAD54 ATPase, and DNA translocation, in stimulation of DNA pairing as compared with its role in BM of Holliday junctions. This result seemingly contradicts the known inability of the RAD54 ATPase-deficient mutants to stimulate DNA strand exchange. We suggest that the RAD54-K189R mutant may form excessively stable complexes with dsDNA, which can be inhibitory for the homology search performed by the RAD51-ssDNA-RAD54 complex. We tested this hypothesis by forming RAD54 nucleoprotein complexes with 32P-labeled dsDNA fragments (an equimolar mixture of 1313-bp and 1373-bp fragments) and challenging them with the same but unlabeled dsDNA competitor (Fig. 11a). Indeed, we found that RAD54-K189R showed an approximately 16-fold decrease (after 30 min of incubation) in its ability to dissociate from a 32P-DNA fragment in the presence of the competitor compared with RAD54 (Fig. 11, b and c). We also found that 50 μm SN caused an 8-fold decrease in RAD54 dissociation from 32P-labeled dsDNA (Fig. 11, b and c). This result is in line with the hypothesis that RAD54-K189R forms intrinsically more stable complexes than wild-type RAD54, even in the presence of SN.
FIGURE 11.
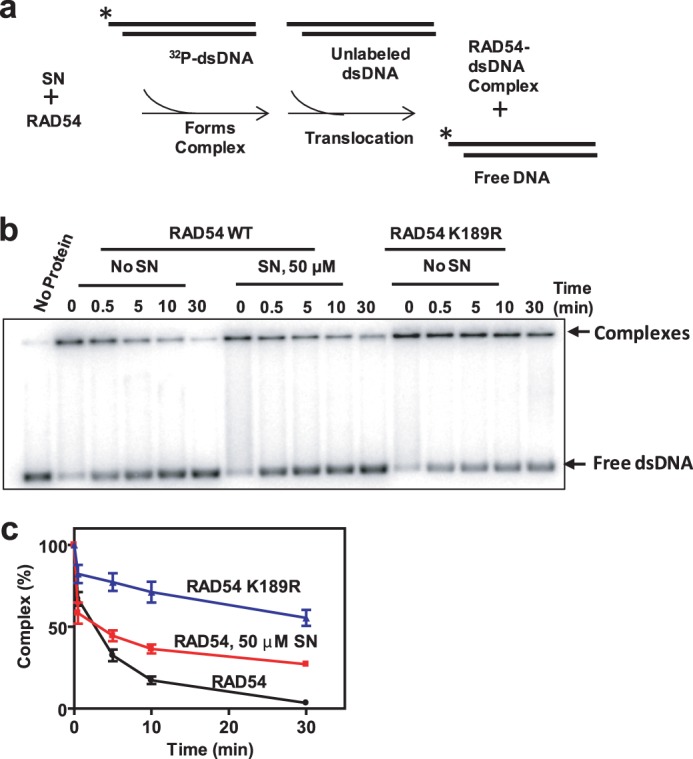
RAD54-K189R forms more stable complexes with dsDNA than RAD54. a, schematic of the dsDNA dissociation assay. The asterisk indicates the 32P-label at the DNA 5′ end. b, 16.5 nm RAD54 or RAD54 K189R was incubated with 2% DMSO or 50 μm SN for 5 min at 30 °C. 32P-labeled pUC19 dsDNA fragments (1313 bp and 1373 bp, 2 μm, nts) were added to form protein-DNA complexes for 10 min. Addition of identical but unlabeled dsDNA fragments (148 μm, nts) initiated the exchange of RAD54 and RAD54 K189R between dsDNA molecules after the protein-DNA complexes were cross-linked by 0.25% glutaraldehyde and analyzed by electrophoresis in 1.5% agarose gels. c, the data presented as a graph. Error bars represent the S.E.
The K189R Mutant Inhibits RAD51-promoted D loop Formation
Formation of excessively stable complexes with dsDNA by RAD54 K189R could be inhibitory for the search for homology during the RAD51-mediated DNA strand exchange. We tested the effect of RAD54-K189R on RAD51-mediated DNA pairing. We modified the D loop assay conditions by omitting Mg2+ so that RAD51 DNA pairing was enhanced, even in the absence of RAD54 (34). We found that RAD54-K189R inhibited the extent of D loop formation in a concentration-dependent manner (Fig. 12, a and b). Under the same conditions, wild-type RAD54 showed a significant stimulation in the extent of D loop formation. This result is in accord with the previous report that showed inhibition of DNA strand exchange activity of yeast Rad51 by the yeast RAD54 K341R ATPase-deficient mutant (26).
This result is also consistent with our hypothesis that the RAD54-K189R mutant that forms non-optimally stable complexes could hinder the search for homology by RAD51. In contrast, SN does not completely block the ATPase activity of RAD54, reducing it 9-fold (at 50 μm SN) (Fig. 10a), which may not prevent RAD54 dissociation from dsDNA, allowing RAD51-ssDNA-RAD54 to perform the search for homology in DNA strand exchange.
DISCUSSION
RAD54 is an HR protein that belongs to the SWI2/SNF2 family of ATPase-dependent DNA translocases. Important activities of RAD54 include stimulation of RAD51-mediated DNA strand exchange and BM of Holliday junctions, a key HR intermediate. However, the mechanisms responsible for these two activities remain to be elucidated. Here, to gain an insight into these mechanisms, we employed a specific small-molecule inhibitor of RAD54, SN.
SN was first isolated from cultures of S. flocculus and was found to have antitumor activity on a broad range of cancers, particularly malignant lymphomas, squamous cell carcinoma of the cervix, breast cancer, malignant melanoma, and head/neck cancers (35, 36). However, SN also showed the toxic side effect of bone marrow depression that halted further clinical studies (36, 56). Nevertheless, it was found that, in some resistant cases, SN was effective where other chemotherapeutics had failed (36). Furthermore, in cases where toxic side effects were minimized, tumor regression and associated clinical benefits occurred in advanced cancer patients (57, 58). Although an exact mechanism for how SN exerts its effect in vivo is unknown, it is proposed that the antitumor activity of SN can be attributed to its quinone group and its ability to generate ROS (59). In the presence of reducing agents and oxygen, a quinone group can become reduced by accepting one or two electrons, resulting in the formation of the semiquinone radical and dihydroquinone, respectively. The semiquinone radical can donate its extra electron to oxygen, generating ROS that can damage DNA and other cellular components (60, 61).
We found that SN inhibits RAD54 BM activity. The inhibition required generation of ROS by SN. In the presence of catalase or high DTT concentrations that can neutralize or scavenge ROS, the inhibition was alleviated. Conversely, the transition metal Cu2+ that increases the rate of SN reduction and ROS generation enhances the inhibitory effect of SN. However, inhibition of RAD54 BM also required physical binding of SN to RAD54, consistent with the short lifetime of ROS and relatively small distances that they can travel from the source. Thus, SN did not affect the DNA strand exchange activity of RAD51 and the BM activity of E. coli RuvAB, proteins that show no structural homology to RAD54. Similarly, two other tested quinone-containing compounds, lapachol and a toxoflavin analog, that are capable of generating ROS did not inhibit RAD54 BM.
We showed previously that RAD54 binds specifically to Holliday junctions, forms multiprotein complexes, and promotes BM by translocating along DNA (17, 18). Here we analyzed the mechanism of RAD54 BM inhibition by SN and found that SN primarily targets the ATPase activity of RAD54. Examination of RAD54 by electrophoresis in SDS-polyacrylamide gels showed that SN causes aggregation of RAD54 as well as its fragmentation, which was further increased by Cu2+ that enhances generation of ROS (51). The major fragmentation site was mapped using terminus-specific antibodies to the conserved helicase motif III located in the central portion of the protein. These data indicate that SN interacts near the central cleft of RAD54 and causes local protein damage by generating ROS (Fig. 8c). The central cleft of RAD54 sits between the two RecA-like domains that contain the seven conserved helicase motifs I, Ia, II, III, IV, V, and VI (54, 55) that bind ATP and DNA. Motif III of the N-terminal RecA-like domain is in close proximity to the central cleft of RAD54 and interacts with the γ-phosphate group of ATP, DNA, and the C-terminal RecA-like domain (54, 55). This motif is likely responsible for bringing the two RecA-like domains together to close up and bind ATP for hydrolysis. Disruption of motif III by SN would be expected to decrease the ATPase and DNA-translocase activity of RAD54, both of which are crucial for BM activity. Also, the proximity of the DNA binding site to this motif III may be responsible for the mild decline in DNA binding by RAD54 caused by SN.
RAD54 performs its functions in a close association with RAD51. These proteins interact both physically and functionally, mutually enhancing their activities (11–13, 25, 62). Thus, RAD51 stimulates ATP hydrolysis and BM by RAD54. Importantly, RAD54 stimulates DNA strand exchange promoted by RAD51, a salient activity of HR. The mechanism of DNA strand exchange stimulation by RAD54 has been studied extensively. It has been shown that stimulation of RAD51-promoted DNA strand exchange requires ATP hydrolysis by RAD54 because RAD54 mutants lacking the ATPase activity did not show stimulation (14, 16, 26). Because ATP hydrolysis is also required for RAD54 translocation on DNA, it is commonly thought that stimulation of DNA strand exchange involves RAD54-promoted ATPase-dependent dsDNA translocation in the proximity of the RAD51-ssDNA filament. Surprisingly, we found that SN has a significantly stronger inhibitory effect on both ATPase and BM activities of RAD54 than on its ability to stimulate DNA strand exchange promoted by RAD51. These results indicate differences in the role that the RAD54 ATPase and DNA translocation play in RAD54 BM and stimulation of DNA strand exchange. We also found that the RAD54-K189R ATPase-deficient mutant formed non-optimally stable complexes with dsDNA. Furthermore, RAD54 K189R inhibited D loop formation by RAD51, consistent with the hypothesis that formation of excessively stable RAD54-dsDNA complexes interferes with the search for homology by the RAD51-ssDNA-RAD54 complex, which requires rapid cycles of dsDNA dissociation and reassociation. In contrast, 50 μm SN caused significant but partial inhibition (89%) of the ATPase, which still allowed for the dissociation of RAD54-dsDNA complexes.
The role of the ATPase-dependent DNA translocation of RAD54 in stimulation of the RAD51 pairing activity cannot be entirely ruled out by our results because SN, under tested concentrations, does not block it completely. During this limited DNA translocation, RAD54 can still cause local unwinding in dsDNA which, as has been proposed previously (27, 29, 63), can facilitate homology recognition by RAD51. However, our data may argue against extensive translocation as a mean of dsDNA tracking along the RAD51-ssDNA filament during the search for homology. In addition, none of the known proteins (30–34) or ions (38, 64) that stimulate RAD51 DNA strand exchange activity act by promoting dsDNA translocation, indicating against the essential role of dsDNA translocation during the search for homology.
It is also possible that RAD54 induces conformational changes in RAD51 that activate its DNA strand exchange activity. It has been shown previously that RAD54 has an ATPase-independent mode of RAD51 stimulation. It stabilizes the RAD51-ssDNA filament independently of ATP hydrolysis (22, 25). Although the molecular mechanism of this stabilization remains to be elucidated, it is possible that RAD54, through physical interactions, induces conformational changes in RAD51 that lead to the filament stabilization. It is known from previous studies that RAD51 is prone to conformational changes that have a strong effect on DNA strand exchange activity. Various factors, like nucleotides or monovalent salts, were shown to induce these changes.
In vivo, SN can cause DNA breaks and other lesions in the presence of metal ions like Fe2+ or Cu2+ (65, 66). In addition, it was shown that SN also interacts with topoisomerase II (37, 67). It is thought that by stabilizing the cleavage complexes of topoisomerase II with DNA and inhibiting DNA rejoining, SN may further increase formation of DNA breaks. Our results demonstrate that SN can specifically inhibit RAD54 ATPase activity with approximately the same efficiency. Thus, both induction of DNA breaks and inhibition of RAD54, one of the key proteins of DNA double strand break repair, may contribute to the therapeutic efficacy of SN against cancer cells.
Compounds that either generate ROS directly or induce endogenous ROS by disrupting cell metabolism are an important tool of anticancer therapy (68). Specifically, quinone derivatives today are one of the largest classes of antitumor agents approved for clinical use in the United States (69, 70). Further progress in developing ROS-generating anticancer agents may involve the identification of specific cellular targets that are responsible for killing cancer cells and are less toxic to normal cells. Structural modifications of these compounds, including SN, will enhance their affinities for specific targets and eliminate harmful side effects because of interactions with physiologically important macromolecules.
Here we used SN to analyze the mechanisms of RAD54 BM activity and its ability to stimulate DNA strand exchange promoted by RAD51. Our results demonstrate important distinctions between these two activities. Although BM strictly depends on protein ATPase and on DNA translocation, stimulation of DNA strand exchange shows less of a dependence. This study demonstrates the power of specific small-molecule inhibitors to dissect the mechanisms of protein activities.
Acknowledgments
We thank Dr. Robert Sammelson (Ball State University) and Dr. Patrick Sung (Yale) for providing lavendamycin analogs and the pMJ5 plasmid, respectively. We also thank Dr. Olga Mazina, Dr. Marilyn Jorns, Dr. Michael Jackson, and Nadish Goyal (all of Drexel University College of Medicine) for comments and discussion.
This work was supported, in whole or in part, by National Institutes of Health Grants CA100839, DA033981, and MH097512 (to A. V. M.) and 1R21AI087388 (to S. C.). This work was also supported by a KECK Foundation award and by Leukemia and Lymphoma Society Scholar Award 1054-09 (to A. V. M.).

This article contains supplemental Fig. S1 and Table S1.
F. Huang, J. Negri, N. Tolliday, and A. V. Mazin, manuscript in preparation.
- HR
- homologous recombination
- ssDNA
- single-stranded DNA
- BM
- branch migration
- SN
- streptonigrin
- ROS
- reactive oxygen species
- DMSO
- dimethyl sulfoxide
- TFO
- triplex-forming oligonucleotide
- PX
- partial X-junction.
REFERENCES
- 1. Moynahan M. E., Jasin M. (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 11, 196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sung P., Klein H. (2006) Mechanism of homologous recombination. Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 7, 739–750 [DOI] [PubMed] [Google Scholar]
- 3. Kowalczykowski S. C. (2008) Structural biology. Snapshots of DNA repair. Nature 453, 463–466 [DOI] [PubMed] [Google Scholar]
- 4. Liu Y., West S. C. (2004) Happy Hollidays. 40th anniversary of the Holliday junction. Nat. Rev. Mol. Cell Biol. 5, 937–944 [DOI] [PubMed] [Google Scholar]
- 5. Holliday R. (1964) A mechanism for gene conversion in fungi. Genet. Res. 5, 282–304 [DOI] [PubMed] [Google Scholar]
- 6. Mazin A. V., Mazina O. M., Bugreev D. V., Rossi M. J. (2010) Rad54, the motor of homologous recombination. DNA Repair 9, 286–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Game J. C. (1993) DNA double-strand breaks and the RAD50-RAD57 genes in Saccharomyces. Semin. Cancer Biol. 4, 73–83 [PubMed] [Google Scholar]
- 8. Essers J., van Steeg H., de Wit J., Swagemakers S. M., Vermeij M., Hoeijmakers J. H., Kanaar R. (2000) Homologous and non-homologous recombination differentially affect DNA damage repair in mice. EMBO J. 19, 1703–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmuckli-Maurer J., Rolfsmeier M., Nguyen H., Heyer W. D. (2003) Genome instability in rad54 mutants of Saccharomyces cerevisiae. Nucleic Acids Res. 31, 1013–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Matsuda M., Miyagawa K., Takahashi M., Fukuda T., Kataoka T., Asahara T., Inui H., Watatani M., Yasutomi M., Kamada N., Dohi K., Kamiya K. (1999) Mutations in the RAD54 recombination gene in primary cancers. Oncogene 18, 3427–3430 [DOI] [PubMed] [Google Scholar]
- 11. Golub E. I., Kovalenko O. V., Gupta R. C., Ward D. C., Radding C. M. (1997) Interaction of human recombination proteins Rad51 and Rad54. Nucleic Acids Res. 25, 4106–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang H., Xie Y., Houston P., Stemke-Hale K., Mortensen U. H., Rothstein R., Kodadek T. (1996) Direct association between the yeast Rad51 and Rad54 recombination proteins. J. Biol. Chem. 271, 33181–33186 [DOI] [PubMed] [Google Scholar]
- 13. Clever B., Interthal H., Schmuckli-Maurer J., King J., Sigrist M., Heyer W. D. (1997) Recombinational repair in yeast. Functional interactions between Rad51 and Rad54 proteins. EMBO J. 16, 2535–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Petukhova G., Stratton S., Sung P. (1998) Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature 393, 91–94 [DOI] [PubMed] [Google Scholar]
- 15. Sigurdsson S., Van Komen S., Petukhova G., Sung P. (2002) Homologous DNA pairing by human recombination factors Rad51 and Rad54. J. Biol. Chem. 277, 42790–42794 [DOI] [PubMed] [Google Scholar]
- 16. Mazina O. M., Mazin A. V. (2004) Human Rad54 protein stimulates DNA strand exchange activity of hRad51 protein in the presence of Ca2+. J. Biol. Chem. 279, 52042–52051 [DOI] [PubMed] [Google Scholar]
- 17. Mazina O. M., Rossi M. J., Thomaä N. H., Mazin A. V. (2007) Interactions of human rad54 protein with branched DNA molecules. J. Biol. Chem. 282, 21068–21080 [DOI] [PubMed] [Google Scholar]
- 18. Bugreev D. V., Mazina O. M., Mazin A. V. (2006) Rad54 protein promotes branch migration of Holliday junctions. Nature 442, 590–593 [DOI] [PubMed] [Google Scholar]
- 19. Pâques F., Haber J. E. (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63, 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bugreev D. V., Hanaoka F., Mazin A. V. (2007) Rad54 dissociates homologous recombination intermediates by branch migration. Nat. Struct. Mol. Biol. 14, 746–753 [DOI] [PubMed] [Google Scholar]
- 21. Solinger J. A., Lutz G., Sugiyama T., Kowalczykowski S. C., Heyer W. D. (2001) Rad54 protein stimulates heteroduplex DNA formation in the synaptic phase of DNA strand exchange via specific interactions with the presynaptic Rad51 nucleoprotein filament. J. Mol. Biol. 307, 1207–1221 [DOI] [PubMed] [Google Scholar]
- 22. Wolner B., Peterson C. L. (2005) ATP-dependent and ATP-independent roles for the Rad54 chromatin remodeling enzyme during recombinational repair of a DNA double strand break. J. Biol. Chem. 280, 10855–10860 [DOI] [PubMed] [Google Scholar]
- 23. Solinger J. A., Kiianitsa K., Heyer W. D. (2002) Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51:dsDNA filaments. Mol. Cell 10, 1175–1188 [DOI] [PubMed] [Google Scholar]
- 24. Alexeev A., Mazin A., Kowalczykowski S. C. (2003) Rad54 protein possesses chromatin-remodeling activity stimulated by the Rad51-ssDNA nucleoprotein filament. Nat. Struct. Biol. 10, 182–186 [DOI] [PubMed] [Google Scholar]
- 25. Mazin A. V., Alexeev A. A., Kowalczykowski S. C. (2003) A novel function of Rad54 protein. Stabilization of the Rad51 nucleoprotein filament. J. Biol. Chem. 278, 14029–14036 [DOI] [PubMed] [Google Scholar]
- 26. Petukhova G., Van Komen S., Vergano S., Klein H., Sung P. (1999) Yeast Rad54 promotes Rad51-dependent homologous DNA pairing via ATP hydrolysis-driven change in DNA double helix conformation. J. Biol. Chem. 274, 29453–29462 [DOI] [PubMed] [Google Scholar]
- 27. Mazin A. V., Bornarth C. J., Solinger J. A., Heyer W. D., Kowalczykowski S. C. (2000) Rad54 protein is targeted to pairing loci by the Rad51 nucleoprotein filament. Mol. Cell 6, 583–592 [DOI] [PubMed] [Google Scholar]
- 28. Tan T. L., Essers J., Citterio E., Swagemakers S. M., de Wit J., Benson F. E., Hoeijmakers J. H., Kanaar R. (1999) Mouse Rad54 affects DNA conformation and DNA-damage-induced Rad51 foci formation. Curr. Biol. 9, 325–328 [DOI] [PubMed] [Google Scholar]
- 29. Van Komen S., Petukhova G., Sigurdsson S., Stratton S., Sung P. (2000) Superhelicity-driven homologous DNA pairing by yeast recombination factors Rad51 and Rad54. Mol. Cell 6, 563–572 [DOI] [PubMed] [Google Scholar]
- 30. Chi P., San Filippo J., Sehorn M. G., Petukhova G. V., Sung P. (2007) Bipartite stimulatory action of the Hop2-Mnd1 complex on the Rad51 recombinase. Genes Dev. 21, 1747–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pezza R. J., Voloshin O. N., Vanevski F., Camerini-Otero R. D. (2007) Hop2/Mnd1 acts on two critical steps in Dmc1-promoted homologous pairing. Genes Dev. 21, 1758–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wiese C., Dray E., Groesser T., San Filippo J., Shi I., Collins D. W., Tsai M. S., Williams G. J., Rydberg B., Sung P., Schild D. (2007) Promotion of homologous recombination and genomic stability by RAD51AP1 via RAD51 recombinase enhancement. Mol. Cell 28, 482–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Modesti M., Budzowska M., Baldeyron C., Demmers J. A., Ghirlando R., Kanaar R. (2007) RAD51AP1 is a structure-specific DNA binding protein that stimulates joint molecule formation during RAD51-mediated homologous recombination. Mol. Cell 28, 468–481 [DOI] [PubMed] [Google Scholar]
- 34. Bugreev D. V., Mazina O. M., Mazin A. V. (2009) Bloom syndrome helicase stimulates RAD51 DNA strand exchange activity through a novel mechanism. J. Biol. Chem. 284, 26349–26359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bolzán A. D., Bianchi M. S. (2001) Genotoxicity of streptonigrin. A review. Mutat. Res. 488, 25–37 [DOI] [PubMed] [Google Scholar]
- 36. Harris M. N., Medrek T. J., Golomb F. M., Gumport S. L., Postel A. H., Wright J. C. (1965) Chemotherapy with streptonigrin in advanced cancer. Cancer 18, 49–57 [DOI] [PubMed] [Google Scholar]
- 37. Yamashita Y., Kawada S., Fujii N., Nakano H. (1990) Induction of mammalian DNA topoisomerase II dependent DNA cleavage by antitumor antibiotic streptonigrin. Cancer Res. 50, 5841–5844 [PubMed] [Google Scholar]
- 38. Sigurdsson S., Trujillo K., Song B., Stratton S., Sung P. (2001) Basis for avid homologous DNA strand exchange by human Rad51 and RPA. J. Biol. Chem. 276, 8798–8806 [DOI] [PubMed] [Google Scholar]
- 39. Bugreev D. V., Mazina O. M., Mazin A. V. (2006) Analysis of branch migration activities of proteins using synthetic DNA substrates. Nat. Protoc. 10.1038/nprot.2006.217 [DOI] [Google Scholar]
- 40. Rossi M. J., Mazina O. M., Bugreev D. V., Mazin A. V. (2010) Analyzing the branch migration activities of eukaryotic proteins. Methods 51, 336–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bravman T., Bronner V., Lavie K., Notcovich A., Papalia G. A., Myszka D. G. (2006) Exploring “one-shot” kinetics and small molecule analysis using the ProteOn XPR36 array biosensor. Anal. Biochem. 358, 281–288 [DOI] [PubMed] [Google Scholar]
- 42. Kowalczykowski S. C., Krupp R. A. (1987) Effects of Escherichia coli SSB protein on the single-stranded DNA-dependent ATPase activity of Escherichia coli RecA protein. Evidence that SSB protein facilitates the binding of RecA protein to regions of secondary structure within single-stranded DNA. J. Mol. Biol. 193, 97–113 [DOI] [PubMed] [Google Scholar]
- 43. Firman K., Szczelkun M. D. (2000) Measuring motion on DNA by the type I restriction endonuclease EcoR124I using triplex displacement. EMBO J. 19, 2094–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jaskelioff M., Van Komen S., Krebs J. E., Sung P., Peterson C. L. (2003) Rad54p is a chromatin remodeling enzyme required for heteroduplex DNA joint formation with chromatin. J. Biol. Chem. 278, 9212–9218 [DOI] [PubMed] [Google Scholar]
- 45. Iype L. E., Wood E. A., Inman R. B., Cox M. M. (1994) RuvA and RuvB proteins facilitate the bypass of heterologous DNA insertions during RecA protein-mediated DNA strand exchange. J. Biol. Chem. 269, 24967–24978 [PubMed] [Google Scholar]
- 46. Ariyoshi M., Nishino T., Iwasaki H., Shinagawa H., Morikawa K. (2000) Crystal structure of the Holliday junction DNA in complex with a single RuvA tetramer. Proc. Natl. Acad. Sci. U.S.A. 97, 8257–8262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Whitehouse I., Stockdale C., Flaus A., Szczelkun M. D., Owen-Hughes T. (2003) Evidence for DNA translocation by the ISWI chromatin-remodeling enzyme. Mol. Cell. Biol. 23, 1935–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Davies K. J. (1987) Protein damage and degradation by oxygen radicals. I. General aspects. J. Biol. Chem. 262, 9895–9901 [PubMed] [Google Scholar]
- 49. Latuasan H. E., Berends W. (1961) On the origin of the toxicity of toxoflavin. Biochim. Biophys. Acta 52, 502–508 [DOI] [PubMed] [Google Scholar]
- 50. Goulart M. O., Falkowski P., Ossowski T., Liwo A. (2003) Electrochemical study of oxygen interaction with lapachol and its radical anions. Bioelectrochemistry 59, 85–87 [DOI] [PubMed] [Google Scholar]
- 51. Hajdu J., Armstrong E. C. (1981) Interaction of metal ions with streptonigrin. 1. Formation of copper(II) and zinc(II) complexes of the antitumor antibiotic. J. Am. Chem. Soc. 103, 232–234 [Google Scholar]
- 52. Anderberg P. I., Harding M. M., Lay P. A. (2004) The effect of metal ions on the electrochemistry of the antitumor antibiotic streptonigrin. J. Inorg. Biochem. 98, 720–726 [DOI] [PubMed] [Google Scholar]
- 53. Godson G. N., Schoenich J., Sun W., Mustaev A. A. (2000) Identification of the magnesium ion binding site in the catalytic center of Escherichia coli primase by iron cleavage. Biochemistry 39, 332–339 [DOI] [PubMed] [Google Scholar]
- 54. Thomä N. H., Czyzewski B. K., Alexeev A. A., Mazin A. V., Kowalczykowski S. C., Pavletich N. P. (2005) Structure of the SWI2/SNF2 chromatin-remodeling domain of eukaryotic Rad54. Nat. Struct. Mol. Biol. 12, 350–356 [DOI] [PubMed] [Google Scholar]
- 55. Dürr H., Körner C., Müller M., Hickmann V., Hopfner K. P. (2005) X-ray structures of the Sulfolobus solfataricus SWI2/SNF2 ATPase core and its complex with DNA. Cell 121, 363–373 [DOI] [PubMed] [Google Scholar]
- 56. Smith G. M., Gordon J. A., Sewell I. A., Ellis H. (1967) A trial of streptonigrin in the treatment of advanced malignant disease. Br. J. Cancer 21, 295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sullivan R. D., Miller E., Zurek W. Z., Rodriguez F. R. (1963) Clinical effects of prolonged (continuous) infusion of streptonigrin (NSC-45383) in advanced cancer. Cancer Chemother. Rep. 33, 27–40 [PubMed] [Google Scholar]
- 58. Pittillo R. F., Woolley C. (1974) Biological assay of streptonigrin (NSC 45383) in body fluids and tissues of mice. Antimicrob. Agents Chemother. 5, 82–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kremer W. B., Laszlo J. (1967) Comparison of biochemical effects of isopropylidine azastreptonigrin (NSC-62709) with streptonigrin (NSC-45383). Cancer Chemother. Rep. 51, 19–24 [PubMed] [Google Scholar]
- 60. Miller D. S., Laszlo J., McCarty K. S., Guild W. R., Hochstein P. (1967) Mechanism of action of streptonigrin in leukemic cells. Cancer Res. 27, 632–638 [PubMed] [Google Scholar]
- 61. Lown J. W. (1983) The mechanism of action of quinone antibiotics. Mol. Cell Biochem. 55, 17–40 [DOI] [PubMed] [Google Scholar]
- 62. Haseltine C. A., Kowalczykowski S. C. (2009) An archaeal Rad54 protein remodels DNA and stimulates DNA strand exchange by RadA. Nucleic Acids Res. 37, 2757–2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ristic D., Wyman C., Paulusma C., Kanaar R. (2001) The architecture of the human Rad54-DNA complex provides evidence for protein translocation along DNA. Proc. Natl. Acad. Sci. U.S.A. 98, 8454–8460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bugreev D. V., Mazin A. V. (2004) Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc. Natl. Acad. Sci. U.S.A. 101, 9988–9993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sugiura Y., Kuwahara J., Suzuki T. (1984) DNA interaction and nucleotide sequence cleavage of copper-streptonigrin. Biochim. Biophys. Acta 782, 254–261 [DOI] [PubMed] [Google Scholar]
- 66. Long G. V., Harding M. M., Fan J. Y., Denny W. A. (1997) Interaction of the antitumour antibiotic streptonigrin with DNA and oligonucleotides. Anticancer Drug Des. 12, 453–472 [PubMed] [Google Scholar]
- 67. Capranico G., Palumbo M., Tinelli S., Zunino F. (1994) Unique sequence specificity of topoisomerase II DNA cleavage stimulation and DNA binding mode of streptonigrin. J. Biol. Chem. 269, 25004–25009 [PubMed] [Google Scholar]
- 68. Wondrak G. T. (2009) Redox-directed cancer therapeutics. Molecular mechanisms and opportunities. Antioxid. Redox Signal. 11, 3013–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Powis G. (1989) Free radical formation by antitumor quinones. Free Radic. Biol. Med. 6, 63–101 [DOI] [PubMed] [Google Scholar]
- 70. Sanchez-Cruz P., Alegría A. E. (2009) Quinone-enhanced reduction of nitric oxide by xanthine/xanthine oxidase. Chem. Res. Toxicol. 22, 818–823 [DOI] [PMC free article] [PubMed] [Google Scholar]