

Background: A novel gene, fad104, regulates adipocyte and osteoblast differentiation in vitro.
Results: fad104 disruption caused craniosynostosis-like premature ossification of the calvarial bone.
Conclusion: fad104 has an essential role for calvarial bone formation through regulation of BMP/Smad signaling.
Significance: This study provides new insight into the molecular mechanism of calvarial ossification.
Keywords: Adipogenesis, Bone, Bone Morphogenetic Protein (BMP), Osteoblasts, SMAD Transcription Factor, Calvarial Bone Formation
Abstract
Osteogenesis is a complex process that is orchestrated by several growth factors, extracellular cues, signaling molecules, and transcriptional factors. Understanding the mechanisms of bone formation is pivotal for clarifying the pathogenesis of bone diseases. Previously, we reported that fad104 (factor for adipocyte differentiation 104), a novel positive regulator of adipocyte differentiation, negatively regulated the differentiation of mouse embryonic fibroblasts into osteocytes. However, the physiological role of fad104 in bone formation has not been elucidated. Here, we clarified the role of fad104 in bone formation in vivo and in vitro. fad104 disruption caused craniosynostosis-like premature ossification of the calvarial bone. Furthermore, analyses using primary calvarial cells revealed that fad104 negatively regulated differentiation and BMP/Smad signaling pathway. FAD104 interacted with Smad1/5/8. The N-terminal region of FAD104, which contains a proline-rich motif, was capable of binding to Smad1/5/8. We demonstrated that down-regulation of Smad1/5/8 phosphorylation by FAD104 is dependent on the N-terminal region of FAD104 and that fad104 functions as a novel negative regulator of BMP/Smad signaling and is required for proper development for calvarial bone. These findings will aid a comprehensive description of the mechanism that controls normal and premature calvarial ossification.
Introduction
Bone mass and strength are balanced by bone formation by osteoblasts and bone resorption by osteoclasts (1). Numerous growth factors, hormones, cytokines, and extracellular matrix proteins trigger several signaling cascades and transcriptional networks to regulate osteogenesis (2, 3). Among them, bone morphogenetic proteins (BMPs),3 which are members of the transforming growth factor β (TGF-β) superfamily, strongly potentiate osteoblast differentiation and bone formation (4).
Previously, we isolated several novel up-regulated genes in the early stages of adipocyte differentiation using the polymerase chain reaction (PCR) subtraction method (5, 6) and demonstrated that they positively regulated adipocyte differentiation in mouse 3T3-L1 cells (7–10). The expression of fad104, also called fndc3b, is temporarily increased 3 h after adipogenic induction. The FAD104 protein has a proline-rich region, nine fibronectin type III domains, and a transmembrane domain. Our laboratory previously reported that fad104 positively regulated adipocyte differentiation but negatively regulated osteoblast differentiation in mouse embryonic fibroblasts (MEFs) (11).
We also demonstrated that fad104-deficient mice died at birth because of lung abnormalities, such as immature alveolar epithelial type II cells (12, 13). However, the skeletal phenotype of fad104-deficient mice was not examined adequately. In this paper, we characterized the function of fad104 in bone formation both in vivo and in vitro. Skeletal staining of fad104-deficient embryos revealed that fad104 disruption caused craniosynostosis-like premature calvarial ossification. Furthermore, fad104 negatively regulated primary calvarial cell differentiation through the inhibition of the BMP/Smad signaling cascade and interacted with Smad1/5/8. The N terminus of FAD104 was important for inhibition of Smad1/5/8 phosphorylation. These findings indicate that fad104 suppressed BMP/Smad signaling and controlled normal calvarial bone formation.
EXPERIMENTAL PROCEDURES
Animal Care
Animal experiments were performed with approval from the Committee on the Ethics of Animal Experiments in Nagoya City University. The generation of mice with targeted disruption of fad104 has been described (12). fad104-deficient mice were generated by crossing fad104 heterozygous knock-out mice maintained in the C57BL/6J strain background. All heterozygous knock-out mice were backcrossed onto the C57BL/6J background for more than 13 generations.
Alizarin Red S/Alcian Blue Skeletal Staining
Wild-type and fad104−/− embryos at E18.5 were killed under anesthesia and frozen. These specimens were skinned, and the viscera were removed. The samples were fixed in ethanol for 3 days; delipidated in acetone for 4 days; stained with Alizarin red S/Alcian blue solution for 7 days; destained with 1% KOH, 50% glycerol until completely cleared; and then photographed. The anteroposterior length, transversal length of the calvaria, and the anterior fontanel areas were measured by NIH Image software (National Institutes of Health). The femur widths of femur are presented as the mean values of the middle and ossified ends, as measured using NIH Image.
Histology
Femurs from wild-type and fad104-deficient embryo at E18.5 were fixed overnight at 4 °C in 4% paraformaldehyde, embedded in paraffin, and sectioned 20 μm thick. Sections were stained with Alizarin red S to visualize the mineralized tissue and counterstained with hematoxylin. The morphometric analysis was performed with images of Alizarin red S-stained femurs. We measured the femur width of all sections and selected the thickest of femurs. Then the central width of cortical bone in these sections was measured.
Preparation of Primary Calvarial Cells and Osteogenesis
Primary calvarial cells were isolated from wild-type and fad104−/− embryos at E18.5 according a method described by Kanamoto et al., (14). Cells were isolated by five sequential 10-min digestions in 0.1% collagenase, 0.2% dispase in minimum essential medium at 37 °C. Fractions 3–5 were collected and resuspended in α-minimum essential medium (α-MEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS) and plated. The fractions were used at low passage numbers (<2 passages) throughout the experiments. Cells were cultured in α-MEM supplemented with 10% FBS. Osteogenesis was induced according to methods described by Kishimoto et al. (11). Calvarial cells were grown to confluence, and the medium was changed to α-MEM supplemented with 10% FBS, 50 μg/ml l-ascorbic acid, 10 mm β-glycerophosphate, and 100 ng/ml BMP-2 (Peprotech), which was renewed every other day.
Preparation of MEFs and Adipocyte Differentiation
Wild-type and fad104−/− MEFs were isolated from decapitated embryonic day 13.5 embryos (12). Cells were cultured in α-MEM supplemented with 10% FBS. For the differentiation using potent adipogenic inducers, the medium was changed to α-MEM supplemented with 1 μm dexamethasone, 0.5 mm 3-isobutyl-1-methylxantine, 5 μg/ml insulin, and 10% FBS at 2 days postconfluence (12). For the adipocyte differentiation using osteogenic inducer containing BMP2, the medium was changed to α-MEM supplemented with 100 ng/ml BMP2, 50 μg/ml l-ascorbic acid, 10 mm β-glycerophosphate, and 10% FBS at 2 days postconfluence. These media were renewed every other day (11).
Adenoviral Infection
An adenoviral vector carrying mouse fad104 cDNA was constructed using the Adenovirus Expression Vector Kit (dual version) version 2 (TaKaRa). The fad104 open reading frame was blunted and subcloned into the SmiI site of the pAxCAwtit2 cosmid vector. To obtain recombinant adenoviruses, pAxCAwtit2-fad104 and the control vector pAxCAiLacZit were transfected into human embryonic kidney (HEK293T) cells by Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Subsequently, adenoviruses were propagated in HEK293T cells, and the viral supernatant was collected. The viral titers were determined by 50% tissue culture infective dose analysis. Primary calvarial cells and HeLa cells were infected with recombinant adenoviruses by incubating with adenoviruses at a multiplicity of infection of 200.
Alizarin Red S Staining
Cells were rinsed with phosphate-buffered saline (PBS (−); Ca2+/Mg2+-free PBS), fixed with 4% paraformaldehyde, and stained with 1% Alizarin red S (Sigma).
Real-time Quantitative PCR (qRT-PCR)
qRT-PCR was performed using an ABI PRISM 7000 sequence detection system (Applied Biosystems) with predesigned primers and probe sets for fad104, runx2, Ocn, Id1, and 18 S rRNA (Applied Biosystems) and TaqMan Universal PCR mixture.
Western Blotting
Cells were washed with PBS (−) and lysed in radioimmunoprecipitation assay buffer supplemented with a protease inhibitor mixture and a phosphatase inhibitor mixture (Nacalai Tesque). After centrifugation, the supernatant was harvested. Equal protein amounts were resolved using SDS-PAGE. The resolved proteins were transferred to a nitrocellulose membrane and probed using primary antibody and subsequently secondary antibody conjugated with horseradish peroxidase (1:10,000; Jackson ImmunoResearch Laboratories). Specific proteins were detected using an enhanced chemiluminescence system (GE Healthcare). Primary antibodies recognizing mouse FAD104 (1:200) (13), phospho-Smad1/5/8 (1:500; Cell Signaling), Smad1/5/8 (1:200; Santa Cruz Biotechnology, Inc.), β-actin (1:100,000; Sigma), FLAG (1:500; Sigma), and Myc (1:500; Roche Applied Science) were used.
Plasmid Construction
p3xFLAG-fad104, a FLAG-tagged FAD104 expression plasmid, has been described previously (12). p3xFLAG-fad104ΔN, a FLAG-tagged FAD104 mutant expression plasmid lacking the N-terminal proline-rich motif (Δ1–212) (FAD104ΔN), was constructed by subcloning the fragment amplified by PCR with the primers 5′-ACGCGTCGACTTACTGTTGTAACATGAAAG-3′ and 5′-GGTGTCTGGACCATGATAAACG-3′ into p3xFLAG-CMVTM-7.1 (Sigma). pCMV-Myc-fad104N, a Myc-tagged N-terminal region (amino acids 1–277) of the FAD104 expression plasmid, and pGEX-4T2-fad104N, a glutathione S-transferase (GST)-tagged N-terminal region (amino acids 1–277) of the FAD104 expression plasmid, were constructed by subcloning the fragment amplified by PCR with the primers (5′-AGAGAATTCGAATGTACGTCACCATGATGATG-3′) and (5′-TTTCTCGAGTTAGTCTTGCACCCTCTTTACTT-3′) into the pCMV-Myc (BD Biosciences) or pGEX-4T2 (GE Healthcare) plasmids, respectively. pCMV-Myc-fad104 was constructed by subcloning the fragment digested from p3xFLAG-fad104 into the pCMV-Myc vector.
Cell Cultures and BMP2 Stimulation
C2C12 and HeLa cells were cultured as described previously (7). For BMP2 treatment, C2C12 cells were starved for 6 h and then treated with 100 ng/ml recombinant human BMP2.
Immunoprecipitation
The p3xFLAG-fad104, p3xFLAG-fad104ΔN, or pCMV-Myc-fad104N plasmid was introduced into HeLa cells using the calcium phosphate co-precipitation method (15). The cells were harvested 48 h post-transfection and lysed with Nonidet-P40 lysis buffer (50 mm Tris-HCl, pH 7.5, 0.5% Nonidet P-40, and 0.15 m NaCl) containing the protease inhibitor mixture. Lysates were incubated with the anti-Smad1/5/8 antibody overnight on ice. The mixtures were rotated with protein G-coupled Sepharose beads (GE Healthcare) for 3 h at 4 °C. Normal rabbit IgG (Santa Cruz Biotechnology) was used as a negative control. After extensive washing, the bound proteins were detected via Western blotting.
For detection of interaction between FAD104 and Smad1/5/8 under physiological conditions, C2C12 cells were plated and lysed with Nonidet-P40 lysis buffer. Lysates were incubated with the anti-Smad1/5/8 antibody overnight on ice. The mixtures were rotated with protein G-coupled Sepharose beads overnight at 4 °C. Normal rabbit IgG was used as a negative control. The bound proteins were detected via Western blotting using anti-FAD104 antibody.
GST Pull-down Assay
The pGEX-4T2 and pGEX-4T2-fad104N plasmids were each transformed into BL21 Escherichia coli cells. Cultures (100 ml) were grown to an Aλ = 600 nm = 1.0, and protein expression was induced with 0.01 mm isopropyl β-d-1-thiogalactopyranoside for 1 h at 30 °C. The BL21 cells were centrifuged at 6,000 × g for 10 min at 4 °C, and the proteins were extracted by resuspending the pellets in 5 ml of PBS-G supplemented with proteinase inhibitor mixture. Cells were lysed by sonication on ice. Soluble extracts were collected by centrifugation at 10,000 × g for 10 min at 4 °C. GST or GST-FAD104N lysate was bound to GST-Sepharose beads overnight at 4 °C. The beads were then washed three times with Nonidet P-40 lysis buffer at 4 °C. Equal amounts of C2C12 cell lysates were added to GST- or GST-FAD104N-bound beads and subsequently rotated overnight at 4 °C. After washing, the bound proteins were detected by Western blotting. To analyze direct interaction between FAD104 and Smad1 or Smad5, FLAG-tagged Smad1 and Smad5 were expressed in BL21 E. coli cells and purified using FLAG M2 affinity gel (Sigma). After washing, the proteins were eluted with FLAG peptide (Sigma) and incubated with GST- or GST-FAD104N-bound beads overnight at 4 °C. The bound proteins were detected by Western blotting.
Immunofluorescence
HeLa cells were plated onto cell disks (SUMITOMO BAKELITE) 1 day before transfection. The cells were transfected with FLAG-tagged Smad1 expression plasmid (p3xFLAG-Smad1) and Myc-tagged FAD104 expression plasmid (pCMV-Myc-fad104) or Myc-empty vector using Lipofectamine 2000. 24 h after transfection, the cells were starved for 6 h and then treated with 100 ng/ml recombinant human BMP2 for 2 h. Each cell disk was fixed and incubated with mouse monoclonal anti-FLAG antibody (Sigma) for 1 h at room temperature. After washing five times, TRITC-conjugated goat anti-mouse IgG (Sigma) and fluorescein isothiocyanate (FITC)-conjugated anti-Myc antibody (Sigma) were incubated for 1 h at room temperature. The signals for FITC and TRITC were detected by fluorescence microscopy (BX51, Olympus).
Statistical Tests
Analyses were performed using Excel 2010 (Microsoft Corp.). Statistically significant differences were evaluated using Student's t test.
RESULTS
Disruption of fad104 Caused Skeletal Abnormalities at E18.5
As reported previously, fad104 negatively regulates the differentiation of MEFs into osteocytes (11). However, the role of fad104 in bone formation remains unclear. Therefore, we carefully observed the appearance of fad104-deficient mice at E18.5 and found that the overall cranial shapes were more rounded when compared with that of wild-type mice (Fig. 1A). To further characterize these cranial malformations, we visualized the skulls using Alizarin red S/Alcian blue skeletal staining, which indicated that both the anteroposterior and transversal lengths of the calvaria were slightly, but significantly, decreased in fad104-deficient mice compared with the wild type (Fig. 1, B and C). Notably, the anterior fontanels were markedly closed in fad104-deficient mice; nevertheless, anterior fontanels are usually open at E18.5, as shown in the wild-type mice (Fig. 1, B and C). These phenotypes indicate that disruption of fad104 causes premature calvarial ossification like craniosynostosis.
FIGURE 1.

Disruption of fad104 causes skeletal deformities in the anterior fontanel and femurs. A, lateral views of the skulls from wild-type and fad104-deficient mice at E18.5. B, a superior view of the stained calvaria in wild-type and fad104-deficient mice at E18.5. C, the anteroposterior length, transversal length, and the areas of anterior fontanel of wild-type and fad104-deficient calvaria. Each length and area was measured using NIH Image (wild type, n = 3; fad104−/−, n = 6). D, skeletal staining of wild-type and fad104-deficient mice without the skull at E18.5. E, skeletal staining of wild-type and fad104-deficient legs at E18.5. Femur widths were calculated using NIH Image (wild type, n = 3; fad104−/−, n = 6). The data are presented as the means with S.E. F, Arizalin red S staining of wild-type and fad104-deficient femur section. Sections were counterstained by hematoxylin staining. The red color indicates the mineralized tissues. The thickness of cortical bone is indicated by the means with S.E (wild-type, n = 8; fad104−/−, n = 7).
Next, the skeleton besides the skull was evaluated. In appearance, the constitution of bones in fad104-deficient mice was comparable with the wild type. Although marked skeletal abnormalities were not observed in both ossified and cartilage region, we noticed that the femurs of fad104-deficient mice were slightly wider (Fig. 1, D and E). To further characterize the femur abnormality, we performed histological examination with Alizarin red S staining. Importantly, the thickness of cortical bone was significantly increased, whereas abnormalities were not observed in chondrocytes (Fig. 1F). Collectively, these results suggested that fad104 played a role in bone formation, at least in the calvarium and femur cortical bones.
The bone abnormalities in fad104-deficient mice were quite restricted in calvaria and femur. To determine whether the distribution of fad104 is different, depending on each bone, we tried to compare the expression levels of fad104 at E18.5 in each bone by in situ hybridization and immunohistochemistry. However, we unfortunately failed to show the precise expression pattern of fad104 in these experiments, due in part to the susceptibility of bones to peeling.
Hence, we next examined in situ hybridization patterns of fad104 using the GenePaint database. According to this database, the expression of fad104 was particularly high in calvarium and lung epithelium at E14.5. In addition, fad104 expression was also detected in ribs and axial skeleton. Then we examined the expression of fad104 in calvaria, femur, humerus, clavicle, and rib and found that the expression levels of fad104 were comparable between these bone tissues; likewise, levels of runx2, a master regulator for osteoblast differentiation, and ocn (osteocalcin), a marker of osteocytes (Fig. 2), were comparable. These data imply that the restricted bone phenotype in fad104-deficient mice is not due to the expression pattern of fad104 in each bone.
FIGURE 2.
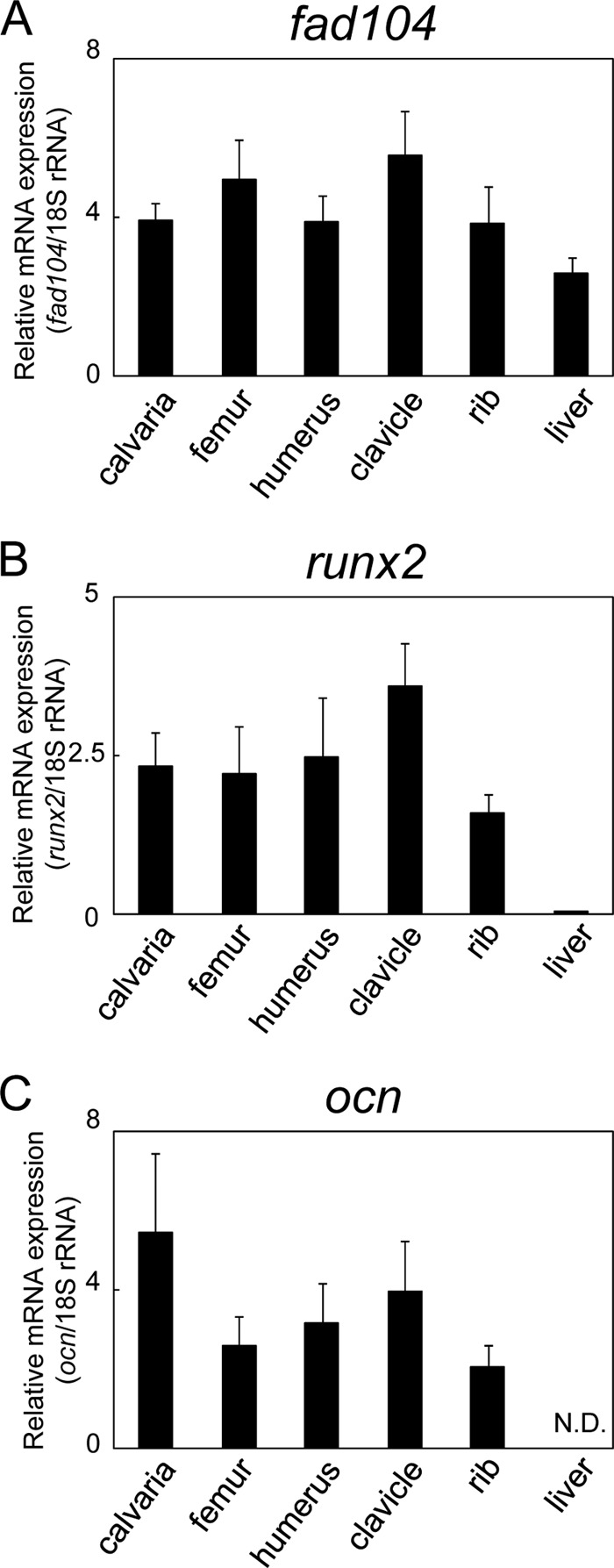
The expression level of fad104, runx2, and ocn in various bone tissues and liver. The expression levels of fad104 (A), runx2 (B), and ocn (C) mRNA in calvaria, femur, humerus, clavicle, ribs, and liver at E18.5 were determined by qRT-PCR and normalized with 18 S rRNA expression. The data represent means with S.E. (error bars) (n = 5).
fad104 Negatively Regulates Calvarial Cell Differentiation
The significant calvarial abnormalities in fad104-deficient mice prompted us to investigate the function of fad104 in calvarial osteoblasts and evaluate the role of fad104 in calvarial cell differentiation. As reported previously (11), fad104 expression was decreased in early stages of osteoblast differentiation in MEFs. However, its expression in the differentiation of primary calvarial cells remains unknown. Therefore, we first examined fad104 mRNA expression and found that during calvarial cell differentiation, it was decreased at 6 h and remained low up to 48 h (Fig. 3A). Similarly, FAD104 protein levels were decreased at 6 h and further down-regulated until day 12 (Fig. 3B), indicating that fad104 expression was inhibited during the early phases of calvarial cell differentiation.
FIGURE 3.
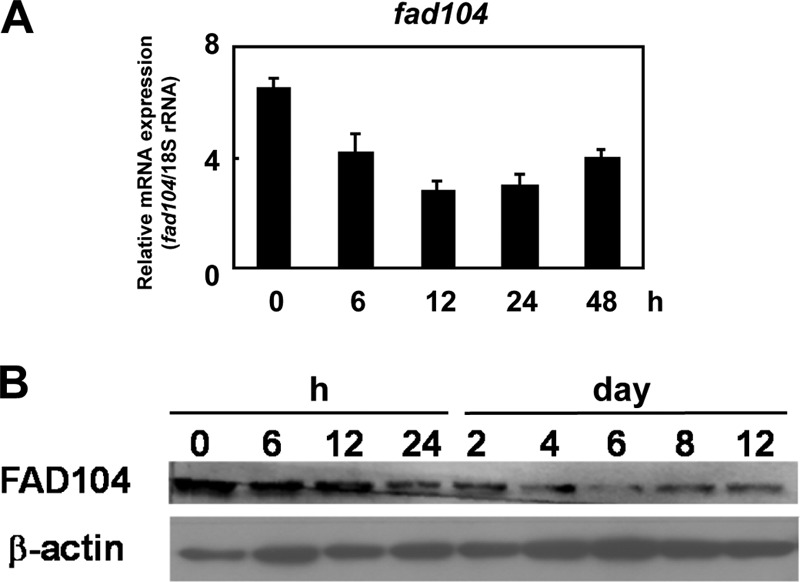
The expression of fad104 decreases in the early stage of calvarial cell differentiation. A, fad104 mRNA expression during calvarial cell differentiation. fad104 mRNA expression at the indicated time points was examined and normalized with 18 S rRNA expression by qRT-PCR. Each column represents the mean and S.D. (error bars) (n = 3). B, FAD104 protein levels during calvarial cell differentiation. The level of FAD104 at the indicated time points was examined by Western blotting.
Next, to evaluate the impact of fad104 on calvarial cell differentiation, wild-type and fad104-deficient cells were induced to differentiate in osteogenic media containing BMP2. Disruption of fad104 enhanced matrix mineralization, as visualized by Alizarin red S staining, and the expression of ocn, a marker of osteoblast differentiation (Fig. 4, A and B). To further define the role of fad104 in calvarial cell differentiation, we generated an adenoviral vector carrying fad104 to obtain greater gene transduction efficiency into primary calvarial cells. The adenovirus-mediated FAD104 expression in fad104-deficient calvarial cells was almost comparable with endogenous FAD104 expression in wild-type calvarial cells on differentiation days 0 and 4 (Fig. 4C). The adenoviral FAD104 expression suppressed the mineralization and the expression of ocn enhanced by fad104 deletion (Fig. 4, D and E). Furthermore, FAD104 overexpression in wild-type cells inhibited calvarial cell differentiation, as assessed by mineralization and ocn expression analysis (Fig. 4, F–H). These results indicated that fad104 negatively regulated calvarial cell differentiation.
FIGURE 4.

fad104 negatively regulates calvarial cell differentiation. A, the mineralization of wild-type and fad104-deficient calvarial cells. Confluent calvarial cells were incubated in osteogenic medium for 12 days and assessed by Alizarin red S staining. B, ocn mRNA expression in wild-type and fad104-deficient calvarial cells at the indicated time points after osteogenic induction. ocn levels were determined and normalized with 18 S rRNA expression by qRT-PCR. The data are presented as means with S.D. (error bars) (n = 3). C, reconstitution of FAD104 expression in fad104-deficient calvarial cells by adenoviral infection during calvarial cell differentiation. Cells were grown to confluence. Wild-type calvarial cells were infected with LacZ (multiplicity of infection = 200), and fad104-deficient calvarial cells were infected with LacZ or FAD104 (multiplicity of infection = 200). The next day (day 0), medium was replaced with fresh medium containing an osteogenic inducer. On days 0 and 4, cells were lysed, and FAD104 expression was determined by Western blotting. β-Actin expression was used as a control. virus (−), uninfected cells. D, the mineralization of wild-type calvarial cells infected with LacZ and fad104-deficient calvarial cells infected with LacZ or FAD104. The cells were infected and cultured under the same conditions as in C and assessed by Alizarin red S staining on day 12. E, mRNA expression of ocn in wild-type calvarial cells infected with LacZ and fad104-deficient calvarial cells infected with LacZ or FAD104 on day 12 after osteogenic induction. The ocn levels were determined and normalized with 18 S rRNA expression by qRT-PCR. The data are presented as means and S.D. (n = 3). F, FAD104 overexpression in wild-type calvarial cells during differentiation. Wild-type calvarial cells were infected with LacZ or FAD104. The next day (day 0), medium was replaced with fresh medium containing an osteogenic inducer. On days 0 and 4, cells were lysed, and FAD104 expression was determined by Western blotting. β-Actin expression was used as a control. G, the mineralization of wild-type calvarial cells infected with LacZ or FAD104. The cells were infected and cultured under the same conditions as in F and assessed by Alizarin red S staining on day 12. H, the ocn mRNA expression in wild-type calvarial cells infected with LacZ or FAD104 on day 12 after osteogenesis induction. The ocn levels were determined and normalized with 18 S rRNA expression by qRT-PCR. The data are presented as means with S.D. (n = 3). *, p < 0.05. C and F show typical results. Similar results were obtained in at least two independent experiments.
FAD104 Negatively Regulates Smad1/5/8 Phosphorylation during Calvarial Cell Differentiation
BMP2 is one of the most potent osteogenic inducers, and the canonical BMP signaling mechanism depends heavily on the Smad pathway (16, 17). BMP2 binds to type I and II BMP receptors and activates downstream molecules Smad1/5/8. Smad1/5/8 phosphorylated by the BMP receptor form a complex with Smad4, which is then translocated to the nucleus, where it regulates the expression of various genes (16, 17). Therefore, we examined Smad1/5/8 phosphorylation during calvarial cell differentiation. In wild-type calvarial cells, Smad1/5/8 phosphorylation was increased 6 and 12 h postinduction and decreased at 24 h postinduction. In contrast, the phosphorylation of Smad1/5/8 was greater in fad104-deficient calvarial cells during the differentiation process; nevertheless, the levels of total Smad1/5/8 protein were only marginally increased in fad104-deficient calvarial cells (Fig. 5A). Moreover, adenovirus-mediated FAD104 expression decreased Smad1/5/8 phosphorylation enhanced by fad104 deletion (Fig. 5B). Additionally, FAD104 overexpression in wild-type cells decreased the phosphorylation levels of Smad1/5/8 during differentiation (Fig. 5C).
FIGURE 5.
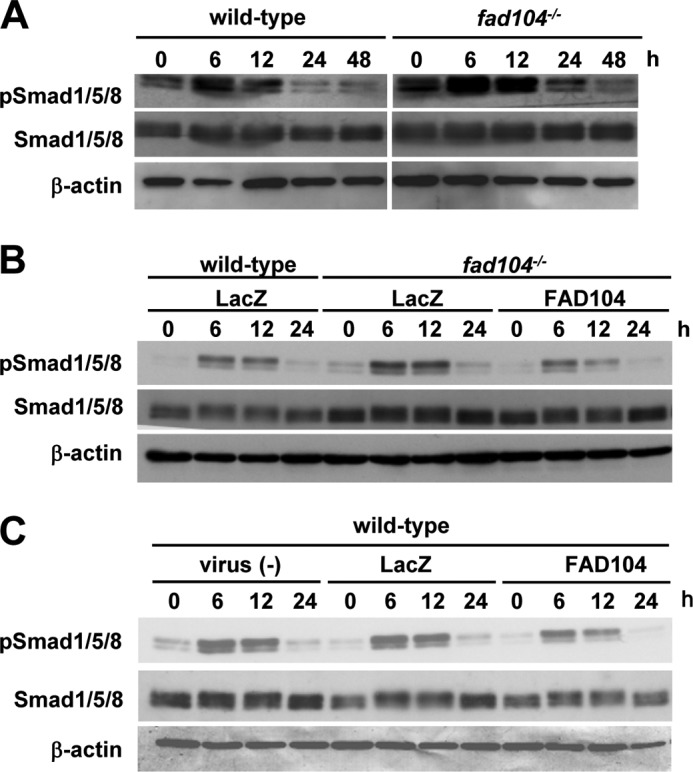
fad104 negatively regulates Smad1/5/8 phosphorylation during calvarial cell differentiation. A, the phosphorylation levels of Smad1/5/8 in wild-type and fad104-deficient calvarial cells during differentiation. At the indicated time points after osteogenic induction, the phosphorylation and protein levels of Smad1/5/8 were determined by Western blotting. β-Actin expression was used as a control. B, Smad1/5/8 phosphorylation levels in wild-type calvarial cells infected with LacZ and fad104-deficient calvarial cells infected with LacZ or FAD104 during differentiation. At the indicated time points after osteogenesis induction, the Smad1/5/8 phosphorylation and protein levels were determined by Western blotting. β-Actin expression was used as a control. C, the phosphorylation levels of Smad1/5/8 in wild-type calvarial cells infected with LacZ or FAD104 during differentiation. At the indicated time points after osteogenesis induction, the Smad1/5/8 phosphorylation levels and protein levels were determined by Western blotting. β-Actin expression was used as a control. These figures show typical results. Similar results were obtained in at least two independent experiments.
Furthermore, we examined whether FAD104 negatively regulated the phosphorylation level of Smad1/5/8 in vivo. The calvarias prepared from wild-type and fad104-deficient mice were incubated with serum-free medium and treated with BMP2. The phosphorylation level of Smad1/5/8 in fad104-deficient calvarias was slightly increased in contrast to those in wild-type calvarias in starved and BMP2-treated states. Thus, basically the same result was obtained in vivo. However, the differences are relatively small, probably due to the lower population of phosphorylated Smad1/5/8 positive cells, maybe progenitors, in whole calvarial cells, including progenitors and differentiated osteocytes (Fig. 6). Taken together, these results suggested that fad104 negatively regulated Smad1/5/8 phosphorylation during calvarial cell differentiation.
FIGURE 6.

Disruption of fad104 enhanced the phosphorylation level of Smad1/5/8 in calvaria. Calvarias prepared from wild-type and fad104−/− embryos at E18.5 were incubated with serum-free medium for 5 h and treated with BMP2 for 1 h. After washing, calvarias were lysed with radioimmunoprecipitation assay buffer. The Smad1/5/8 phosphorylation level and the protein levels of total Smad1/5/8 and FAD104 were determined by Western blotting. β-Actin was used as an expression control. The data show a typical result using two pairs of calvarias (wild-type and fad104−/−).
fad104 Directly Suppresses the BMP/Smad Signaling Pathway
Our results demonstrated that fad104 negatively regulated Smad1/5/8 phosphorylation after osteogenic induction. However, it remained unclear whether fad104 directly regulated BMP/Smad signaling, because the osteogenic medium contained ascorbic acid, β-glycerophosphate, and fetal bovine serum in addition to BMP2. To solve this problem, we evaluated whether fad104 directly influenced Smad1/5/8 phosphorylation induced solely by BMP2. Wild-type and fad104-deficient calvarial cells were cultured in serum-free medium and then treated with BMP2. In the wild-type cells, Smad1/5/8 phosphorylation was increased at 15 and 120 min following BMP2 treatment. Disruption of fad104 markedly increased BMP2-induced phosphorylation of Smad1/5/8 (Fig. 7A).
FIGURE 7.

fad104 negatively regulates BMP/Smad1/5/8 signaling. A, the Smad1/5/8 phosphorylation levels in wild-type and fad104-deficient calvarial cells after BMP2 treatment. At the indicated time points after BMP2 treatment, the Smad1/5/8 phosphorylation and protein levels were determined by Western blotting. β-Actin expression was used as a control. N, not starved. B, mRNA levels of Id1 in wild-type and fad104-deficient calvarial cells after BMP2 treatment. The Id1 levels were determined and normalized with 18 S rRNA expression by qRT-PCR. The data are presented as the means and S.D. (error bars) (n = 3). C, the Smad1/5/8 phosphorylation levels after BMP2 treatment in wild-type calvarial cells infected with LacZ and fad104-deficient calvarial cells infected with LacZ or FAD104. Nearly confluent cells were infected with each adenovirus. After 24 h, the cells were starved for 6 h and then treated with 100 ng/ml BMP2. At the indicated time points after BMP2 treatment, the Smad1/5/8 phosphorylation and protein levels were determined by Western blotting. β-Actin expression was used as a control. D, the mRNA levels of Id1 after BMP2 treatment in wild-type calvarial cells infected with LacZ and fad104-deficient calvarial cells infected with LacZ or FAD104. Nearly confluent cells were infected with each adenovirus. After 24 h, the cells were starved for 6 h and then treated with 100 ng/ml of BMP2. The levels of Id1 were determined and normalized with 18 S rRNA expression by qRT-PCR. The data are presented as the means and S.D. (n = 3). E, Smad1/5/8 phosphorylation levels after BMP2 treatment in wild-type calvarial cells infected with LacZ or FAD104. Nearly confluent cells were infected with each adenovirus. After 24 h, the cells were starved for 6 h and then treated with 100 ng/ml BMP2. At the indicated time points after BMP2 treatment, the Smad1/5/8 phosphorylation and protein levels were determined by Western blotting. β-Actin expression was used as a control. F, mRNA levels of Id1 after BMP2 treatment in wild-type calvarial cells infected with LacZ or FAD104. Nearly confluent cells were infected with each adenovirus. After 24 h, the cells were starved for 6 h and then treated with 100 ng/ml BMP2. The Id1 levels were determined and normalized with 18 S rRNA expression by qRT-PCR. The data are presented as the means and S.D. (n = 3). *, p < 0.05. A, C, and E show typical results. Similar results were obtained in at least two independent experiments.
Furthermore, the mRNA level of Id1, a representative BMP-responsive gene, was significantly increased in fad104-deficient calvarial cells (Fig. 7B). Moreover, enhanced BMP/Smad signaling in fad104-deficient calvarial cells was normalized by adenoviral reconstitution of FAD104 (Fig. 7, C and D). These results indicated that fad104 disruption leads to excessive activation of the BMP/Smad signaling pathway. Furthermore, adenovirus-mediated FAD104 overexpression in wild-type cells significantly inhibited BMP/Smad signaling, as assessed by Smad1/5/8 phosphorylation and Id1 expression (Fig. 7, E and F). These results demonstrated that fad104 negatively regulated the BMP/Smad signaling pathway.
FAD104 Negatively Regulated Smad1/5/8 Phosphorylation during Adipocyte Differentiation
Our previous studies showed that FAD104 potentiated the differentiation of MEFs into adipocytes (12). Seemingly, this is inconsistent with the fact that FAD104 negatively regulates BMP/Smad signaling because BMP/Smad signaling also potentiates adipogenesis. To address this issue, we examined whether FAD104 suppress BMP/Smad signaling during adipogenesis of MEFs. MEFs can be effectively stimulated to undergo adipogenesis by adipogenic mixture containing 3-isobutyl-1-methylxantine, insulin, and dexamethasone (12). In this condition, Smad1/5/8 phosphorylation was transiently up-regulated at 1 h after induction and decreased until 6 h. Disruption of fad104 enhanced the Smad1/5/8 phosphorylation until 6 h after induction compared with wild type (Fig. 8A). In addition, the enhancement of Smad1/5/8 phosphorylation by fad104 disruption was observed during BMP2-induced adipogenesis (Fig. 8B). These results indicate that fad104 suppresses BMP/Smad signaling in the process of adipogenesis.
FIGURE 8.
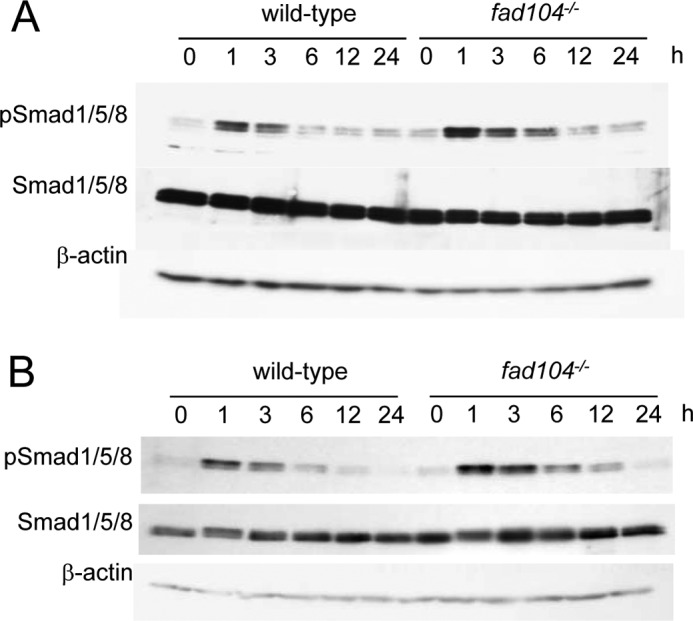
The phosphorylation level of Smad1/5/8 during adipocyte differentiation of MEFs. The MEFs prepared from wild-type and fad104−/− embryos were brought to confluence in 10% FBS. After 2 days of incubation, the medium was changed to the differentiation medium containing dexamethasone, 3-isobutyl-1-methylxantine, and insulin (A) or containing BMP2, l-ascorbic acid, and β-glycerophosphate (B). At the indicated time points after induction, the phosphorylation and protein levels of Smad1/5/8 were determined by Western blotting. β-Actin expression was used as a control. A and B show typical results, and similar results were obtained in an independent experiment using another pair of MEFs.
fad104 Prevents the Translocation of Smad1 to the Nucleus
Smad1/5/8 activated by BMP2 binds to Smad4 and translocates to the nucleus. Therefore, we next tested whether FAD104 inhibited the BMP2-induced translocation of Smad1 to the nucleus using HeLa cells. It is known that HeLa cells are responsive to BMP2 (18). Indeed, we observed the increase of Smad1/5/8 phosphorylation by BMP2 treatment in HeLa cells (Fig. 9A, left). FLAG-tagged Smad1 expression plasmid was introduced into HeLa cells with Myc-tagged FAD104 expression plasmid or empty vector. In the absence of Myc-FAD104, FLAG-Smad1 was observed in the nucleus by BMP2 treatment (Fig. 9B, top). On the other hand, in Myc-FAD104 overexpression cells, FLAG-Smad1 remained localized in the cytoplasm and could not translocate to the nucleus even after BMP2 treatment (Fig. 9B, bottom). Moreover, the cytoplasmic localization of FLAG-Smad1 was partially merged with Myc-FAD104 (Fig. 9C). These results indicated that FAD104 inhibited the BMP2-induced nuclear translocation of Smad1. Also, it was noteworthy that Smad1/5/8 phosphorylation was impaired by FAD104 expression in HeLa cells as well (Fig. 9A, right).
FIGURE 9.

fad104 inhibits the translocation of Smad1 to the nucleus. A, the Smad1/5/8 phosphorylation levels after BMP2 treatment in HeLa cells infected with FAD104 or LacZ. Nearly confluent cells were infected with each adenovirus. After 24 h, the cells were starved for 6 h and then treated with 100 ng/ml BMP2. At 30 and 120 min after BMP2 treatment, the phosphorylation and protein levels of Smad1/5/8 were determined by Western blotting. β-Actin expression was used as a control. The figures show typical results, and similar results were obtained in at least two independent experiments. B, FLAG-tagged Smad1 expression plasmid was transiently introduced into HeLa cells with Myc-tagged FAD104 expression plasmid or empty vector. After transfection, the cells were starved for 6 h and treated BMP2 for 2 h. After conducting immunofluorescent staining, the signals of FLAG-Smad1 (red), Myc-FAD104 (green), and DAPI (blue) were detected with fluorescence microscopy. C, FLAG-tagged Smad1 expression plasmid and Myc-tagged FAD104 expression plasmid were transiently introduced into HeLa cells. After transfection, the cells were starved for 6 h and treated with BMP2 for 2 h. The signals of FLAG-Smad1 (red), Myc-FAD104 (green), and DAPI (blue) were detected with fluorescence microscopy.
FAD104 Interacts with Smad1/5/8, and the N-terminal Region of FAD104 Is Required for Suppression of BMP-induced Smad1/5/8 Phosphorylation
The N terminus of FAD104 is proline-rich and contains several canonical binding sites for SH3 (PXXP) and type I WW domains (PPLP and PXXY), which are mediated by protein-protein interactions and signal transduction (19–21). First, we investigated whether FAD104 could bind Smad1/5/8 using FLAG-tagged FAD104 expression plasmids transfected into HeLa cells via immunoprecipitation (IP) analyses and observed that binding occurs (Fig. 10A).
FIGURE 10.

FAD104 interacts with Smad1/5/8, and the N terminus of FAD104 is required for inhibition of Smad1/5/8 phosphorylation. A, the interactions of FAD104 with Smad1/5/8 by an IP assay. HeLa cells were transfected with 3xFLAG-FAD104 expression plasmids. IP experiments were performed using anti-Smad1/5/8 antibody. Normal rabbit IgG was used as a negative control. Immunoprecipitates and 0.5% input were resolved and detected by Western blotting with anti-FLAG antibody. B, interaction between FAD104 and Smad1/5/8 under physiological conditions. Lysates from intact C2C12 cells were immunoprecipitated using an antibody against Smad1/5/8. Immunoprecipitates and 0.5% input were resolved and detected by Western blotting with anti-FAD104 antibody. C, interaction of FAD104ΔN with Smad1/5/8 by IP assay. HeLa cells in a 10-cm culture dish were transfected with FLAG-FAD104 (15 μg) or FLAG-FAD104ΔN (1.875 μg) expression plasmids to equalize the amount of FLAG-FAD104 and FLAG-FAD104ΔN. IP experiments were performed by anti-Smad1/5/8 antibody. Normal rabbit IgG was used as a negative control. Immunoprecipitates and 0.2% input were resolved and detected by Western blotting with anti-FLAG antibody. D, interaction of the N terminus of FAD104 with Smad1/5/8 by IP assay. HeLa cells were transfected with Myc-FAD104N expression plasmids. IP experiments were performed by anti-Smad1/5/8 antibody. Normal rabbit IgG was used as a negative control. Immunoprecipitates and 0.2% input were resolved and detected by Western blotting with anti-Myc antibody. E, interaction of the N terminus of FAD104 with Smad1/5/8 via the GST pull-down assay. Cell lysates prepared from C2C12 cells were used for a GST pull-down assay with GST or GST-FAD104N. Bottom, Coomassie Blue staining of the GST proteins. Bound protein samples were immunoblotted with anti-Smad1/5/8 antibody. The input volume was 5% of that of the cell lysates for the pull-down assay. F, interaction of the N terminus of FAD104 with phospho-Smad1/5/8 via the GST pull-down assay. The cell lysates prepared from C2C12 cells treated BMP2 were used for a GST pull-down assay with GST or GST-FAD104N. Bottom, indicates Coomassie Blue staining of the GST proteins. Bound protein samples were immunoblotted with anti-phospho-Smad1/5/8 antibody. The input volume was 5% of that of the cell lysates for the pull-down assay. G, direct interaction between the N terminus of FAD104 and Smad1/5/8. FLAG-tagged Smad1 and Smad5 expressed in E. coli cells were purified using FLAG M2 affinity gel and eluted. The eluates were used for a GST pull-down assay with GST or GST-FAD104N. Bottom, Coomassie Blue staining of the GST proteins. Bound protein samples were immunoblotted with anti-FLAG antibody. The input volume was 5% of that of the eluates for the pull-down assay. H, Smad1/5/8 phosphorylation levels in C2C12 cells introduced 3xFLAG, 3xFLAG FAD104, or 3xFLAG FAD104ΔN expression plasmids. C2C12 cells transiently transfected with each plasmid were starved for 6 h and treated with BMP2 for 1 h. The Smad1/5/8 phosphorylation and protein levels of total Smad1/5/8 were determined by Western blotting. β-Actin expression was used as a control. A–H show typical results. Similar results were obtained in at least two independent experiments.
Importantly, we also found that FAD104 interacted with Smad1/5/8 under physiological conditions (Fig. 10B). To further characterize this binding, we examined whether the N terminus of FAD104 was involved in binding to Smad1/5/8. FAD104 and its deletion mutant lacking the N-terminal region and proline-rich motif (FAD104ΔN) was introduced into HeLa cells and analyzed via IP. The efficacy of binding between FAD104ΔN and Smad1/5/8 was lower than that between FAD104 full-length and Smad1/5/8 (Fig. 10C).
We further tested whether the N terminus of FAD104 is alone sufficient to interact with Smad1/5/8. Expression plasmids containing Myc-tagged FAD104 N termini (amino acids 1–277) (Myc-FAD104N) were introduced into HeLa cells and analyzed via IP, which showed that Myc-FAD104N interacted with Smad1/5/8 (Fig. 10D). Furthermore, a GST pull-down assay revealed that GST-FAD104N bound to both the unphosphorylated and phosphorylated form of Smad1/5/8 (Fig. 10, E and F). In addition, a GST pull-down assay using recombinant FLAG-tagged Smad1 and Smad5 purified from E. coli showed that the N-terminal region of FAD104 directly interacted with Smad1 and Smad5 (Fig. 10G). These results suggested that the N-terminal region of FAD104 was important for binding to Smad1/5/8.
Next, we investigated the importance of the N-terminal region of FAD104 in Smad1/5/8 phosphorylation. FAD104 and FAD104ΔN were transfected into C2C12 myoblasts, which are capable of BMP2-induced trans-differentiation to form osteoblasts. Our results showed that FAD104 expression decreased BMP2-induced Smad1/5/8 phosphorylation, but FAD104ΔN did not, indicating that the N terminus of FAD104 was required for suppression of Smad1/5/8 phosphorylation (Fig. 10H).
DISCCUSION
In this study, we demonstrated that fad104 was required for normal calvarial bone formation and negatively regulated calvarial cell differentiation through inhibition of BMP/Smad signaling. Most importantly, disruption of fad104 caused premature ossification of the anterior fontanel, closely resembling craniosynostosis, which is a congenital developmental disorder involving the premature fusion of cranial sutures and fontanels and results in cranial dysmorphism (22, 23). Reportedly, craniosynostosis is correlated to several gene mutations, including FGF receptor (24, 25). BMP signaling was also closely correlated to cranial bone formation (25, 26). Mutations of the msx2 (msh homeobox 2) gene, which is a target of BMP signaling, can induce craniosynostosis (26, 27). Considering the phenotype of fad104-deficient mice and the inhibitory role of fad104 in BMP/Smad signaling, fad104 mutations are probably the cause of unexplained skull malformations, as in craniosynostosis. To clarify this issue, future single nucleotide polymorphism (SNP) analyses should be performed to investigate the role of fad104 in cranial deformities.
Bones are derived from two different mechanisms, endochondral and intramembranous ossification. Endochondral ossification is involved in the replacement of cartilage by bone and is necessary for almost all bone formation in vertebrates, whereas in intramembranous ossification, bones are formed by direct condensation of mesenchymal cells without cartilage involvement, particularly in the skull, clavicles, and cortical bones (27, 28). In fad104-deficient mice, excessive ossification was observed mainly in the calvaria and femur cortical bones. Although significant defects were not seen in clavicles, these phenotypes imply that fad104 affects particularly intramembranous ossification. We wondered if this specificity is caused by the expression pattern of fad104, but the expression levels of fad104 were comparable between some bone tissues. Another approach using the GenePaint database also showed that fad104 is also expressed in ribs and axial bone at E14.5, suggesting that the expression pattern of fad104 could not explain the specificity of the phenotype in fad104-deficient calvaria. Although we could not definitely show the reason why the bone phenotypes in fad104-deficient mice were restricted to calvaria, the abnormal bones were all formed by intramembranous ossification. Analyses of the relationship between fad104 and regulatory factors, such as MSX2 and TWIST, that contribute to intramembranous ossification might help to elucidate the specificity of bone phenotype in fad104-deficient mice.
It was reported that BMP signaling positively regulates the commitment/differentiation of mesenchymal progenitors into adipocyte (29). Here, we showed that fad104 negatively regulates calvarial osteoblast differentiation through the BMP/Smad pathway. On the contrary, we previously reported that fad104 positively regulates adipocyte differentiation (10). Furthermore, FAD104 negatively regulated Smad1/5/8 phosphorylation during adipogenesis as well. This raises the question of why the adipogenesis induced by BMP2 was inhibited in fad104-deficient MEFs although BMP2 potentiates adipogenesis. We think there are several possibilities. At first, BMP2 has a crucial role in commitment of mesenchymal progenitors, such as MEFs, to adipogenic lineage. On the other hand, it was reported that exogenous BMP did not affect lipid accumulation after adipogenic induction in 3T3-L1, which is a preadipocyte cell line committed to adipocyte lineage, suggesting that BMP particularly contributes to the commitment process (30). In contrast, fad104 positively regulates adipogenesis not only in MEFs but also in 3T3-L1 cells (9, 11, 12). These results suggest that fad104 might regulate the differentiation of committed progenitor cells toward adipocytes. Therefore, even if BMP2-induced commitment to adipogenic lineage were enhanced in fad104-deficient MEFs, subsequent differentiation might be inhibited by fad104 disruption likewise in the case of 3T3-L1 cells. Second, FAD104 might also affect other unidentified signaling molecules that regulate adipogenesis in addition to Smad1/5/8. In either case, the detailed mechanisms by which FAD104 regulates adipogenesis need to be elucidated in the future.
The expression of FAD104 began to decrease at the beginning (6–12 h) of calvarial cell differentiation. We believe that this time course is parallel to up-regulation of Smad1/5/8. From these results, FAD104 might keep the phosphorylation level of Smad1/5/8 low before osteogenic induction. Once differentiation was induced, down-regulation of Smad1/5/8 phosphorylation by FAD104 was gradually diminished with the reduction of FAD104 expression.
Runx2 (Runt-related transcription factor 2) functions as a master regulator for activating the program of osteogenesis (16). Because FAD104 negatively regulates the phosphorylation level of Smad1/5/8 and osteoblast differentiation, we tested whether FAD104 repressed the transactivation activity of Runx2 using a reporter assay. The overexpression of FAD104 slightly decreased Runx2 activity, but did not have a significant effect on the transactivation activity of Runx2 (data not shown). The activity of Runx2 is regulated in a complicated manner. In addition to Smad1/5/8, various factors, such as p300 and Hdac1, were involved in its activity (16). Therefore, it is possible that this complexity might make it difficult to understand the contribution of FAD104 to the transactivation of Runx2. We previously demonstrated that the expression level of runx2 was increased in MEFs prepared from fad104-deficient mice compared with that in wild-type mice (11). It is possible that FAD104 contributes more to the regulation of the expression level of Runx2 than to the regulation of the transactivation activity of Runx2 during osteogenesis. It is necessary to examine the mechanism by which FAD104 regulates the expression level of Runx2.
We observed that FAD104 interacts with Smad1/5/8 and that the proline-rich N terminus of FAD104 is essential for suppression of BMP2-induced Smad1/5/8 phosphorylation, although the detailed mechanism by which fad104 regulates the phosphorylation of Smad1/5/8 remains unclear. Smad1/5/8 phosphorylation is tightly regulated by various molecules, such as Smad phosphatase (e.g. pyruvate hydroxylase phosphatase), E3 ubiquitin-ligase (e.g. Smurf (SMAD-specific E3 ubiquitin protein ligase)), and inhibitory Smads (e.g. Smad6/7) (16, 31). In our preliminary experiments, FAD104 interactions with pyruvate hydroxylase phosphatase, Smurf1/2, and Smad6/7 were undetected (data not shown). Although more detailed analyses are needed, these results indicated fad104 regulation of Smad1/5/8 phosphorylation via distinct mechanisms.
We hypothesized that FAD104, which localizes to the endoplasmic reticulum, might mask the Smad1/5/8 phosphorylation sites and traps unphosphorylated Smad1/5/8 within the endoplasmic reticulum. To clarify these possibilities, we examined which state of Smad1/5/8, the phosphorylated or unphosphorylated form, could bind to FAD104. Contrary to our hypothesis, FAD104 could bind to both phosphorylated and unphosphorylated Smad1/5/8, suggesting that FAD104 did not mask the phosphorylation sites of Smad1/5/8.
In addition to the binding of FAD104 to Smad1/5/8, we also found that FAD104 inhibited the BMP2-induced translocation of Smad into the nucleus. Because the translocation of Smad1/5/8 into nuclei requires its phosphorylation, the cytoplasmic localization of Smad1/5/8 might be due to the decline of Smad1/5/8 phosphorylation by FAD104 overexpression. Although the mechanisms, by which FAD104 regulates Smad1/5/8 phosphorylation, are unclear, we could provide one possibility about this phenomenon. We found that (i) FAD104 interacted with phosphorylated Smad1/5/8, and (ii) the Smad1/5/8 localization was maintained in the cytoplasm by FAD104 expression even after BMP2 treatment, resulting in the decrease of Smad1/5/8 in nuclei. The reduction of the Smad1/5/8 phosphorylation level in FAD104-overexpressing cells, as shown in Fig. 9A, might be caused by the instability of the cytoplasmic phosphorylated Smad1/5/8. Future studies are definitely needed to evaluate these possibilities.
Here, we demonstrated the significance of fad104 in embryonic bone formation; however, the roles of fad104 in the aging process remain unresolved. The imbalance between bone formation and bone resorption with aging leads to metabolic bone disorders, such as osteoporosis and poor fracture healing (32). To test whether fad104 is involved in the pathological processes of these disorders, it is necessary to generate and analyze osteoblast-specific fad104 conditional knock-out and/or transgenic mice. In conclusion, our present findings provide evidence that fad104 is closely involved in the negative regulation of the BMP/Smad signaling pathway and is required for proper anterior fontanel ossification.
Acknowledgments
We thank Drs. Takashi Ueda and Shinya Ugawa at Nagoya City University for assistance in skeletal staining. We also thank Dr. Mitsuru Morimoto at the RIKEN Center for Developmental Biology for suggestions in femur histological analysis. We are grateful to Dr. Toshihisa Komori (Nagasaki University) and Dr. Shingo Maeda (Kagoshima University) for useful discussion.
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, the Japan Society for the Promotion of Science (JSPS), JSPS Research Fellow, the Mitsui Sumitomo Insurance Welfare Foundation, and the Nakatomi Foundation.
- BMP
- bone morphogenetic protein
- MEF
- mouse embryonic fibroblast
- TRITC
- tetramethylrhodamine
- IP
- immunoprecipitation
- α-MEM
- α-minimum essential medium
- qRT-PCR
- quantitative RT-PCR
- En
- embryonic day n.
REFERENCES
- 1. Karsenty G., Kronenberg H. M., Settembre C. (2009) Genetic control of bone formation. Annu. Rev. Cell Dev. Biol. 25, 629–648 [DOI] [PubMed] [Google Scholar]
- 2. Canalis E. (2009) Growth factor control of bone mass. J. Cell Biochem. 108, 769–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang W., Yang S., Shao J., Li Y. P. (2007) Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front. Biosci. 12, 3068–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Canalis E., Economides A. N., Gazzerro E. (2003) Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr. Rev. 24, 218–235 [DOI] [PubMed] [Google Scholar]
- 5. Imagawa M., Tsuchiya T., Nishihara T. (1999) Identification of inducible genes at the early stage of adipocyte differentiation of 3T3-L1 cells. Biochem. Biophys. Res. Commun. 254, 299–305 [DOI] [PubMed] [Google Scholar]
- 6. Nishizuka M., Tsuchiya T., Nishihara T., Imagawa M. (2002) Induction of Bach1 and ARA70 gene expression at an early stage of adipocyte differentiation of mouse 3T3-L1 cells. Biochem. J. 361, 629–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tominaga K., Johmura Y., Nishizuka M., Imagawa M. (2004) Fad24, a mammalian homolog of Noc3p, is a positive regulator in adipocyte differentiation. J. Cell Sci. 117, 6217–6226 [DOI] [PubMed] [Google Scholar]
- 8. Hishida T., Eguchi T., Osada S., Nishizuka M., Imagawa M. (2008) A novel gene, fad49, plays a crucial role in the immediate early stage of adipocyte differentiation via involvement in mitotic clonal expansion. FEBS J. 275, 5576–5588 [DOI] [PubMed] [Google Scholar]
- 9. Tominaga K., Kondo C., Johmura Y., Nishizuka M., Imagawa M. (2004) The novel gene fad104, containing a fibronectin type III domain, has a significant role in adipogenesis. FEBS Lett. 577, 49–54 [DOI] [PubMed] [Google Scholar]
- 10. Tominaga K., Kondo C., Kagata T., Hishida T., Nishizuka M., Imagawa M. (2004) The novel gene fad158, having a transmembrane domain and leucine-rich repeat, stimulates adipocyte differentiation. J. Biol. Chem. 279, 33840–33848 [DOI] [PubMed] [Google Scholar]
- 11. Kishimoto K., Kato A., Osada S., Nishizuka M., Imagawa M. (2010) Fad104, a positive regulator of adipogenesis, negatively regulates osteoblast differentiation. Biochem. Biophys. Res. Commun. 397, 187–191 [DOI] [PubMed] [Google Scholar]
- 12. Nishizuka M., Kishimoto K., Kato A., Ikawa M., Okabe M., Sato R., Niida H., Nakanishi M., Osada S., Imagawa M. (2009) Disruption of the novel gene fad104 causes rapid postnatal death and attenuation of cell proliferation, adhesion, spreading and migration. Exp. Cell Res. 315, 809–819 [DOI] [PubMed] [Google Scholar]
- 13. Kishimoto K., Nishizuka M., Ueda T., Kajita K., Ugawa S., Shimada S., Osada S., Imagawa M. (2011) Indispensable role of factor for adipocyte differentiation 104 (fad104) in lung maturation. Exp. Cell Res. 317, 2110–2123 [DOI] [PubMed] [Google Scholar]
- 14. Kanamoto T., Mizuhashi K., Terada K., Minami T., Yoshikawa H., Furukawa T. (2009) Isolation and characterization of a novel plasma membrane protein, osteoblast induction factor (obif), associated with osteoblast differentiation. BMC Dev. Biol. 9, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen C., Okayama H. (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell Biol. 7, 2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen G., Deng C., Li Y. P. (2012) TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 8, 272–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wan M., Cao X. (2005) BMP signaling in skeletal development. Biochem. Biophys. Res. Commun. 328, 651–657 [DOI] [PubMed] [Google Scholar]
- 18. Chen H. B., Shen J., Ip Y. T., Xu L. (2006) Identification of phosphatases for Smad in the BMP/Dpp pathway. Genes Dev. 20, 648–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Obholz K. L., Akopyan A., Waymire K. G., MacGregor G. R. (2006) FNDC3A is required for adhesion between spermatids and Sertoli cells. Dev. Biol. 298, 498–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Macias M. J., Wiesner S., Sudol M. (2002) WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 513, 30–37 [DOI] [PubMed] [Google Scholar]
- 21. Musacchio A., Gibson T., Lehto V. P., Saraste M. (1992) SH3. An abundant protein domain in search of a function. FEBS Lett. 307, 55–61 [DOI] [PubMed] [Google Scholar]
- 22. Tagariello A., Heller R., Greven A., Kalscheuer V. M., Molter T., Rauch A., Kress W., Winterpacht A. (2006) Balanced translocation in a patient with craniosynostosis disrupts the SOX6 gene and an evolutionarily conserved non-transcribed region. J. Med. Genet. 43, 534–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marie P. J., Debiais F., Haÿ E. (2002) Regulation of human cranial osteoblast phenotype by FGF-2, FGFR-2 and BMP-2 signaling. Histol. Histopathol. 17, 877–885 [DOI] [PubMed] [Google Scholar]
- 24. Passos-Bueno M. R., Serti Eacute A. E., Jehee F. S., Fanganiello R., Yeh E. (2008) Genetics of craniosynostosis. Genes, syndromes, mutations and genotype-phenotype correlations. Front. Oral Biol. 12, 107–143 [DOI] [PubMed] [Google Scholar]
- 25. Rice D. P., Rice R., Thesleff I. (2003) Molecular mechanisms in calvarial bone and suture development, and their relation to craniosynostosis. Eur. J. Orthod. 25, 139–148 [DOI] [PubMed] [Google Scholar]
- 26. Maxson R., Ishii M. (2008) The Bmp pathway in skull vault development. Front. Oral Biol. 12, 197–208 [DOI] [PubMed] [Google Scholar]
- 27. de Crombrugghe B., Lefebvre V., Nakashima K. (2001) Regulatory mechanisms in the pathways of cartilage and bone formation. Curr. Opin. Cell Biol. 13, 721–727 [DOI] [PubMed] [Google Scholar]
- 28. Olsen B. R., Reginato A. M., Wang W. (2000) Bone development. Annu. Rev. Cell Dev. Biol. 16, 191–220 [DOI] [PubMed] [Google Scholar]
- 29. Tang Q. Q., Lane M. D. (2012) Adipogenesis. From stem cell to adipocyte. Annu. Rev. Biochem. 81, 715–736 [DOI] [PubMed] [Google Scholar]
- 30. Suenaga M., Kurosawa N., Asano H., Kanamori Y., Umemoto T., Yoshida H., Murakami M., Kawachi H., Matsui T., Funaba M. (2013) Bmp4 expressed in preadipocytes is required for the onset of adipocyte differentiation. Cytokine 64, 138–145 [DOI] [PubMed] [Google Scholar]
- 31. Bruce D. L., Sapkota G. P. (2012) Phosphatases in SMAD regulation. FEBS Lett. 586, 1897–1905 [DOI] [PubMed] [Google Scholar]
- 32. Karsenty G. (2003) The complexities of skeletal biology. Nature 423, 316–318 [DOI] [PubMed] [Google Scholar]