

Background: Bovine lactoferricin (LfcinB) promotes anti-catabolism and anti-inflammation in articular cartilage.
Results: LfcinB induces IL-11 via AP-1, which in turn induces TIMP-1 via STAT3.
Conclusion: LfcinB sequentially regulates IL-11 and TIMP-1 expression through distinct mechanisms in articular chondrocytes.
Significance: These findings further suggest the potential of LfcinB as a novel therapeutic agent in osteoarthritis.
Keywords: Articular Cartilage, Chondrocytes, Gene Regulation, Interleukin, Osteoarthritis, Signal Transduction
Abstract
Bovine lactoferricin (LfcinB), a multifunctional peptide, was recently demonstrated to be anti-catabolic and anti-inflammatory in human articular cartilage. LfcinB blocks IL-1-mediated proteoglycan depletion, matrix-degrading enzyme expression, and pro-inflammatory mediator induction. LfcinB selectively activates ERK1/2, p38 (but not JNK), and Akt signaling. However, the relationship between these pathways and LfcinB target genes has never been explored. In this study, we uncovered the remarkable ability of LfcinB in the induction of an anti-inflammatory cytokine, IL-11. LfcinB binds to cell surface heparan sulfate to initiate ERK1/2 signaling and activate AP-1 complexes composed of c-Fos and JunD, which transactivate the IL-11 gene. The induced IL-11 functions as an anti-inflammatory and chondroprotective cytokine in articular chondrocytes. Our data show that IL-11 directly attenuates IL-1-mediated catabolic and inflammatory processes ex vivo and in vitro. Moreover, IL-11 activates STAT3 signaling pathway to critically up-regulate TIMP-1 expression, as a consecutive secondary cellular response after IL-11 induction by LfcinB-ERK-AP-1 axis in human adult articular chondrocytes. The pathological relevance of IL-11 signaling to osteoarthritis is evidenced by significant down-regulation of its cognate receptor expression in osteoarthritic chondrocytes. Together, our results suggest a two-step mechanism, whereby LfcinB induces TIMP-1 through an IL-11-dependent pathway involving transcription factor AP-1 and STAT3.
Introduction
Homeostatic chondrocytes maintain a delicate balance between catabolic and anabolic processes, which is characterized by dynamic and steady turnover of molecules in its extracellular matrix (ECM).2 Such a balance, however, is susceptible to disruption by various noxious stimuli, such as pro-inflammatory cytokines. In particular, IL-1 is considered as a prominent cytokine that perturbs cartilage homeostasis and results in ECM degradation. IL-1 has been shown to inhibit ECM synthesis (1), induce cartilage-degrading proteases and inflammatory mediators (2), and enhance chondrocyte apoptosis (3). Pathological ramifications from aberrant IL-1 signaling include amplification of inflammatory responses in chondrocytes by supernormally induced pro-inflammatory mediators, such as IL-6, IL-8, and Toll-like receptor 2 (TLR2) (2, 4). ECM fragments as the result of protease-mediated degeneration also promote catabolic effects through TLR2/TLR4 (5), thus helping perpetuate cartilage degradation. Pro-inflammatory mediators including IL-1 have been implicated in several degenerative joint diseases, such as osteoarthritis (OA). Pharmaceutical targeting of these inflammatory mediators is being actively explored in OA therapy, and the anti-IL-1 strategy using IL-1 receptor antagonist serves as a typical example.
Anti-inflammatory cytokines have been demonstrated to attenuate inflammatory responses and thus joint damage in OA and rheumatoid arthritis conditions. IL-1 receptor antagonist represents the best understood cytokine in this category in cartilage biology. IL-1 receptor antagonist directly dampens IL-1 signaling, thus protecting chondrocytes from excessive catabolic and inflammatory activities. In experimental models, IL-1 receptor antagonist effectively inhibits IL-1-mediated cartilage destruction (6). The other proposed anti-inflammatory cytokines, by contrast, are inadequately characterized in the context of OA pathogenesis. IL-4 and IL-10 appear to be chondroprotective in vivo, and a negative correlation between their expression and that of TNF has been reported in OA cartilage (7). IL-11 generated by chondrocytes stimulates the expression of tissue inhibitor of metalloproteinase 1 (TIMP-1), an endogenous inhibitor of matrix metalloproteinase (MMP) (8). In rheumatoid arthritis synovium, IL-11 directly inhibits MMP-1 and MMP-3 production, up-regulates TIMP-1, and inhibits TNF-α production in the presence of soluble IL-11 receptor (9). Another cytokine IL-13 also blocks collagenolysis in the presence of IL-1 and increases TIMP activity (10). Despite a lack of in vivo assessments, these findings suggest that these anti-inflammatory cytokines could be candidates for effective OA therapy.
Previously we characterized bovine lactoferricin (LfcinB), a 25-amino acid peptide derived from the glycoprotein bovine lactoferrin, in human cartilage and synovium. LfcinB blocks IL-1-mediated catabolic and inflammatory processes in vitro and ex vivo, possibly through a heparan sulfate-dependent mechanism (11). In nucleus pulposus cells, LfcinB inhibits the detrimental activities of IL-1 and LPS and synergizes with bone morphogenetic protein-7 in promoting anabolic processes (12, 13). Gene expression analyses indicated that LfcinB down-regulates several MMPs, aggrecanases, and pro-inflammatory mediators (11). LfcinB also up-regulates two anti-inflammatory cytokines, IL-4 and IL-10, as well as TIMP-3 (11, 14). LfcinB specifically triggers ERK1/2, p38, and Akt signaling, with the ERK1/2 response being the most robust (11, 14). However, it still remained undefined how these pathways regulate LfcinB target genes. In this current study, we aimed to (i) further define the anti-inflammatory property of LfcinB and (ii) provide a link between LfcinB-specific signaling pathways and its chondroprotective activity.
EXPERIMENTAL PROCEDURES
Materials
Anti-MMP-1 and anti-MMP-13 antibodies were sent from Dr. Gillian Murphy's laboratory. Phospho-STAT3 (Tyr705) and STAT3 antibodies were purchased from Cell Signaling Technology (Danvers, MA); TIMP-1 and GAPDH antibodies were purchased from Abcam (Cambridge, MA). Anti-IL-11, anti-c-Fos, anti-c-Jun, and anti-JunD antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). IL-11Rα (interleukin 11 receptor α subunit) antibody was purchased from R&D Systems (Minneapolis, MN). PD98059, SB203580, LY294002, and Akt inhibitor IV were purchased from EMD Chemicals (Gibbstown, NJ). Recombinant human IL-1β and IL-11 were purchased from PeproTech (Rocky Hill, NJ). LfcinB was purchased from BioSynthesis (Lewisville, TX). Heparin, heparan sulfate, and heparinase III were purchased from Sigma. siRNA targeting IL11, FOS, and STAT3 were acquired from Invitrogen.
Tissue Acquisition and Chondrocyte Isolation
Post-mortem human femoral cartilage (age ranging from 40 to 70) was obtained through the Gift of Hope Organ and Tissue Donor Network (Elmhurst, IL) within 72 h. Prior approval by the local ethics committee and consent from donor families were acquired. Before processing, each specimen was graded following a 5-point scale modified from that of Collins (15). Cartilage for this study was graded 0 or 1, unless otherwise specified. Osteoarthritic femoral and tibial cartilage was obtained from patients (age ranging from 40 to 70) through the Orthopedic Tissue and Implant Repository Study (Chicago, IL) with prior consent from the patients. Human tissues were handled based on the guidelines of the Human Investigation Committee of Rush University Medical Center. After aseptic dissection, cartilage was digested in DMEM/Ham's F-12 (1:1) medium with 0.2% Pronase for 1 h, followed by overnight digestion with 0.025% Collagenase P supplemented with 5% FBS in a humidified atmosphere with 5% CO2 and continuous agitation. Chondrocytes released from enzymatic digestion were resuspended to a density of 3 × 106 cells/ml in DMEM/Ham's F-12 medium (1:1) supplemented with 10% FBS (complete medium). For short term monolayer culture, the cells were then plated onto 6-well (2 ml/well), 12-well (1 ml/well), or 24-well (0.5 ml/well) plates. After 3-day culture, the media were replaced with serum-free DMEM/Ham F-12 medium (1:1). After another 24 h, culture media were replaced with fresh serum-free medium again, 2 h prior to treatments. LfcinB concentrations used in monolayer culture were 50 and 100 μg/ml. FGF-2 (100 ng/ml) or IL-1β (5 ng/ml) was also applied wherever appropriate. The cells were harvested after 24-h stimulation and subjected to downstream analyses as detailed below.
Immunoblotting
Cell lysates were prepared using a modified radioimmune precipitation buffer (16). Total protein concentrations were determined by a BCA protein assay (Pierce). Equal amounts of protein were resolved by 10% SDS-PAGE and transferred to nitrocellulose membrane for immunoblotting. Immunoreactivity was visualized using the ECL system (Amersham Biosciences) and the Signal Visual Enhancer system (Pierce) to magnify the signal.
Reverse Transcription and Real Time Polymerase Chain Reaction
Total RNA from normal and osteoarthritic articular chondrocytes was isolated using TRIzol reagent (Invitrogen) according to the instructions provided by the manufacturer. RT was carried out with 1 μg of total RNA using ThermoScriptTM RT-PCR system (Invitrogen) for first strand cDNA synthesis. For real time PCR, cDNA was amplified using a MyiQ real time PCR detection system (Bio-Rad). Relative gene expression was determined using the ΔΔCT method, as detailed by manufacturer guidelines (Bio-Rad). 18 S rRNA and GAPDH were used as internal controls in the reactions for normalization. The standard deviations in samples represent at least five different donors from independent experiments. The primer sequences used in this study are listed in Table 1.
TABLE 1.
Primers used in real time PCR analyses
| Gene | Primer sequence (5′ → 3′) | NCBI reference no. |
|---|---|---|
| GAPDH | NM_002046.4 | |
| Forward: | TCGACAGTCAGCCGCATCTTCTTT | |
| Reverse: | GCCCAATACGACCAAATCCGTTGA | |
| 18 S rRNA | NR_003286.2 | |
| Forward: | CGGCTACCACATCCAAGGAA | |
| Reverse: | GCTGGAATTACCGCGGCT | |
| ACAN | NM_001135.3 | |
| Forward: | TCTTGGAGAAGGGAGTCCAACTCT | |
| Reverse: | ACAGCTGCAGTGATGACCCTCAGA | |
| MMP1 | NM_002421.3 | |
| Forward: | AGTGACTGGGAAACCAGATGCTGA | |
| Reverse: | GCTCTTGGCAAATCTGGCGTGTAA | |
| MMP13 | NM_002427.3 | |
| Forward: | ACCCTGGAGCACTCATGTTTCCTA | |
| Reverse: | TGGCATCAAGGGATAAGGAAGGGT | |
| IL1B | NM_000576.2 | |
| Forward: | ATGACCTGAGCACCTTCTTTCCCT | |
| Reverse: | GCATCGTGCACATAAGCCTCGTTA | |
| IL6 | NM_000600.3 | |
| Forward: | AAGCCAGAGCTGTGCAGATGAGTA | |
| Reverse: | TTCGTCAGCAGGCTGGCATTTGT | |
| IL8 | NM_000584.3 | |
| Forward: | TCTTGGCAGCCTTCCTGATTTCTG | |
| Reverse: | GGGTGGAAAGGTTTGGAGTATGTC | |
| IL11 | NM_000641.3 | |
| Forward: | TACCCGTATGGGACAAAGCTGCAA | |
| Reverse: | TGCACCATGTTGCTTAACCCTCAC | |
| TIMP1 | NM_003254.2 | |
| Forward: | CATCCTGTTGTTGCTGTGGCTGAT | |
| Reverse: | AAGGTGGTCTGGTTGACTTCTGGT | |
| IL11RA | NM_001142784.2 | |
| Forward: | CTCAAGTTCCGTTTGCAGTACCGT | |
| Reverse: | TCCAGGTGCCAGCATCTAGAAAGT | |
| FOS | NM_005252.3 | |
| Forward: | AGATTGCCAACCTGCTGAAGGAGA | |
| Reverse: | AAGCCACAGACATCTCTTCTGGGA | |
| STAT3 | NM_139276.2 | |
| Forward: | ATGGAAGAATCCAACAACGGCAGC | |
| Reverse: | TCCTCAGTCACAATCAGGGAAGCA | |
IL-8 ELISA
The IL-8 ELISA kit used in this study has a sensitivity of <2 pg/ml and an assay range of 25.6–1000 pg/ml, according to the manufacturer's descriptions (Thermo Fisher Scientific). This assay does not cross-react with other interleukins. After adding standards and samples in duplicates, multiwell plates were covered and incubated at room temperature for 1 h. Then wells were washed three times, followed by the addition of 50 μl of biotinylated antibody reagent into each well. The plates were incubated for another hour at room temperature. After washing the wells three times, 100 μl of streptavidin-HRP working solution was added into each well, followed by 30-min incubation at room temperature. Plates were then washed three times, and 100 μl of TMB substrate was added to each well. The reactions were allowed to proceed in the dark for 30 min at room temperature before stop solution was added. Absorbance of each well was measured at 450 and 550 nm.
Preparation of Nuclear Extract
A nuclear extraction kit (Affymetrix, Santa Clara, CA) was used based on the manufacturer's instructions. Upon the completion of stimulation, media were removed, and washed cells were lysed in 1 ml of buffer A. After incubation on ice for 10 min, lysates were centrifuged at 14,000 × g at 4 °C for 3 min. Supernatants were discarded, and cell pellets were resuspended in 40 μl of buffer B. Then samples were incubated on ice for 1 h before centrifugation at 14,000 × g, 4 °C for 5 min. The supernatants were aliquoted and stored at −80 °C. Total protein concentration in each sample was determined by BCA assay (Pierce) right before EMSA.
EMSA
The nuclear extracts were prepared after stimulation as described above. The EMSA kit (Affymetrix) was used according to the manufacturer's instructions. EMSA was performed by incubating labeled biotin-conjugated probes with 5 μg of nuclear extract. Samples were resolved in 6% nondenaturing polyacrylamide gels. Then proteins were transferred to Pall Biodyne B membrane. The membrane was blocked with 1× blocking buffer and then incubated with streptavidin-HRP conjugate. After proper washing, the signals were visualized in a chemiluminescence imaging system. In each experiment, a separate reaction using unlabeled double-stranded DNA (cold probe) was set up to demonstrate binding specificity.
Transfection of siRNA
Nucleofection was optimized for human articular chondrocytes based on the manual of the NucleofectorTM kit (Lonza, Walkersville, MD) as described previously (16, 17). Chondrocytes were cultivated for 3 days before transfection. For knockdown experiments, siRNA at a concentration of 200 nm (20 pmol/sample) was used for transfection. After 48 h, cell lysates were subjected to quantitative PCR (qPCR) and immunoblotting for validation of successful knockdown. In parallel, stimulations were performed 48 h after transfection. Cell lysates, total RNA, and conditioned media were collected for downstream analyses.
Cartilage Explant Culture
Full-thickness explants of 4-mm diameters were prepared from freshly isolated, healthy femoral cartilage strips. Explants then recovered in DMEM/F-12 (1:1, supplemented with 10% FBS) for 48 h. Culture media were changed to DMEM/F-12 (1:1) supplemented with 1% mini-ITS+ premix (BD Biosciences, San Jose, CA) 48 h before treatments started. The explants were treated with FGF-2 (100 ng/ml) or IL-1β (5 ng/ml), in the presence or absence of LfcinB (50 and 100 μg/ml).
Histology
After dissection, human articular cartilage explants were fixed in 4% paraformaldehyde. After embedding the explants in paraffin, serial sections of 8-μm thickness were prepared and placed onto slides. Then sections were deparaffinized in xylene, followed by stepwise rehydration in ethanol and distilled water. For Safranin-O Fast Green staining, sections were immersed in 0.1% Fast Green for 3 min, followed by 1% Safranin-O for 15 min.
Statistical Analyses
All experiments were performed with 3–5 biological replicates, without pooling samples from different donors. Statistical significance was determined by Student's t test or one-way repeated measures analysis of variance followed by Sidak post hoc test, using the SPSS 17 software (IBM Corporation, Somers, NY). p values lower than 0.05 were considered to be statistically significant in each test. Each value in the figures is presented as the mean ± standard deviation.
RESULTS
LfcinB Potently Induces IL-11, an Anti-inflammatory Cytokine, in Human Articular Chondrocytes
Our previous studies revealed that LfcinB represses the expression of cartilage-degrading proteases (e.g., MMP-13 and ADAMTS-5) and pro-inflammatory mediators (e.g., IL-1β, IL-8, and TLR2). Simultaneously, LfcinB significantly induces multiple anti-inflammatory cytokines (IL-4 and IL-10), providing a molecular basis on which LfcinB counteracts the catabolic and inflammatory activities promoted by IL-1β and FGF-2 in articular cartilage (11). Our previous promising results motivated us to further investigate LfcinB-controlled signaling pathways and its downstream target genes that may mediate its chondroprotective effects in human primary chondrocytes. When surveying the mRNA expression of other potential anti-inflammatory targets, we observed a striking 40- and 120-fold induction of IL-11 upon stimulation with two different concentrations of LfcinB (50 and 100 μg/ml, respectively) (Fig. 1A; p < 0.01 and p < 0.001, respectively). This finding was confirmed by concurrent increases in IL-11 protein production in both intracellular and extracellular compartments (Fig. 1B). In both compartments, the molecular weight of IL-11 is ∼19 kDa, corresponding to the mature form of IL-11 with its signal peptide removed. To determine whether such an induction is temporally robust, we stimulated chondrocytes with LfcinB (100 μg/ml) for different durations (24, 48, and 72 h). Our data show that IL-11 induction by LfcinB peaked at the 24-h time point and was sustained over 72 h (Fig. 1C; p < 0.001 and p < 0.01).
FIGURE 1.

LfcinB induces IL-11 expression in human articular chondrocytes. A, chondrocytes were incubated with LfcinB (50 and 100 μg/ml) for 24 h. Transcripts of IL-11 were quantified by qPCR. B, the abundance of intracellular and extracellular IL-11 was analyzed by immunoblotting using cell lysates and conditioned media, respectively. C, chondrocytes were stimulated with LfcinB (100 μg/ml) for 24, 48, and 72 h. IL-11 mRNA expression was measured by qPCR. **, p < 0.01; ***, p < 0.001.
IL-11 Antagonizes IL-1β-induced Proteoglycan Depletion in Human Articular Cartilage ex Vivo Organ Culture
IL-11 was previously shown to be an anti-inflammatory cytokine and chondroprotective (18). Nevertheless, its biological roles in joint homeostasis had never been examined in detail. We first determined the potency of IL-11 using an ex vivo culture model, in which full thickness human articular cartilage explants were incubated with IL-1β (5 ng/ml) in the presence or absence of IL-11 (100 and 200 ng/ml) for 11 days. Our histological analyses revealed that IL-1β-elicited proteoglycan (PG) depletion was dose-dependently counteracted by IL-11, as indicated by the PG retention in the presence of IL-1β challenge (Fig. 2A, panel b versus panels c and d). This finding suggests that LfcinB-mediated anti-inflammatory action is, at least in part, achieved through the induction of IL-11 in human articular cartilage.
FIGURE 2.

IL-11 counteracts IL-1β-induced PG depletion. A, full thickness cartilage explants were maintained in DMEM/F-12 medium supplemented with 1% mini-ITS+ premix and stimulated with IL-1β (5 ng/ml) in the presence or absence of IL-11 (100 and 200 ng/ml). After 11 days, sections from each group were stained with Safranin-O Fast Green dyes to reveal gross PG content. B, chondrocytes in monolayer were stimulated with IL-1β (5 ng/ml) in the presence or absence of IL-11 (50 and 100 ng/ml) or IL-11 alone (50 and 100 ng/ml). Aggrecan (ACAN) transcripts were quantified by qPCR. *, p < 0.05; **, p < 0.01. Ctrl, control.
Spurred by this finding, we next characterized whether such PG preservation by IL-11 is due to its biological effects on aggrecan gene expression. Consistent with our previous observation, IL-1β caused marked suppression of aggrecan gene expression (Fig. 2B; p < 0.01). This effect, however, was significantly attenuated by IL-11 co-treatment at the concentration of 100 ng/ml (Fig. 2B; p < 0.05). IL-11 alone did not enhance aggrecan expression, suggesting chondroprotection exerted by IL-11 is mainly via anti-inflammatory, but not pro-anabolic action. In addition, IL-11 did not elicit cytotoxicity in chondrocytes (data not shown).
IL-11 Acts as a Chondroprotective Cytokine Antagonizing Autocrine and Paracrine Catabolic Action of IL-1β in Human Articular Chondrocytes
IL-1β acts in autocrine and paracrine manners by inducing IL-1β itself as well as multiple pro-inflammatory cytokines and chemokines (e.g., IL-6 and IL-8) in many cell types, including human articular chondrocytes (19). We next determined whether IL-11 also attenuates IL-1β-stimulated pro-inflammatory cytokine expression in human primary articular chondrocytes. Our results revealed that IL-1β-mediated up-regulation of IL-1β, IL-6, and IL-8 was significantly compromised by IL-11 co-treatment (100 ng/ml) on mRNA levels (Fig. 3, A and B; p < 0.05). Consequently, the highly induced IL-8 level in conditioned medium was significantly down-regulated by co-treatment with IL-11, as assessed by ELISA (Fig. 3C; p < 0.001).
FIGURE 3.

IL-11 moderately antagonizes IL-1β-mediated inflammatory and catabolic effects. A, chondrocytes in monolayer were stimulated with IL-1β (5 ng/ml) in the presence or absence of IL-11 (50 and 100 ng/ml) or IL-11 alone (50 and 100 ng/ml). IL-1β and IL-6 transcripts were quantified by qPCR. B, chondrocytes were stimulated with IL-1β (1 ng/ml) in the presence or absence of IL-11 (50 and 100 ng/ml) or IL-11 alone (50 and 100 ng/ml). IL-8 transcripts were measured by qPCR. C, in parallel, IL-8 concentrations in the conditioned media were determined by ELISA. Samples were properly diluted to fit the assay range. Dilution factors were used to calculate the original concentrations. D, chondrocytes treated as described above were subjected to qPCR quantification of MMP-1 and MMP-13 transcripts. E, conditioned media were collected from chondrocytes treated as described above. Secreted MMP-1 and MMP-13 were assessed by immunoblotting. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
IL-1β significantly induces cartilage-degrading enzymes such as MMP-1 and MMP-13 in human adult articular chondrocytes. Thus, we tested whether IL-11 also mitigates IL-1β-induced ECM protease expression. The presence of IL-11 at 100 ng/ml effectively attenuated IL-1β-stimulated collagenase expression at mRNA level (Fig. 3D; p < 0.05). Correspondingly at the protein levels, the enhanced expression of MMP-1 and MMP-13 by IL-1β was notably reversed by co-incubation with IL-11 in a concentration-dependent manner (Fig. 3E).
LfcinB-induced Anti-inflammatory Effects Are Mediated, in Part, via Up-regulation of IL-11 in Human Articular Chondrocytes
Because LfcinB markedly up-regulates IL-11 and IL-11 antagonizes IL-1β-induced catabolism, we then hypothesized that the induction of IL-11 by LfcinB may at least partially account for LfcinB-induced anti-inflammation. To directly evaluate the link between the biological impact of LfcinB and IL-11, we knocked down IL-11 gene by siRNA followed by stimulation with IL-1β (5 ng/ml) in the presence or absence of LfcinB (50 μg/ml) for 24 h. We were able to achieve efficient knockdown of IL-11, as reflected by the levels of IL-11 in untreated and LfcinB-stimulated cells (Fig. 4A; p < 0.01). Importantly, the antagonistic effects of LfcinB on IL-1β-induced targets (i.e., MMP-1 and MMP-13) were diminished by IL-11 knockdown on both mRNA (Fig. 4B; p < 0.05) and protein levels (Fig. 4C). These data provide direct evidence that LfcinB-induced anti-inflammatory effects are mediated, in part, via up-regulation of IL-11 in human primary articular chondrocytes.
FIGURE 4.

IL-11 acts as a secondary molecule in LfcinB-mediated responses. A, chondrocytes transfected with siRNA targeting IL-11 were cultured in the presence or absence of LfcinB (50 μg/ml). IL-11 transcripts were quantified by qPCR to reveal knockdown efficiency. B, chondrocytes transfected with IL-11 siRNA were incubated with IL-1β (5 ng/ml) and LfcinB (50 μg/ml) for 24 h. MMP-1 and MMP-13 transcripts were measured by qPCR. C, in parallel, secreted MMP-1 and MMP-13 were analyzed using conditioned media by immunoblotting. D, chondrocytes were first incubated with LfcinB (100 μg/ml) for 24 h and then stimulated with IL-1β (5 ng/ml) for another 24 h. MMP-1 and MMP-13 transcripts were quantified by qPCR. E, IL-1β mRNA level was quantified by qPCR. F, chondrocytes were primed with IL-1β (5 ng/ml) for 48 h before LfcinB (100 μg/ml) stimulation for 24 h. MMP-1 and MMP-13 mRNA levels were quantified by qPCR. G, likewise, IL-1β transcript was quantified by qPCR. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Based on our previous studies, LfcinB can directly antagonize IL-1 within 24 h (11), possibly independent of secondary IL-11 actions. To further examine how LfcinB exerts anti-inflammatory effects in chondrocytes, we primed cells with LfcinB (100 μg/ml, 24 h) or IL-1β (5 ng/ml, 48 h). We then stimulated LfcinB-primed cells with IL-1β (5 ng/ml), or IL-1β-primed cells with LfcinB (100 μg/ml), for 24 h. Strikingly, IL-1β-mediated induction of MMP-1, MMP-13, and IL-1β was essentially abrogated in LfcinB-primed chondrocytes (Fig. 4, D and E; p < 0.01). In IL-1β-primed cells, LfcinB was still able to significantly down-regulate MMP-13 (Fig. 4F; p < 0.01) and IL-1β (Fig. 4G; p < 0.05). These data further confirm that LfcinB promotes anti-inflammatory processes through primary and secondary mechanisms in chondrocytes.
LfcinB-mediated IL-11 Induction Is Heparan Sulfate-dependent
LfcinB contains a heparan sulfate (HS)-binding motif, and it has been demonstrated that this motif mediates some of its biological effects (20). Recently, we showed that LfcinB is capable of antagonizing two HS-dependent factors, FGF-2 and IL-1β (11), suggesting that LfcinB-mediated antagonism against catabolic cytokines is possibly through competitive bindings to HS on cell surface. Hence, we hypothesized that LfcinB requires HS on the plasma membrane of chondrocytes to elicit downstream responses. To test this hypothesis, chondrocytes were pretreated with heparinase, which removed cell surface HS, before they were stimulated with LfcinB (50 μg/ml) for 24 h. Heparinase pretreatment resulted in significant attenuation of LfcinB-mediated effects, such as IL-11 induction (Fig. 5, A and B; p < 0.01) and MMP repression (Fig. 5, C and D; p < 0.05), on mRNA and protein levels. To determine whether LfcinB requires cell-bound HS to exert its functions, LfcinB was preincubated with exogenous heparin or HS before it was applied onto chondrocytes. Exogenous heparin and HS not only dramatically attenuated LfcinB-mediated ERK1/2, p38 and Akt signaling (Fig. 5E), but also abolished LfcinB-mediated IL-11 induction and MMP repression on mRNA and protein levels (Fig. 5, F–I), suggesting that LfcinB forms a complex with membrane-bound HS to trigger cellular responses. Together, these findings strongly suggest that LfcinB critically depends on cell surface HS to exert its bioactivities.
FIGURE 5.

Heparan sulfate is required for LfcinB signaling and downstream effects. A, chondrocytes were incubated with heparinase III (1 Sigma unit/ml) in serum-free DMEM overnight. Then chondrocytes were stimulated with LfcinB (50 μg/ml) in the presence of heparinase III (1 Sigma unit/ml) in serum-free DMEM for 24 h. IL-11 transcripts were quantified by qPCR. B, IL-11 in conditioned media and whole cell lysates were examined by immunoblotting. C, transcripts of MMP-1, MMP-3, and MMP-13 were quantified by qPCR. D, MMP-1, MMP-3, and MMP-13 in the conditioned media were analyzed by immunoblotting. E, LfcinB (50 μg/ml) was preincubated with heparin (50 μg/ml) or HS (50 μg/ml) for 40 min and then used to stimulate chondrocytes for 30 min and 60 min. The levels of activated and total ERK1/2, p38, and Akt were analyzed by immunoblotting. F, LfcinB (50 μg/ml) was preincubated with heparin (50 μg/ml) or HS (50 μg/ml) for 40 min and then used to stimulate chondrocytes for 24 h. IL-11 transcripts were quantified by qPCR. G, IL-11 in conditioned media, and whole cell lysates were examined by immunoblotting. H, transcripts of MMP-1, MMP-3, and MMP-13 were quantified by qPCR. I, MMP-1, MMP-3, and MMP-13 in the conditioned media were analyzed by immunoblotting. Hep, heparin. *, p < 0.05; **, p < 0.01.
LfcinB Induces IL-11 via the ERK1/2-c-Fos/JunD Axis
Previously, we reported robust activation of ERK1/2 and Akt by LfcinB, which sustains for >60 min in human articular chondrocytes (11). Unlike ERK1/2 and Akt, LfcinB induced p38 activation only in a transient manner (less than 60 min; data not shown). In addition, we did not observe activation of JNK, NFκB, SMAD1/5/8, SMAD2/3, or STAT3 pathway within 2 h after LfcinB stimulation (data not shown). Our results suggest that the major signaling pathways induced by LfcinB are ERK1/2, Akt, and potentially p38 in human articular chondrocytes.
Because the IL-11 induction is the most dramatic and robust cellular response elicited by LfcinB in articular chondrocytes (maximal 120-fold), we wished to determine the responsible signaling pathway. Human articular chondrocytes were preincubated with individual pathway inhibitors of ERK1/2, p38, and Akt, followed by LfcinB stimulation for 24 h. Our qPCR results demonstrate that only ERK1/2 inhibition led to a prominent reversal of the IL-11 expression (Fig. 6A; p < 0.01), suggesting ERK1/2 is the key regulatory pathway. ERK1/2 is an upstream regulatory kinase of AP-1. Two functional proximal activator protein 1 (AP-1) elements residing side by side within the −82 to −92 region of IL-11 promoter had been reported in human IL-11 transcription (Fig. 6B) (21). To directly examine AP-1 activation status, we analyzed AP-1 protein-DNA interaction by EMSA using nuclear extracts from chondrocytes stimulated with LfcinB (1 h) in the presence or absence of individual pathway inhibitors. We observed enhanced binding of AP-1 to its consensus DNA sequence after stimulation, indicating that LfcinB effectively activates AP-1 (Fig. 6C, lane 2). The enhanced AP-1 binding was specifically abolished upon ERK1/2 inhibition (Fig. 6C, lane 3) but not by inhibitors of p38 (lane 4) or Akt (lane 5). Our data suggest that the ERK1/2 signaling pathway plays a primary role in regulating AP-1 activity after LfcinB stimulation.
FIGURE 6.

LfcinB potently induces IL-11 expression via the ERK1/2-c-Fos/JunD axis. A, chondrocytes in monolayer were preincubated with individual pharmacological inhibitors for 1 h, before stimulation with LfcinB (50 μg/ml) for 24 h. IL-11 transcripts were quantified by qPCR. B, structure of IL-11 promoter. Two adjacent AP-1 elements are present between −80 and −100. C, chondrocytes were preincubated with individual pharmacological inhibitors for 1 h before LfcinB (50 μg/ml) stimulation for another hour. A binding reaction using unlabeled (cold) AP-1 probe was set up for each EMSA assay to demonstrate binding specificity. The AP-1-DNA complex was visualized using a chemiluminescence imaging system. D, nuclear extracts from articular chondrocytes were sequentially incubated with AP-1 probe and individual antibodies against c-Fos, JunD, and c-Jun. A control reaction using IgG was set up to demonstrate antibody specificity. Another binding reaction using unlabeled (cold) AP-1 probe was also performed to demonstrate probe binding specificity. E, chondrocytes were incubated with LfcinB (50 and 100 μg/ml) for 24 h. Fos transcripts and protein expression were assessed by qPCR. F, in parallel, conditioned media were analyzed by immunoblotting for IL-11 expression. G, chondrocytes were transfected with siRNA targeting fos. Fos protein expression was then examined by immunoblotting to validate knockdown efficiency. Chondrocytes were also transfected with siRNA with scrambled sequences as a control. H, chondrocytes transfected with fos siRNA were stimulated with LfcinB (50 μg/ml) for 24 h. IL-11 transcripts were measured by qPCR. I, after treating chondrocytes as described above, IL-11 protein expression was examined using cell lysates. GAPDH was used as loading control. Scr, scrambled. **, p < 0.01.
The AP-1 complex can be either a homo- or a heterodimer, depending on stimuli and cellular context (22). Thus, next we further defined the components of LfcinB-induced AP-1 complex. Human primary chondrocytes were incubated with LfcinB for 1 h, and supershift assays were performed using nuclear extracts that were sequentially incubated with AP-1 probes and individual antibodies against potential AP-1 components (i.e., c-Fos, c-Jun, and JunD). Our results demonstrated that only antibodies against c-Fos and JunD specifically caused supershift of the protein-DNA complexes, suggesting that LfcinB-induced AP-1 heterodimer primarily consists of c-Fos and JunD (Fig. 6D, lanes 3 and 4).
It has been reported that elevated c-fos expression can lead to augmented AP-1 transcriptional activity (23). Therefore, we also determined whether LfcinB regulates the expression of AP-1 component c-fos, which may influence gross AP-1 activity. Cells were cultured with LfcinB (50 and 100 μg/ml) for 24 h followed by quantification of c-fos transcripts. Our qPCR data revealed that c-fos mRNA level was increased by 3-fold in the presence of LfcinB (Fig. 6E; p < 0.01). Correspondingly, c-Fos protein level was notably augmented upon LfcinB stimulation in a concentration-dependent manner (Fig. 6F). LfcinB-mediated induction of AP-1 components appeared to be limited to c-Fos, because we did not observe similar stimulation of JunD, the other component of AP-1 heterodimers, by LfcinB (data not shown).
Because c-fos expression was specifically up-regulated by LfcinB, we further determined the role of c-fos in LfcinB-mediated IL-11 expression. Nucleofection of siRNA successfully knocked down c-fos at both mRNA (data not shown) and protein levels, in contrast with scrambled siRNA (Fig. 6G). Our data show that reduced c-fos by siRNA significantly impaired the extent of IL-11 induction by LfcinB on mRNA level (Fig. 6H; p < 0.01). Correspondingly, intra- and extracellular levels of IL-11 protein were also reversed upon c-fos knockdown (Fig. 6I). Collectively, our results evidence that LfcinB-induced c-Fos plays a critical role in the transcriptional regulation of IL-11 expression.
LfcinB Up-regulates TIMP-1 via IL-11/STAT3-dependent Manner as a Secondary Stimulatory Event
TIMP family members (TIMP-1–4) are the key natural chondroprotective inhibitors of cartilage-degrading enzyme activities (i.e., MMPs and ADAMTS). Although stimulation with IL-11 significantly up-regulates TIMP-1 at both mRNA and protein levels within 24 h (Fig. 7, A and B; p < 0.01), in our initial experiments we were unable to detect significant TIMP-1 induction by LfcinB during the same time frame in human articular chondrocytes. This apparent discrepancy led us to conjecture that LfcinB stimulates TIMP-1 through IL-11 production as a secondary event only when extracellular IL-11 concentration reaches a critical level capable of triggering notable TIMP-1 expression at a later time point. To test this notion, articular chondrocytes were treated with LfcinB for different durations (24, 48, and 72 h), followed by qPCR analyses of TIMP-1 expression. Indeed, we observed significant TIMP-1 induction at the 48-h point, and the induced TIMP-1 mRNA and protein levels were sustained until 72 h after LfcinB stimulation (Fig. 7, C and D; p < 0.05). Our finding suggests that LfcinB-mediated TIMP-1 expression is a secondary stimulatory event and is highly likely to be mediated by the induced IL-11.
FIGURE 7.

LfcinB up-regulates IL-11, which in turn induces TIMP-1 expression via STAT3 signaling pathway. A, chondrocytes in monolayer were incubated with IL-11 (50 ng/ml) for 24 h. TIMP-1 transcripts were quantified by qPCR. B, extracellular and intracellular TIMP-1 protein was analyzed by immunoblotting. C, chondrocytes were incubated with LfcinB (100 μg/ml) for different durations (24, 48, and 72 h). TIMP-1 transcripts were quantified by qPCR. D, chondrocytes were treated as described above. TIMP-1 protein expression in both intracellular and extracellular fractions was examined by immunoblotting. E, chondrocytes in monolayer were treated with LfcinB (100 μg/ml) in the presence or absence of IL-11 neutralization antibody (10 μg/ml). TIMP-1 mRNA expression was examined by qPCR. F, TIMP-1 protein expression was examined by immunoblotting. G, chondrocytes were treated with either IL-11 (50 ng/ml) or LfcinB (50 μg/ml) for various durations (0.5, 1, 24, 48, and 72 h). STAT3 activation was examined by immunoblotting using an anti-phospho-STAT3 antibody. Total STAT3 levels were used as loading controls. H, naive chondrocytes and chondrocytes transfected with STAT3 siRNA were incubated with LfcinB (100 μg/ml) for 24 h. TIMP-1 mRNA levels were measured by qPCR. I, extracellular and intracellular TIMP-1 protein levels were analyzed by immunoblotting. J and K, IL-11 mRNA (J) and protein levels (K) were assessed by qPCR and immunoblotting using the same set of samples as described above. Scr, scrambled. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We next sought to determine the contribution of induced IL-11 in such delayed TIMP-1 up-regulation by LfcinB. Chondrocytes were incubated with LfcinB in the presence or absence of IL-11 neutralization antibody for 48 h, followed by analyses of expression levels of TIMP-1. Our qPCR and immunoblotting results indicate that blockage of IL-11 action essentially abolished TIMP-1 induction at the mRNA and protein levels after LfcinB stimulation (Fig. 7, E and F; p < 0.05). This finding highlights the importance of induced IL-11 in mediating secondary cellular responses to LfcinB in chondrocytes.
Activation of STAT3 has been demonstrated to transactivate TIMP-1 (24, 25). In our initial experiments, treatment of articular chondrocytes with LfcinB showed no activation of STAT3 within 2 h. Cells stimulated with IL-11, however, rapidly activate STAT3 signaling, as represented by the phosphorylation of Tyr705 within 30 min, and the activation sustained for 24 h (Fig. 7G, upper panel). Although we did not observe immediate STAT3 activation upon LfcinB stimulation, it remained possible that STAT3 can be activated after a sufficient amount of IL-11 is produced. To test this notion, chondrocytes were incubated with LfcinB for different time periods (24, 48, and 72 h), and STAT3 phosphorylation was analyzed by immunoblotting. To our surprise, STAT3 was markedly phosphorylated after 24 h, and the activation sustained till >72 h after LfcinB stimulation (Fig. 7G, lower panel). Such kinetics clearly demonstrates sequential activation profile of STAT3 in which LfcinB activates STAT3 via IL-11 stimulation as a secondary signaling event.
To directly ascertain the participation of STAT3 in TIMP-1 regulation, STAT3 was knocked down in human primary chondrocytes by nucleofection using siRNA targeting STAT3, and then the cells were stimulated with LfcinB for 24 and 48 h. Analyses were conducted with qPCR for mRNA level and immunoblotting for protein level of TIMP-1. Knockdown of STAT3 significantly impaired TIMP-1 induction by LfcinB at the 48-h time point (Fig. 7, H and I; p < 0.01). By contrast, LfcinB-mediated IL-11 induction was not affected by STAT3 knockdown at the 24-h time point (Fig. 7, J and K), suggesting that STAT3 specifically mediates TIMP-1 expression in response to LfcinB via induced IL-11. Thus, our data together demonstrate that LfcinB primarily utilizes the ERK1/2-AP-1 axis to potently induce IL-11 expression, which in turn subsequently up-regulates TIMP-1 expression through the STAT3 signaling pathway in articular chondrocytes (summarized in Fig. 9).
FIGURE 9.
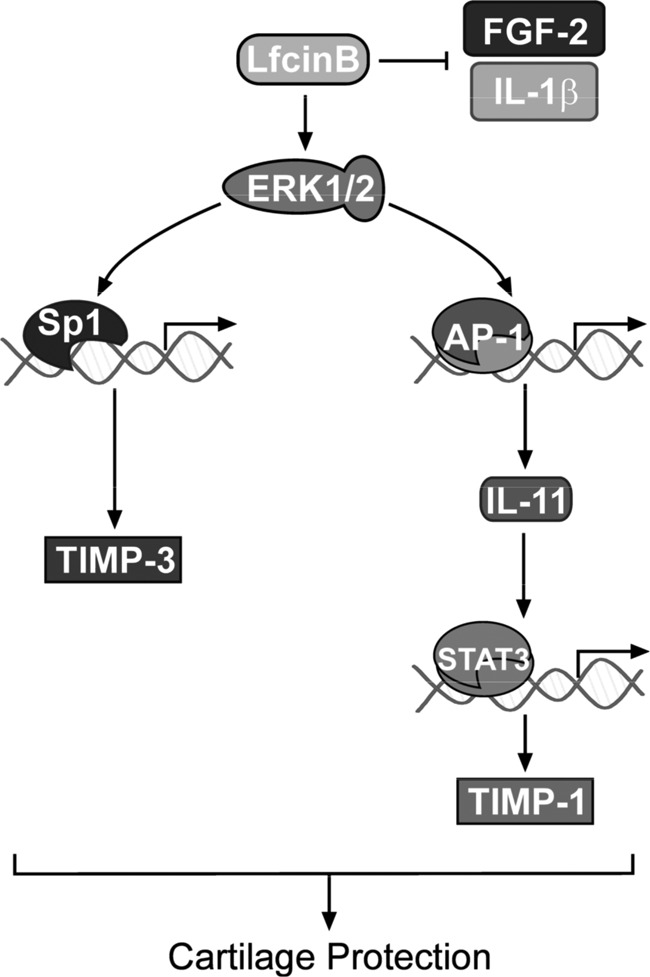
Hypothetical model of LfcinB-mediated chondroprotection. Evidence suggests that LfcinB exerts its chondroprotective activities through multiple mechanisms, including (i) competitive binding to the co-receptors of IL-1 and FGF-2; (ii) induction of anti-inflammatory cytokines, such as IL-11; and (iii) induction of anti-catabolic TIMP-3.
IL-11 and Its Cognate Receptor IL-11Rα Are Dysregulated in Human OA Chondrocytes
Our observations that LfcinB strikingly induces IL-11 inspired the question of whether such modulated genetic responses in fact bring benefits to OA therapy. Using total RNA and cell lysates prepared from age- and grade-matched healthy (grades 0 and 1) and OA chondrocytes, we observed statistically significant up-regulation of IL-11 transcripts in OA cells (Fig. 8A; p < 0.05). This change may represent a reparative effort in OA chondrocytes to curb inflammatory processes. Nonetheless, when we analyzed the level of IL-11Rα (a specific cognate receptor of IL-11), we found markedly reduced IL-11Rα in OA chondrocytes compared with healthy donor samples at both mRNA (Fig. 8B; p < 0.01) and protein levels (Fig. 8C), suggesting that IL-11 and IL-11Rα dysregulation in chondrocytes are associated with OA.
FIGURE 8.
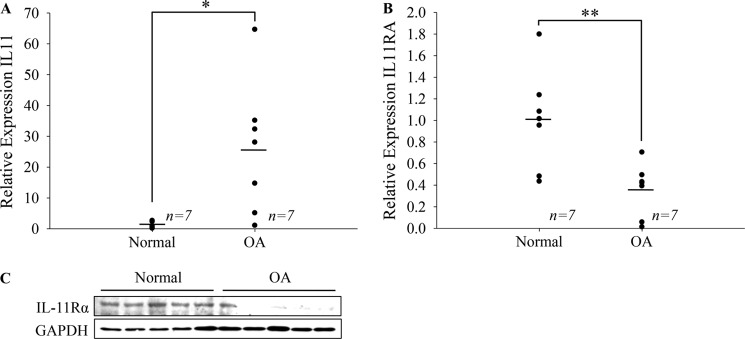
IL-11 signaling is disrupted in OA chondrocytes. A, IL-11 transcripts were quantified using age-matched (40–70 years old) normal femoral and OA femoral/tibial chondrocytes (n = 7). B, likewise, IL-11Rα transcripts were quantified by qPCR. C, IL-11Rα protein expression in these individuals was analyzed by immunoblotting. GAPDH was used as a loading control. * p < 0.05; ** p < 0.01.
DISCUSSION
As previously shown, LfcinB bears potential as a therapeutic peptide for OA intervention. In human articular cartilage and synovium, LfcinB effectively antagonized IL-1 and FGF-2, thus conferring chondroprotection (11). Here, we uncovered another important mechanism indicating that LfcinB-mediated anti-catabolic and chondroprotective actions are in part through IL-11 induction. LfcinB robustly activates ERK1/2, which in turn dramatically elevates IL-11 expression (>100-fold induction by 100 μg/ml LfcinB; p < 0.001) in a c-Fos/JunD-dependent manner. Induced IL-11 serves as a secondary anti-inflammatory mediator in chondrocytes that are stimulated with LfcinB. IL-11 also promotes anti-catabolism via induction of TIMP-1, hence contributing to protection of cartilage from degradation. Although STAT3 signaling is not required for LfcinB-mediated IL-11 expression, it is essential to IL-11-mediated TIMP-1 expression. Moreover, we show that IL-11 signaling may be suppressed as a result of IL-11Rα down-regulation in OA chondrocytes. Evidence from a handful of studies suggests the utility of IL-11 as a means of reducing arthritis inflammation or diseases with an arthritic component (9, 26). The beneficial bioactivities of IL-11 observed in these studies may be linked to its modulatory function in macrophage (27, 28), which represent one of the predominant cell populations in synovium in the pathological joint condition. Our findings not only corroborate the protective role of IL-11 in synovial joint but also unveiled cartilage as a target tissue of this anti-inflammatory cytokine. IL-11 exhibits moderate yet significant antagonism against IL-1 in PG metabolism, collagenase expression, and inflammatory mediator expression. It is worth noting that our findings from in vitro experiments are not in perfect accordance with results acquired from ex vivo experiments using cartilage explants, where striking chondroprotection was achieved with IL-11. Such an apparent discrepancy may arise from at least two sources. First, prolonged treatment with IL-11 in ex vivo culture may allow for delayed cellular responses, which could not be assessed in the monolayer short term culture model. The delayed responses possibly cooperate with the immediate transcriptional events to promote stronger protective activities. Second, chondrocytes reside in very different microenvironments between monolayer culture and ex vivo culture. In monolayer, chondrocytes synthesize their ECM de novo, whereas in cartilage explants, chondrocytes are surrounded by preformed intact ECM. We conjecture that there may be components inside the native ECM that can potentiate IL-11-mediated responses. It is also worth mentioning that data from two studies do not support the anti-inflammatory role of IL-11 in synovial joint (29, 30). However, caveats exist in these studies. The randomized trial of recombinant human IL-11 in rheumatoid arthritis treatment suffered from considerable placebo effects, which confounded the possible therapeutic benefit of IL-11 (29). The other study, which utilized an acute arthritic model, adopted an experimental scheme with frequent intra-articular injections of supranormal amounts of IL-11 (30). Such an ultra high dose of IL-11 may not generate a translatable outcome. Therefore, further in vivo studies using arthritic animal models are warranted to elucidate the beneficial effects mediated by IL-11.
The HS binding affinity of LfcinB allows it to interact with chondrocytes via cell surface heparan sulfate proteoglycans (HSPGs). Here, we directly demonstrate that cell surface HS is critical to LfcinB-mediated biological effects in human primary chondrocytes. The binding of LfcinB to HS appears to be the furthest upstream event, because disruption of this interaction profoundly inhibits LfcinB-mediated intracellular signaling and target gene regulation. Together with our previous findings (11, 14), we propose that the interactions between LfcinB and cell-bound HSPGs are essential to its antagonism against FGF-2 and IL-1β, as well as its self-contained activities. Although challenging, it is of great interest to identify which HSPGs specifically bind to LfcinB in chondrocytes. Among known HSPGs, perhaps syndecans are the most probable binding partners of LfcinB, because their expression has been reported in chondrocytes (31) and involved in OA pathogenesis (32). LfcinB binding to syndecans may generate a chondroprotective effect (e.g., induction of TIMPs and IL-11) while minimizing FGF-2 and IL-1β binding opportunity leading to reduction of catabolism. Future studies are warranted to elucidate this aspect of LfcinB biology.
Our results indicate that IL-11 induction takes place during the first phase after LfcinB stimulation, and then an elevation of extracellular IL-11 level will trigger secondary responses to sustain chondroprotection. IL-11 induction represents the most dramatic response triggered by LfcinB. The robustness of ERK1/2 activation by LfcinB can explain why IL-11 is up-regulated to this magnitude. The transcriptional cascade governing IL-11 induction and subsequent TIMP-1 expression represents an interesting model of LfcinB action. The immediate IL-11 induction by LfcinB is achieved via the ERK1/2-AP-1 axis. Our EMSA data unequivocally indicate that ERK1/2, not p38 or Akt, determines AP-1 activity. A given AP-1 heterodimer consists of components from Fos, Jun, activating transcription factor, or Jun dimerization protein families (33). Our supershift data revealed that c-Fos and JunD, but not c-Jun, comprise the AP-1 complex upon LfcinB stimulation. This finding recapitulates the specificity of MAP kinases in AP-1 activation. From a structural standpoint, c-Jun only possesses a D domain necessary for JNK targeting, whereas JunD contains a D domain and a DEF motif, rendering itself a favored substrate for both JNK and ERK. Fos is also a phosphorylation substrate of ERK (34). The fact that LfcinB activates ERK1/2 but not JNK corresponds to the finding that JunD rather than c-Jun is incorporated into the AP-1 dimer. The c-Fos/JunD model in LfcinB-mediated IL-11 induction is also supported by two more documented phenomena: (i) JunD occupies IL-11 promoter, although not exclusively (21), and (ii) c-Fos/JunD dimer is able to result in strong transactivation (35). The recruitment of JunD instead of c-Jun may impart transcriptional specificity to LfcinB-induced AP-1, hence not producing the same detrimental effects as c-Fos/c-Jun complex does in chondrocytes (36–39).
The induced IL-11 initiates STAT3 pathway to up-regulate TIMP-1. Prominent STAT3 activation was noted 24 h after LfcinB stimulation, yet the increase in TIMP-1 expression could not be detected until 24 h later. Such a delay suggests that persistent STAT3 signaling is required for TIMP-1 induction by IL-11. Active STAT3 may not solely account for TIMP-1 induction, but it appears to be essential. Knockdown of STAT3 results in complete reversal of TIMP-1 levels, suggesting STAT3 does not act as a transcription co-factor in this context. Thus, our data provide a mechanistic explanation for IL-11-mediated TIMP-1 expression in chondrocytes.
Recently, we reported that LfcinB stimulates other anti-inflammatory cytokines such as IL-4 and IL-10 (11). In addition, we also found that LfcinB markedly up-regulates TIMP-3 expression during the first phase of cellular response in chondrocytes via activation of Sp1 (14). Combined with our previous findings (11), we propose that LfcinB promotes anti-catabolic and anti-inflammatory activities via three routes: (i) competitive binding to heparan sulfate PGs on chondrocyte plasma membrane, thus blocking IL-1 and FGF-2 actions; (ii) immediate induction of multiple anti-inflammatory cytokines, including IL-11, which directly antagonizes IL-1-mediated inflammation and up-regulates TIMP-1; and (iii) early induction of TIMP-3 to further limit endogenous protease activities. A proposed model is illustrated in Fig. 9 to summarize the mode of action of LfcinB in human articular chondrocytes. In keeping with this hypothesis, our experiments using LfcinB-pretreated chondrocytes show that when LfcinB-triggered primary and secondary mechanisms are fully operating, IL-1β fails to launch characteristic catabolic and inflammatory programs in these cells. By contrast, 24-h administration of LfcinB partially inhibits IL-1β effects in IL-1β-primed chondrocytes, suggesting that secondary actions of LfcinB-induced targets are indeed important. Interestingly, LfcinB-mediated genetic responses appear to compensate for some deficiencies in OA chondrocytes. The dedicated receptor of IL-11, IL-11Rα, is significantly down-regulated in OA chondrocytes, which probably leads to partial loss of IL-11 anti-inflammatory signaling. The down-regulation of IL-11Rα, however, does not alter the response of OA chondrocytes to exogenous IL-11, because we observed that OA chondrocytes highly up-regulate TIMP-1 expression upon IL-11 stimulation (data not shown). Importantly, OA chondrocytes remain sensitive to LfcinB stimulation in terms of IL-11 induction (data not shown), suggesting that both primary and secondary mechanisms utilized by LfcinB still produce beneficial biological effects in OA chondrocytes. We also observed repression of TIMP-3 expression in OA, suggesting a lack of control of proteolytic activities in this disease state (14). Therefore, LfcinB conduces to the restoration of chondrocyte homeostasis through replenishing IL-11Rα signaling and TIMP-3. Taken altogether, our findings again demonstrate the potential of LfcinB as a chondroprotective molecule, and our current understanding of its mode of action provides a mechanistic basis for future in vivo investigations.
Acknowledgments
We thank the Gift of Hope Organ Tissue Donor Network, as well as Drs. Chubinskaya and Margulis, for making human tissues available, and we also extend our appreciation to the tissue donor families who made it possible. We thank Dr. Gabriella Cs-Szabo and David Gerard for time and efforts in OA tissue acquisition.
This work was supported, in whole or in part, by National Institutes of Health Grants AR053220 (to H.-J. I.), AR062136 (to H.-J. I.), and AR055915 (to D. C.).
- ECM
- extracellular matrix
- TLR
- Toll-like receptor
- OA
- osteoarthritis
- TIMP
- tissue inhibitor of metalloproteinase
- MMP
- matrix metalloproteinase
- qPCR
- quantitative PCR
- PG
- proteoglycan
- HS
- heparan sulfate
- AP-1
- activator protein 1
- HSPG
- heparan sulfate proteoglycan.
REFERENCES
- 1. Malfait A. M., Verbruggen G., Veys E. M., Lambert J., De Ridder L., Cornelissen M. (1994) Comparative and combined effects of interleukin 6, interleukin 1β, and tumor necrosis factor alpha on proteoglycan metabolism of human articular chondrocytes cultured in agarose. J. Rheumatol. 21, 314–320 [PubMed] [Google Scholar]
- 2. Goldring M. B. (2000) Osteoarthritis and cartilage. The role of cytokines. Curr. Rheumatol. Rep. 2, 459–465 [DOI] [PubMed] [Google Scholar]
- 3. Csaki C., Mobasheri A., Shakibaei M. (2009) Synergistic chondroprotective effects of curcumin and resveratrol in human articular chondrocytes. Inhibition of IL-1β-induced NF-κB-mediated inflammation and apoptosis. Arthritis Res. Ther. 11, R165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Su S. L., Tsai C. D., Lee C. H., Salter D. M., Lee H. S. (2005) Expression and regulation of Toll-like receptor 2 by IL-1β and fibronectin fragments in human articular chondrocytes. Osteoarthritis Cartilage 13, 879–886 [DOI] [PubMed] [Google Scholar]
- 5. Liu-Bryan R., Terkeltaub R. (2010) Chondrocyte innate immune myeloid differentiation factor 88-dependent signaling drives procatabolic effects of the endogenous Toll-like receptor 2/Toll-like receptor 4 ligands low molecular weight hyaluronan and high mobility group box chromosomal protein 1 in mice. Arthritis Rheum. 62, 2004–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Calich A. L., Domiciano D. S., Fuller R. (2010) Osteoarthritis. Can anti-cytokine therapy play a role in treatment? Clin. Rheumatol. 29, 451–455 [DOI] [PubMed] [Google Scholar]
- 7. Moos V., Fickert S., Müller B., Weber U., Sieper J. (1999) Immunohistological analysis of cytokine expression in human osteoarthritic and healthy cartilage. J. Rheumatol. 26, 870–879 [PubMed] [Google Scholar]
- 8. Maier R., Ganu V., Lotz M. (1993) Interleukin-11, an inducible cytokine in human articular chondrocytes and synoviocytes, stimulates the production of the tissue inhibitor of metalloproteinases. J. Biol. Chem. 268, 21527–21532 [PubMed] [Google Scholar]
- 9. Hermann J. A., Hall M. A., Maini R. N., Feldmann M., Brennan F. M. (1998) Important immunoregulatory role of interleukin-11 in the inflammatory process in rheumatoid arthritis. Arthritis Rheum. 41, 1388–1397 [DOI] [PubMed] [Google Scholar]
- 10. Cleaver C. S., Rowan A. D., Cawston T. E. (2001) Interleukin 13 blocks the release of collagen from bovine nasal cartilage treated with proinflammatory cytokines. Ann. Rheum. Dis. 60, 150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yan D., Chen D., Shen J., Xiao G., van Wijnen A. J., Im H. J. (2013) Bovine lactoferricin is anti-inflammatory and anti-catabolic in human articular cartilage and synovium. J. Cell. Physiol. 228, 447–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim J. S., Ellman M. B., Yan D., An H. S., Kc R., Li X., Chen D., Xiao G., Cs-Szabo G., Hoskin D. W., Buechter D. D., Van Wijnen A. J., Im H. J. (2013) Lactoferricin mediates anti-inflammatory and anti-catabolic effects via inhibition of IL-1 and LPS activity in the intervertebral disc. J. Cell. Physiol. 228, 1884–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ellman M. B., Kim J., An H. S., Chen D., Kc R., Li X., Xiao G., Yan D., Suh J., van Wjnen A. J., Wang J. H., Kim S. G., Im H. J. (2013) Lactoferricin enhances BMP7-stimulated anabolic pathways in intervertebral disc cells. Gene 524, 282–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yan D., Chen D., Hawse J. R., van Wijnen A. J., Im H. J. (2013) Bovine lactoferricin induces TIMP-3 via the ERK1/2-Sp1 axis in human articular chondrocytes. Gene 517, 12–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muehleman C., Bareither D., Huch K., Cole A. A., Kuettner K. E. (1997) Prevalence of degenerative morphological changes in the joints of the lower extremity. Osteoarthritis Cartilage 5, 23–37 [DOI] [PubMed] [Google Scholar]
- 16. Muddasani P., Norman J. C., Ellman M., van Wijnen A. J., Im H. J. (2007) Basic fibroblast growth factor activates the MAPK and NFκB pathways that converge on Elk-1 to control production of matrix metalloproteinase-13 by human adult articular chondrocytes. J. Biol. Chem. 282, 31409–31421 [DOI] [PubMed] [Google Scholar]
- 17. Loeser R. F., Yammani R. R., Carlson C. S., Chen H., Cole A., Im H. J., Bursch L. S., Yan S. D. (2005) Articular chondrocytes express the receptor for advanced glycation end products. Potential role in osteoarthritis. Arthritis Rheum. 52, 2376–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walmsley M., Butler D. M., Marinova-Mutafchieva L., Feldmann M. (1998) An anti-inflammatory role for interleukin-11 in established murine collagen-induced arthritis. Immunology 95, 31–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Im H. J., Muddasani P., Natarajan V., Schmid T. M., Block J. A., Davis F., van Wijnen A. J., Loeser R. F. (2007) Basic fibroblast growth factor stimulates matrix metalloproteinase-13 via the molecular cross-talk between the mitogen-activated protein kinases and protein kinase Cδ pathways in human adult articular chondrocytes. J. Biol. Chem. 282, 11110–11121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andersen J. H., Jenssen H., Sandvik K., Gutteberg T. J. (2004) Anti-HSV activity of lactoferrin and lactoferricin is dependent on the presence of heparan sulphate at the cell surface. J. Med. Virol. 74, 262–271 [DOI] [PubMed] [Google Scholar]
- 21. Tang W., Yang L., Yang Y. C., Leng S. X., Elias J. A. (1998) Transforming growth factor-β stimulates interleukin-11 transcription via complex activating protein-1-dependent pathways. J. Biol. Chem. 273, 5506–5513 [DOI] [PubMed] [Google Scholar]
- 22. Hess J., Angel P., Schorpp-Kistner M. (2004) AP-1 subunits. Quarrel and harmony among siblings. J. Cell Sci. 117, 5965–5973 [DOI] [PubMed] [Google Scholar]
- 23. Nakakuki T., Birtwistle M. R., Saeki Y., Yumoto N., Ide K., Nagashima T., Brusch L., Ogunnaike B. A., Okada-Hatakeyama M., Kholodenko B. N. (2010) Ligand-specific c-Fos expression emerges from the spatiotemporal control of ErbB network dynamics. Cell 141, 884–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bugno M., Graeve L., Gatsios P., Koj A., Heinrich P. C., Travis J., Kordula T. (1995) Identification of the interleukin-6/oncostatin M response element in the rat tissue inhibitor of metalloproteinases-1 (TIMP-1) promoter. Nucleic Acids Res. 23, 5041–5047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen X., Wang J., Zhou F., Wang X., Feng Z. (2003) STAT proteins mediate angiotensin II-induced production of TIMP-1 in human proximal tubular epithelial cells. Kidney Int. 64, 459–467 [DOI] [PubMed] [Google Scholar]
- 26. Anguita J., Barthold S. W., Samanta S., Ryan J., Fikrig E. (1999) Selective anti-inflammatory action of interleukin-11 in murine Lyme disease. Arthritis decreases while carditis persists. J. Infect. Dis. 179, 734–737 [DOI] [PubMed] [Google Scholar]
- 27. Trepicchio W. L., Wang L., Bozza M., Dorner A. J. (1997) IL-11 regulates macrophage effector function through the inhibition of nuclear factor-κB. J. Immunol. 159, 5661–5670 [PubMed] [Google Scholar]
- 28. Trepicchio W. L., Bozza M., Pedneault G., Dorner A. J. (1996) Recombinant human IL-11 attenuates the inflammatory response through down-regulation of proinflammatory cytokine release and nitric oxide production. J. Immunol. 157, 3627–3634 [PubMed] [Google Scholar]
- 29. Moreland L., Gugliotti R., King K., Chase W., Weisman M., Greco T., Fife R., Korn J., Simms R., Tesser J., Hillson J., Caldwell J., Schnitzer T., Lyons D., Schwertschlag U. (2001) Results of a phase-I/II randomized, masked, placebo-controlled trial of recombinant human interleukin-11 (rhIL-11) in the treatment of subjects with active rheumatoid arthritis. Arthritis Res. 3, 247–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wong P. K., Campbell I. K., Robb L., Wicks I. P. (2005) Endogenous IL-11 is pro-inflammatory in acute methylated bovine serum albumin/interleukin-1-induced (mBSA/IL-1) arthritis. Cytokine 29, 72–76 [DOI] [PubMed] [Google Scholar]
- 31. Knudson C. B., Knudson W. (2001) Cartilage proteoglycans. Semin. Cell Dev. Biol. 12, 69–78 [DOI] [PubMed] [Google Scholar]
- 32. Echtermeyer F., Bertrand J., Dreier R., Meinecke I., Neugebauer K., Fuerst M., Lee Y. J., Song Y. W., Herzog C., Theilmeier G., Pap T. (2009) Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nat. Med. 15, 1072–1076 [DOI] [PubMed] [Google Scholar]
- 33. Karin M., Liu Z., Zandi E. (1997) AP-1 function and regulation. Curr. Opin. Cell Biol. 9, 240–246 [DOI] [PubMed] [Google Scholar]
- 34. Monje P., Marinissen M. J., Gutkind J. S. (2003) Phosphorylation of the carboxyl-terminal transactivation domain of c-Fos by extracellular signal-regulated kinase mediates the transcriptional activation of AP-1 and cellular transformation induced by platelet-derived growth factor. Mol. Cell. Biol. 23, 7030–7043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McBride K., Nemer M. (1998) The C-terminal domain of c-fos is required for activation of an AP-1 site specific for jun-fos heterodimers. Mol. Cell. Biol. 18, 5073–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hwang S. G., Yu S. S., Poo H., Chun J. S. (2005) c-Jun/activator protein-1 mediates interleukin-1β-induced dedifferentiation but not cyclooxygenase-2 expression in articular chondrocytes. J. Biol. Chem. 280, 29780–29787 [DOI] [PubMed] [Google Scholar]
- 37. Chiu Y. C., Yang R. S., Hsieh K. H., Fong Y. C., Way T. D., Lee T. S., Wu H. C., Fu W. M., Tang C. H. (2007) Stromal cell-derived factor-1 induces matrix metalloprotease-13 expression in human chondrocytes. Mol. Pharmacol. 72, 695–703 [DOI] [PubMed] [Google Scholar]
- 38. Healy Z. R., Zhu F., Stull J. D., Konstantopoulos K. (2008) Elucidation of the signaling network of COX-2 induction in sheared chondrocytes. COX-2 is induced via a Rac/MEKK1/MKK7/JNK2/c-Jun-C/EBPβ-dependent pathway. Am. J. Physiol. Cell Physiol. 294, C1146–C1157 [DOI] [PubMed] [Google Scholar]
- 39. Litherland G. J., Elias M. S., Hui W., Macdonald C. D., Catterall J. B., Barter M. J., Farren M. J., Jefferson M., Rowan A. D. (2010) Protein kinase C isoforms ζ and ι mediate collagenase expression and cartilage destruction via STAT3- and ERK-dependent c-fos induction. J. Biol. Chem. 285, 22414–22425 [DOI] [PMC free article] [PubMed] [Google Scholar]