

Background: Localization of the drug transporter P-glycoprotein (Pgp) to the plasma membrane is thought to be the only contributor of Pgp-mediated multidrug resistance (MDR).
Results: Lysosomal Pgp sequesters ionizable chemotherapeutics into lysosomes to prevent interaction with molecular targets, resulting in drug resistance.
Conclusion: Lysosomal Pgp mediates drug resistance.
Significance: Pgp-mediated sequestration of chemotherapeutics into lysosomes can be exploited pharmacologically.
Keywords: Chemical Biology, Chemoresistance, Drug Resistance, Drug Transport, Lysosomes, P-glycoprotein, Chloroquine, Doxorubicin, Multidrug Resistance
Abstract
Localization of the drug transporter P-glycoprotein (Pgp) to the plasma membrane is thought to be the only contributor of Pgp-mediated multidrug resistance (MDR). However, very little work has focused on the contribution of Pgp expressed in intracellular organelles to drug resistance. This investigation describes an additional mechanism for understanding how lysosomal Pgp contributes to MDR. These studies were performed using Pgp-expressing MDR cells and their non-resistant counterparts. Using confocal microscopy and lysosomal fractionation, we demonstrated that intracellular Pgp was localized to LAMP2-stained lysosomes. In Pgp-expressing cells, the Pgp substrate doxorubicin (DOX) became sequestered in LAMP2-stained lysosomes, but this was not observed in non-Pgp-expressing cells. Moreover, lysosomal Pgp was demonstrated to be functional because DOX accumulation in this organelle was prevented upon incubation with the established Pgp inhibitors valspodar or elacridar or by silencing Pgp expression with siRNA. Importantly, to elicit drug resistance via lysosomes, the cytotoxic chemotherapeutics (e.g. DOX, daunorubicin, or vinblastine) were required to be Pgp substrates and also ionized at lysosomal pH (pH 5), resulting in them being sequestered and trapped in lysosomes. This property was demonstrated using lysosomotropic weak bases (NH4Cl, chloroquine, or methylamine) that increased lysosomal pH and sensitized only Pgp-expressing cells to such cytotoxic drugs. Consequently, a lysosomal Pgp-mediated mechanism of MDR was not found for non-ionizable Pgp substrates (e.g. colchicine or paclitaxel) or ionizable non-Pgp substrates (e.g. cisplatin or carboplatin). Together, these studies reveal a new mechanism where Pgp-mediated lysosomal sequestration of chemotherapeutics leads to MDR that is amenable to therapeutic exploitation.
Introduction
Cellular mechanisms of cancer multidrug resistance (MDR)5 have been studied extensively because they constitute a major factor to the reduced efficacy of many chemotherapeutics (1). One of the best-characterized mechanisms of MDR occurs via drug pumps that actively efflux various cytotoxic compounds from cells for cytoprotection (1).
These latter molecules include the well studied P-glycoprotein (Pgp) (1). Although the mechanism of action of Pgp on the plasma membrane is well characterized (1), drug resistance mediated by intracellular Pgp has not been comprehensively investigated. Indeed, there are conflicting reports on the distribution and roles of intracellular Pgp in MDR (2–4).
In this study, we demonstrate that Pgp in lysosomes actively sequesters the Pgp substrate doxorubicin (DOX) into these organelles. Furthermore, incubation of cells with the potent Pgp inhibitors valspodar (Val) or elacridar (Ela) or silencing of Pgp with siRNA inhibited sequestration of DOX in lysosomes and led to its redistribution to its primary target, namely DNA in the nucleus. Significantly, lysosomal Pgp conferred drug resistance, and this could be overcome by lysosomotropic weak bases that induced a marked increase in DOX cytotoxicity, an effect only observed in Pgp-positive cells. We also showed that only Pgp substrates that become charged at acidic lysosomal pH were capable of conferring lysosomal Pgp-dependent drug resistance. For the first time, we present a novel mechanism of MDR mediated by intracellular Pgp in lysosomes.
EXPERIMENTAL PROCEDURES
Cell Culture
The human cervical carcinoma derived KB-3-1 (KB31) cell line was obtained from the ATCC, and the vinblastine (VBL)-resistant variant KB-V-1 (KBV1, grown in VBL at 1 μg/ml) was a gift from Dr. Maria Kavallaris (Children's Cancer Institute Australia, Sydney, Australia). The 2008 human ovarian carcinoma cell line and the paclitaxel (PAC)-resistant 2008/P200 cell line (grown in PAC at 200 ng/ml) were provided by Dr. John Allen (Centenary Institute, Sydney, Australia). All cell lines were grown in DMEM (Invitrogen) under standard growth conditions (5).
Western Blot Analysis and Antibodies
Established methods were implemented for Western blotting (5) using monoclonal antibodies for Pgp (catalog no. P7965, Sigma-Aldrich); lysosomal-associated membrane protein 2 (LAMP2) (catalog no. ab25631, Abcam, Cambridge, UK); histone deacetylase 1 (HDAC1) (catalog no. sc-7872, Santa Cruz Biotechnology, Santa Cruz, CA); and succinate dehydrogenase complex, subunit A (SDHA) (catalog no. ab14715, Mitosciences, Eugene, OR).
Flow Cytometry
For extracellular Pgp staining, cells were incubated with FITC-conjugated Pgp antibody (1:100, BD Biosciences, East Rutherford, NJ) for 30 min on ice and washed four times with ice-cold PBS. The cells were then removed from the plate using 1 mm EDTA/PBS (pH 7.4), centrifuged, and resuspended in ice-cold PBS for flow cytometric analysis. Notably, the cells were not permeabilized to prevent intracellular staining of Pgp.
For the rhodamine 123 (Rh123, Sigma-Aldrich) accumulation assay, cells were preincubated with either control medium or the well known Pgp inhibitors Val (1 μm, Novartis) or Ela (0.1 μm, GlaxoSmithKline) for 30 min. The medium was then aspirated, and the cells were loaded with Rh123 (1 μg/ml) using a 15-min/37 °C incubation in the presence of control medium alone or the Pgp inhibitors. The overlying medium was subsequently removed, and the cells were detached using 1 mm EDTA/PBS (pH 7.4) and centrifuged. Then the cell pellet was resuspended in ice-cold PBS for flow cytometric analysis.
FITC-conjugated Pgp and Rh123 were detected with a FACSCanto flow cytometer (BD Biosciences), and 10,000 events were acquired for every sample. Data analysis was performed using FlowJo software, version 7.5.5 (Tree Star Inc., Ashland, OR).
[14C]DOX Uptake and Efflux
For [14C]DOX uptake studies, each plate containing 5 × 105 cells in 35-mm culture dishes was preincubated with either control medium or the Pgp inhibitors Val (1 μm) or Ela (0.1 μm) for 30 min/37 °C. Then, [14C]DOX (1 μCi, PerkinElmer Life Sciences, Waltham, MA) was added in the presence or absence of these inhibitors, and the incubation continued for 30 min at 37 °C. The cells were subsequently washed four times on ice with ice-cold PBS and collected for analysis using a β-scintillation counter (PerkinElmer Life Sciences).
For [14C]DOX efflux studies, cells were preincubated with [14C]DOX (1 μCi) for 30 min at 37 °C, washed four times on ice with ice-cold PBS, and then reincubated for up to 60 min at 37 °C with medium alone or medium containing Val (1 μm) or Ela (0.1 μm). Both medium and cells were collected separately into β-counting vials and analyzed using the β-counter described above. The efflux of [14C]DOX into the overlying medium was expressed as a percentage of the total [14C]DOX present in the cells.
Cellular Proliferation
Proliferation was examined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (6). Cells (3 × 103/well) were seeded in 96-well plates and allowed to adhere overnight. The next day, Pgp substrates were added in combination with the Pgp inhibitors Val (1 μm) or Ela (0.1 μm) or the lysosomotropic weak bases ammonium chloride (NH4Cl, 3 mm), chloroquine (CLQ, 1 μm), or methylamine (MA, 100 μm) at non-toxic concentrations (data not shown). Cells were then incubated with these agents for 72 h at 37 °C and processed using standard procedures (6). Results were validated using viable cell counts (6).
Immunofluorescence, Confocal Microscopy, and Image Analysis
For assessment of intracellular Pgp colocalization and function using the Pgp substrate DOX (Pfizer), cells (1 × 105 cells/ml) were grown on coverslips and incubated with DOX (10 μm, 2 h, 37 °C) followed by paraformaldehyde fixation (4%, 15 min, 20 °C) and digitonin permeabilization (100 μm, 10 min, 20 °C). Importantly, the mild detergent digitonin was utilized to specifically avoid dissolving the lysosomal membrane (7). After blocking with 5% BSA, immunofluorescence was performed by incubation (16 h, 4 °C) with FITC-conjugated anti-Pgp (1:100, catalog no. 557002, BD Biosciences) and anti-LAMP2 (1:20, catalog no. ab25631, Abcam) antibodies, MitoTracker® Deep Red (50 nm, Invitrogen), and DAPI (0.5 μm) Invitrogen). In the case of the primary incubation with anti-LAMP2, this was followed by treatment (1 h, 4 °C) with Alexa Fluor-conjugated secondary antibodies (1:1000, catalog nos. A-21200 and A-21201, Invitrogen).
For experiments using lysosomotropic weak bases (Sigma-Aldrich), a 30 min pretreatment of cells with NH4Cl (3 mm), CLQ (1 μm), or MA (100 μm) was performed, followed by incubation with DOX (10 μm, 2 h, 37 °C) in the continued presence of the lysosomotropic bases. Stained samples were examined with a Zeiss LSM 510 Meta confocal microscope (Zeiss, Oberkochen, Germany) equipped with FITC (excitation, 495 nm; emission, 516 nm) and Texas Red (excitation, 577 nm; emission, 592 nm) filters and captured with Zeiss LSM 510 META software (Zeiss). Pearson's correlation coefficient and Mander's overlap coefficient were determined, and scatter plots were generated using Imaris (Bitplane, Zurich, Switzerland) and Zeiss Axiovision colocalization software (Zeiss) from the intracellular compartments of cells.
Lysosomal Fractionation
Lysosomally enriched fractions were prepared from cultured cells using the lysosome enrichment kit (Pierce Biotechnology, Waltham, MA). The lysosome-specific enzyme marker acid phosphatase (Sigma-Aldrich) (8) showed that this fraction was lysosome-enriched, as determined by spectrophotometric assessment at 405 nm. The purity of the fractions was also assessed using antibodies (described under “Western Blot and Antibodies”) against LAMP2 (lysosomes), SDHA (mitochondria), and HDAC1 (nuclei).
Transient Silencing of Pgp (MDR1) Using siRNA
For all siRNA treatments against MDR1 (MDR1 siRNA, catalog nos. 4123 and 3933, Ambion, Carlsbad, CA), a siRNA-Lipofectamine mixture (50 nm MDR1 siRNA and 1:400 Lipofectamine 2000) was added to the cells (at 30% confluency) and incubated for 72 h at 37 °C prior to further experiments. The effectiveness of Pgp silencing was assessed using both Western blotting and the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. As a control, scrambled siRNA (Scr siRNA, Invitrogen) was used at the same concentration as MDR1 siRNA.
Calculation of Speciation Plots
Speciation plots were prepared using published pKa values derived from potentiometric titration data (9–12). Hyperquad2008 software (Protonic Software, Leeds, UK) was used to generate speciation plots from these pKa values.
Statistics
Data were compared using Student's t test. Results were expressed as mean ± S.D. (number of experiments) and considered to be statistically significant when p < 0.05.
RESULTS
Pgp Protects Cells from Cytotoxic Pgp Substrates
To understand the role of intracellular Pgp in MDR, Pgp expression and functionality were initially assessed in the well known KBV1 (+Pgp)/KB31 (−Pgp) drug resistance cell model (13, 14). We showed that KBV1 (+Pgp) cells cultured with VBL (13) expressed high Pgp levels compared with parental KB31 (−Pgp) cells without VBL selection, as shown by Western blotting (Fig. 1A) and flow cytometry (Fig. 1B).
FIGURE 1.
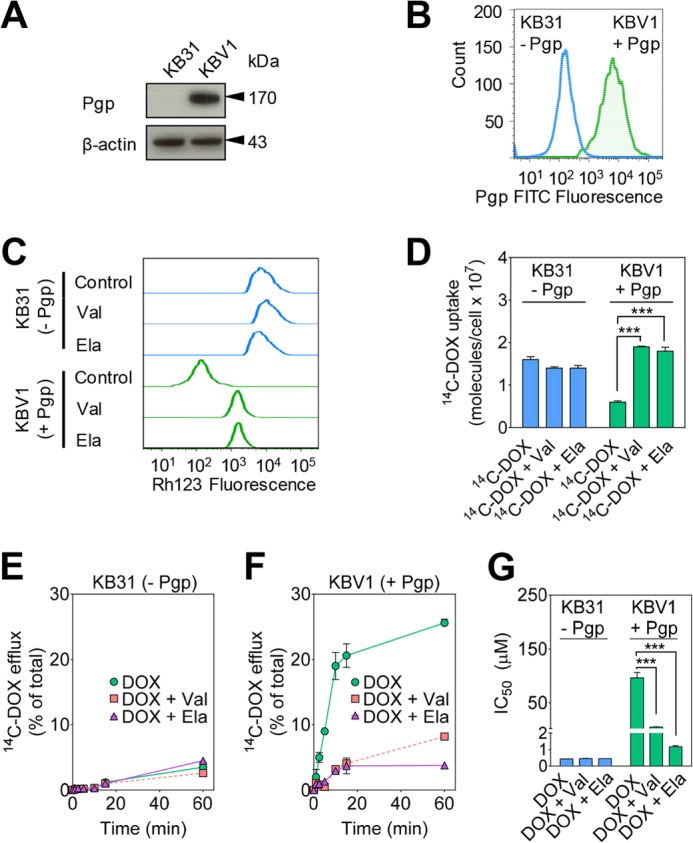
Functional Pgp causes DOX resistance. A, Western blot analysis showing that Pgp is only expressed in KBV1 (+Pgp) and not in KB31 (−Pgp) cells. β-Actin was used as a loading control. B, flow cytometry demonstrates higher plasma membrane expression of Pgp in KBV1 (+Pgp) cells than KB31 (−Pgp) cells. Cells were not permeabilized to prevent intracellular staining of Pgp prior to flow cytometric analysis. C, Rh123 accumulation is decreased in KBV1 (+Pgp) control cells compared with KB31 (−Pgp) control cells, whereas Pgp inhibition increases Rh123 accumulation only in KBV1 (+Pgp) cells. Briefly, cells were preincubated with either control medium or the Pgp inhibitors Val (1 μm) or Ela (0.1 μm) for 30 min at 37 °C. Cells were then loaded with Rh123 (1 μg/ml) for 15 min at 37 °C in the presence or absence of these inhibitors. D, Pgp inhibitors increase [14C]DOX uptake only in KBV1 (+Pgp) cells. Briefly, KBV1 (+Pgp) or KB31 (−Pgp) cells were preincubated with either control medium or the Pgp inhibitors Val (1 μm) or Ela (0.1 μm) for 30 min at 37 °C. [14C]DOX was then added, the incubation continued for 30 min at 37 °C, and then the cells were washed. E and F, Pgp inhibitors block efflux of [14C]DOX only from KBV1 (+Pgp) cells. Briefly, cells were labeled with [14C]DOX for 30 min at 37 °C, washed, and then reincubated for up to 60 min at 37 °C in the presence or absence of the Pgp inhibitors Val (1 μm) or Ela (0.1 μm). G, DOX cytotoxicity (IC50/72 h) is potentiated by the Pgp inhibitors Val (1 μm) or Ela (0.1 μm) in KBV1 (+Pgp) cells but not KB31 (−Pgp) cells. The results in A–C are representative of three experiments, whereas those in D–G are mean ± S.D. (three experiments with at least four replicates in each experiment). ***, p < 0.001 versus control.
To assess Pgp function, flow cytometric studies then progressed to examining the cellular accumulation of the fluorescent Pgp substrate Rh123 (13) in KBV1 (+Pgp) cells relative to KB31 (−Pgp) cells (Fig. 1C). In these studies, KB31(−Pgp) and KBV1 (+Pgp) cells were either preincubated for 30 min at 37 °C with control medium or medium containing the well characterized Pgp inhibitors Val (1 μm) or Ela (0.1 μm) (15). The cells were then loaded with Rh123 for 15 min at 37 °C in the presence or absence of these inhibitors. These experiments demonstrated significantly (p < 0.001) greater Rh123 accumulation (as measured by Rh123 fluorescence) in control KB31 (−Pgp) cells relative to control KBV1 (+Pgp) cells (Fig. 1C). This observation was in accordance with the expression of Pgp in KBV1 (+Pgp) cells (Fig. 1, A and B), which actively effluxes Rh123 from cells (13). Hence, high Pgp levels in KBV1 cells lead to pronounced Rh123 efflux, resulting in lower cellular accumulation of this substrate. Notably, incubation with Ela or Val had no effect on Rh123 fluorescence in KB31 (−Pgp) cells but caused a marked increase in Rh123 in KBV1 (+Pgp) cells (Fig. 1C). This finding is consistent with the efficacy of these inhibitors at preventing Rh123 efflux from KBV1 (+Pgp) cells via Pgp, leading to its cellular accumulation (Fig. 1C).
The functionality of Pgp in KBV1 (+Pgp) cells was further substantiated by the reduced uptake (Fig. 1D) and increased efflux (E and F) of the 14C-labeled Pgp substrate DOX (16) in KBV1 (+Pgp) cells relative to KB31 (−Pgp) cells. In [14C]DOX uptake studies, the Pgp inhibitors Ela or Val had no significant (p > 0.05) effect on the uptake of [14C]DOX by KB31 (−Pgp) cells relative to the control over a 30-min/37 °C incubation (Fig. 1D). In contrast, in KBV1 (+Pgp) cells, both Val and Ela resulted in a marked and significant (p < 0.001) increase in cellular [14C]DOX levels relative to incubation with [14C]DOX alone (Fig. 1D). This finding was consistent with the Pgp inhibitors preventing release of [14C]DOX from the cell, leading to its accumulation. It is also notable that [14C]DOX levels in control KB31 (−Pgp) cells were significantly (p < 0.001) higher than those in KBV1 (+Pgp) cells (Fig. 1D), and, again, this was consistent with Pgp mediating [14C]DOX release from KBV1 (+Pgp) cells.
In experiments examining [14C]DOX efflux, KB31 (−Pgp) and KBV1 (+Pgp) cells were preincubated with [14C]DOX for 30 min at 37 °C, washed four times, and then reincubated for up to 60 min at 37 °C in the presence or absence of Val or Ela (Fig. 1, E and F). In KB31 (−Pgp) cells, the efflux of [14C]DOX was less than 5% of the total cellular [14C]DOX after 60 min and was not significantly (p > 0.05) affected by the Pgp inhibitors Val (1 μm) or Ela (0.1 μm) (Fig. 1E). In contrast, using KBV1 (+Pgp) cells, the efflux of [14C]DOX in control cells was increased markedly, leading to the release of 25% of total cellular [14C]DOX after a 60 min of incubation (Fig. 1F). Furthermore, Val or Ela significantly (p < 0.001) reduced [14C]DOX efflux by KBV1 (+Pgp) cells (Fig. 1F). Collectively, these studies demonstrate the functionality of Pgp transport activity in KBV1 (+Pgp) cells relative to KB31 (−Pgp) cells.
Considering the decreased uptake of [14C]DOX (Fig. 1D) because of the Pgp-mediated increase in its efflux from KBV1 (+Pgp) cells (Fig. 1F), studies then examined the cytotoxicity of DOX in the presence and absence of the Pgp inhibitors Val and Ela in KB31 (−Pgp) and KBV1 (+Pgp) cells over 72 h at 37 °C (Fig. 1G). Because of the absence of Pgp in KB31 (−Pgp) cells, the cytotoxic activity of DOX was marked in either the presence or absence of the Pgp inhibitors, leading to an IC50 of < 0.5 μm (Fig. 1G). In contrast, KBV1 (+Pgp) cells were significantly (p < 0.001) more resistant to DOX than KB31 (−Pgp) cells, with KBV1 (+Pgp) cells having an IC50 of 96.4 ± 10.0 μm because of Pgp expression (Fig. 1G). Additionally, both Pgp inhibitors significantly (p < 0.001) sensitized KBV1 (+Pgp) cells to DOX, resulting in a marked decrease in the IC50 value (Fig. 1G). These data show that Pgp is functional and protects KBV1 (+Pgp) cells from DOX cytotoxicity.
Intracellular Localization of Pgp in Lysosomes
Having carefully established Pgp functionality in KBV1 (+Pgp) cells, we then investigated mechanisms of MDR involving intracellular Pgp. The subcellular localization of Pgp was initially examined using confocal microscopy implementing well characterized organelle-specific antibodies or dyes, namely LAMP2 for lysosomes (17, 18), DAPI for nuclei (19), and MitoTracker® Deep Red (Mito DRed) for mitochondria (18). In agreement with the [14C]DOX efflux studies described above (Fig. 1F), Pgp was identified on the plasma membrane of KBV1 (+Pgp) cells (Fig. 2, A–C). In addition, intracellular Pgp showed a punctate pattern of Pgp staining (Fig. 2A). This Pgp staining colocalized with the lysosomal marker LAMP2, leading to a yellow punctate pattern in the merge (Fig. 2A, arrows).
FIGURE 2.

Intracellular Pgp is localized to lysosomes in KBV1 (+Pgp) cells. A, Pgp colocalizes with lysosomal-associated membrane protein 2 (LAMP2, a well characterized lysosomal marker, arrows), whereas no colocalization was observed with DAPI (nuclear marker) (B) or MitoTracker® Deep Red (Mito Dred, mitochondrial marker) (C). The scatter plots, Pearson's correlation coefficient, and Mander's overlap coefficient in A–C were calculated using Imaris and Axiovision software from intracellular compartments of all cells. Photographs are typical from three experiments (A–C). Scale bars = 50 μm.
Analysis of these images using scatter plots, Pearson's correlation coefficient (20) (0.936), and Mander's overlap coefficient (20) (0.925) also validated the colocalization of Pgp and LAMP2 (Fig. 2A). No significant (p > 0.05) colocalization of Pgp was observed with nuclei (Fig. 2B, DAPI) or the mitochondria (C, Mito DRed). Indeed, scatter plots, Pearson's correlation coefficient (0.086 and 0.38 for nuclei and mitochondria, respectively), and Mander's overlap coefficient (0.377 and 0.493 for nuclei and mitochondria, respectively) further verified these observations (Fig. 2, B and C). These investigations suggested that intracellular Pgp was localized to lysosomes and not to mitochondria or nuclei.
To further assess Pgp localization in lysosomes, subcellular fractionation was performed using KBV1 (+Pgp) cells. The activity of lysosome-specific acid phosphatase (21) was used as a marker to assess the purity of the KBV1 (+Pgp) lysosomal fraction (Fig. 3). Assessment of the total cell and lysosome-enriched fractions demonstrated that the lysosome-enriched fraction possessed > 20-fold acid phosphatase activity than the total lysate (Fig. 3A). Western blotting demonstrated that the lysosomal fraction was enriched with Pgp and lysosomes (LAMP2, Ref. 18) but did not contain nuclei (HDAC1, Ref. 22), and had a similar level of mitochondria (SDHA, Ref. 23) as the total lysates (Fig. 3B). Hence, the marked enrichment of Pgp in the lysosomal fraction is not from mitochondria because the SDHA level was similar between the lysosomal and total fractions (Fig. 3B). Collectively, these Pgp localization studies and fractionation results indicate that intracellular Pgp is localized to lysosomes but not mitochondria or nuclei.
FIGURE 3.

Lysosomal KBV1 (+Pgp) cell fraction is enriched with Pgp. A, lysosome-enriched fractions of KBV1 (+Pgp) cells demonstrate significantly (p < 0.001) higher acid phosphatase activity compared with total cell lysates. B, Western blot analysis of the enriched lysosomal fraction in A demonstrating that the lysosomal fraction was highly enriched with Pgp. However, this lysosomal fraction did not contain histone deacetylase 1 (HDAC1) from the nucleus and had a similar level of mitochondrial SDHA as the total cell lysate. Western blot analyses in A and B are mean ± S.D. (three experiments). ***, p < 0.001 versus control.
Functional Assessment of Pgp in Lysosomes
To assess the functionality of lysosomal Pgp, intracellular trafficking and localization of the intrinsically fluorescent Pgp substrate DOX (24) was examined by confocal microscopy. Incubation of DOX with KB31 (−Pgp) cells led to its nuclear accumulation, as demonstrated by merging DOX (Fig. 4A) with DAPI (Fig. 4C), forming a purple colocalization pattern in the merge (Fig. 4D). This is expected because DOX intercalates within its primary molecular target, DNA (25, 26). However, no DOX was found to colocalize with lysosomal LAMP2 in KB31 (−Pgp) cells (Fig. 4, A, B, and D), and the Pgp inhibitors Val or Ela did not alter DOX localization in the nuclei (Fig. 4, E–L). In clear contrast, DOX colocalized (Fig. 4M) primarily with LAMP2 in KBV1 (+Pgp) cells (Fig. 4N), leading to yellow fluorescence in the merge (Fig. 4P, arrows). This observation demonstrates that the pronounced Pgp expression in KBV1 (+Pgp) cells results in marked accumulation of DOX in LAMP2-stained lysosomes. Interestingly, in KBV1 (+Pgp) cells, the redistribution of DOX (Fig. 4, Q and U) to nuclei (Fig. 4, S and W) was observed in the presence of the Pgp inhibitors, as shown by the purple nuclear pattern in the merge (Fig. 4, T and X). These results indicate that inhibition of Pgp activity using Val or Ela prevented the uptake of DOX into the lysosomal compartment and resulted in the redirection of DOX to nuclei.
FIGURE 4.
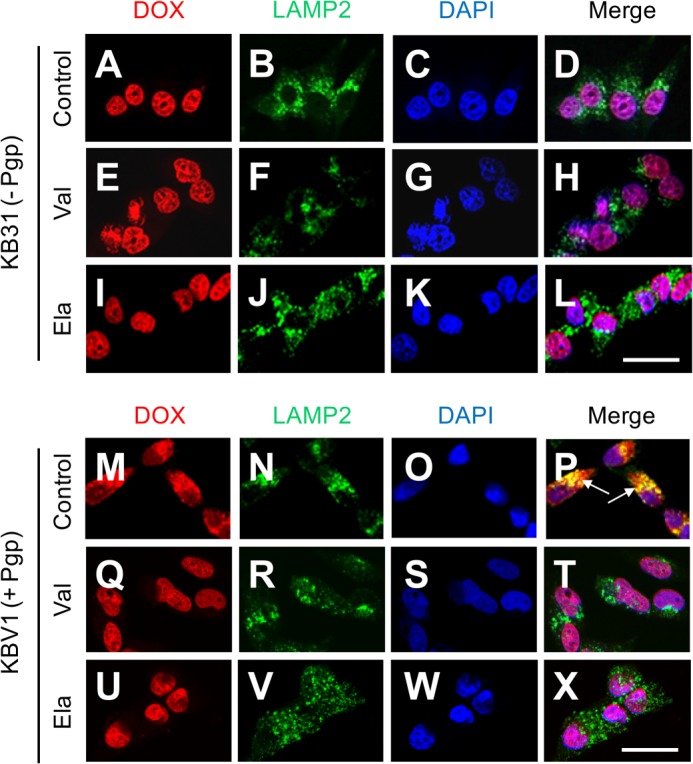
Colocalization of DOX with LAMP2-stained lysosomes is Pgp-dependent. A–D, in KB31 (−Pgp) cells, colocalization of DOX with the nuclear marker DAPI was demonstrated under control conditions. E–L, the Pgp inhibitors Val (E–H) and Ela (I–L) did not affect DOX distribution relative to the control in KB31 (−Pgp) cells. M–P, incubation of KBV1 (+Pgp) cells expressing functional Pgp with DOX leads to colocalization of this drug with the marker for lysosomes, LAMP2 (P, arrows), whereas the Pgp inhibitors Val (Q–T) and Ela (U–X) result in DOX redistribution to the nucleus. The results in A–X are typical from three experiments. Scale bars = 50 μm.
Critically, Val and Ela have been suggested to neutralize lysosomal pH and induce lysosomal swelling (27). To assess whether this effect may be relevant to our results, the size of the cell (forward scatter) and its granularity (side scatter) were analyzed by flow cytometry because changes in side scatter denote lysosomal swelling and lysosomotropism (27, 28). Importantly, no change in side scatter in KB31 (−Pgp) and KBV1 (+Pgp) cells was observed at the concentrations of Val and Ela utilized in our experiments (data not shown). Hence, under the conditions implemented in this investigation, Val and Ela inhibited Pgp without exhibiting lysosomotropic properties.
To further determine the functionality of lysosomal Pgp, transient silencing of Pgp in KBV1 (+Pgp) cells with two different types of Pgp siRNA was employed (Fig. 5). Silencing of Pgp by siRNA significantly (p < 0.001) decreased Pgp expression (Fig. 5A) and also significantly (p < 0.001) sensitized KBV1 (+Pgp) cells to DOX relative to the DOX-treated Scr siRNA control (Fig. 5B). Notably, the sensitization of KBV1 (+Pgp) cells to DOX by Pgp siRNA is due to the loss of Pgp that actively effluxes this cytotoxic agent.
FIGURE 5.

Functional silencing of Pgp in KBV1 (+Pgp) cells results in redistribution of DOX from LAMP2-stained lysosomes to DAPI-stained nuclei with concomitant toxicity. A, Western blot analysis showing transient silencing of Pgp in KBV1 (+Pgp) cells incubated with two types of Pgp siRNA (72 h, 37 °C) relative to KBV1 (+Pgp) cells treated with Scr siRNA. B, Pgp silencing in KBV1 (+Pgp) cells using a 72-h incubation with siRNA resulted in increased cytotoxicity (IC50/72 h) of DOX relative to KBV1 (+Pgp) cells treated with Scr siRNA. The Pgp inhibitor Ela (0.1 μm) increased cytotoxicity of DOX only in Scr siRNA-treated KBV1 (+Pgp) cells relative to Pgp siRNA-treated (Pgp-silenced) cells (72 h, 37 °C). C, siRNA silencing of Pgp (72 h, 37 °C) resulted in redistribution of DOX to nuclei, as indicated by DAPI staining, whereas Scr siRNA-treated KBV1 (+Pgp) cells resulted in accumulation of DOX in LAMP2-stained lysosomes (arrow). The results in A and C are typical from three experiments. The graph in B is mean ± S.D. ***, p < 0.001 versus control (i.e. drug alone). Scale bar = 50 μm.
These Pgp siRNA- and Scr siRNA-treated cells were then used to assess the Pgp-dependent sequestration of DOX into lysosomes (Fig. 5C). In Scr siRNA-treated cells that markedly express Pgp (Fig. 5A), DOX was found to primarily colocalize with LAMP2-stained lysosomes, leading to a yellow punctate pattern in the merge (C, arrow). However, in contrast, Pgp silencing did not result in colocalization of DOX and LAMP2 (Fig. 5C). Instead, Pgp siRNA-treated cells led to nuclear localization of DOX, resulting in a purple nuclear pattern in the merge (Fig. 5C). Together, the studies in Figs. 4 and 5 demonstrate that lysosomal sequestration of DOX requires both Pgp expression and function, which can be prevented by using Pgp inhibitors or Pgp siRNA.
Pgp in Lysosomes Confers Drug Resistance
Next, we assessed whether lysosomal Pgp can confer drug resistance via DOX sequestration. From analysis of DOX pKa data (9), it was found that DOX was fully charged at the acidic pH of the lysosome (29) (i.e. pH ∼5, Fig. 6A) and, consequently, would become trapped in acidic lysosomes (16). This trapping occurs because the charged drug cannot cross the lysosomal membrane. This observation is in accordance with the general principle that the passage of charged molecules across membranes is impeded relative to their neutral counterparts (16). Hence, to disrupt lysosomal DOX trapping, lysosomal pH was neutralized with the well known lysosomotropic weak bases ammonium chloride (NH4Cl), CLQ, or MA (30). Thus, after neutralization of lysosomes using these agents (30), a significant proportion of DOX would become neutral, as shown by the speciation plot (Fig. 6A). Hence, the neutral species of DOX should then pass through the lysosomal membrane to access the nuclei and become associated with one of its major molecular targets, namely DNA (25, 26).
FIGURE 6.

Pgp in lysosomes confers drug resistance to DOX. A, speciation plot of DOX derived from pKa values showing that 100% of DOX is charged at lysosomal pH (pH ∼5) (9). B and C, the lysosomotropic weak bases ammonium chloride (NH4Cl, 3 mm), CLQ (1 μm), and MA (100 μm) increase sensitivity (IC50/72 h) to DOX in KBV1 (+Pgp) cells but not in KB31 (−Pgp) cells. D, Western blot demonstrating significantly (p < 0.001) higher expression of Pgp in 2008/P200 (+Pgp) cells relative to 2008 (−Pgp) cells. E, flow cytometry was conducted to confirm the higher expression of plasma membrane Pgp in 2008/P200 (+Pgp) cells relative to 2008 (−Pgp) cells. F, Rh123 accumulation is decreased in control 2008/P200 (+Pgp) cells compared with control 2008 (−Pgp) cells, whereas Pgp inhibition increases Rh123 accumulation only in 2008/P200 (+Pgp) cells. Cells were preincubated with either control medium or the Pgp inhibitors Val (1 μm) or Ela (0.1 μm) for 30 min at 37 °C. Cells were then loaded with Rh123 (1 μg/ml) for 15 min at 37 °C in the presence or absence of these inhibitors. G and H, the lysosomotropic weak bases used in B and C also sensitized 2008/P200 (+Pgp) cells to DOX but not 2008 (−Pgp) cells (IC50/72 h). The plot in A was generated using Hyperquad2008. The results in B–H are from three experiments. ***, p < 0.001 versus control (i.e. DOX alone).
Incubation with these lysosomotropic weak bases did not significantly (p > 0.05) alter DOX toxicity in KB31 (−Pgp) cells (Fig. 6B). In clear contrast, lysosomotropic weak bases significantly (p < 0.001) increased DOX cytotoxicity in KBV1 (+Pgp) cells by 27- to 50-fold (Fig. 6C). These observations suggest that, in cells without Pgp, DOX does not accumulate in lysosomes and that, hence, the lysosomotropic weak bases have no effect on the cytotoxicity of this drug. In contrast, in cells expressing Pgp, neutralization of lysosomal pH using lysosomotropic weak bases leads to relocalization of DOX from lysosomes to DNA in the nucleus (25, 26), leading to cell death.
To ensure that the above effect was not cell line-specific, another set of cells, i.e. 2008 cells and PAC-selected, Pgp-expressing 2008/P200 cells, were used after characterization of their Pgp expression and function (Fig. 6, D–F). Indeed, 2008/P200 (+Pgp) cells showed significantly (p < 0.001) Pgp expression than their 2008 (−Pgp) counterparts, as shown by Western blot analysis (Fig. 6D) and flow cytometry (Fig. 6E). Moreover, Rh123 accumulation assays showed that, as found for KB31 (−Pgp) and KBV1 (+Pgp) cells (Fig. 1C), significantly (p < 0.001) greater Rh123 accumulation occurred in control 2008 (−Pgp) cells relative to control 2008/P200 (+Pgp) cells (Fig. 6F). Notably, incubation with Ela or Val had no effect on Rh123 accumulation in 2008 (−Pgp) cells but caused a marked increase in Rh123 accumulation in 2008/P200 (+Pgp) cells (Fig. 1C) because of the ability of the inhibitors to block cellular release of Rh123.
Examining the effect of lysosomotropic weak bases on the 2008 (−Pgp) cells demonstrated that they did not significantly (p > 0.05) affect the cytotoxicity of DOX (Fig. 6G). In contrast, the three lysosomotropic weak bases significantly (p < 0.001) increased cytotoxicity in 2008/P200 (+Pgp) cells (Fig. 6H), as found for KBV1 (+Pgp) cells (Fig. 6C). Hence, the lysosomotropic weak bases sensitized both Pgp-expressing cell types to DOX but not their non-Pgp-expressing counterparts.
Lysosomal Pgp Increases Lysosomal Trapping of the Pgp Substrate DOX, Preventing This Cytotoxic Agent from Reaching Its Nuclear Targets
The potential Pgp-dependent sequestration of DOX into lysosomes was then further assessed using confocal microscopy (Fig. 7). In KBV1 (+Pgp) cells treated with DOX alone, colocalization of DOX with LAMP2 was observed, leading to yellow fluorescence in the merged image (Fig. 7, arrows). This observation suggested the functional role of Pgp in lysosomes, as also shown in Figs. 4, M–P, and 5C. Interestingly, incubation with lysosomotropic weak bases (i.e. NH4Cl, CLQ, or MA) effectively inhibited DOX accumulation in LAMP2-stained lysosomes, as shown by the green fluorescence in the merge (Fig. 7). Instead, DOX localized within nuclei, as shown by the purple nuclear pattern in the merge (Fig. 7). Together with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay cytotoxicity data (Fig. 6, B, C, G, and H), these results demonstrate that Pgp-dependent accumulation of DOX in LAMP2-stained lysosomes (Fig. 7) contributes to drug resistance.
FIGURE 7.
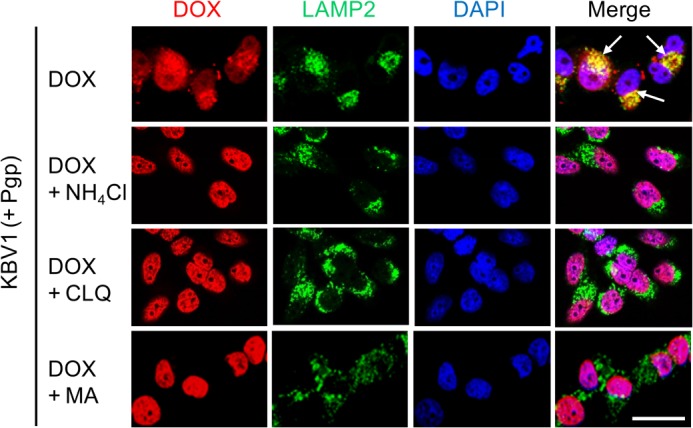
Lysosomal Pgp increases lysosomal trapping of the Pgp substrate DOX, thereby preventing DOX from reaching its nuclear targets. KBV1 (+Pgp) cells expressing functional Pgp led to colocalization of DOX (red) with the marker for lysosomes, LAMP2 (green), leading to yellow fluorescence after the image merge (arrows). The addition of lysosomotropic weak bases to KBV1 (+Pgp) cells redistributed DOX (red) from the lysosomes (green, LAMP2) to the nucleus (blue, DAPI), leading to purple fluorescence of the nuclei in the merged image. Confocal microscopy results are typical from three experiments. Scale bar = 50 μm.
Drug Resistance Conferred by Pgp Is Dependent on the Charge of Pgp Substrates at Acidic Lysosomal pH
The results in Figs. 6 and 7 indicate that the charged species of DOX are important for lysosomally mediated cytoprotection. Considering this, we assessed whether this cytoprotective effect occurs with other common Pgp substrates (Fig. 8). This was examined by assessing the ionization of a range of cytotoxic drugs and then determining whether the lysosomotropic weak bases could overcome Pgp-mediated drug resistance.
FIGURE 8.

Drug resistance conferred by Pgp is dependent on the charge of Pgp substrates at acidic pH. A, lysosomal trapping ability of a range of Pgp substrates as shown by speciation plots (derived from pKa values of each substrate (9, 11, 12, 29)). At lysosomal pH (pH ∼5), DOX (100%), DNR (100%), and VBL (71%) are charged, whereas COL and PAC are not. Incubation (IC50/72 h) of KB31 (−Pgp) cells (B) or 2008 (−Pgp) cells (C) with the lysosomotropic weak bases ammonium chloride (NH4Cl, 3 mm), CLQ (1 μm), or MA (100 μm) did not significantly change cytotoxicity to the Pgp substrates DOX, DNR, VBL, COL, or PAC. However, incubation of Pgp-expressing KBV1 (+Pgp) cells (B) or 2008/P200 (+Pgp) cells (C) with the lysosomotropic weak bases markedly increased sensitivity to the Pgp substrates DOX, DNR, and VBL but not to COL or PAC. The graphs (B and C) show mean ± S.D. ***, p < 0.001 versus control (i.e. drug alone).
First, the distribution of ionized species derived from pKa data (9, 11, 12) was plotted as a function of pH for the cytotoxic drugs DOX (pKa, 8.2), daunorubicin (DNR) (pKa, 8.4), VBL (pKa, 5.4, 7.4), colchicine (COL) (pKa, 12.3), and PAC (pKa, 11.9) (Fig. 8A). At the lysosomal pH of 5 (29), DOX, DNR, and VBL were 100, 100, and 71% charged, respectively, whereas COL and PAC (10) were 0% charged (Fig. 8A). Second, the cytotoxicity of Pgp substrates in the presence of lysosomotropic weak bases was examined in KB31 (−Pgp) and KBV1 (+Pgp) cells (Fig. 8B). Although no significant difference in cytotoxicity was observed for KB31 (−Pgp) cells (Fig. 8B), DOX, DNR, and VBL displayed significantly (p < 0.001) increased cytotoxicity in the presence of the lysosomotropic weak bases in KBV1 (+Pgp) cells (Fig. 8B). In contrast, the other Pgp substrates, namely COL and PAC (31), did not display increased cytotoxicity in the presence of lysosomotropic weak bases (Fig. 8B). Because both of these latter drugs are Pgp substrates (31), the lack of effect of lysosomotropic weak bases on their cytotoxicity could be explained by the fact that they do not become charged and trapped in the acidic pH of lysosomes (11, 12). Similar results to those presented for KBV1 (+Pgp) and KB31 (−Pgp) (Fig. 8B) were also demonstrated in 2008 (−Pgp) and 2008/P200 cells (Fig. 8C). Finally, it is notable that no significant alteration in cytotoxicity of the non-Pgp substrates cisplatin or carboplatin (32) were observed by lysosomotropic weak bases, regardless of Pgp expression (Fig. 9).
FIGURE 9.
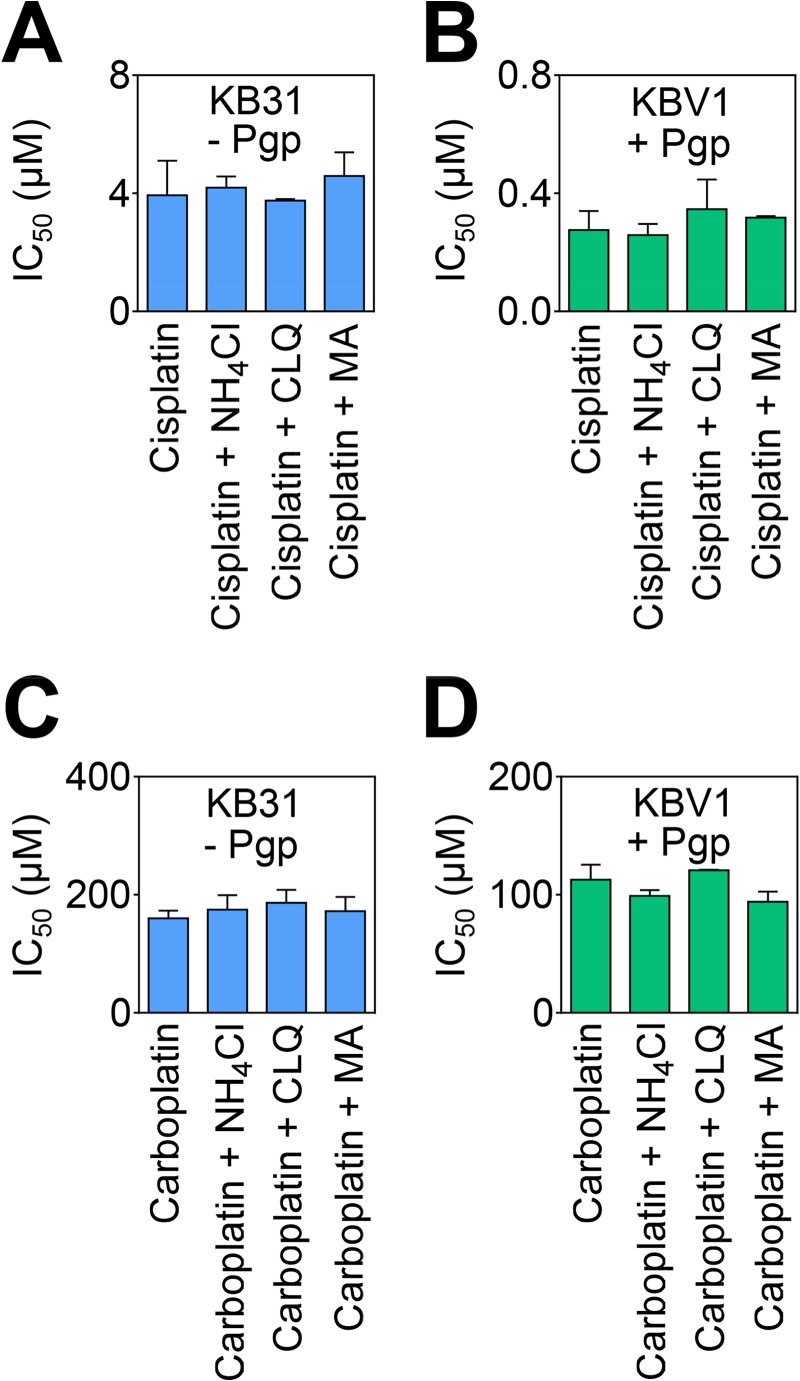
Lysosomotropic weak bases have no effect on the cytotoxicity of drugs that are not Pgp substrates. Incubation of KB31 (−Pgp) or KBV1 (+Pgp) cells with lysosomotropic weak bases has no effect on the cytotoxicity of cisplatin (A and B) or carboplatin (C and D), both of which are not Pgp substrates (32). Cells were incubated for 72 h at 37 °C with cisplatin or carboplatin in the presence or absence of the lysosomotropic weak bases ammonium chloride (NH4Cl, 3 mm), CLQ (1 μm), or MA (100 μm). Proliferation was then assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Results shown are mean ± S.D. (three experiments).
In summary, resistance to cytotoxic agents conferred by lysosomal Pgp is dependent on the charge of these drugs at acidic lysosomal pH (i.e. pH 5). Ionizable Pgp substrates that become charged under acidic lysosomal conditions become trapped in lysosomes. Hence, this leads to cytoprotection because these agents are prevented from accessing their molecular targets, e.g. in the case of DOX, DNA in the nucleus (25, 26).
DISCUSSION
It is well established that plasma membrane Pgp actively effluxes cytotoxic substrates such as DOX, resulting in MDR (1). However, intracellular localization of Pgp and its contribution to MDR have not been comprehensively investigated, particularly in relation to drug ionization properties. For the first time, this investigation demonstrates that lysosomal Pgp plays an important role in conferring drug resistance and offers a novel strategy of targeting lysosomes to overcome MDR.
This study focused on three key organelles for intracellular Pgp localization, namely the nucleus (4, 33), mitochondrion (34, 35), and lysosome (36), where Pgp expression has been suggested (3, 4, 35, 36) but not fully characterized. We showed that Pgp was present in lysosomes but not in nuclei or mitochondria (Fig. 2, A–C). Considering that the lysosomal membrane originates from the plasma membrane via endocytosis (37), Pgp should be detected in lysosomes. Consistent with our observations of Pgp being localized to lysosomes and not mitochondria, several studies have reported the absence of Pgp in mitochondria of KBV1 (+Pgp) cells (19) and Pgp-transfected cells (38). Furthermore, two independent groups reported Pgp to be present in lysosomes of cells transfected with Pgp (36, 38). However, the functionality of lysosomal Pgp in these latter studies was not elucidated.
Because of inversion of the topological orientation of Pgp during endocytosis and the formation of lysosomes (Fig. 10), lysosomal Pgp should be active. Because the catalytic sites for ATP hydrolysis and the drug-binding sites on Pgp remain on the cytoplasmic surface of vesicles (39, 40), this would enable the transport of Pgp substrates into the vesicle lumen (Fig. 10A). In this study, the functionality of lysosomal Pgp was demonstrated by investigations showing that only Pgp-expressing cells accumulated DOX (24) in lysosomes (Fig. 4); that inhibition of Pgp prevented lysosomal uptake of DOX and resulted in its nuclear redistribution (Fig. 4); and that siRNA-mediated silencing of Pgp also prevented lysosomal uptake of DOX, resulting in its accumulation in nuclei (Fig. 5). Hence, for the first time, these results clearly demonstrate the functionality of lysosomal Pgp.
FIGURE 10.
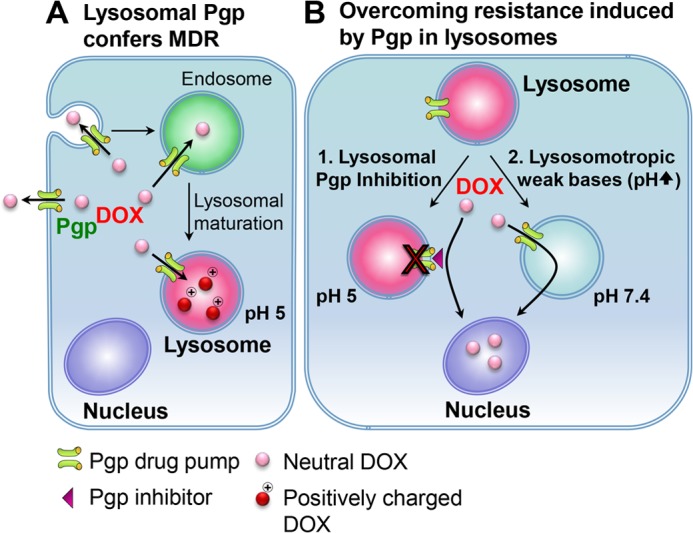
Lysosomal trapping by Pgp as an alternative multidrug resistance mechanism. A, as part of endocytosis, the plasma membrane containing Pgp buds inwards to form early endosomes. During endocytosis, the topology of Pgp will be inverted, as shown for other membrane proteins (45), leading to the transport of substrates into the vesicle lumen. As the endosome matures into a lysosome, it becomes increasingly acidified. The Pgp on the lysosomal membrane remains functional because its catalytic active sites and ATP-binding domains are still exposed in the cytosol (39, 40). When a Pgp substrate, such as DOX, enters the cell, the drug is not only effluxed out of the cell by Pgp on the plasma membrane but also sequestered into the acidic lysosomes by lysosomal Pgp pumps. If the Pgp substrate is charged at acidic pH (such as DOX), then lysosomal trapping occurs. The trapping of charged drugs will prevent Pgp substrates from reaching their molecular targets (e.g. the nucleus for DOX), leading to increased resistance in Pgp-expressing cells. B, this study describes two mechanisms to overcome lysosomal Pgp-dependent multidrug resistance: direct blocking of Pgp by Pgp inhibitors, leading to prevention of increased uptake of Pgp substrates into lysosomes (1), or the combination of cytotoxic drugs (e.g. DOX) with lysosomotropic weak bases (e.g. CLQ) to prevent lysosomal trapping by raising lysosomal pH (2). These two approaches offer Pgp substrates an opportunity to overcome multidrug resistance by reaching their targets instead of becoming trapped in Pgp-containing lysosomes.
We next assessed whether functional lysosomal Pgp can confer MDR. It is generally known that neutral species of drugs can permeate membranes, whereas charged species are far less permeable (41–43). As a weak base, DOX can be charged and trapped in the acidic pH of the lysosome (11) (Fig. 10A). Considering this, raising lysosomal pH with well characterized lysosomotropic weak bases (37) allows the neutral species of DOX to transverse the lysosomal membrane and relocate to the nucleus (Figs. 7 and 10B). Furthermore, this incubation with lysosomotropic weak bases resulted in increased sensitivity to DOX only in Pgp-expressing cells (Fig. 6, C and H), demonstrating the importance of lysosomal Pgp in conferring drug resistance.
Similarly, speciation plots showed that other chemotherapeutics that are Pgp substrates (i.e. DNR and VBL) could be charged at the acidic pH of the lysosome (Fig. 8), leading to entrapment within this organelle. Intriguingly, our studies also demonstrated that this entrapment was abrogated using lysosomotropic weak bases that increase the acidic pH of the lysosome (37). This effect leads to the neutral cytotoxic drugs passing through the lysosomal membrane, enabling access to their targets, which, for DOX, is DNA in the nucleus (Fig. 10B). These observations explain the increased sensitivity of Pgp-expressing cells to DOX, DNR, and VBL caused by lysosomotropic weak bases (Fig. 8, B and C). In contrast, the cytotoxicity of PAC and COL was not affected by lysosomotropic weak bases because they remain neutral at the acidic pH of lysosomes. Hence, PAC and COL remained membrane-permeable, exploiting their molecular targets regardless of lysosomal pH. Additionally, no alteration in cytotoxicity of the non-Pgp substrates cisplatin and carboplatin was observed by lysosomotropic weak bases, irrespective of Pgp expression (Fig. 9). Collectively, these data indicate that there are three essential properties for lysosomal trapping and lysosomal Pgp-mediated drug resistance of chemotherapeutics. The drug must be a Pgp substrate, this agent must become charged at lysosomal pH, and the key molecular targets must lie outside the lysosome.
In summary, this investigation has dissected the role of lysosomal Pgp in drug resistance. We demonstrate that Pgp is not only functional on the plasma membrane but also in lysosomes. Indeed, lysosomal Pgp increases lysosomal trapping of Pgp substrates, thereby preventing such drugs from reaching their targets. This investigation offers a novel strategy for overcoming MDR by targeting lysosomes using specific Pgp inhibitors or a combination of lysosomotropic weak bases with anticancer drugs. In fact, CLQ has been demonstrated previously to overcome resistance to cytotoxics that are Pgp substrates in cancer cells, although the molecular mechanism involved remained unknown (44). This study provides the first detailed explanation that elucidates this earlier observation.
This work was supported by a National Health and Medical Research Council of Australia project grant.
- MDR
- multidrug resistance
- Pgp
- P-glycoprotein
- DOX
- doxorubicin
- Val
- valspodar
- Ela
- elacridar
- VBL
- vinblastine
- PAC
- paclitaxel
- SDHA
- succinate dehydrogenase complex, subunit A
- CLQ
- chloroquine
- MA
- methylamine
- Scr
- scrambled
- DNR
- daunorubicin
- COL
- colchicine
- Rh123
- rhodamine 123.
REFERENCES
- 1. Higgins C. F. (2007) Multiple molecular mechanisms for multidrug resistance transporters. Nature 446, 749–757 [DOI] [PubMed] [Google Scholar]
- 2. Fu D., Bebawy M., Kable E. P., Roufogalis B. D. (2004) Dynamic and intracellular trafficking of P-glycoprotein-EGFP fusion protein. Implications in multidrug resistance in cancer. Int. J. Cancer 109, 174–181 [DOI] [PubMed] [Google Scholar]
- 3. Solazzo M., Fantappiè O., Lasagna N., Sassoli C., Nosi D., Mazzanti R. (2006) P-GP localization in mitochondria and its functional characterization in multiple drug-resistant cell lines. Exp. Cell Res. 312, 4070–4078 [DOI] [PubMed] [Google Scholar]
- 4. Baldini N., Scotlandi K., Serra M., Shikita T., Zini N., Ognibene A., Santi S., Ferracini R., Maraldi N. M. (1995) Nuclear immunolocalization of P-glycoprotein in multidrug-resistant cell lines showing similar mechanisms of doxorubicin distribution. Eur. J. Cell Biol. 68, 226–239 [PubMed] [Google Scholar]
- 5. Yuan J., Lovejoy D. B., Richardson D. R. (2004) Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity. In vitro and in vivo assessment. Blood 104, 1450–1458 [DOI] [PubMed] [Google Scholar]
- 6. Richardson D. R., Tran E. H., Ponka P. (1995) The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents. Blood 86, 4295–4306 [PubMed] [Google Scholar]
- 7. Ivanova S., Repnik U., Bojic L., Petelin A., Turk V., Turk B. (2008) Chapter Nine Lysosomes in Apoptosis. Methods Enzymol. 442, 183–199 [DOI] [PubMed] [Google Scholar]
- 8. Braun M., Waheed A., von Figura K. (1989) Lysosomal acid phosphatase is transported to lysosomes via the cell surface. EMBO J. 8, 3633–3640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raghunand N., Gillies R. J. (2000) pH and drug resistance in tumors. Drug Resist. Updates 3, 39–47 [DOI] [PubMed] [Google Scholar]
- 10. Straubinger R. M. (1995) in Taxol. Science and Applications (Suffness M., ed) pp. 237–254, CRC Press, New York [Google Scholar]
- 11. Mahoney B. P., Raghunand N., Baggett B., Gillies R. J. (2003) Tumor acidity, ion trapping and chemotherapeutics. I. Acid pH affects the distribution of chemotherapeutic agents in vitro. Biochem. Pharmacol. 66, 1207–1218 [DOI] [PubMed] [Google Scholar]
- 12. Molad Y. (2002) Update on colchicine and its mechanism of action. Curr. Rheumatol. Rep. 4, 252–256 [DOI] [PubMed] [Google Scholar]
- 13. Abulrob A. N., Mason M., Bryce R., Gumbleton M. (2000) The effect of fatty acids and analogues upon intracellular levels of doxorubicin in cells displaying P-glycoprotein mediated multidrug resistance. J. Drug Target. 8, 247–256 [DOI] [PubMed] [Google Scholar]
- 14. Lavie Y., Cao H., Bursten S. L., Giuliano A. E., Cabot M. C. (1996) Accumulation of glucosylceramides in multidrug-resistant cancer cells. J. Biol. Chem. 271, 19530–19536 [DOI] [PubMed] [Google Scholar]
- 15. Akhtar N., Ahad A., Khar R. K., Jaggi M., Aqil M., Iqbal Z., Ahmad F. J., Talegaonkar S. (2011) The emerging role of P-glycoprotein inhibitors in drug delivery. A patent review. Expert. Opin. Ther. Pat. 21, 561–576 [DOI] [PubMed] [Google Scholar]
- 16. Jaffrézou J. P., Levade T., Chatelain P., Laurent G. (1992) Modulation of subcellular distribution of doxorubicin in multidrug-resistant P388/ADR mouse leukemia cells by the chemosensitizer ((2-isopropyl-1-(4-[3-N-methyl-N-(3,4-dimethoxy-β- phenethyl)amino]propyloxy)-benzenesulfonyl))indolizine. Cancer Res. 52, 6440–6446 [PubMed] [Google Scholar]
- 17. Huynh K. K., Eskelinen E. L., Scott C. C., Malevanets A., Saftig P., Grinstein S. (2007) LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 26, 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pezzini F., Gismondi F., Tessa A., Tonin P., Carrozzo R., Mole S. E., Santorelli F. M., Simonati A. (2011) Involvement of the mitochondrial compartment in human NCL fibroblasts. Biochem. Biophys. Res. Commun. 416, 159–164 [DOI] [PubMed] [Google Scholar]
- 19. Paterson J. K., Gottesman M. M. (2007) P-Glycoprotein is not present in mitochondrial membranes. Exp. Cell Res. 313, 3100–3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bolte S., Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 [DOI] [PubMed] [Google Scholar]
- 21. Lovejoy D. B., Jansson P. J., Brunk U. T., Wong J., Ponka P., Richardson D. R. (2011) Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res. 71, 5871–5880 [DOI] [PubMed] [Google Scholar]
- 22. Dokmanovic M., Clarke C., Marks P. A. (2007) Histone deacetylase inhibitors. Overview and perspectives. Mol. Cancer Res. 5, 981–989 [DOI] [PubMed] [Google Scholar]
- 23. Bardella C., Pollard P. J., Tomlinson I. (2011) SDH mutations in cancer. Biochim. Biophys. Acta 1807, 1432–1443 [DOI] [PubMed] [Google Scholar]
- 24. Shen F., Chu S., Bence A. K., Bailey B., Xue X., Erickson P. A., Montrose M. H., Beck W. T., Erickson L. C. (2008) Quantitation of doxorubicin uptake, efflux, and modulation of multidrug resistance (MDR) in MDR human cancer cells. J. Pharmacol. Exp. Ther. 324, 95–102 [DOI] [PubMed] [Google Scholar]
- 25. Taatjes D. J., Koch T. H. (2001) Nuclear targeting and retention of anthracycline antitumor drugs in sensitive and resistant tumor cells. Curr. Med. Chem. 8, 15–29 [DOI] [PubMed] [Google Scholar]
- 26. van Rosmalen A., Cullinane C., Cutts S. M., Phillips D. R. (1995) Stability of adriamycin-induced DNA adducts and interstrand crosslinks. Nucleic Acids Res. 23, 42–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kannan P., Brimacombe K. R., Kreisl W. C., Liow J. S., Zoghbi S. S., Telu S., Zhang Y., Pike V. W., Halldin C., Gottesman M. M., Innis R. B., Hall M. D. (2011) Lysosomal trapping of a radiolabeled substrate of P-glycoprotein as a mechanism for signal amplification in PET. Proc. Natl. Acad. Sci. U.S.A. 108, 2593–2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barranco W. T., Eckhert C. D. (2006) Cellular changes in boric acid-treated DU-145 prostate cancer cells. Br. J. Cancer 94, 884–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Altan N., Chen Y., Schindler M., Simon S. M. (1998) Defective acidification in human breast tumor cells and implications for chemotherapy. J. Exp. Med. 187, 1583–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Antoine J. C., Goud B., Jouanne C., Maurice M., Feldmann G. (1985) Ammonium chloride, methylamine and chloroquine reversibly inhibit antibody secretion by plasma cells. Biol. Cell 55, 41–54 [DOI] [PubMed] [Google Scholar]
- 31. Wang Y. H., Li Y., Yang S. L., Yang L. (2005) Classification of substrates and inhibitors of P-glycoprotein using unsupervised machine learning approach. J. Chem. Inf. Model. 45, 750–757 [DOI] [PubMed] [Google Scholar]
- 32. Stordal B., Hamon M., McEneaney V., Roche S., Gillet J. P., O'Leary J. J., Gottesman M., Clynes M. (2012) Resistance to paclitaxel in a cisplatin-resistant ovarian cancer cell line is mediated by P-glycoprotein. PLoS ONE 7, e40717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maraldi N. M., Zini N., Santi S., Scotlandi K., Serra M., Baldini N. (1999) P-glycoprotein subcellular localization and cell morphotype in MDR1 gene-transfected human osteosarcoma cells. Biol. Cell 91, 17–28 [PubMed] [Google Scholar]
- 34. Solazzo M., Fantappiè O., D'Amico M., Sassoli C., Tani A., Cipriani G., Bogani C., Formigli L., Mazzanti R. (2009) Mitochondrial expression and functional activity of breast cancer resistance protein in different multiple drug-resistant cell lines. Cancer Res. 69, 7235–7242 [DOI] [PubMed] [Google Scholar]
- 35. Munteanu E., Verdier M., Grandjean-Forestier F., Stenger C., Jayat-Vignoles C., Huet S., Robert J., Ratinaud M. H. (2006) Mitochondrial localization and activity of P-glycoprotein in doxorubicin-resistant K562 cells. Biochem. Pharmacol. 71, 1162–1174 [DOI] [PubMed] [Google Scholar]
- 36. Rajagopal A., Simon S. M. (2003) Subcellular localization and activity of multidrug resistance proteins. Mol. Biol. Cell 14, 3389–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Styrt B., Klempner M. S. (1986) Inhibition of neutrophil oxidative metabolism by lysosomotropic weak bases. Blood 67, 334–342 [PubMed] [Google Scholar]
- 38. Fu D., Roufogalis B. D. (2007) Actin disruption inhibits endosomal traffic of P-glycoprotein-EGFP and resistance to daunorubicin accumulation. Am. J. Physiol. Cell Physiol. 292, C1543–1552 [DOI] [PubMed] [Google Scholar]
- 39. Sarkadi B., Price E. M., Boucher R. C., Germann U. A., Scarborough G. A. (1992) Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated membrane ATPase. J. Biol. Chem. 267, 4854–4858 [PubMed] [Google Scholar]
- 40. Shapiro A. B., Ling V. (1994) ATPase activity of purified and reconstituted P-glycoprotein from Chinese hamster ovary cells. J. Biol. Chem. 269, 3745–3754 [PubMed] [Google Scholar]
- 41. de Duve C., de Barsy T., Poole B., Trouet A., Tulkens P., Van Hoof F. (1974) Commentary. Lysosomotropic agents. Biochem. Pharmacol. 23, 2495–2531 [DOI] [PubMed] [Google Scholar]
- 42. Seelig A. (2007) The role of size and charge for blood-brain barrier permeation of drugs and fatty acids. J. Mol. Neurosci. 33, 32–41 [DOI] [PubMed] [Google Scholar]
- 43. Saparov S. M., Antonenko Y. N., Pohl P. (2006) A new model of weak acid permeation through membranes revisited. Does Overton still rule? Biophys. J. 90, L86–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shiraishi N., Akiyama S., Kobayashi M., Kuwano M. (1986) Lysosomotropic agents reverse multiple drug resistance in human cancer cells. Cancer Lett. 30, 251–259 [DOI] [PubMed] [Google Scholar]
- 45. Mukherjee S., Ghosh R. N., Maxfield F. R. (1997) Endocytosis Physiol. Rev. 77, 759–803 [DOI] [PubMed] [Google Scholar]