

Results: Gremlin-1 binds with high affinity to macrophage migration inhibitory factor and attenuates the progression of atherosclerosis.
Conclusion: We describe a novel mechanism that regulates foam cell formation and plaque growth.
Significance: The findings disclose a new mechanism for the regulation of plaque growth and may open novel therapeutic strategies to control the progression of atherosclerosis.
Keywords: Atherosclerosis, Chemokines, Inflammation, Monocytes, Vascular Biology
Abstract
Monocyte infiltration and macrophage formation are pivotal steps in atherosclerosis and plaque vulnerability. Gremlin-1/Drm is crucial in embryo-/organogenesis and has been shown to be expressed in the adult organism at sites of arterial injury and to inhibit monocyte migration. The purpose of the present study was to evaluate and characterize the role of Gremlin-1 in atherosclerosis. Here we report that Gremlin-1 is highly expressed primarily by monocytes/macrophages in aortic atherosclerotic lesions of ApoE−/− mice and is secreted from activated monocytes and during macrophage development in vitro. Gremlin-1 reduces macrophage formation by inhibiting macrophage migration inhibitory factor (MIF), a cytokine critically involved in atherosclerotic plaque progression and vulnerability. Gremlin-1 binds with high affinity to MIF (KD = 54 nm), as evidenced by surface plasmon resonance analysis and co-immunoprecipitation, and reduces MIF-induced release of TNF-α from macrophages. Treatment of ApoE−/− mice with a dimeric recombinant fusion protein, mGremlin1-Fc, but not with equimolar control Fc or inactivated mGremlin1-Fc, reduced TNF-α expression, the content of monocytes/macrophages of atherosclerotic lesions, and attenuated atheroprogression. The present data disclose that Gremlin-1 is an endogenous antagonist of MIF and define a role for Gremlin-1/MIF interaction in atherosclerosis.
Introduction
Atherosclerosis is a chronic disease of the arteries characterized by inflammation of the vessel wall (1–3). Endothelial activation, monocyte recruitment, and the formation of macrophages are critical events in the development of atherosclerosis (4). Macrophages secrete cytokines/chemokines and growth factors that promote atheroprogression and contribute substantially to plaque vulnerability and acute complications of the disease such as acute coronary syndromes (5). The cytokine macrophage migration inhibitory factor (MIF)2 is a noncognate ligand of CXC chemokine receptors and regulates monocyte recruitment toward atherosclerotic lesions (6). Blocking or genetic deletion of MIF reduces macrophage and T-cell content of atherosclerotic plaques and attenuates the progression of atherosclerosis in ApoE−/− mice (7).
Gremlin-1 and its rat homolog Drm (down-regulated by v-mos) are highly conserved 20.7-kDa glycoproteins (8, 9). Gremlin-1 belongs to the DAN/Cerberus protein family, which is a member of the cysteine knot superfamily that includes TGF-β and VEGF (10). Gremlin-1 is a bone morphogenetic protein (BMP) antagonist and binds to BMP-2, -4, and -7 (9). It exists in secreted and cell-associated forms (9). The Gremlin-1 gene encodes a 23- and 28-kDa protein that is glycosylated before secretion (9). Gremlin-1-dependent inhibition of BMPs is important for embryogenesis and development of organs such as limbs, kidney, and lungs (11, 12). Gremlin-1 knock-out mice are neonatally lethal with significant renal and lung defects (13). Transgenic mice overexpressing Gremlin-1 under the control of the osteocalcin promoter show reduced bone formation (14). Gremlin-1 is expressed in endothelial cells exposed to disturbed flow in mouse aorta and in human coronary arteries, suggesting a role in inflammation and atherosclerosis (15). In the adult system, Gremlin-1 regulates cell proliferation and stem cell differentiation (16). Secreted Gremlin-1 binds to BMPs and prevents ligand/receptor interaction and subsequent downstream signaling (11). Gremlin-1 interacts with Slit-1/-2 proteins and inhibits monocyte migration induced by SDF-1-α or fMLP (17). Gremlin-1 is expressed and secreted by tumor cells and activated endothelial cells (18, 19), binds VEGF receptor-2 (VEGFR-2), and acts as a pro-angiogenic agonist (10) implying a role in angiogenesis, vascular development, and neovascularization. Further, heparan sulfate proteoglycans act as functional Gremlin-1 co-receptors in endothelial cells and affect its interaction with VEGFR-2 and its angiogenic activity (20). Gremlin-1 is expressed in the liver of mice developing liver fibrosis (21), is up-regulated in pericytes in response to elevated glucose levels suggesting a role in diabetic retinopathy (22), and has been implicated in tubulointerstitial fibrosis in diabetic nephropathy (23).
Given these functions of Gremlin-1, we hypothesized that Gremlin-1 plays a role in vascular inflammation and atherosclerosis. Here we show that Gremlin-1 regulates monocyte/macrophage function in vitro, is an endogenous antagonist of MIF, and binds with high affinity to MIF. Administration of a dimeric recombinant fusion protein, mGremlin-1-Fc, reduces the content of macrophages in atherosclerotic plaques and limits atheroprogression and lesion instability. Thus, Gremlin-1 is an important factor in the process of vascular inflammation and atherosclerosis and might be used to treat atherosclerosis and to improve plaque stability in patients at risk for acute coronary syndromes.
MATERIALS AND METHODS
The methods used in the present work are summarized below. Reagents and all methods are presented in detail under the supplemental “Methods.”
Cell Isolation and Cell Lines
Human monocytes were isolated from peripheral venous blood samples by adherence after Ficoll-Paque purification of peripheral blood mononuclear cells as described (24). Flp-InTM-CHO cells were grown following the recommendations of the manufacturer (Invitrogen).
MIF-induced TNF-α Secretion from Macrophages
Macrophages were differentiated from human monocytes isolated from buffy coats by density gradient centrifugation and differentiated as described (25). Human macrophages were stimulated with 0.25 μg/ml MIF, 0.5 μg/ml Gremlin-1, or MIF and Gremlin-1 for 12 h in RPMI 1640 with 2% FCS and 0.5 μg/ml polymyxin B. When MIF and Gremlin-1 were added together, they were preincubated with each other for 10 min before being added to the culture. TNF-α concentration was determined in cell supernatants using ELISA (R&D Systems, Minneapolis, MN).
Immunohistochemistry
For immunostaining and confocal microscopy, human cells were fixed with 2% formaldehyde and permeabilized with 0.2% Triton X-100. Human cells were stained with rabbit polyclonal anti-Gremlin-1 (polyclonal, clone RB2060, Abnova), rabbit polyclonal CD68 antibody (polyclonal, Abbiotec), anti-MIF antibody (polyclonal, R&D Systems, Wiesbaden, Germany), or isotype control antibodies for 1 h. For immunohistochemical analysis, aortic tissue samples were embedded in paraffin. The 5-μm-thick paraffin sections were immunostained either by the avidin-biotin complex (ABC) method (LSAB+ system-HRP, Dako, Heverlee, Belgium) or by immunofluorescence using primary antibodies and secondary Alexa Fluor-labeled antibodies (Molecular Probes). Mouse aortic tissue was stained using the following primary antibodies: rabbit polyclonal anti-Gremlin-1 (polyclonal, clone RB2060, Abnova, Germany), rabbit polyclonal mouse anti-CD68 (Abbiotec), rat monoclonal macrophage antibody (monoclonal, clone M3/84, Mac-3, BD Biosciences), goat anti-MIF (polyclonal, R&D Systems), anti-TNF-α, and isotype control antibodies according to the standard protocol. Corresponding biotinylated secondary antibodies (Dako) were used. Adjacent sections were stained with hematoxylin and eosin to visualize the corresponding structures of the plaques. To detect binding of the recombinant fusion protein mGremlin-1-Fc or control Fc, goat monoclonal antibodies were used against human IgG (Vector Laboratories). Unspecific binding was prevented by bovine serum albumin (3%, 1 h). Samples were covered with mounting medium (Dako) and analyzed by compound microscopy (Axiovert 200, Zeiss, and Nikon Digital Sight DS-U1, Nikon, Japan) or confocal microscopy (LSM510, Zeiss, and Leica TCSSP, Leica Microsystems).
Mouse Model of Atherosclerosis and Disease Progression
Male ApoE−/− mice (B6.129P2-ApoetmUnc) were purchased from The Jackson Laboratory. Starting at the age of 4 weeks the mice received a cholesterol-rich diet (1.25% cholesterol, Harlan research diets, 0.2% cholate) throughout the experiments. C57BL/6J wild-type mice (Charles River Laboratories) served as controls. To assess Gremlin-1 expression in atherosclerotic arteries, mice received this diet for 4 or 12 weeks. At the end of this period the mice were sacrificed, and the main arteries (aortic root, aortic arch, aorta thoracica, and aorta abdominalis) were collected, conserved in TRIzol (Sigma), and frozen at −80 °C for later total RNA extraction or embedded in paraffin for immunohistochemistry.
To study the effect of mGremlin-1-Fc, 4-week-old ApoE−/− mice were fed with a cholesterol-rich diet. At the age of 10 weeks, mGremlin-1-Fc (1 μg/g body weight, n = 8) or control Fc (equimolar, n = 8) was administered intraperitoneally three times/week for a further 4 weeks. Further, inactivated mGremlin-1-Fc (1 μg/g body weight, n = 3) was used and compared with active mGremlin-1-Fc (1 μg/g body weight, n = 3). mGremlin-1-Fc was inactivated by incubating the fusion protein for 10 min at 95 °C. Thereafter, the mice were sacrificed in general anesthesia. All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH publication No. 85-23, revised 1996) and the German law for the welfare of animals. Animal studies were approved by the local authorities (regional board, Tübingen).
After euthanasia of the animals, the vessels were perfused with saline in situ followed by perfusion with 4% paraformaldehyde through the left ventricle. Subsequently, the vessels were transferred into 4% paraformaldehyde for fixation. Arteries from mice with longtime mGremlin-1-Fc treatment were immediately stained with oil red O. Afterward the vessels were photographed with a Zeiss Axiovert 200 and the Axiocam MRc5 (Zeiss) using the AxioVision software. Plaque areas and the total vessel area were determined, and the relative plaque extension was expressed as a percentage of the total vessel area. Afterward, vessels were embedded in paraffin, cut in 5-μm sections, and stained with H&E or immunostained for the specific visualization of macrophages, foam cells, and T-lymphocytes using anti-CD68, anti-Gremlin-1, anti-MIF, anti-Mac3, anti-TNF-α, and anti-Fc.
After staining and immunostaining, sections were viewed under a Nikon compound microscope (Digital Sight DS-U1, Nikon), and digital images were taken with a Nikon camera (Digital Sight DS-5M, Nikon) at a resolution of 2592 × 1944 pixels. The imaging software NIS-Elements Basic Research (Nikon) was used. Macrophage and T-cell numbers were determined by counting blue nuclei in positively stained cells within lesions. On tissue sections of the aortic arch, nine plaques on random sections were analyzed per mouse at ×10 magnification regarding macrophage/foam cell number (CD68-positive cells) per total cell number and macrophage/foam cell number per total plaque area.
Statistical Analysis
Data are presented as mean ± S.E. Statistical analyses on continuous data were performed using unpaired two-tailed Student's t test. Correlations were analyzed using Spearman's rank correlation coefficient. A p value of 0.05 or less was regarded as significant.
RESULTS
Gremlin-1 Is Expressed in Atherosclerotic Lesions in ApoE−/− Mice
To evaluate the expression of Gremlin-1 in atherogenesis we used the ApoE−/− mouse, which spontaneously develops atherosclerotic lesions (26). Four-week-old ApoE−/− mice were fed for 4 and 12 weeks with a cholesterol-rich diet, and Gremlin-1 mRNA expression in aortic tissue was analyzed by RT-PCR (Fig. 1a). Formation of atherosclerotic plaques was verified by Sudan red III staining of the aorta (not shown). We found that Gremlin-1 mRNA expression in aortic tissue was significantly increased in ApoE−/− mice 12 weeks after the beginning of the cholesterol-rich diet compared with ApoE−/− mice fed for 4 weeks or wild type control animals (Fig. 1a). No dynamic mRNA expression in atherosclerotic tissue was found for another DAN protein, Gremlin-2/PRDC (data not shown). To locate the expression in atherosclerotic lesions, we further studied Gremlin-1 mRNA expression using in situ hybridization on atherosclerotic plaques and wild type aorta (Fig. 1b and supplemental Fig. 1). Gremlin-1 expression was very low in the aortic tissue of wild type mice (Fig. 1b and supplemental Fig. 1).
FIGURE 1.

Gremlin-1 is expressed in atherosclerotic lesions in ApoE−/− mice. Aortic tissue was obtained from ApoE−/− (n = 10) and wild type (n = 7) mice at the age of 8 and 16 weeks, respectively. ApoE−/− mice were fed with a cholesterol-rich diet for 4 or 12 weeks beginning at the age of 4 weeks. a, mRNA expression of Gremlin-1 or aldolase was determined by RT-PCR, and mRNA expression was quantified by densitometry and calculated in the same manner as Gremlin-1/aldolase. Data represent mean ± S.E. (**, indicates statistical significance of p < 0.01). b, Gremlin-1 expression was analyzed in aortic tissue derived from 16-week-old ApoE−/− and wild type mice by in situ hybridization using specific probes. In situ hybridization revealed enhanced Gremlin-1 mRNA expression in atherosclerotic lesions. Arrows indicate Gremlin-1-positive cells. Representative images of three in situ hybridization analyses are shown (50). c, protein expression of Gremlin-1 in aortic tissue from 16-week-old ApoE−/− and wild type mice was assessed by immunostaining using a mAb directed against human Gremlin-1 (upper left panels). Parallel sections were stained with anti-CD68 to detect monocytes/macrophages (upper right panels). Immunostaining with anti-Gremlin-1 showed that Gremlin-1 protein expression in aortic tissue of ApoE−/− mice was substantially enhanced compared with the aortic specimen derived from wild type mice. Anti-CD68 staining revealed that Gremlin-1 expression in aortic lesions from ApoE−/− mice was found primarily in monocytes/macrophages as shown by confocal colocalization studies (lower panel). Representative images of seven immunostainings are shown altogether. d, Gremlin-1 is expressed in isolated monocytes/macrophages and is released upon activation. RT-PCR analysis and immunoblotting with anti-Gremlin-1 showed that Gremlin-1 is expressed on the protein level in monocytes, macrophages/foam cells, and human aortic endothelial cells. The two immunoreactive bands represent the 23- and 28-kDa form of Gremlin-1 as also shown in the recombinant Gremlin-1 control. Representative results of five independently performed experiments are shown. e, Gremlin-1 protein expression was further analyzed by confocal microscopy after staining of permeabilized monocytes or macrophages/foam cells with fluorochrome-conjugated mAb directed against Gremlin-1 (red, phycoerythrin) or CD14 or CD68 (green, fluorescein isothiocyanate), respectively. f, isolated monocytes were stimulated with 1 μg/ml LPS for the indicated times. Intracellular protein expression of Gremlin-1 was determined in cell lysates by immunoblotting using an anti-human Gremlin-1 mAb. g, secreted Gremlin-1 was quantified in the cell supernatant by ELISA. Data represent the mean ± S.E. of five independent experiments. h, isolated monocytes were stimulated with vehicle control, LPS (1 μg/ml), oxidized LDL (oxLDL) (50 μg/ml), and TNF-α (100 ng/ml). Gremlin-1 was determined in the supernatant by ELISA after 6 h.
To study Gremlin-1 expression at the protein level, aortic tissue derived from ApoE−/− and wild type mice, respectively, was analyzed with a mAb directed against Gremlin-1. Protein expression of Gremlin-1 was increased in aortic atherosclerotic lesions obtained from 16-week-old ApoE−/− mice but not in aortic tissue from wild type mice as shown by immunohistology (Fig. 1c, left panel). Gremlin-1 expression was preferentially found in advanced atherosclerotic lesions and in large cells accumulating in plaques (supplemental Fig. 1). The specificity of the Gremlin-1 signal was demonstrated by the absence of staining signals when an idiotypic irrelevant primary mAb was used or when staining was performed only with the second conjugated antibody (data not shown). Gremlin-1 protein expression was found predominantly in large and round-shaped cells within the atherosclerotic plaque. These cells were found to be primarily CD68-positive (Fig. 1c, lower panel). Thus, enhanced Gremlin-1 expression in atherosclerotic plaques is preferentially found in CD68-positive monocyte/macrophages, inflammatory cells that play a critical role in atherogenesis (5).
Gremlin-1 Is Expressed in Monocytes and Released upon Activation
High levels of Gremlin-1 expression were described in non-dividing and terminally differentiated cells such as tumor endothelial cells (18, 19), neurons, or alveolar epithelial cells (27) but not in monocytes/macrophages thus far. Our in vivo findings imply that enhanced Gremlin-1 expression in atherosclerotic lesions is preferentially found in CD68-positive monocytes/macrophages (Fig. 1c). Thus, we asked whether Gremlin-1 is expressed in isolated monocytes and cultivated macrophages/foam cells in vitro. As shown by RT-PCR, Gremlin-1 mRNA is expressed in isolated monocytes and macrophages/foam cells (Fig. 1d). Furthermore, immunoblotting and immunohistochemistry studies showed that Gremlin-1 protein is highly expressed in monocytes/macrophages (Fig. 1, d and e). Immunoblotting revealed two anti-Gremlin-1 immunoreactive bands with an estimated molecular weight of ∼23 and 28 kDa in monocytes/macrophages and in human aortic endothelial cells indicating the presence of a nonglycosylated and a glycosylated form of Gremlin-1 as described previously (9). Gremlin-1 has been described as occurring in a cellular and a secreted form (9). Next, we asked whether monocytes secrete Gremlin-1 upon activation. Isolated monocytes were stimulated with lipopolysaccharide (LPS) for up to 48 h before the cellular and secreted forms of Gremlin-1 were determined by immunoblotting (cellular) and ELISA (secreted), respectively. No significant change upon monocyte activation was observed for intracellular Gremlin-1 protein expression (Fig. 1f). In contrast, secretion of Gremlin-1 of LPS-activated monocytes was increased ∼6-fold over time (Fig. 1g). Stimulation of monocytes with oxidized LDL or TNF-α also resulted in a comparable Gremlin-1 secretion (Fig. 1h).
Gremlin-1 Inhibits Monocyte Differentiation into Macrophages
The mechanisms of monocyte differentiation into macrophages play a critical role in atherogenesis and plaque vulnerability (28). Thus, we evaluated the effect of Gremlin-1 on macrophage and foam cell development using a previously described in vitro assay (29–32). In the presence of Gremlin-1 (Drm), but not of Gremlin-2 (PRDC) or control diluent, the formation of macrophages/foam cells was substantially reduced by ∼50–70% (Fig. 2a). The differentiation of monocytes to macrophages/foam cells was verified using phase contrast images, Sudan red III staining marking large granular and lipid-rich cells, May-Gruenwald staining revealing a nonsegmented nucleus surrounded by a large cytoplasm with enhanced granularity, naphtyl acetate esterase and CD68 immunostaining indicating macrophage/foam cells, and transmission electron microscopy showing large vacuoles typical for foam cells (supplemental Fig. 2a).
FIGURE 2.

Gremlin-1 inhibits monocyte differentiation into macrophages. a, macrophages/foam cells were differentiated from monocytes as described (30). The formation of macrophages/foam cells was evaluated over time through cell counting in the presence of Gremlin-1/Drm (1 μg/ml), Gremlin-2/PRDC (1 μg/ml), or the unspecific IgG (1 μg/ml) control. Representative photomicrographs are shown from five independent experiments. Data represent the mean ± S.E. (**, p < 0.01). b, macrophages/foam cells were differentiated from monocytes as described (30). 36 cytokines and chemokines were analyzed in the supernatant of this culture when most monocytes had differentiated into macrophages and foam cells (human cytokine array kit, R&D Systems). Expressions are shown as the density of the immunoreactive spots minus the base-line density of the gel. Among the analyzed factors MIF was prominently expressed in the macrophage/foam cell culture, indicating the central function of MIF in the differentiation of monocytes to macrophages and foam cells. c, the effect of MIF (10 ng/ml) compared with control diluent on monocyte/macrophage differentiation was evaluated. Data represent the mean ± S.E. of three independent experiments (**, p < 0.01). d, anti-MIF mAb (25 μg/ml) and IgG control (25 μg/ml) were analyzed regarding their effect on monocyte/macrophage differentiation. Data represent the mean ± S.E. of three independent experiments (*, p < 0.05).
Further, we evaluated the effect of Gremlin-1 on lipoprotein uptake and cholesterol efflux in macrophages (33). Primary mouse peritoneal macrophages were used and loaded with 50 μg/ml acetylated LDL and 3 μCi/ml 3H-labeled cholesterol. We found that Gremlin-1 significantly reduced the cholesterol uptake of macrophages compared with controls (p < 0.05) (supplemental Fig. 2e). Further, cholesterol efflux was slightly reduced in the presence of Gremlin-1 compared with controls (supplemental Fig. 2f).
Using a capture antibody-based array kit (R&D Systems, ARY005) we found that several cytokines and chemokines are up-regulated during macrophage development (Fig. 2b). Most prominently, the level of MIF was substantially enhanced in the culture supernatant (Fig. 2b). Further, when recombinant MIF was added to human monocytes, macrophage development was significantly enhanced compared with vehicle control (Fig. 2c). Moreover, a blocking anti-human MIF mAb significantly reduced macrophage/foam cell differentiation compared with control IgG (Fig. 2d), indicating that MIF plays a critical role in human monocyte/macrophage development. Murine monocytes, isolated from MIF −/− mice, were not able to differentiate into macrophages/foam cells, whereas murine monocytes from wild type mice formed macrophages/foam cells in vitro in the presence of acetylated LDL. The addition of recombinant murine MIF protein restored the ability of murine MIF−/− monocytes to form macrophages/foam cells, which in turn was inhibited in the presence of Gremlin-1 (p < 0.01) (supplemental Fig. 2, c and d).
Gremlin-1 Inhibits MIF-induced Secretion of TNF-α from Monocytes and Binds to MIF
MIF is a 12.5-kDa cytokine that is expressed in monocytes/macrophages and is secreted in response to proinflammatory and mitogenic stimuli (34, 35). MIF plays a central role in the regulation of monocyte function in atherosclerosis and plaque cellularity (6, 7). Thus, we asked whether MIF-dependent monocyte function is regulated by Gremlin-1. Macrophages were differentiated from human monocytes as described (25). Macrophages were treated with MIF (0.25 μg/ml), Gremlin-1 (0.5 μg/ml), or MIF plus Gremlin-1 for 12 h, and TNF-α was determined in cell supernatants. We found that MIF but not Gremlin-1 induced secretion of TNF-α (Fig. 3a). However, when macrophages were incubated with both MIF and Gremlin-1, secretion of TNF-α was almost completely abolished (Fig. 3a). These data imply that Gremlin-1 interacts with MIF and inhibits MIF-induced macrophage release of TNF-α from macrophages.
FIGURE 3.

Gremlin-1 inhibits MIF-induced secretion of TNF-α of monocytes and is a MIF-binding protein. a, macrophages were stimulated with MIF (0.25 μg/ml), Gremlin-1 (0.5 μg/ml), or a combination of both for 12 h. The concentration of TNF-α in the cell supernatant was determined by ELISA. Data represent the mean ± S.E. of three experiments. (**, indicates statistical significance of p < 0.01). b, computational modeling of the protein structures of MIF and Gremlin-1 and their predicted interaction with an estimated binding affinity of KD = 65 nm. Molecular modes were generated with Molegro Virtual Docker 5. The surface charge distribution is shown; negative charge is in red and positive charge in blue. c, surface plasmon resonance analysis. The binding of Gremlin-1 (100 nm), Gremlin-1 (100 nm) that has been preincubated with an excess of MIF (800 nm) for 10 min, and MIF alone (control) to the sensor surface coated with MIF (51–57) is shown. d, co-immunoprecipitation of the Gremlin-1/MIF complexes of monocyte cell extracts. Anti-Gremlin-1 immunoprecipitation (IP) followed by anti-MIF Western blotting (WB, lower panel) or anti-MIF immunoprecipitation followed by anti-gremlin-1 Western blotting (upper panel). Control: immunoprecipitation with an irrelevant idiotypic IgG. Representative results of five independently performed IP experiments are shown. e, co-localization of Gremlin-1 and MIF in aortic atherosclerotic lesions of ApoE−/− mice. Paraffin-embedded aortic tissue from 16-week-old ApoE−/− mice was stained with mAb directed against Gremlin-1 or MIF, respectively. Immunostainings were analyzed by confocal microscopy.
To further characterize the binding of Gremlin-1 to MIF, we first created the protein structures by in silico molecular modeling. Consistent with the reported x-ray crystal structure of human MIF (36, 37), the computational modeling data indicated that MIF forms a homotrimer as shown in Fig. 3b. Using LOMETS (38) homology modeling, we predicted the three-dimensional structure of the Gremlin-1 protein with I-TASSER (39). Further, applying a flexible blind docking approach, we modeled the protein-protein interactions between the MIF homotrimer and Gremlin-1. These computational docking studies indicated that Gremlin-1 binds with high affinity to MIF (Fig. 3b). Moreover, we calculated a dissociation constant (KD) of ∼65 nm for the binding of Gremlin-1 to the MIF homotrimer. To confirm the predicted interaction between MIF and Gremlin-1, we used surface plasmon resonance analysis allowing label-free measurement of molecular interactions. Indeed, analysis of the binding kinetics of Gremlin-1 to MIF that was immobilized on the sensor chip surface analysis revealed specific high affinity binding with a KD of 54 nm (Fig. 3c). Accordingly, MIF or MIF plus Gremlin-1 showed only low interactions with the sensor chip surface (Fig. 3c). The interaction between MIF and Gremlin-1 was further investigated by co-immunoprecipitation. As verified by immunoblotting, human monocytes expressed significant amounts of Gremlin-1 (Fig. 1d) and MIF (not shown). Anti-MIF mAb co-immunoprecipitated Gremlin-1 and anti-Gremlin-1 mAb also co-precipitated MIF (Fig. 3d). In addition, we also found colocalization of Gremlin-1 and MIF in atherosclerotic lesions in ApoE−/− mice (Fig. 3e). Taken together, these results document a totally unexpected molecular interaction between Gremlin-1 and MIF and define Gremlin-1 as an antagonist of MIF.
Prolonged Administration of Recombinant Gremlin-1 Reduces the Content of Monocytes/Macrophages in Atherosclerotic Plaques and Attenuates Atheroprogression in ApoE−/− Mice
MIF plays a pivotal role in atherosclerosis by promoting monocyte/macrophage activation (6, 7). MIF is highly expressed in atherosclerotic tissue (6, 7) (Fig. 3e). Inhibition or genetic deletion of MIF in mice results in marked reduction of inflammation and atheroprogression (6, 7). As shown above, Gremlin-1 is strongly overexpressed in aortic atherosclerotic tissue (Fig. 1, a–c), binds to MIF (Fig. 3), and inhibits MIF-induced TNF-α secretion of macrophages (Fig. 3a). Therefore, we hypothesized that the interaction of Gremlin-1 and MIF modulates monocyte/macrophage function and accumulation in atherosclerotic plaques and thus regulates atheroprogression and -stability in vivo.
To test whether Gremlin-1 regulates the development of atherosclerosis in vivo, we designed and generated a fusion protein with an enhanced plasma half-life, mGremlin-1-Fc, consisting of murine Gremlin-1 fused to a human Fc domain as described previously for other fusion proteins (33, 40, 41) (Fig. 4a). Fc-containing fusion proteins have been shown to have a favorable pharmacokinetic profile allowing for the achievement of a sustained and stable bioavailability in mice (42). As shown in Fig. 4, b and c, mGremlin-1-Fc was generated in significant amounts and high purity using the Flp-In CHO eukaryotic expression system (33, 40). Applying a flexible blind docking approach, we modeled the protein/protein interactions between the mGremlin-1-Fc and MIF homotrimers. These computational docking studies indicated that mGremlin-1-Fc binds with high affinity to MIF (KD = 85 nm) (Fig. 4c). Measurements made by dynamic light scattering revealed that mGremlin-1-Fc is a dimer (Fig. 4d), and analysis of the binding of mGremlin-1-Fc to MIF revealed a KD of 76 nm (Fig. 4e). As described for human Gremlin-1, the recombinant fusion protein mGremlin-1-Fc significantly inhibits MIF-induced secretion of TNF-α from macrophages concentration-dependently by 50 to 90% (Fig. 4f).
FIGURE 4.

Design, generation, and characterization of a recombinant fusion protein mGremlin-1-Fc as MIF antagonist. a, design and schematic drawing of mGremlin-1-Fc. The recombinant fusion protein mGremlin-1-Fc, consisting of the murine Gremlin-1 domain and the fragment crystallizable region of human IgG2 (Fc), and a corresponding IgG2 Fc control protein without the Gremlin-1 domain were designed, cloned, expressed in Flp-In CHO expression cell lines and purified via protein G affinity chromatography as described (32, 49). b, mGremlin-1-Fc was characterized by immunoblotting with anti-IgG (detection of Fc domain) and anti-Gremlin-1 (detection of Gremlin-1 domain). c, computational modeling of the protein structures of mGremlin-1-Fc and MIF and their predicted interaction with an estimated binding affinity of KD = 85 nm. d, measurements taken by dynamic light scattering revealed that mGremlin-1-Fc is a dimer. e, specific binding curves of MIF to Gremlin-1 (10 nm) and mGremlin-1-Fc (10 nm) as analyzed by dynamic light scattering. mGremlin-1-Fc binds with high affinity to MIF (KD = 76 nm) comparable to Gremlin-1 (KD = 54 nm). f, mGremlin-1-Fc inhibits MIF-induced TNF-α secretion of macrophages to an extent similar to Gremlin-1. Data represent the mean ± S.E. of three experiments (**, indicates statistical significance of p < 0.01).
To assess the effect of systemic and prolonged administration of mGremlin-1-Fc on atheroprogression, 10-week-old ApoE−/− mice fed with a cholesterol-rich diet were treated with mGremlin-1-Fc (1 μg/g body weight) or an equimolar amount of control Fc intraperitoneally three times/week for 4 weeks (Fig. 5a). Thereafter, the mice were sacrificed, and plaque cellularity, morphology, and extension were assessed by immunohistochemistry or Sudan red lipid staining, respectively (41, 43, 44). We found that CD68- and Mac3-positive cells were substantially reduced in atherosclerotic lesions in mice treated with mGremlin-1-Fc compared with control Fc (Fig. 5, b and c, and supplemental Fig. 3). Furthermore, the size of CD68-positive cells was significantly reduced in the mGremlin-1-Fc group (Fig. 5b, right micrographs). In addition, we found that the TNF-α and MIF expression was reduced remarkably in mGremlin-1-Fc-treated mice compared with control Fc-treated mice (Fig. 5c), indicating that mGremlin-1-Fc antagonizes MIF-induced secretion of TNF-α in macrophages in vivo as shown above on a cellular level in vitro (Fig. 3). In addition, MIF expression was also decreased in mGremlin-1-Fc-treated ApoE−/− mice compared with Fc-treated ApoE−/− mice (Fig. 5c), suggesting that Gremlin-1 attenuates MIF release from monocytes/macrophages within the atherosclerotic plaque.
FIGURE 5.
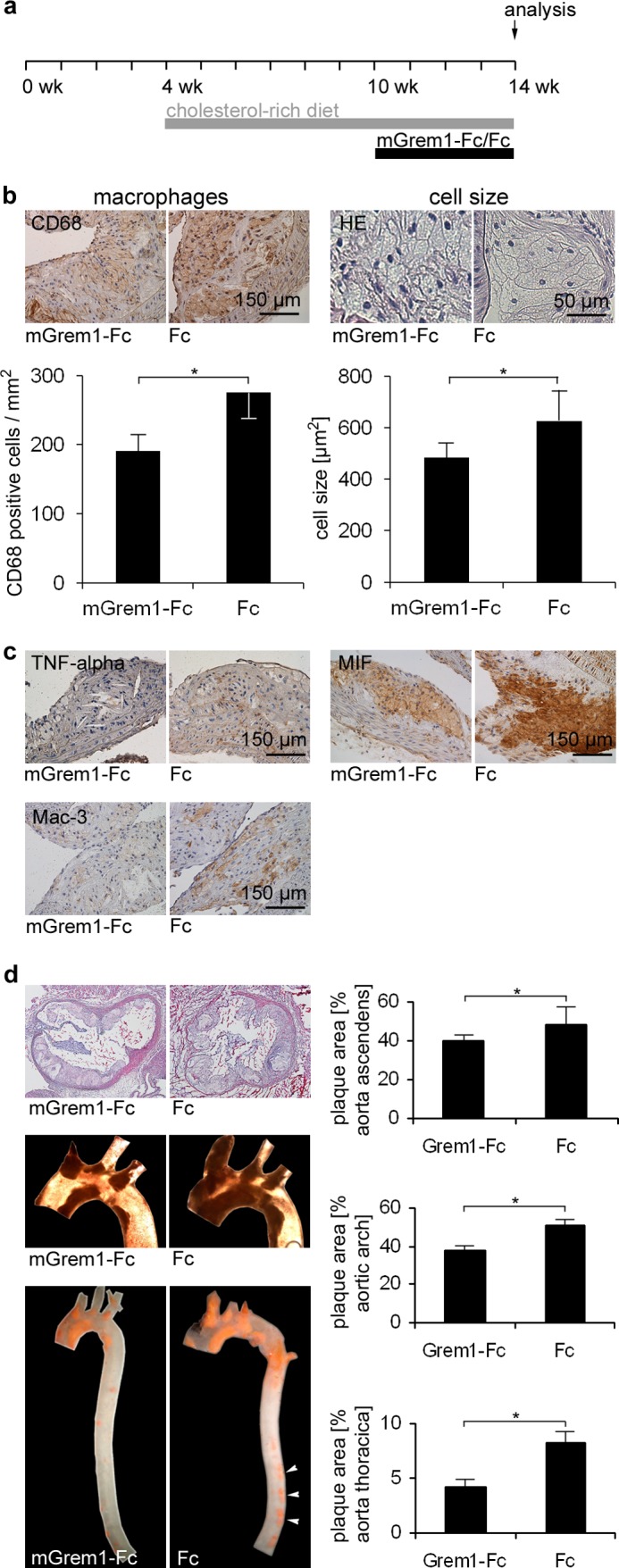
Gremlin-1 reduces the number of monocytes/macrophages in atherosclerotic plaques and attenuates atheroprogression in ApoE−/− mice. a, experimental protocol for the treatment and feeding of ApoE−/− mice. mGremlin-1-Fc (1 μg/g body weight) (n = 8) or equimolar Fc control (0.6 μg/g body weight) (n = 8) was injected intraperitoneally 3 times/week into 10-week-old mice for 4 weeks. b, mGremlin-1-Fc substantially reduced the number and cell size of CD68-positive cells in atherosclerotic lesions. Representative immunostainings of CD68 expression and H&E staining of atherosclerotic aortic tissue of mGremlin-1-Fc and Fc-treated ApoE−/− mice are shown. Data represent the mean ± S.E. of eight mice (*, p < 0.05). c, administration of mGremlin-1-Fc reduced TNF-α, MIF, and Mac-3 expression in aortic plaques. Representative immunostainings of TNF-α, MIF, and Mac-3 of atherosclerotic aortic tissue in mGremlin-1-Fc and Fc-treated ApoE−/− mice are shown. d, mGremlin-1-Fc reduces atherosclerotic plaque extension. Representative photographs of H&E staining of aortic root sections (two upper left micrographs) and Sudan red III-stained aortic arches and thoracic aortas (two central and lower left macroscopic photographs) as well as quantitative analysis of the plaque extension (right panel) are shown. The mean percentages of plaque areas in relation to total vessel areas of all mice are shown with mean ± S.E. (n = 8 for each group). (*, p < 0.05, statistical significance).
The effect of mGremlin-1-Fc on plaque formation in the aortic root, aortic arch, and descending thoracic aorta was determined macroscopically on the basis of Sudan red III-stained aortic segments of mice. In the 14-week-old ApoE−/− mice, plaques covered on average 50.1 ± 2.5% of the aortic arch in the Fc control group and 37.9 ± 2.4% in the mGremlin-1-Fc treatment group (p < 0.05). Similarly, plaque extension in the descending thoracic aorta was significantly (p < 0.05) reduced in the mGremlin-1-Fc-treated group (4.1 ± 0.7%) compared with the Fc control group (8.2 ± 0.8%) (Fig. 5d). Thus, systemic and prolonged administration of Gremlin-1 results in a change in morphology and reduced cellularity of atherosclerotic plaques (plaque vulnerability) and a decrease in plaque extension of the thoracic aorta. Further, we tested the effect of inactivated mGremlin-1-Fc in a second independent experiment on atheroprogession (supplemental Fig. 3a). We found that atherosclerotic lesion formation was significantly reduced in ApoE−/− mice treated with mGremlin-1-Fc compared with mice receiving inactivated mGremlin-1-Fc (plaques covered on average 49.9 ± 4.6% of the aortic arch in the inactivated mGremlin-1-Fc control group and 28.3 ± 3.2% in the mGremlin-1-Fc treatment group (p < 0.05). In the descending thoracic aorta plaque extension was 24.6 ± 1.2% in the mGremlin-1-Fc versus 34.4 ± 0.9% in the inactivated mGremlin-1-Fc-treated group (p < 0.01) (supplemental Fig. 3a).
DISCUSSION
The major finding of the present study is that Gremlin-1 is involved in atherosclerotic lesion formation in a mouse model of atherosclerosis, and administration of Gremlin-1 attenuated atheroprogression and plaque cellularity. In particular, we found that (i) Gremlin-1 is highly expressed in monocytes/macrophages in atherosclerotic plaques; (ii) Gremlin-1 inhibits monocyte differentiation into macrophages and inhibits TNF-α secretion of macrophages stimulated by MIF via direct binding to MIF with high affinity; and (iii) prolonged administration of recombinant Gremlin-1 reduces the monocyte/macrophage contents of atherosclerotic plaques and attenuates the lesion burden in ApoE−/− mice.
Atherosclerosis and related diseases such as acute coronary syndromes, myocardial infarction, ischemic stroke, and heart failure are a major health burden not only in Western society. The formation of unstable plaques in coronary or cerebral arteries is a critical stage of the atherosclerotic disease, which is associated with a high risk for acute myocardial or cerebral infarction (45). Unstable or vulnerable plaques are characterized by high inflammatory activity, reflected by a high content of inflammatory cells such as monocytes/macrophages (45).
Gremlin-1 is a glycoprotein that was first described as a major factor in the regulation of embryonic development due to its binding to BMPs (8). More recently, the role of Gremlin-1 in the function of differentiated cells in the adult organism became apparent. Gremlin-1 is a proangiogenic agonist of the VEGF receptor-2 (VEGFR-2) (10) and an antagonist of BMPs (9). Gremlin-1 interacts directly with cell surface proteins such as members of the Slit protein family (17) and heparan sulfate proteoglycans (20), indicating a role for Gremlin-1 in cell function independent of BMP antagonism. Gremlin-1 has been shown to be overexpressed in endothelial cells stressed by abnormal flow (15) and in the neointima after mechanical vessel injury (46), indicating a role for this protein in the pathophysiology of arterial lesions.
Thus, we speculated that Gremlin-1 plays a role in atherosclerosis and plaque instability. We found that Gremlin-1 was highly expressed in monocytes and macrophages and thus in inflammatory cells that are critically involved in atherogenesis (47) and plaque vulnerability (45). Furthermore, we found that Gremlin-1 is present and secreted in substantial amounts in monocytes and that Gremlin-1 inhibits monocyte-derived formation of macrophages in culture (Fig. 2). This implies that secreted and monocyte-derived Gremlin-1 acts as an autocrine and/or paracrine factor to regulate monocyte function and differentiation into macrophages during atherosclerotic plaque formation. Macrophage development is orchestrated by chemokines and their receptors (48). Here we have demonstrated that Gremlin-1 reduced the release of MIF during macrophage development in vitro and in atherosclerotic plaques in ApoE−/− mice treated with recombinant Gremlin-1. MIF plays an important role in the mechanisms of inflammation and in the pathophysiology of inflammatory diseases such as sepsis and atherosclerosis (49). Interestingly, we found that the activation and release of TNF-α in macrophages induced by MIF is inhibited by Gremlin-1, implying interference of this protein with the MIF/monocyte interaction. Totally unexpectedly we found that Gremlin-1 binds with high affinity to MIF (KD = 54 nm) (Fig. 3), and we have provided evidence for the first time that an endogenous inhibitor of MIF exists in vivo. By surface plasmon resonance analysis, the binding of Gremlin-1 to MIF could only be detected when MIF was bound to the sensor surface and when Gremlin-1 was added in the fluid phase. In contrast, no specific interaction was detectable when Gremlin-1 was bound to the sensor surface either via amino or carboxyl groups. This observation is consistent with modeling indicating that the binding region of the Gremlin-1 molecule consists of the CTCK (C-terminal cystine knot-like) domain, which carries critical positive and negative charges that would be abrogated by cross-linkers targeting either amino or carboxylic groups.
Previous studies have shown that blockade and genetic deletion of MIF result in a marked reduction of atherosclerotic lesion formation and macrophage infiltration (6). Supportive of this idea, our present data demonstrate that the administration of a recombinant fusion molecule, mGremlin-1-Fc, which binds with high affinity (KD = 76 nm) to MIF, substantially reduces the cellularity of atherosclerotic lesions in ApoE−/− mice, most prominently due to a decrease in macrophage infiltration. The fact that our fusion molecule binds MIF implies a role of Gremlin-1/MIF interaction in the pathogenesis of atherosclerosis and plaque growth. Thus, the administration of Gremlin-1 might be a promising strategy to limit atheroprogression and plaque growth. Although our present data indicate that the Gremlin-1/MIF interaction is critically involved in plaque biology and progression, we cannot exclude that interaction of Gremlin-1 with other proteins such as BMPs or receptors such as VEGFR-2 may interfere with the mechanisms of atherosclerosis. However, the substantially reduced TNF-α expression in aortic plaques obtained from mGremlin-1-Fc-treated mice indicates that Gremlin-1/MIF interaction is the predominant mechanism attenuating plaque cellularity and atheroprogression.
Acknowledgments
We thank Ingrid Epple, Hanna Schnell, and Jadwiga Kwiatkowska for excellent technical assistance. We thank R. Bucala and L. Leng for originally supplying the NIH/III.D9 clone.
This study was supported in part by grants from the Deutsche Forschungsgemeinschaft (DFG) (Transregio-SFB-19 and Klinische Forschergruppe KFO-274, “Platelets-Molecular Mechanisms and Translational Implications”) and by DFG Grant MU 2928/2-1 to Iris Müller, the University of Tübingen (Iris Müller Fortüne Research Program; H.L. Interdisciplinary Center for Clinical Research (IZKF)).

This article contains supplemental “Methods” and Figs. 1–3.
- MIF
- macrophage migration inhibitory factor
- BMP
- bone morphogenetic protein
- VEGFR-2
- vascular endothelial growth factor receptor-2.
REFERENCES
- 1. Hansson G. K., Libby P. (2006) The immune response in atherosclerosis: a double-edged sword. Nat. Rev. Immunol. 6, 508–519 [DOI] [PubMed] [Google Scholar]
- 2. Lusis A. J. (2000) Atherosclerosis. Nature 407, 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ross R. (1999) Atherosclerosis: an inflammatory disease. N. Engl. J. Med. 340, 115–126 [DOI] [PubMed] [Google Scholar]
- 4. Libby P., Ridker P. M., Hansson G. K. (2011) Progress and challenges in translating the biology of atherosclerosis. Nature 473, 317–325 [DOI] [PubMed] [Google Scholar]
- 5. Weber C., Zernecke A., Libby P. (2008) The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat. Rev. Immunol. 8, 802–815 [DOI] [PubMed] [Google Scholar]
- 6. Bernhagen J., Krohn R., Lue H., Gregory J. L., Zernecke A., Koenen R. R., Dewor M., Georgiev I., Schober A., Leng L., Kooistra T., Fingerle-Rowson G., Ghezzi P., Kleemann R., McColl S. R., Bucala R., Hickey M. J., Weber C. (2007) MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 13, 587–596 [DOI] [PubMed] [Google Scholar]
- 7. Weber C., Kraemer S., Drechsler M., Lue H., Koenen R. R., Kapurniotu A., Zernecke A., Bernhagen J. (2008) Structural determinants of MIF functions in CXCR2-mediated inflammatory and atherogenic leukocyte recruitment. Proc. Natl. Acad. Sci. U.S.A. 105, 16278–16283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hsu D. R., Economides A. N., Wang X., Eimon P. M., Harland R. M. (1998) The Xenopus dorsalizing factor, Gremlin, identifies a novel family of secreted proteins that antagonize BMP activities. Mol. Cell 1, 673–683 [DOI] [PubMed] [Google Scholar]
- 9. Topol L. Z., Bardot B., Zhang Q., Resau J., Huillard E., Marx M., Calothy G., Blair D. G. (2000) Biosynthesis, post-translation modification, and functional characterization of Drm/Gremlin. J. Biol. Chem. 275, 8785–8793 [DOI] [PubMed] [Google Scholar]
- 10. Mitola S., Ravelli C., Moroni E., Salvi V., Leali D., Ballmer-Hofer K., Zammataro L., Presta M. (2010) Gremlin is a novel agonist of the major proangiogenic receptor VEGFR2. Blood 116, 3677–3680 [DOI] [PubMed] [Google Scholar]
- 11. Michos O., Gonçalves A., Lopez-Rios J., Tiecke E., Naillat F., Beier K., Galli A., Vainio S., Zeller R. (2007) Reduction of BMP4 activity by Gremlin 1 enables ureteric bud outgrowth and GDNF/WNT11 feedback signalling during kidney branching morphogenesis. Development 134, 2397–2405 [DOI] [PubMed] [Google Scholar]
- 12. Shi W., Zhao J., Anderson K. D., Warburton D. (2001) Gremlin negatively modulates BMP-4 induction of embryonic mouse lung branching morphogenesis. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L1030–L1039 [DOI] [PubMed] [Google Scholar]
- 13. Khokha M. K., Hsu D., Brunet L. J., Dionne M. S., Harland R. M. (2003) Gremlin is the BMP antagonist required for maintenance of Shh and Fgf signals during limb patterning. Nat. Genet. 34, 303–307 [DOI] [PubMed] [Google Scholar]
- 14. Gazzerro E., Pereira R. C., Jorgetti V., Olson S., Economides A. N., Canalis E. (2005) Skeletal overexpression of Gremlin impairs bone formation and causes osteopenia. Endocrinology 146, 655–665 [DOI] [PubMed] [Google Scholar]
- 15. Chang K., Weiss D., Suo J., Vega J. D., Giddens D., Taylor W. R., Jo H. (2007) Bone morphogenic protein antagonists are coexpressed with bone morphogenic protein 4 in endothelial cells exposed to unstable flow in vitro in mouse aortas and in human coronary arteries: role of bone morphogenic protein antagonists in inflammation and atherosclerosis. Circulation 116, 1258–1266 [DOI] [PubMed] [Google Scholar]
- 16. Diecke S., Quiroga-Negreira A., Redmer T., Besser D. (2008) FGF2 signaling in mouse embryonic fibroblasts is crucial for self-renewal of embryonic stem cells. Cells Tissues Organs 188, 52–61 [DOI] [PubMed] [Google Scholar]
- 17. Chen B., Blair D. G., Plisov S., Vasiliev G., Perantoni A. O., Chen Q., Athanasiou M., Wu J. Y., Oppenheim J. J., Yang D. (2004) Cutting edge: bone morphogenetic protein antagonists Drm/Gremlin and Dan interact with Slits and act as negative regulators of monocyte chemotaxis. J. Immunol. 173, 5914–5917 [DOI] [PubMed] [Google Scholar]
- 18. Namkoong H., Shin S. M., Kim H. K., Ha S. A., Cho G. W., Hur S. Y., Kim T. E., Kim J. W. (2006) The bone morphogenetic protein antagonist Gremlin 1 is overexpressed in human cancers and interacts with YWHAH protein. BMC Cancer 6, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sneddon J. B., Zhen H. H., Montgomery K., van de Rijn M., Tward A. D., West R., Gladstone H., Chang H. Y., Morganroth G. S., Oro A. E., Brown P. O. (2006) Bone morphogenetic protein antagonist Gremlin 1 is widely expressed by cancer-associated stromal cells and can promote tumor cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 103, 14842–14847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chiodelli P., Mitola S., Ravelli C., Oreste P., Rusnati M., Presta M. (2011) Heparan sulfate proteoglycans mediate the angiogenic activity of the vascular endothelial growth factor receptor-2 agonist gremlin. Arterioscler. Thromb. Vasc. Biol. 31, e116–e127 [DOI] [PubMed] [Google Scholar]
- 21. Boers W., Aarrass S., Linthorst C., Pinzani M., Elferink R. O., Bosma P. (2006) Transcriptional profiling reveals novel markers of liver fibrogenesis: gremlin and insulin-like growth factor-binding proteins. J. Biol. Chem. 281, 16289–16295 [DOI] [PubMed] [Google Scholar]
- 22. Kane R., Stevenson L., Godson C., Stitt A. W., O'Brien C. (2005) Gremlin gene expression in bovine retinal pericytes exposed to elevated glucose. Br. J. Ophthalmol. 89, 1638–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dolan V., Murphy M., Sadlier D., Lappin D., Doran P., Godson C., Martin F., O'Meara Y., Schmid H., Henger A., Kretzler M., Droguett A., Mezzano S., Brady H. R. (2005) Expression of Gremlin, a bone morphogenetic protein antagonist, in human diabetic nephropathy. Am. J. Kidney Dis. 45, 1034–1039 [DOI] [PubMed] [Google Scholar]
- 24. Schmidt R., Bültmann A., Ungerer M., Joghetaei N., Bülbül O., Thieme S., Chavakis T., Toole B. P., Gawaz M., Schömig A., May A. E. (2006) Extracellular matrix metalloproteinase inducer regulates matrix metalloproteinase activity in cardiovascular cells: implications in acute myocardial infarction. Circulation 113, 834–841 [DOI] [PubMed] [Google Scholar]
- 25. Colognato R., Slupsky J. R., Jendrach M., Burysek L., Syrovets T., Simmet T. (2003) Differential expression and regulation of protease-activated receptors in human peripheral monocytes and monocyte-derived antigen-presenting cells. Blood 102, 2645–2652 [DOI] [PubMed] [Google Scholar]
- 26. Nakashima Y., Plump A. S., Raines E. W., Breslow J. L., Ross R. (1994) ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler. Thromb. 14, 133–140 [DOI] [PubMed] [Google Scholar]
- 27. Topol L. Z., Marx M., Laugier D., Bogdanova N. N., Boubnov N. V., Clausen P. A., Calothy G., Blair D. G. (1997) Identification of drm, a novel gene whose expression is suppressed in transformed cells and which can inhibit growth of normal but not transformed cells in culture. Mol. Cell. Biol. 17, 4801–4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ley K., Miller Y. I., Hedrick C. C. (2011) Monocyte and macrophage dynamics during atherogenesis. Arterioscler. Thromb. Vasc. Biol. 31, 1506–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stellos K., Langer H., Gnerlich S., Panagiota V., Paul A., Schönberger T., Ninci E., Menzel D., Mueller I., Bigalke B., Geisler T., Bültmann A., Lindemann S., Gawaz M. (2010) Junctional adhesion molecule A expressed on human CD34+ cells promotes adhesion on vascular wall and differentiation into endothelial progenitor cells. Arterioscler. Thromb. Vasc. Biol. 30, 1127–1136 [DOI] [PubMed] [Google Scholar]
- 30. Daub K., Langer H., Seizer P., Stellos K., May A. E., Goyal P., Bigalke B., Schönberger T., Geisler T., Siegel-Axel D., Oostendorp R. A., Lindemann S., Gawaz M. (2006) Platelets induce differentiation of human CD34+ progenitor cells into foam cells and endothelial cells. FASEB J. 20, 2559–2561 [DOI] [PubMed] [Google Scholar]
- 31. Seizer P., Schiemann S., Merz T., Daub K., Bigalke B., Stellos K., Müller I., Stöckle C., Müller K., Gawaz M., May A. E. (2010) CD36 and macrophage scavenger receptor a modulate foam cell formation via inhibition of lipid-laden platelet phagocytosis. Semin. Thromb. Hemost. 36, 157–162 [DOI] [PubMed] [Google Scholar]
- 32. Daub K., Siegel-Axel D., Schönberger T., Leder C., Seizer P., Müller K., Schaller M., Penz S., Menzel D., Büchele B., Bültmann A., Münch G., Lindemann S., Simmet T., Gawaz M. (2010) Inhibition of foam cell formation using a soluble CD68-Fc fusion protein. J. Mol. Med. 88, 909–920 [DOI] [PubMed] [Google Scholar]
- 33. Nijstad N., de Boer J. F., Lagor W. R., Toelle M., Usher D., Annema W., van der Giet M., Rader D. J., Tietge U. J. (2011) Overexpression of apolipoprotein O does not impact on plasma HDL levels or functionality in human apolipoprotein A-I transgenic mice. Biochim. Biophys. Acta 1811, 294–299 [DOI] [PubMed] [Google Scholar]
- 34. Calandra T., Bernhagen J., Mitchell R. A., Bucala R. (1994) The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J. Exp. Med. 179, 1895–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Merk M., Baugh J., Zierow S., Leng L., Pal U., Lee S. J., Ebert A. D., Mizue Y., Trent J. O., Mitchell R., Nickel W., Kavathas P. B., Bernhagen J., Bucala R. (2009) The Golgi-associated protein p115 mediates the secretion of macrophage migration inhibitory factor. J. Immunol. 182, 6896–6906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sun H. W., Bernhagen J., Bucala R., Lolis E. (1996) Crystal structure at 2.6-Å resolution of human macrophage migration inhibitory factor. Proc. Natl. Acad. Sci. U.S.A. 93, 5191–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sugimoto H., Suzuki M., Nakagawa A., Tanaka I., Nishihira J. (1996) Crystal structure of macrophage migration inhibitory factor from human lymphocyte at 2.1 Å resolution. FEBS Lett. 389, 145–148 [DOI] [PubMed] [Google Scholar]
- 38. Wu S., Zhang Y. (2007) LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375–3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Massberg S., Konrad I., Bültmann A., Schulz C., Münch G., Peluso M., Lorenz M., Schneider S., Besta F., Müller I., Hu B., Langer H., Kremmer E., Rudelius M., Heinzmann U., Ungerer M., Gawaz M. (2004) Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J. 18, 397–399 [DOI] [PubMed] [Google Scholar]
- 41. Zeibig S., Li Z., Wagner S., Holthoff H. P., Ungerer M., Bültmann A., Uhland K., Vogelmann J., Simmet T., Gawaz M., Münch G. (2011) Effect of the oxLDL-binding protein Fc-CD68 on plaque extension and vulnerability in atherosclerosis. Circ. Res. 108, 695–703 [DOI] [PubMed] [Google Scholar]
- 42. Schönberger T., Siegel-Axel D., Bussl R., Richter S., Judenhofer M. S., Haubner R., Reischl G., Klingel K., Münch G., Seizer P., Pichler B. J., Gawaz M. (2008) The immunoadhesin glycoprotein VI-Fc regulates arterial remodelling after mechanical injury in ApoE-/- mice. Cardiovasc. Res. 80, 131–137 [DOI] [PubMed] [Google Scholar]
- 43. Massberg S., Gawaz M., Grüner S., Schulte V., Konrad I., Zohlnhöfer D., Heinzmann U., Nieswandt B. (2003) A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J. Exp. Med. 197, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schulz C., Schäfer A., Stolla M., Kerstan S., Lorenz M., von Brühl M. L., Schiemann M., Bauersachs J., Gloe T., Busch D. H., Gawaz M., Massberg S. (2007) Chemokine fractalkine mediates leukocyte recruitment to inflammatory endothelial cells in flowing whole blood: a critical role for P-selectin expressed on activated platelets. Circulation 116, 764–773 [DOI] [PubMed] [Google Scholar]
- 45. Naghavi M., Libby P., Falk E., Casscells S. W., Litovsky S., Rumberger J., Badimon J. J., Stefanadis C., Moreno P., Pasterkamp G., Fayad Z., Stone P. H., Waxman S., Raggi P., Madjid M., Zarrabi A., Burke A., Yuan C., Fitzgerald P. J., Siscovick D. S., de Korte C. L., Aikawa M., Juhani Airaksinen K. E., Assmann G., Becker C. R., Chesebro J. H., Farb A., Galis Z. S., Jackson C., Jang I. K., Koenig W., Lodder R. A., March K., Demirovic J., Navab M., Priori S. G., Rekhter M. D., Bahr R., Grundy S. M., Mehran R., Colombo A., Boerwinkle E., Ballantyne C., Insull W., Jr., Schwartz R. S., Vogel R., Serruys P. W., Hansson G. K., Faxon D. P., Kaul S., Drexler H., Greenland P., Muller J. E., Virmani R., Ridker P. M., Zipes D. P., Shah P. K., Willerson J. T. (2003) From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies. Part I. Circulation 108, 1664–1672 [DOI] [PubMed] [Google Scholar]
- 46. Maciel T. T., Melo R. S., Schor N., Campos A. H. (2008) Gremlin promotes vascular smooth muscle cell proliferation and migration. J. Mol. Cell. Cardiol. 44, 370–379 [DOI] [PubMed] [Google Scholar]
- 47. Gawaz M., Langer H., May A. E. (2005) Platelets in inflammation and atherogenesis. J. Clin. Invest. 115, 3378–3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weber C., Noels H. (2011) Atherosclerosis: current pathogenesis and therapeutic options. Nat. Med. 17, 1410–1422 [DOI] [PubMed] [Google Scholar]
- 49. Zernecke A., Shagdarsuren E., Weber C. (2008) Chemokines in atherosclerosis: an update. Arterioscler. Thromb. Vasc. Biol. 28, 1897–1908 [DOI] [PubMed] [Google Scholar]
- 50. Thisse C., Thisse B., Schilling T. F., Postlethwait J. H. (1993) Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail, and no tail mutant embryos. Development 119, 1203–1215 [DOI] [PubMed] [Google Scholar]
- 51. Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang Y. (2009) I-TASSER: fully automated protein structure prediction in CASP8. Proteins 77, Suppl. 9, 100–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ritchie D. W., Kozakov D., Vajda S. (2008) Accelerating and focusing protein-protein docking correlations using multi-dimensional rotational FFT generating functions. Bioinformatics 24, 1865–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ritchie D. W., Venkatraman V. (2010) Ultra-fast FFT protein docking on graphics processors. Bioinformatics 26, 2398–2405 [DOI] [PubMed] [Google Scholar]
- 55. Hetényi C., van der Spoel D. (2002) Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 11, 1729–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Thomsen R., Christensen M. H. (2006) MolDock: a new technique for high-accuracy molecular docking. J. Med. Chem. 49, 3315–3321 [DOI] [PubMed] [Google Scholar]
- 57. Hanlon A. D., Larkin M. I., Reddick R. M. (2010) Free-solution, label-free protein-protein interactions characterized by dynamic light scattering. Biophys. J. 98, 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]