

Background: Typical phytochromes include a protonated chromophore in the parent states (Pr and Pfr) that transiently deprotonates during photoconversion.
Results: In Agp2, the pKa of the chromophore is lowered from >11 to 7.6 during the conversion from Pfr to Pr.
Conclusion: Chromophore protonation affects light-induced and thermal Pr to Pfr conversion.
Significance: Agp2 can act as integrated light and pH sensor.
Keywords: Bacterial Protein Kinases, Bacterial Signal Transduction, Photobiology, Photoreceptors, Raman Spectroscopy, UV Spectroscopy, Bilin, Biliprotein, Domain Swap Mutants, Flash Photolysis
Abstract
Phytochromes are widely distributed photoreceptors with a bilin chromophore that undergo a typical reversible photoconversion between the two spectrally different forms, Pr and Pfr. The phytochrome Agp2 from Agrobacterium tumefaciens belongs to the group of bathy phytochromes that have a Pfr ground state as a result of the Pr to Pfr dark conversion. Agp2 has untypical spectral properties in the Pr form reminiscent of a deprotonated chromophore as confirmed by resonance Raman spectroscopy. UV/visible absorption spectroscopy showed that the pKa is >11 in the Pfr form and ∼7.6 in the Pr form. Unlike other phytochromes, photoconversion thus results in a pKa shift of more than 3 units. The Pr/Pfr ratio after saturating irradiation with monochromatic light is strongly pH-dependent. This is partially due to a back-reaction of the deprotonated Pr chromophore at pH 9 after photoexcitation as found by flash photolysis. The chromophore protonation and dark conversion were affected by domain swapping and site-directed mutagenesis. A replacement of the PAS or GAF domain by the respective domain of the prototypical phytochrome Agp1 resulted in a protonated Pr chromophore; the GAF domain replacement afforded an inversion of the dark conversion. A reversion was also obtained with the triple mutant N12S/Q190L/H248Q, whereas each single point mutant is characterized by decelerated Pr to Pfr dark conversion.
Introduction
Phytochromes are photoreceptors with a bilin chromophore that were discovered in plants (1) where they control various developmental processes; they also exist in bacteria and fungi. The basic principle of phytochrome photochemistry is the photoconversion between two spectrally different forms, the red-absorbing form Pr and the far-red-absorbing form Pfr. In typical phytochromes, Pr is thermostable, and Pfr is either thermostable or metastable, i.e. it converts slowly into Pr in darkness. In so-called bathy phytochromes, Pfr is the thermostable form, whereas Pr is metastable and converts slowly into Pfr in darkness (2–4). Upon irradiation with red light, Pr is transformed to Pfr via a series of intermediates termed lumi-R, meta-Ra, and meta-Rc. Pfr to Pr photoconversion, induced by far-red light, involves the intermediates lumi-F and meta-F (5, 6). All phytochromes have a common tridomain N terminus with the so-called PAS, GAF, and PHY domains (see Fig. 1 as example). The C terminus is most often a histidine (His) kinase, which in some cases is fused to a C-terminal response regulator. In some phytochromes, the His kinase is replaced by a GGDEF domain (3). Another group of biliprotein photoreceptors, restricted to cyanobacteria, is the group of cyanobacteriochromes (7). These proteins have one or more bilin-binding GAF domains and a highly variable domain arrangement. The N-terminal PAS, GAF, and PHY domains of phytochromes are required for chromophore insertion and photoconversion (6). Spectral properties of truncated phytochromes that consist of PAS, GAF, and PHY domains are usually very similar to those of the full-length proteins. Structural studies show that the major part of the chromophore pocket is formed by the GAF domain, which is connected with the PAS domain by an unusual “figure of eight” knot (8). The PHY domain folds back onto the GAF domain, providing additional chromophore contact sites (9, 10). The C-terminal end of the PHY domain probably forms a long helix with the N terminus of the His kinase. Regarding the nature of the chromophore and the chromophore binding site, two major groups of phytochromes are distinguished, biliverdin (BV)3-binding and phycocyanobilin/phytochromobilin-binding phytochromes. Plant phytochromes belong to the latter group, and cyanobacteria have representatives in both groups, whereas other bacterial and fungal phytochromes are BV-binding phytochromes. Quite interestingly, the chromophore-binding Cys residue of BV-binding phytochromes is located in the N terminus of the PAS domain, whereas the Cys binding site in phycocyanobilin- and phytochromobilin-binding phytochromes is in the center of the GAF domain (11).
FIGURE 1.

Domain arrangements of the A. tumefaciens phytochromes and the domain swap mutants used in the present study. Agp1 has the prototypical domain arrangement of bacterial phytochromes with N-terminal PAS, GAF, and PHY domains and a C-terminal His kinase (HisK). These domains are drawn in dark gray tones. Agp2 has a similar domain arrangement, but the histidine kinase is of the HWE type, and it bears a C-terminal response regulator. Agp2 domains are drawn in light gray tones. The range of each domain is indicated above the Agp1 and Agp2 diagrams. The amino acids that are indicated by vertical bars and numbered under the Agp2 diagram refer to the point mutants that were also used in the present study. The lower four diagrams show the domain arrangements and the denomination of the domain swap mutants. Dark gray color indicates that the respective domain of Agp2 is replaced by that of Agp1.
Vibrational spectroscopy, NMR studies, and pH titrations combined with UV/visible spectroscopy have shown that the chromophore is typically protonated in the Pr and Pfr states and that Pr to Pfr photoconversion is accompanied by a transient proton release (12–14). Free phytochrome chromophores are unprotonated in neutral solution (15), and protonation under acidic conditions occurs at the nitrogen atom of ring B or ring C; i.e. a positive charge is distributed over both central rings. In the biliprotein phycocyanin, an Asp side chain serves as the counter ion for the protonated bilin chromophore (16). A comparable scenario with an Asp facing toward the chromophore has recently been observed in structures of two cyanobacteriochromes (17). Phytochrome crystal structures show that a highly conserved Asp residue within the conserved Pro-Ala-Ser-Asp-Ile-Pro motif is central in the chromophore pocket (8). In the structure of the bathy phytochrome PaBphP, the carboxylic acid side chain of this Asp residue is hydrogen-bonded with the ring D nitrogen of the Pfr chromophore (9). The same interaction has been confirmed by NMR measurements on cyanobacterial Cph1 (18). The Asp side chain could compensate for the positive charge of the chromophore in the Pfr state, although an interaction with the central pyrrole rings would have been expected. In the Pr form, this hydrogen bond is lost due to the Z to E isomerization around the methine bridge connecting rings C and D as shown by the crystallographic analyses in the Pr form (8, 10, 19) and temperature scan crystallography for the Pfr to Pr photoconversion of PaBphP (20). It is postulated that charge compensation is distributed over several groups including the propionic side chain and a conserved His residue. In phytochrome variants in which this His residue is mutated, the chromophore is preferentially unprotonated at pH 7.8 (21).
The soil bacterium Agrobacterium tumefaciens contains two phytochromes termed Agp1 and Agp2 (22) or AtBphP1 and AtBphP2 (2). Both have been used as model phytochromes for biochemical and biophysical studies. Agp1 may be regarded as a typical phytochrome with a C-terminal His kinase. It has a Pr ground state, and the Pfr form converts slowly to Pr in darkness. Agp2, which is characterized in the present study, has a His kinase of the HWE type. HWE stands for those amino acids that are conserved in this particular group of kinases. Unlike typical bacterial phytochromes, Agp2 has a C-terminal response regulator (see Fig. 1). The domain arrangement is thus comparable with fungal phytochromes (23). The domain arrangement suggests an intramolecular phosphorelay from the His kinase to the C-terminal response regulator. In addition, Agp2 is a bathy phytochrome with a Pfr ground state and a Pr to Pfr dark conversion.
In a recent study, 15 phytochromes with an Agp2-like domain arrangement have been identified in the order Rhizobiales (3). Spectral properties of six members of this group (Agp2, Avp1, Avp2, and phytochromes of Rhizobium leguminosarum, Rhizobium etli, and Azorhizobium caulinodans) have been characterized by in vivo photometry and studies on recombinant proteins (3). All of these phytochromes with the exception of Avp1, which displays unusual spectral properties, are bathy phytochromes. This correlation points to a co-evolution of phytochromes with HWE kinases and the bathy phenotype.
The bathy phytochromes characterized so far have slightly different properties. BphP1 of the photosynthetic rhizobial bacterium Bradyrhizobium ORS278 and a homologous protein of Rhodopseudomonas palustris lack the C-terminal His kinase but carry a PAS/S box at the C terminus (24). Pseudomonas aeruginosa phytochrome PaBphP has the prototypical domain arrangement with a C-terminal His kinase (25).
In addition to the domain arrangement and the bathy phenotype, Agp2 also differs from prototypical phytochromes by its Pr absorption characteristics. Under standard buffer conditions (pH 7.8), the photoconversion of Pfr to Pr is characterized by only a weak absorbance change in the 700-nm region where the absorption maximum of Pr is located. By spectral studies on extracts of Agrobacterium phytochrome mutants, it has been found that compounds of the cell extract modulate the difference spectrum of Agp2; in the presence of cell extract, the difference spectrum has more balanced positive and negative parts. Titration series showed that the unknown interacting factor binds to Agp2 in a specific manner and has a concentration around 20 μm in the cell extract (4, 26).
Here we show that the unusual spectral properties of Agp2 Pr are caused by the chromophore protonation state. Whereas the Pfr chromophore is protonated up to a pH of 11, the Pr chromophore has a pKa value of 7.6 and is thus only partially protonated at neutral pH. This conclusion was confirmed by resonance Raman (RR) spectroscopy. Flash photolysis on the Pr to Pfr half-cycle suggests that after photoisomerization the deprotonated chromophore can undergo a back-conversion from an intermediate state to Pr, resulting in a low efficiency of the net Pr to Pfr photoconversion. We also analyzed the role of protein domains and selected amino acids in the dark conversion and protonation of the Pr chromophore.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
All expression vectors are based on the plasmid pET21b (Novagen). The vector pAG2-M1 encodes for full-length Agp2 with a C-terminal His6 tag, and pAG2-M2 encodes for the truncated protein Agp2-M2 (amino acids 1–501), which lacks the His kinase and the response regulator. Agp2-M2 also has a C-terminal His6 tag. Cloning of pAG2-M1 and pAG2-M2 was described previously (4, 27). Expression vectors for domain swap mutants were generated by PCR of the respective fragments and blunt end ligation; the detailed positions are given in Fig. 1. For the Agp1 domains, the vector pAG1 (22) was used as template. Point mutants were generated according to the QuikChange site-directed-mutagenesis (Agilent) protocol. All expressed proteins have a C-terminal His6 tag for affinity purification. Expression and purification were performed as described (4). Briefly, Escherichia coli BL21(DE3) cells (New England Biolabs) with the desired plasmid were grown at 37 °C in LB medium with ampicillin up to an A600 nm of 0.6. Addition of 20 μg/ml isopropyl 1-thio-β-d-galactopyranoside induced specific protein expression carried out at 16 °C until an A600 nm of 3 was reached. Cells were harvested by centrifugation, washed with basic buffer (50 mm Tris-HCl, 300 mm NaCl, 5 mm EDTA, pH 7.8), centrifuged again, and extracted with a French pressure cell in the same buffer. Soluble proteins were precipitated with ammonium sulfate, suspended in low concentrated imidazole buffer (10 mm imidazole, 50 mm Tris-HCl, 300 mm NaCl, pH 7.8) and purified by Ni2+ affinity chromatography. After elution with highly concentrated imidazole buffer (250 mm imidazole, 50 mm Tris-HCl, 300 mm NaCl, pH 7.8), phytochrome-containing fractions were precipitated with ammonium sulfate. Proteins were usually dissolved in basic buffer containing 50 mm Tris-HCl, 5 mm EDTA, 300 mm NaCl, pH 7.8.
Chromophore Assembly and pH Titrations
All UV/visible spectra were recorded with a Jasco V550 photometer at 20 °C. Photoconversion was performed with light-emitting diodes of 655 nm/300 μmol m−2 s−1 for red light and 780 nm/1000 μmol m−2 s−1 for far-red light with typical irradiation times of 60 s. The biliverdin chromophore was purchased from Frontier Scientific and stored as a 4 mm DMSO stock solution. Purified apoproteins were incubated either with a 0.8:1 or a 2:1 molar ratio of chromophore:protein. The assembly was followed by absorption measurements and proceeded at least until no further absorption changes were detectable. Excess chromophore was removed by using desalting columns (NAP-10 columns, GE Healthcare). For varying the pH, the protein (3–5 mg/ml) was precipitated with ammonium sulfate and suspended in buffer (50 mm Tris, 300 mm NaCl, 5 mm EDTA) with the desired pH (from 4.1 to 11.9). The pH was adjusted with HCl or NaOH.
UV/visible spectra of the pure Pr form were calculated under the following assumptions. (i) Spectra of the pure Pfr form are given by the non-irradiated samples. (ii) The irradiated sample contains only Pr and Pfr. (iii) The Pr form has an absorbance ratio A705 nm/A755 nm of 0.1 (28). The wavelength positions correspond to the absorption maxima of Pr and Pfr. The ratio is based on the spectrum of an Agp2 adduct with a locked 15Za-BV chromophore, which has a Pr-like spectrum. Pure Pr spectra of Agp2 can otherwise not be constructed. With these assumptions, data evaluation gave consistent results. When lower or higher A705 nm/A755 nm values were chosen, the calculated spectra had a more or less aberrant shape.
Thus, pure Pr spectra were calculated according to
 |
where f is the fraction of Pfr/Ptot. This value was adjusted until APr, 705 nm/APr, 755 nm = 0.1.
Calculated maxima of the Pr spectra were plotted versus the pH. To determine the pKa, the Henderson-Hasselbalch equation was fitted to the data (OriginPro 8.5).
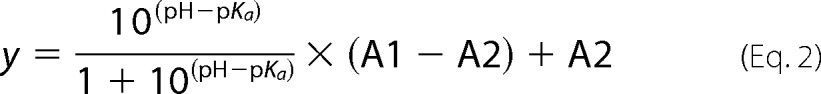 |
Here A1 and A2 refer to the absorbance values of the protonated and deprotonated form, respectively, and y is the measured absorbance.
Flash Photolysis
Flash photolysis experiments on the Pr half-cycle at room temperature were performed using excitation pulses of 10-ns duration and ∼1.5-mJ energy at λ = 660 nm (high energy part of Pr absorption spectrum, i.e. little spectral overlap with Pfr; see Fig. 2, A and C), generated by an optical parametric oscillator (GWU Versa-Scan) pumped by a neodymium-doped yttrium aluminum garnet laser (Quanta-Ray-INDI). The spectrally dispersed continuous wave probe light from a xenon lamp (Hamamatsu, L2273) was detected via a photomultiplier (Hamamatsu, R928). For the experiments, Agp2 was suspended in standard buffer solution (50 mm Tris, 300 mm NaCl, 5 mm EDTA, 6 mm DTT, pH adjusted with HCl or NaOH). The concentration corresponded to ∼0.7 OD units at 750 nm in a cuvette with 1-mm path length at maximum Pfr enrichment. Immediately before each exciting laser shot the sample was pre-illuminated at 785 nm for 10 s. The dose of pre-illumination and the choice of excitation wavelength ensured maximum Pr population and predominantly Pr excitation, respectively. Control experiments with blocked excitation pulse were performed to quantify the contributions of the Pr dark reaction to the photoinduced transient absorption signals within 4 s after photoexcitation.
FIGURE 2.
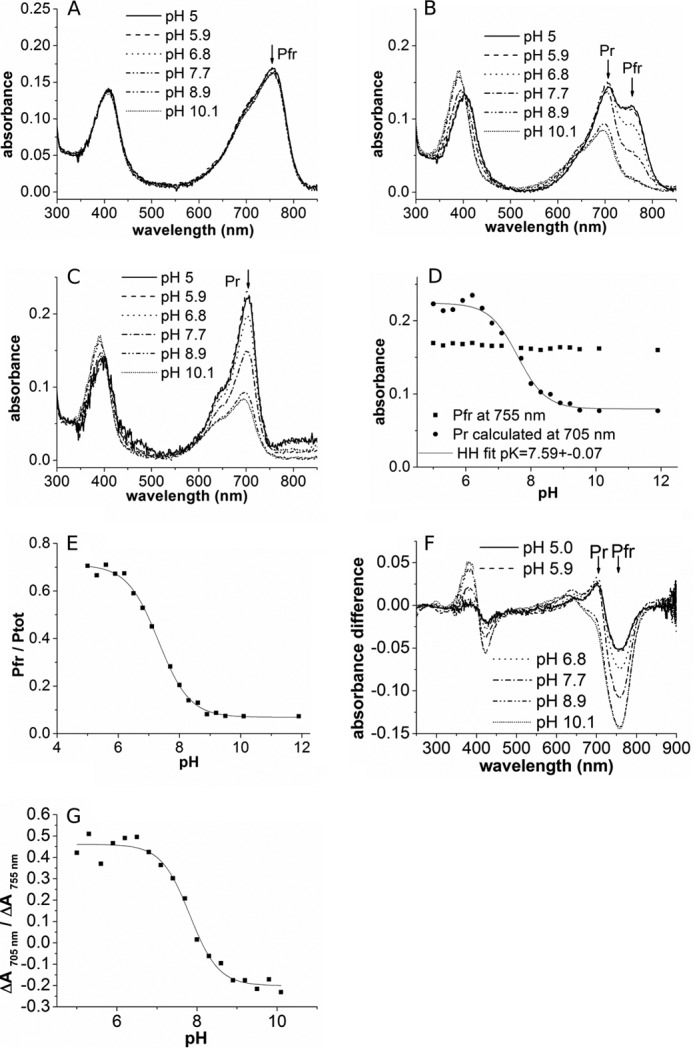
UV/visible spectral properties of full-length Agp2 at different pH values. A, spectra of dark-adapted form (Pfr). B, spectra after far-red irradiation showing a mixture of Pr and Pfr. C, calculated Pr spectra. D, absorption of Pfr and Pr at their respective maximum at different pH values. The Henderson-Hasselbalch (HH) fit to the Pr data yielded a pKa of 7.59. E, estimated fraction (f = Pfr/Ptot) of far-red-irradiated samples as shown in B. F, difference spectra of far-red-irradiated minus dark-adapted Agp2. G, absorbance change ratios from these difference spectra.
Transient absorption data, ΔA(t, λ) (i.e. the time-dependent absorption difference between sample absorbance at time t and the unphotolyzed ground state), were analyzed by a global fit using a sum of exponentials according to Equation 3.
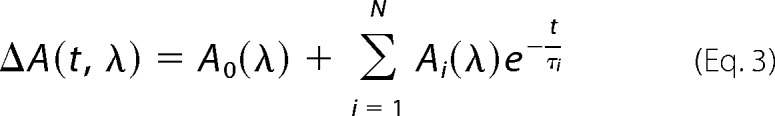 |
yielded time constants τi, decay-associated spectra Ai(λ), and the difference spectrum at long delay times A0(λ). The minimum number of time constants was determined by evaluation of fit residuals and singular value decomposition.
Resonance Raman Spectroscopy
For RR experiments, protein solutions were prepared in Tris-HCl buffer (100 mm Tris, 300 mm NaCl, 5 mm EDTA) at the desired pH. Complementary measurements were performed in D2O (Deutero GmbH) under otherwise identical buffer conditions. The protein concentration corresponded to an A of 50 at 280 nm.
RR spectra were measured at −140 °C either with 1064- or 413-nm excitation using the setups described previously (21, 29). The total accumulation times were 1 h (780 milliwatts) and 5 min (3–4 milliwatts) at 1064 and 413 nm, respectively. The spectrum of the Pfr state was obtained without additional illumination. After 10 s of far-red irradiation with 785 nm (20 milliwatts) at ambient temperature, the RR spectra of the Pr-enriched sample were recorded. Pr to Pfr back-conversion was monitored by measuring the spectrum after red light irradiation (2 min, 644 nm, 3 milliwatts) at ambient temperature prior to the analysis of the protonation equilibrium of the Pr species. The contributions of the Pfr state (and in the case of the 1064-nm measurements also the spectral contribution of the protein matrix) were subtracted on the basis of characteristic marker bands. In all spectra shown in this work, the background was removed by polynomial subtraction. For the determination of the pKa, we have measured both the Pr and Pfr spectrum within pH 6.0 and 10.0 (at 0.5 pH steps). The raw Pr spectra were then subjected to a component analysis (30). Here complete spectra rather than individual bands were fitted to the RR spectra measured as a function of the pH. As only four components (corresponding to four variables) are used, the error of the fitting procedure itself is very low. The four components are the protonated and deprotonated forms of the Pr state, the Pfr state, and the apoprotein. Although the spectra of the latter two components were measured in separate experiments, initial component spectra of the protonated and unprotonated species were obtained by mutual subtraction of the spectra measured at the extreme pH values. These initial spectra were further refined to achieve the best possible global fit to all measured spectra. The relative spectral contributions of the protonated and deprotonated Pr (Ip and Id) as determined from the component analysis are proportional to the relative concentrations (cp and cd) according to
and
where fp and fd are proportionality factors that are proportional to the reciprocal Raman cross-sections.
The relative concentrations were normalized to 1 according to Equation 6.
Then the Henderson-Hasselbalch equation adopts the form
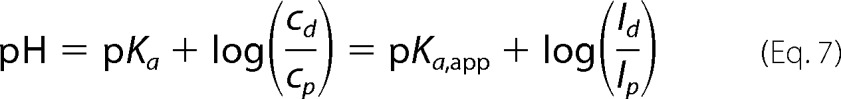 |
where pKa,app is given by Equation 8.
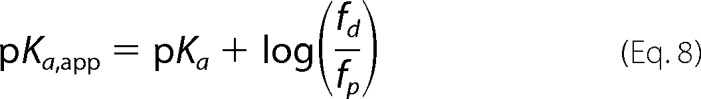 |
The ratio fd/fp is approximated by the reciprocal intensity ratio of the strongest cofactor bands relative to the Phe band at 1005 cm−1 (as an internal standard) of a deprotonated and protonated species that has been determined to be ∼2.
RESULTS
Titration of Agp2: UV/Visible Spectral Properties
It is generally agreed that chromophore deprotonation is indicated by a reduced oscillator strength to the first electronic transition. We thus assumed that the low Pr absorbance of Agp2 as measured at pH 7.8 indicates partial or complete deprotonation of the Pr chromophore, whereas the spectrum of Pfr suggests a protonated chromophore. To determine the pKa of the Pr and Pfr chromophores, we performed titration experiments between pH 5 and 11. Within this range, Pfr spectra were pH-independent (Fig. 2, A and C), indicating that the Pfr chromophore is protonated up to a pH of 11 or more, corresponding to a pKa >11. For the analysis of the Pr form, we irradiated Agp2 with saturating 780 nm light (far-red) before the measurements. After far-red irradiation, the relative Pfr content of prototypical phytochromes is only around 5% Pfr (22). However, this value was much higher in Agp2 (Fig. 2B), and the relative Pfr content was found to be pH-dependent. We estimated that at low pH values around 5 the Pfr/Ptot ratio after saturating far-red irradiation is 0.7 (Fig. 2E). This unexpected high ratio could be due to two effects. (i) Despite the low absorption of Pr at 780 nm, Pr to Pfr photoconversion could contribute to Pfr formation. (ii) Dark conversion occurs already during the ∼40-s delay between the switch off of the actinic light and the beginning of the absorbance measurement. The Pfr/Ptot ratio decreased with increasing pH and reached 0.08 for high pH values of 9 and above (Fig. 2E). This pH-dependent Pfr/Ptot ratio was further characterized by flash photolysis experiments (see below).
From the raw spectra, we calculated pure Pr spectra (Fig. 2C), which show that the extinction coefficient of Pr is pH-dependent. This value is high for low pH and low for high pH (Fig. 2D), indicative for a protonated and deprotonated chromophore, respectively. A Henderson-Hasselbalch fit yielded a pKa of 7.59 ± 0.07. The ratio of absorbance changes that are observed after photoconversion from Pfr to Pr at 705 and 755 nm (−ΔA705 nm/ΔA755 nm) can be taken as a qualitative measure for the protonation state of the Pr chromophore. This “absorbance change ratio” varies from 0.5 for low pH values to −0.2 for high pH values (Fig. 2, F and G).
Titration of Truncated Agp2-M2
The truncated Agp2-M2 contains only the PAS, GAF, and PHY domains and lacks the HWE His kinase and the response regulator (Fig. 1). Usually, spectral properties of such truncated phytochromes are similar to those of the full-length proteins (31). In Agp2, spectra of the truncated Agp2-M2 are considerably different from Agp2 (4). The Pfr spectra of the truncated protein, measured between pH 4 and 12, varied only slightly (Fig. 3, A and D), apparently due to minor scattering effects. Again, the Pfr chromophore appears to be protonated at least up to a pH of 12. After far-red irradiation, the spectral contribution of Pr is not visible (Fig. 3B) because the absorbance of Agp2-M2 Pr is even lower than that of the full-length adduct. Pure Pr spectra were again obtained by calculations (Fig. 3C). These spectra were also pH-dependent (Fig. 3D). However, the pKa of the Pr chromophore was determined to be 6.66 ± 0.08. The fraction Pfr/Ptot after far-red irradiation varied between 0.9 at pH 6 and 0.2 at pH 12. The comparison between the full-length protein and the truncated Agp2-M2, therefore, shows that the His kinase/response regulator affects the protonation state of the Pr chromophore.
FIGURE 3.
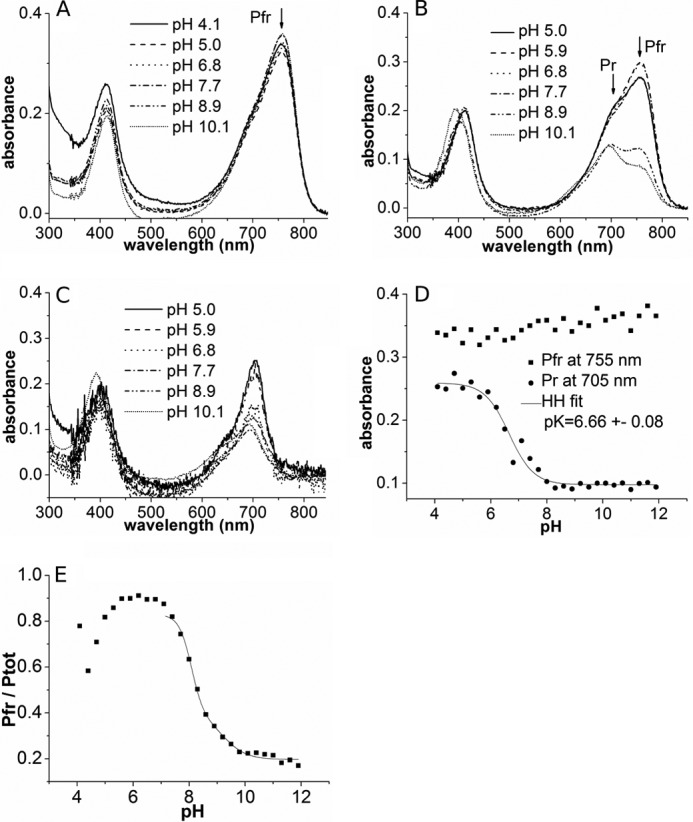
UV/visible spectral properties of Agp2-M2 (PAS-GAF-PHY domains of Agp2 without C-terminal His kinase) at different pH values. A, spectra of dark-adapted form (Pfr). B, spectra after far-red irradiation showing a mixture of Pr and Pfr. C, calculated Pr spectra. D, absorption of Pfr and Pr at their respective maximum at different pH values. The Henderson-Hasselbalch (HH) fit to the Pr data yielded a pKa of 6.68. E, estimated fraction (f = Pfr/Ptot) of far-red-irradiated samples as shown in B.
Flash Photolysis Experiments
The impact of pH on the Pr photoreaction and Pr dark conversion of Agp2 was further characterized by transient absorption experiments on the photoinduced Pr to Pfr conversion. The transient spectra clearly show that the photoconversion patterns at high and low pH differ significantly.
Fig. 4 (left) shows selected transient absorption difference spectra after Pr photoexcitation at pH 6, 7.8, and 9 in the time range up to 4 s after photoexcitation (see “Experimental Procedures”). Negative signals below 725 nm at late times reflect the pump pulse-induced depletion of the Pr state; positive signals above 725 nm display the formation of Pfr on the time scale up to several seconds. The absence of an isosbestic point indicates the presence of more than two molecular species involved as expected for the Pr half-cycle in phytochromes (Agp1 (14), PhyA (32, 33), and Cph1 (12, 34)).
FIGURE 4.

Flash photolysis measurements of the Pr to Pfr photoconversion of Agp2. Left, transient absorption difference spectra of pre-illuminated phytochrome (785 nm) after flash irradiation of Pr (660 nm) at pH 6, 7.8, and 9 at selected times as indicated. Middle, related decay-associated spectra Ai. Right, same as middle but sum of A0 and A4 instead of A0. The thin arrows indicate the time course of signal amplitude. The gray rectangle marks the region of pump pulse artifacts. a.u., absorbance units; mOD, milli-OD units.
The global fit (Equation 3) of the data at each pH revealed four time constants, τ1 to τ4, and associated amplitude spectra A1 to A4 (Fig. 4, middle), the latter describing the spectral absorbance changes associated with the corresponding time constants. A0 stands for the amplitude of total changes, i.e. the difference spectrum of all product states formed 4 s after photoexcitation (including the contributions of the Pr dark conversion) and the spectrum before time 0. At each pH, τ4 is at least 5 times larger than each of the remaining three time constants. By means of control experiments with blocked excitation pulses, it was possible to assign A4 to the dark conversion in terms of amplitude and spectral shape. Thus, the strong amplitude of A0 in the region of Pfr absorption is mainly caused by dark conversion. This separation of time scales and the control experiments allow adding A4 to A0 (Fig. 4, right). Thereby, in good approximation, A0 + A4 reflects the difference spectrum of exclusively the photoinduced amount of Pfr (4 s after photoexcitation) and the initial amount of Pr.
The observed time constants show some variations with increasing pH. In particular, τ3 increases, and τ2 decreases. The value of τ4, the time constant characterizing the dark reaction within the first 4 s (after switching off the pre-illumination at 785 nm), is almost doubled between pH 6 and 9.
The negative amplitude of A3 at 755 nm is assigned to the photoinduced Pfr formation. The similarity of the negative features of A3 at 705 and 650 nm (at pH 7.8 and 9) with the (pure) Pr spectrum is remarkable and is thus assigned to the formation of Pr or a species with a Pr-like spectrum. With increasing pH, the amplitude at 755 nm decreases, whereas the amplitudes at 705 and 650 nm increase. Concomitantly, a change in the final difference spectrum (A0 + A4) is observed: at pH 6, it has the shape of a typical Pfr-Pr difference spectrum, whereas at pH 9, a new feature appears around 775 nm in spectral coincidence with the negative band of A2. A1 does not exhibit significant changes with the pH.
Following the results of similar experiments on Agp1 (35) in terms of observed relaxation time constants and shape of the associated spectra, τ1 is assigned to the decay of meta-Ra and the formation of meta-Rc, and τ3 is assigned to the decay of meta-Rc and, at low pH, the formation of Pfr. The change of A3 with increasing pH (see above) strongly suggests that the deprotonated form undergoes a considerably altered photocycle (Pr half-cycle) with an increased recovery of the Pr state in a shunt reaction and, in consequence, less formation of Pfr. Note, however, that independently of the chromophore protonation state, Pr undergoes a photoisomerization followed by a number of intermediates, i.e. thermally unstable states. The overall amplitude of A2 is very small and difficult to implement into the half-cycle. It could represent a back-reaction within the cycle or a side reaction to another intermediate (5). It is not further discussed here.
Resonance Raman Spectroscopy
RR spectroscopy can be used to monitor the structure and protonation state of phytochrome chromophore. RR spectra of the Pr state of Agp2 were measured in the pH range from 6.0 to 10.0 using 1064- and 413-nm excitation lines, which are in (pre-) resonance with the first and second electronic transitions of the tetrapyrrole cofactor, respectively. The pH-dependent spectral changes correspond to the acid-base reaction of the cofactor as reflected in the UV/visible absorption spectra (Fig. 2C). A selection of RR spectra is shown in Fig. 5A. A careful inspection reveals that several bands, indicated by the vertical dotted lines, are only present at pH 6.0 (e.g. bands at 756, 806, 1295, and 1305 cm−1), whereas others are characteristic features at pH 9.0 (e.g. bands at 711, 1071, 1166, and 1251 cm−1). The Pr spectra at higher pH values (9.5 and 10.0) were reminiscent of deprotonated species at pH 9.0 species (data not shown). The spectrum measured at an intermediate pH of 7.8 includes contributions of both groups of bands, indicating a two-state pH-dependent equilibrium consistent with the UV/visible spectroscopic data (see above).
FIGURE 5.

A, RR spectra of the Pr state of Agp2 at different pH values measured with 1064-nm excitation at 80 K. Residual spectral contributions of the Pfr state and the apoprotein were subtracted. The spectrum at pH 9.0 represents the pure “alkaline” form with a deprotonated chromophore. Minor spectral contributions of this deprotonated species were subtracted from the top spectrum such that it shows only the “acid” form including a protonated chromophore. B, RR spectra of the pure spectra of the protonated (prot.) and deprotonated (deprot.) species in H2O and D2O showing the C=C stretching region. The ring notation of the individual rings is present in C. C, structural formula of the chromophore in the ZZZssa configuration. D, relative contributions of the component spectra of the protonated (gray) and deprotonated species (black) of the Pr state of Agp2 as a function of the pH as determined from the RR spectra by component analysis. Error bars refer to three series of measurements of different samples. The dashed lines represent the fit of the Henderson-Hasselbalch equation (Equation 7) to the data. The vertical dotted line indicates the pKa,app value (7.9), which affords a pKa of 7.6 according to Equation 8.
More information about the structure and protonation state of the chromophore in the two species involved can be obtained from the region between 1500 and 1700 cm−1 that is dominated by the C=C stretching modes of the methine bridges and the individual pyrrole rings (Fig. 5B). In addition, this region includes a unique marker band for the protonation state originating from the in-phase in-plane bending of the N–H groups of rings B and C (N–H ip) (36, 37) (for the notation, see Fig. 5C). In the spectrum of the “acidic” form, this mode is readily assigned to the band at 1571 cm−1 that disappears in D2O (Fig. 5B). Hence, the state prevailing at pH 6.0 includes a protonated chromophore.
For protonated tetrapyrroles in the ZZZssa (Pr) and ZZEssa (Pfr) configuration, vibrational assignments are well established based on theoretical calculations and supported by isotopic labeling (36–41). In general, the vibrational band pattern of the protonated Pr state of Agp2 is quite similar to that of Deinococcus radiodurans, indicating the same ZZZssa geometry of the tetrapyrrole. The strongest RR band originates from the C=C stretching of the C-D methine bridge, which because of the coupling with the N–H ip coordinate of ring C undergoes a downshift in D2O. Hence, this mode is assigned to the band at 1621 cm−1 in H2O (1610 cm−1 in D2O) (40). The nearby hydrogen/deuterium-insensitive band at 1625 cm−1 originates from a mode including the C=C stretching coordinates of both the C-D methine bridge and ring D. The A-B stretching mode appears at higher frequencies and is expected to be slightly hydrogen/deuterium-sensitive due to the admixture of the N–H ip coordinate of ring A. This mode, therefore, is assigned to the weak band at 1651 cm−1 in H2O (1641 cm−1 in D2O). The nearby band at ∼1641 cm−1 in H2O might be due to the C=C stretching of the ring D vinyl substituent, although calculations predict only a very weak Raman activity (39). Alternatively, one may assign this band as well to the A-B stretching, implying a coexistence of two protonated BV conformers with slightly different A-B methine bridge geometries. Such a structural heterogeneity has also been inferred from the RR spectroscopic analysis of the Pr and Pfr states of prototypical phytochromes from D. radiodurans and A. tumefaciens (Agp1) (38, 39).
In the spectrum measured at pH 9.0, a band attributable to the N–H ip mode of rings B and C cannot be detected (Fig. 5B), implying that it represents a neutral species with only ring B or ring C carrying a proton on the pyrrole nitrogen. The overall relatively small hydrogen/deuterium isotopic shifts in the spectra result from minor contributions of the N–H ip coordinates of the remaining three pyrrole rings to the modes of predominantly C=C stretching character. Here we note an additional characteristic feature of the unprotonated species. The relatively broad and partially asymmetric band shapes probably originate from the coexistence of two tautomeric forms in which either ring C or ring B is deprotonated (14, 21, 42).
Fig. 2C shows that the oscillator strength of the second electronic transition at ∼400 nm increases at the expense of the first one (at ∼700 nm) upon increasing the pH. Thus, one would expect a stronger resonance enhancement of the Raman bands of the deprotonated form compared with the protonated species upon 413 nm excitation. This is in fact observed (data not shown), providing further support for the assignment of the spectra measured at pH 6.0 and 9.0 to the protonated and deprotonated species, respectively.
The RR spectra of the Pr state of Agp2, measured as a function of the pH, were disentangled in terms of the relative contributions of the protonated and deprotonated species using the component analysis (see “Experimental Procedures”). Whereas from pH 9.0 to higher pH values the deprotonated species is the only detectable form in the spectrum, the protonated species prevails at pH <7, although the component analysis reveals a small residual contribution of the deprotonated state even at pH 6.0. This finding may most likely reflect the limitations of the two-state acid-base equilibrium model as it assumes that the component spectra of the protonated and deprotonated state do not vary with the pH. However, additional protonation/deprotonation processes that involve amino acid side chains in the vicinity of the tetrapyrrole may alter the electrostatics in the chromophore binding pocket to which RR spectra respond more sensitively than UV/visible absorption spectra. Nevertheless, the present data are sufficient to provide an approximate value for the apparent pKa,app of the cofactor (de)protonation of 7.9. The true pKa, according to Equation 7, is then estimated to be 7.6 (Fig. 5D), which is in good agreement with the value of 7.6 derived from UV/visible absorption experiments.
Domain Swap Mutants and Dark Conversion
To find out which region of the protein is relevant for Pr to Pfr dark conversion and for BV protonation, we generated domain swap mutants in which one domain of Agp2 was replaced by the equivalent domain of Agp1 as outlined in Fig. 1. All proteins were assembled with BV for 15 h and measured after removal of free chromophore at a pH of 7.8 (Fig. 6, A–F). Under these conditions, Agp2 is completely in the Pfr state. A second spectrum was recorded after photoconversion by red or far-red light (Fig. 6, A–F). Fig. 6, G–K, show spectra of irradiated samples that were taken in 10-min intervals during dark conversion. The spectra of Agp2_Gaf1 (Fig. 6C) in which the GAF domain of Agp2 was replaced by that of Agp1 are very similar to those of Agp1 (Fig. 6F). The protein remained in the Pr form during assembly, and after photoconversion into Pfr, it converted back to Pr in darkness (Fig. 6I). The absorbance change ratio was 1.29, which is much higher than the value of 0.24 for Agp2 and almost as high as the value of 1.45 for Agp1. This finding suggests that the Pr chromophore is fully protonated. In Agp2_Pas1, the PAS domain of Agp2 is replaced by that of Agp1. This mutant undergoes Pr to Pfr dark conversion during assembly or after far-red irradiation (Fig. 6, B and H), but dark conversion was very slow and was incomplete even after 15 h such that both Pr and Pfr were present (Fig. 6E). The absorbance change ratio of 0.74 is indicative for a high fraction of the protonated Pr chromophore. Spectra of Agp2_Phy1 in which the PHY domain of Agp2 is replaced by that of Agp1 have also been shown in an earlier study (43). These spectra are reminiscent of a bleached Pr; irradiation induces only minor spectral changes (Fig. 6D). We could not observe dark conversion after irradiation with red or far-red light (Fig. 6J). An absorbance change ratio of 0.06 indicates that only a minor fraction of the Pr chromophore is protonated. When the His kinase/response regulator was replaced by the His kinase of Agp1 (mutant Agp2_Hiskin1), spectra (Fig. 6E) were similar to those of the Agp2 wild type (Fig. 6A) but different from those of the truncated version Agp2-M2 (Fig. 3). Thus, the above effect of the Agp2 His kinase on spectra and chromophore protonation can be replaced by another His kinase and is apparently not specific. Dark conversion of Agp2_Hiskin1 was more rapid than that of Agp2 (Fig. 6K).
FIGURE 6.

Absorption spectra of domain swap mutants. A–F show spectra after dark assembly (solid lines). The protein variants Agp2 (A), Agp2_Pas1 (B), and Agp2_Hiskin1 (F) have a Pfr ground state. These samples were irradiated with far-red (dash-dotted lines). The Agp2_Pas1 dark-adapted sample contains a mixture of Pr and Pfr due to slow dark conversion. Agp2_Gaf1 (C), Agp2_Phy1 (D), and Agp1 (E), which was analyzed in parallel as a control, have a Pr dark state and were irradiated with red light (dashed lines). In G–K, spectra during dark conversion are given. Each sample was first irradiated with saturating red or far-red light. Subsequently, spectra were recorded in 10-min intervals up to 40 min. The direction of absorbance changes is indicated by the arrows. In Agp2 (G), Agp_Pas1 (H), and Agp2_Hiskin1 (K), dark conversion proceeds from Pr to Pfr; in Agp2_Gaf1 (I) it proceeds from Pfr to Pr. No dark conversion was found for Agp2_Phy1 (J).
Point Mutants
To study the role of single amino acids on the protonation status and dark conversion of Agp2, we produced a series of point mutants that were measured by UV/visible spectroscopy. By studying phytochrome sequence homologies, we had noticed that Asn-12 of Agp2 is conserved in ∼50% bathy phytochromes, whereas other phytochromes have different amino acids at that position. Asn-12 is located next to the chromophore-binding Cys-13, and dark conversion depends on the formation of a covalent bond between this Cys residue and the chromophore (44). Therefore, we reasoned that the amino acid could have an impact on the dark conversion of Agp2. We replaced Asn-12 by Ser, the homologous amino acid of Agp1, to yield the mutant N12S. The second mutation, H248Q, was made according to Karniol et al. (45) who proposed that the H248Q mutant of truncated Agp2 has a Pr ground state. His-248 interacts with rings B and C of the chromophore. It is involved in a hydrogen bonding network and in the protonation of the chromophore. The third mutant, Q190L, is equivalent to the mutant Q188L of P. aeruginosa phytochrome PaBphP from Yang et al. (46). According to crystal structure data, the Gln side chain forms ionic interactions with ring D of the Pfr chromophore and stabilizes the C15 E configuration. In that study, the Q188L mutant was reported to have a slower Pr to Pfr dark conversion. Spectra of all mutants measured at pH 7.8 after dark assembly and after photoconversion are given in Fig. 7. Dark conversion after saturating far-red irradiation was followed at 750 nm (close to the Pfr maximum) at pH 6.8, 7.8, and 8.8 for 4000 s. The dark conversion of red pre-irradiated samples was monitored at pH 7.8. The higher initial Pfr level adjusted by red light shows whether the sample undergoes Pfr to Pr dark conversion. All curves were simulated by exponential or linear functions. Time constants and amplitudes of these fit functions are given in Table 1. From the spectra (Fig. 7), it becomes clear that wild type Agp2 is the only variant that is completely in the Pfr form after assembly and dark incubation. All mutants include a residual fraction of Pr, which could be due to either incomplete Pr to Pfr dark conversion or incomplete Pfr to Pr dark conversion (Fig. 7). The dark conversions of Agp2 could well be fitted with a triexponential function (Table 1 and data not shown). As also seen in the flash photolysis measurements (Fig. 4), the time constants increase with increasing pH. This pH dependence was also found for almost all mutants in this study. In Agp2, the pH effect was strongest for the shortest time constant, which increased from 7.8 s at pH 6.8 to 52 s at pH 8.8. Dark conversion of N12S could again be described by triexponential functions. Time constants were slower than those of the wild type, confirming that the amino acid Asn-12, which is close to ring A of the chromophore, has an impact on dark conversion. The rather high absorbance change ratio of 0.8 shows that the chromophore is in the protonated state. The Q190L mutant revealed a mixture of Pr and Pfr after overnight assembly of about equal amounts (Fig. 7). Absorbance measurements after red or far-red irradiation indicate that dark conversion proceeds from Pr to Pfr. However, the time curves could only be fitted with monoexponential functions. All time constants are larger than the corresponding largest time constant of Agp2 (Table 1). The absorbance change ratio of 0.7 again indicates protonation of the Pr chromophore. In the H248Q mutant, the Pfr form dominated after assembly (Fig. 7). Pr to Pfr dark conversion curves could be simulated by biexponential (for pH 6.8) or monoexponential (for pH 7.8 and 8.8) functions; dark conversion was slower than that of N12S, Q190L, and the wild type (Table 1). For the H248Q mutant of the truncated version of Agp2, the Pfr ground state is in contrast to the Pr ground state reported by Karniol et al. (45). After irradiating H248Q with either red or far-red light, the spectral pattern developed in an unusual way as the overall absorbance in the red or far-red wavelength range decreased; the initial ground state recovered slowly in the dark (Fig. 7). We propose that after irradiation the chromophore is deprotonated in the Pr and Pfr forms and that dark conversion switches back to a protonated Pfr chromophore with a higher absorbance.
FIGURE 7.

Absorption spectra of point mutants. Agp2 wild type and mutants were purified as apoproteins and assembled with BV. After a 16-h dark incubation, free chromophore was removed from the protein, and spectra were recorded. Samples were then irradiated with red light (655 nm, 300 μm m−2 s−1) for 60 s, measured, irradiated with far-red light (780 nm, 1000 μm m−2 s−1) for 60 s, and measured again.
TABLE 1.
Time constants of dark conversion
Absorbance was measured continuously at 750 nm (Pfr maximum) for 4000 s after far-red (780 nm) or red (655 nm) irradiation to achieve high initial Pr and Pfr levels, respectively. Far-red pre-irradiation was combined with three different pH values as given above the columns; dark conversion after red pre-irradiation was measured at pH 7.8 only. Exponential decay functions with one, two, or three components were fitted to the data (Origin 8.5). Amplitudes of the fit functions are given in parentheses. The Q190L/H248Q double mutant and the triple mutant could not be fitted with exponential functions. In those cases, linear functions were used to simulate the data. For comparison with the time constants of the exponential fits, the reciprocal slopes are given. Phytochrome concentrations were comparable with those in Fig. 7. AU, absorbance units.
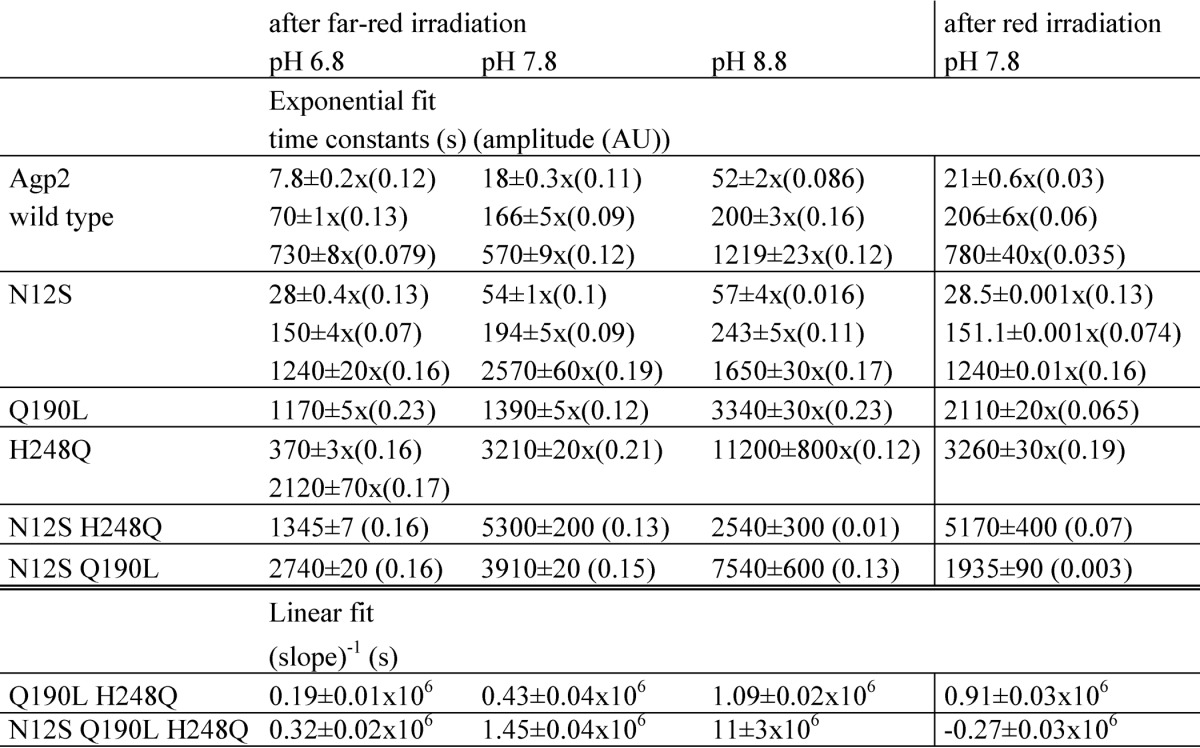
By analyzing double and triple mutants, we found that the mutant effects on dark conversion are largely additive. In all double mutants, the Pr form dominated (Fig. 7), and the triple mutant was completely in the Pr form after overnight assembly (Fig. 7). Dark conversions were always slower than in each single mutant or the wild type protein. Monoexponential functions were fitted to the curves of N12S/Q190L and N12S/H248Q, and the time constants were always above 1000 s (Table 1). Dark conversions of Q190L/H248Q and the N12S/Q190L/H248Q triple mutant seemed to be associated with a linear rather than an exponential time dependence. This indicates that dark conversion in these mutants is so slow that during the measuring time of 4000 s the sample is still in the initial phase of the exponential progression. The pH dependence was no longer given for those two mutants: rates at pH 8.8 were slightly higher than at pH 7.8. The triple mutant converted from Pfr to Pr after red irradiation. The rate constant of 3.7 × 10−6 s−1 was still low but 50 times higher than that of the Pr to Pfr conversion after far-red irradiation. The equilibrium was clearly shifted to the Pr side. A reversion of dark conversion was thus achieved by mutation of one amino acid interacting with ring A of the chromophore, a second amino acid interacting with rings B and C, and a third amino acid interacting with ring D.
DISCUSSION
A. tumefaciens has two phytochromes, Agp1 and Agp2, which have both been investigated biochemically. Agp1 is a prototypical bacterial phytochrome, and Agp2, containing an HWE His kinase and a C-terminal response regulator, is a member of the bathy phytochromes with a Pfr ground state. Titration experiments combined with UV/visible and RR spectroscopy have shown that Agp2 differs from other phytochromes by its untypical chromophore protonation. In Agp2, the pKa of the Pfr chromophore lies above 11, and this value shifts down to 7.6 upon photoconversion into Pr. These observations raise two questions. (a) What is the molecular origin for the deprotonated Pr chromophore and the dramatic pKa change during photoconversion? (b) Is this pKa change associated with a specific biological function?
A homology model for Agp2 (Fig. 8) suggests that Asp-196 interacts with ring D of the Pfr chromophore in the same way as the homolog in Pseudomonas BphP (PaBphP). The Pfr states of PaBphP and Agp2 display nearly identical RR spectra, indicating that even subtle structural details of the chromophore are the same (39). The interaction of the ring D nitrogen of the Pfr chromophore with the carboxylic acid of Asp-196 that is lost in Pr might be the cause for the large pKadifference between Pr and Pfr of Agp2. The carboxylic side chain of Asp-196 may serve as the counterion for the protonated Pfr chromophore with the positive charge of BV on the ring D nitrogen.
FIGURE 8.
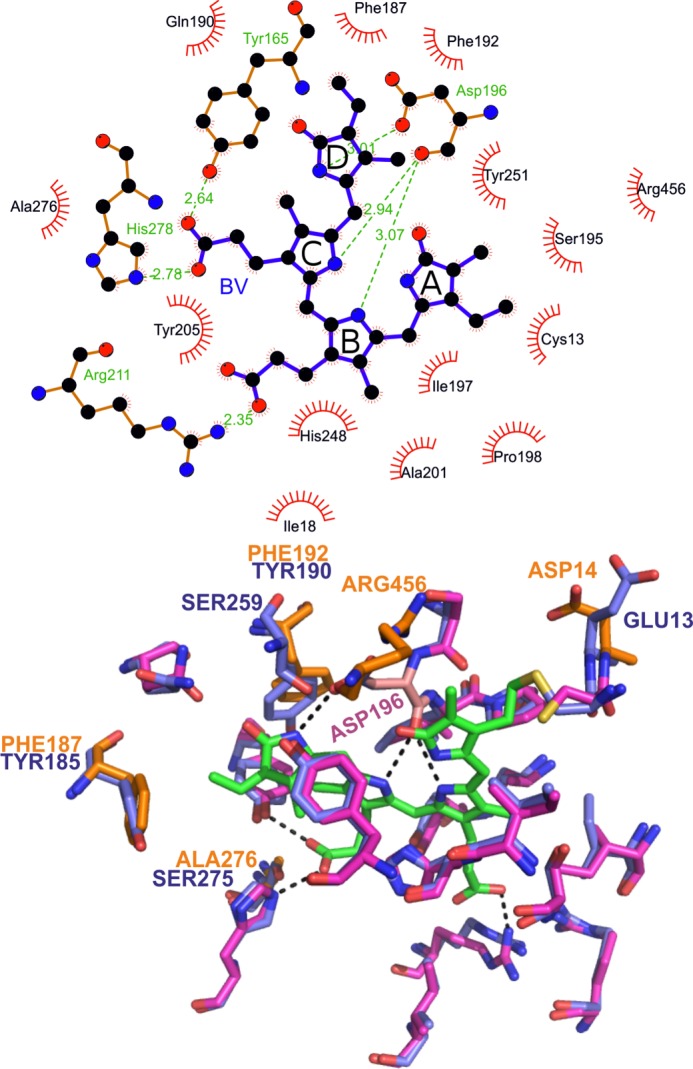
Amino acids in the chromophore pocket of Agp2 based on a homology model generated by SWISS-MODEL. Top, chromophore-interacting amino acids as given by the LIGPLOT program. Bottom, three-dimensional arrangement of the amino acids interacting with the chromophore. Superposition of the Agp2 model with the crystal structure of PaBphP (Protein Data Bank code 3C2W) is shown. The chromophore is drawn with green carbon atoms. Amino acids of PaBphP are drawn with blue carbon atoms. Amino acids of Agp2 are drawn in purple if they are identical with PaBphP or orange if they are different from PaBphP. Asp-196 of Agp2 is drawn with pink carbon atoms. Dashed lines indicate hydrogen bonds.
But why is the pKa of the Agp2 Pr chromophore so low compared with other phytochromes? A comparison of amino acid residues that constitute the chromophore pocket might give a clue to this question. However, 14 of the 18 amino acids of the Agp2 chromophore pocket are identical with PaBphP (Fig. 8), which has a normal Pr spectrum with a protonated chromophore. The four chromophore-interacting amino acids by which Agp2 differs from PaBphP are found in many other phytochromes with normal Pr spectra. Thus, the different protonation of the Pr chromophore cannot be attributed to a single amino acid interacting with the chromophore. A replacement of the GAF domain of Agp2 by the GAF domain of Agp1 resulted in a protonated Pr chromophore. The replacement of the PAS domain yielded also a protonated Pr chromophore (Fig. 6). Substitution of Asp-12 and Glu-190 caused chromophore protonation, whereas the His-248 substitution led to a deprotonated BV. It is also relevant in this context that a compound of the bacterial cellular extract increases the absorbance of the Pr form of Agp2 (4, 26) most likely due to a differential protonation of the Pr chromophore. Finally, the C-terminal part of Agp2 has an impact on the chromophore protonation status (Figs. 2 and 3). It appears that Pr protonation is sensitive to subtle structural changes. Besides Asp-196 and His-248, helix 5 (residues 245–255) within the GAF domain seems to be most relevant for protonation. This helix is located next to the knot between the GAF and PAS domains and might be reoriented by either the GAF or PAS domain swap. This helix contains the highly conserved Tyr-255, which interacts with the chromophore, and a Glu residue next to it facing away from the chromophore.
What is the effect of the deprotonated Pr chromophore on (i) the photocycle and (ii) the capability of photoinduced or thermal chromophore double bond isomerization? Measurements at pH 6 (protonated chromophore) point to a normal conversion to Pfr. At pH 9 where the chromophore in its electronic ground state is deprotonated, a flash induces photoconversion to the intermediate states lumi-R, meta-Ra, and meta-Rc, but from the meta-Rc state, the major fraction seems to convert back to Pr or a Pr-like state. Agp1 mutants with deprotonated chromophore were also unable to complete the transition from meta-Rc to Pfr (29), although in this case, no back-conversion was observed. In Agp2, this futile cycle explains in part why the Pfr/Ptot ratio under far-red illumination is low under high pH (0.08 at pH 9) and high under low pH (0.7 at pH 5.5). A lower extinction coefficient of the Pr chromophore and a slower Pr to Pfr dark conversion at high pH (see flash photolysis results and discussion on sensor properties below) add up to this effect.
Experiments on free bilins suggest that photoisomerization requires a protonated (electronic ground state) chromophore (44), which, however, is in contrast to the results of the present flash photolysis. Likewise, efficient dark conversion is observed at pH 6 and 9. Thus, in Agp2, thermal as well as light-induced double bond isomerization is observed for the deprotonated Pr chromophore. This raises the question of when, at high pH, chromophore reprotonation takes place, which is related to the more general question of when the pKa upshift of the chromophore occurs. Femtosecond transient absorption experiments (47) on Agp24 show that the spectrum of lumi-R at pH 6 is very similar to that at pH 9, suggesting that the lumi-R chromophore is protonated at high pH. However, a protonated precursor (e.g. the Pr excited electronic state) cannot be excluded. On the other hand, quantum chemical calculations on phytochromobilin (48) reported no significant change of pKa upon photoexcitation. This would suggest that a potential photoinduced pKa shift must be facilitated by chromophore-protein interaction.
We consider that Agp2 could function as an integrated light and pH sensor. As a soil bacterium, A. tumefaciens can be exposed to sunlight under many different conditions. A high content of long wavelength far-red light around 750 nm, which results in maximum Pfr to Pr photoconversion, is expected under three different conditions: under green canopy where photosynthetic pigments filter out shorter wavelengths (49, 50), several millimeters below the surface of the soil where long wavelength light transmits more deeply than shorter wavelengths (51), and next to a plant root. Plant stems and roots function as light vessels, and long wavelength light around 750 nm has the highest transmission rates (52). Light mediated via Agp2 has its maximal effect under alkaline conditions. Under acidic conditions with the photoequilibrium of Agp2 on the Pfr side, the kinase activity of Agp2 in the light would differ only slightly from that in darkness. Agrobacterium infects plants through wounded tissue, and Agp2 might play a role in the plant infection process (53). ChvG is a His kinase sensor protein of A. tumefaciens that is required for acid induction of virulence genes (54). At high pH, Agp2 might compensate for ChvG given that the bacteria “see” the plant root.
Besides chromophore protonation, dark conversion of Agp2 has been studied in the present work. In the wild type and in almost all mutants, dark conversion was faster under acidic than under neutral or basic conditions, although also in this case, chromophore C15 Z-E isomerization is possible in the deprotonated state. Our results also show that all three N-terminal domains have an impact on the dark conversion. It is not surprising that a replacement of the GAF domain by that of Agp1 results in a reversion of the dark conversion because the GAF domain forms most of the chromophore contacts. However, a reversion was also found by a replacement of the PHY domain, and a PAS domain replacement caused a slowdown of the dark conversion. The results of single mutations and additive effects of multiple mutations together with the reversed dark conversion in the triple mutant show also that dark behavior of Agp2 is determined by many factors. There is no amino acid that is specifically conserved in bathy phytochromes, and many mutations might lead to an altered Pr to Pfr dark conversion. The overall shape and charge distribution of the chromophore pocket seem to determine direction and speed of dark conversion.
Acknowledgments
We thank Sybille Wörner and Norbert Michael for technical help.
This work was supported in part by Deutsche Forschungsgemeinschaft Sonderforschungsbereiche Sfb 498 (to T. L.) and Sfb 1078 (to P. H. and F. V. E.).
P. Singer, T. Lamparter, and R. Diller, unpublished results.
- BV
- biliverdin
- RR
- resonance Raman
- Ptot
- total phytochrome
- ip
- in-plane.
REFERENCES
- 1. Butler W. L., Norris K. H., Siegelman H. W., Hendricks S. B. (1959) Detection, assay, and preliminary purification of the pigment controlling photoresponsive development of plants. Proc. Natl. Acad. Sci. U.S.A. 45, 1703–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Karniol B., Vierstra R. D. (2003) The pair of bacteriophytochromes from Agrobacterium tumefaciens are histidine kinases with opposing photobiological properties. Proc. Natl. Acad. Sci. U.S.A. 100, 2807–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rottwinkel G., Oberpichler I., Lamparter T. (2010) Bathy phytochromes in rhizobial soil bacteria. J. Bacteriol. 192, 5124–5133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krieger A., Molina I., Oberpichler I., Michael N., Lamparter T. (2008) Spectral properties of phytochrome Agp2 from Agrobacterium tumefaciens are specifically modified by a compound of the cell extract. J. Photochem. Photobiol. B 93, 16–22 [DOI] [PubMed] [Google Scholar]
- 5. Eilfeld P., Rüdiger W. (1985) Absorption spectra of phytochrome intermediates. Z. Naturforsch. C 40, 109–114 [Google Scholar]
- 6. Rockwell N. C., Su Y. S., Lagarias J. C. (2006) Phytochrome structure and signaling mechanisms. Annu. Rev. Plant Biol. 57, 837–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ikeuchi M., Ishizuka T. (2008) Cyanobacteriochromes: a new superfamily of tetrapyrrole-binding photoreceptors in cyanobacteria. Photochem. Photobiol. Sci. 7, 1159–1167 [DOI] [PubMed] [Google Scholar]
- 8. Wagner J. R., Brunzelle J. S., Forest K. T., Vierstra R. D. (2005) A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature 438, 325–331 [DOI] [PubMed] [Google Scholar]
- 9. Yang X., Kuk J., Moffat K. (2008) Crystal structure of Pseudomonas aeruginosa bacteriophytochrome: photoconversion and signal transduction. Proc. Natl. Acad. Sci. U.S.A. 105, 14715–14720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Essen L. O., Mailliet J., Hughes J. (2008) The structure of a complete phytochrome sensory module in the Pr ground state. Proc. Natl. Acad. Sci. U.S.A. 105, 14709–14714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lamparter T., Carrascal M., Michael N., Martinez E., Rottwinkel G., Abian J. (2004) The biliverdin chromophore binds covalently to a conserved cysteine residue in the N-terminus of Agrobacterium phytochrome Agp1. Biochemistry 43, 3659–3669 [DOI] [PubMed] [Google Scholar]
- 12. van Thor J. J., Borucki B., Crielaard W., Otto H., Lamparter T., Hughes J., Hellingwerf K. J., Heyn M. P. (2001) Light-induced proton release and proton uptake reactions in the cyanobacterial phytochrome Cph1. Biochemistry 40, 11460–11471 [DOI] [PubMed] [Google Scholar]
- 13. Strauss H. M., Hughes J., Schmieder P. (2005) Heteronuclear solution-state NMR studies of the chromophore in cyanobacterial phytochrome Cph1. Biochemistry 44, 8244–8250 [DOI] [PubMed] [Google Scholar]
- 14. Borucki B., von Stetten D., Seibeck S., Lamparter T., Michael N., Mroginski M. A., Otto H., Murgida D. H., Heyn M. P., Hildebrandt P. (2005) Light-induced proton release of phytochrome is coupled to the transient deprotonation of the tetrapyrrole chromophore. J. Biol. Chem. 280, 34358–34364 [DOI] [PubMed] [Google Scholar]
- 15. Falk H. (1989) The Chemistry of Linear Oligopyrroles and Bile Pigments, Springer Verlag, Berlin [Google Scholar]
- 16. Duerring M., Schmidt G. B., Huber R. (1991) Isolation, crystallization, crystal structure analysis and refinement of constitutive C-phycocyanin from the chromatically adapting cyanobacterium Fremyella diplosiphon at 1.66 Å resolution. J. Mol. Biol. 217, 577–592 [DOI] [PubMed] [Google Scholar]
- 17. Narikawa R., Ishizuka T., Muraki N., Shiba T., Kurisu G., Ikeuchi M. (2013) Structures of cyanobacteriochromes from phototaxis regulators AnPixJ and TePixJ reveal general and specific photoconversion mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 918–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song C., Psakis G., Lang C., Mailliet J., Gärtner W., Hughes J., Matysik J. (2011) Two ground state isoforms and a chromophore D-ring photoflip triggering extensive intramolecular changes in a canonical phytochrome. Proc. Natl. Acad. Sci. U.S.A. 108, 3842–3847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang X., Stojkovic E. A., Kuk J., Moffat K. (2007) Crystal structure of the chromophore binding domain of an unusual bacteriophytochrome, RpBphP3, reveals residues that modulate photoconversion. Proc. Natl. Acad. Sci. U.S.A. 104, 12571–12576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang X., Ren Z., Kuk J., Moffat K. (2011) Temperature-scan cryocrystallography reveals reaction intermediates in bacteriophytochrome. Nature 479, 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. von Stetten D., Seibeck S., Michael N., Scheerer P., Mroginski M. A., Murgida D. H., Krauss N., Heyn M. P., Hildebrandt P., Borucki B., Lamparter T. (2007) Highly conserved residues D197 and H250 in Agp1 phytochrome control the proton affinity of the chromophore and Pfr formation. J. Biol. Chem. 282, 2116–2123 [DOI] [PubMed] [Google Scholar]
- 22. Lamparter T., Michael N., Mittmann F., Esteban B. (2002) Phytochrome from Agrobacterium tumefaciens has unusual spectral properties and reveals an N-terminal chromophore attachment site. Proc. Natl. Acad. Sci. U.S.A. 99, 11628–11633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blumenstein A., Vienken K., Tasler R., Purschwitz J., Veith D., Frankenberg-Dinkel N., Fischer R. (2005) The Aspergillus nidulans phytochrome FphA represses sexual development in red light. Curr. Biol. 15, 1833–1838 [DOI] [PubMed] [Google Scholar]
- 24. Giraud E., Fardoux J., Fourrier N., Hannibal L., Genty B., Bouyer P., Dreyfus B., Verméglio A. (2002) Bacteriophytochrome controls photosystem synthesis in anoxygenic bacteria. Nature 417, 202–205 [DOI] [PubMed] [Google Scholar]
- 25. Tasler R., Moises T., Frankenberg-Dinkel N. (2005) Biochemical and spectroscopic characterization of the bacterial phytochrome of Pseudomonas aeruginosa. FEBS J. 272, 1927–1936 [DOI] [PubMed] [Google Scholar]
- 26. Oberpichler I., Molina I., Neubauer O., Lamparter T. (2006) Phytochromes from Agrobacterium tumefaciens: difference spectroscopy with extracts of wild type and knockout mutants. FEBS Lett. 580, 437–442 [DOI] [PubMed] [Google Scholar]
- 27. Inomata K., Hammam M. A., Kinoshita H., Murata Y., Khawn H., Noack S., Michael N., Lamparter T. (2005) Sterically locked synthetic bilin derivatives and phytochrome Agp1 from Agrobacterium tumefaciens form photoinsensitive Pr- and Pfr-like adducts. J. Biol. Chem. 280, 24491–24497 [DOI] [PubMed] [Google Scholar]
- 28. Inomata K., Noack S., Hammam M. A., Khawn H., Kinoshita H., Murata Y., Michael N., Scheerer P., Krauss N., Lamparter T. (2006) Assembly of synthetic locked chromophores with Agrobacterium phytochromes Agp1 and Agp2. J. Biol. Chem. 281, 28162–28173 [DOI] [PubMed] [Google Scholar]
- 29. Ly H. K., Utesch T., Diaz-Moreno I., Garcia-Heredia J. M., De La Rosa M. A., Hildebrandt P. (2012) Perturbation of the redox site structure of cytochrome c variants upon tyrosine nitration. J. Phys. Chem. B 116, 5694–5702 [DOI] [PubMed] [Google Scholar]
- 30. Döpner S., Hildebrandt P., Grant Mauk A., Lenk H., Stempfle W. (1996) Analysis of vibrational spectra of multicomponent systems. Application to pH-dependent resonance Raman spectra of ferricytochrome c. Spectrochim. Acta A Mol. Biomol. Spectrosc. 52, 573–584 [Google Scholar]
- 31. Noack S., Michael N., Rosen R., Lamparter T. (2007) Protein conformational changes of Agrobacterium phytochrome Agp1 during chromophore assembly and photoconversion. Biochemistry 46, 4164–4176 [DOI] [PubMed] [Google Scholar]
- 32. Schmidt P., Gensch T., Remberg A., Gärtner W., Braslavsky S. E., Schaffner K. (1998) The complexity of the Pr to Pfr phototransformation kinetics is an intrinsic property of native phytochrome. Photochem. Photobiol. 68, 754–761 [Google Scholar]
- 33. Aramendia P. F., Ruzsicska B. P., Braslavsky S. E., Schaffner K. (1987) Laser flash-photolysis of 124-kilodalton oat phytochrome in H2O and D2O solutions—formation and decay of the I700 intermediates. Biochemistry 26, 1418–1422 [Google Scholar]
- 34. Remberg A., Lindner I., Lamparter T., Hughes J., Kneip C., Hildebrandt P., Braslavsky S. E., Gärtner W., Schaffner K. (1997) Raman spectroscopic and light-induced-kinetic characterization of a recombinant phytochrome of the cyanobacterium Synechocystis. Biochemistry 36, 13389–13395 [DOI] [PubMed] [Google Scholar]
- 35. Borucki B., Otto H., Rottwinkel G., Seibeck S., Lamparter T., Heyn M. P. (2005) Selfassembly and kinetics of photoconversion of Agp1 phytochrome. Biophys. J. 88, 508A [Google Scholar]
- 36. Mroginski M. A., von Stetten D., Kaminski S., Escobar F. V., Michael N., Daminelli-Widany G., Hildebrandt P. (2011) Elucidating photoinduced structural changes in phytochromes by the combined application of resonance Raman spectroscopy and theoretical methods. J. Mol. Struct. 993, 15–25 [Google Scholar]
- 37. Kneip C., Hildebrandt P., Schlamann W., Braslavsky S. E. (1999) Protonation state and structural changes of the tetrapyrrole chromophore during the Pr → Pfr phototransformation of phytochrome: a resonance Raman spectroscopic study. Biochemistry 38, 15185–15192 [DOI] [PubMed] [Google Scholar]
- 38. von Stetten D., Günther M., Scheerer P., Murgida D. H., Mroginski M. A., Krauss N., Lamparter T., Zhang J., Anstrom D. M., Vierstra R. D., Forest K. T., Hildebrandt P. (2008) Chromophore heterogeneity and photoconversion in phytochrome crystals and solution studied by resonance Raman spectroscopy. Angew. Chem. Int. Ed. Engl. 47, 4753–4755 [DOI] [PubMed] [Google Scholar]
- 39. Salewski J., Escobar F. V., Kaminski S., von Stetten D., Keidel A., Rippers Y., Michael N., Scheerer P., Piwowarski P., Bartl F., Frankenberg-Dinkel N., Ringsdorf S., Gärtner W., Lamparter T., Mroginski M. A., Hildebrandt P. (2013) The structure of the biliverdin cofactor in the Pfr state of bathy and prototypical phytochromes. J. Biol. Chem. 288, 16800–16814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mroginski M. A., Kaminski S., von Stetten D., Ringsdorf S., Gärtner W., Essen L. O., Hildebrandt P. (2011) Structure of the chromophore binding pocket in the Pr state of plant phytochrome PhyA. J. Phys. Chem. B 115, 1220–1231 [DOI] [PubMed] [Google Scholar]
- 41. Schwinté P., Foerstendorf H., Hussain Z., Gärtner W., Mroginski M. A., Hildebrandt P., Siebert F. (2008) FTIR study of the photoinduced processes of plant phytochrome phyA using isotope-labeled bilins and density functional theory calculations. Biophys. J. 95, 1256–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matysik J., Hildebrandt P., Smit K., Mark F., Gärtner W., Braslavsky S. E., Schaffner K., Schrader B. (1997) Raman spectroscopic analysis of isomers of biliverdin dimethyl ester. J. Pharm. Biomed. Anal. 15, 1319–1324 [DOI] [PubMed] [Google Scholar]
- 43. Zienicke B., Chen L. Y., Khawn H., Hammam M. A., Kinoshita H., Reichert J., Ulrich A. S., Inomata K., Lamparter T. (2011) Fluorescence of phytochrome adducts with synthetic locked chromophores. J. Biol. Chem. 286, 1103–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Inomata K., Khawn H., Chen L.-Y., Kinoshita H., Zienicke B., Molina I., Lamparter T. (2009) Assembly of Agrobacterium phytochromes Agp1 and Agp2 with doubly locked bilin chromophores. Biochemistry 48, 2817–2827 [DOI] [PubMed] [Google Scholar]
- 45. Karniol B., Wagner J. R., Walker J. M., Vierstra R. D. (2005) Phylogenetic analysis of the phytochrome superfamily reveals distinct microbial subfamilies of photoreceptors. Biochem. J. 392, 103–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang X. J., Kuk J., Moffat K. (2009) Conformational differences between the Pfr and Pr states in Pseudomonas aeruginosa bacteriophytochrome. Proc. Natl. Acad. Sci. U.S.A. 106, 15639–15644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schumann C., Gross R., Michael N., Lamparter T., Diller R. (2007) Sub-picosecond mid-infrared spectroscopy of phytochrome Agp1 from Agrobacterium tumefaciens. ChemPhysChem 8, 1657–1663 [DOI] [PubMed] [Google Scholar]
- 48. Borg O. A., Durbeej B. (2007) Relative ground and excited-state pKa values of phytochromobilin in the photoactivation of phytochrome: a computational study. J. Phys. Chem. B 111, 11554–11565 [DOI] [PubMed] [Google Scholar]
- 49. Franklin K. A., Whitelam G. C. (2005) Phytochromes and shade-avoidance responses in plants. Ann. Bot. 96, 169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smith H. (1982) Light quality, photoperception, and plant strategy. Annu. Rev. Plant Physiol. Plant Mol. Biol. 33, 481–518 [Google Scholar]
- 51. Woolley J. T., Stoller E. W. (1978) Light penetration and light-induced seed-germination in soil. Plant Physiol. 61, 597–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sun Q., Yoda K., Suzuki H. (2005) Internal axial light conduction in the stems and roots of herbaceous plants. J. Exp. Bot. 56, 191–203 [DOI] [PubMed] [Google Scholar]
- 53. Rottwinkel G. (2011) Studien zu Verbreitung, Charakteristika und Funktionen der Bakteriophytochrome in Rhizobiales. Ph.D. thesis/dissertation, Karlsruhe Institute of Technology [Google Scholar]
- 54. Li L., Jia Y., Hou Q., Charles T. C., Nester E. W., Pan S. Q. (2002) A global pH sensor: Agrobacterium sensor protein ChvG regulates acid-inducible genes on its two chromosomes and Ti plasmid. Proc. Natl. Acad. Sci. U.S.A. 99, 12369–12374 [DOI] [PMC free article] [PubMed] [Google Scholar]