

Background: Jak3 is a tyrosine kinase, and its role in mucosal differentiation is not known.
Results: Jak3, expressed in colonic mucosa, was essential for differentiation, goblet cell surface localization, muc2 expression, and epithelial barrier functions.
Conclusion: Jak3 plays a protective role against predisposition to colitis by promoting mucosal differentiation.
Significance: Understanding mucosal functions of Jak3 has important implications for patients with inflammatory bowel disease.
Keywords: β-Catenin, Colitis, Differentiation, Inflammatory Bowel Disease, Innate Immunity, Jak Kinase
Abstract
Janus kinase 3 (Jak3) is a nonreceptor tyrosine kinase expressed in both hematopoietic and nonhematopoietic cells. Previously, we characterized the functions of Jak3 in cytoskeletal remodeling, epithelial wound healing, and mucosal homeostasis. However, the role of Jak3 in mucosal differentiation and inflammatory bowel disease was not known. In this report, we characterize the role of Jak3 in mucosal differentiation, basal colonic inflammation, and predisposition toward colitis. Using the Jak3 knock-out (KO) mouse model, we show that Jak3 is expressed in colonic mucosa of mice, and the loss of mucosal expression of Jak3 resulted in reduced expression of differentiation markers for the cells of both enterocytic and secretory lineages. Jak3 KO mice showed reduced expression of colonic villin, carbonic anhydrase, secretory mucin muc2, and increased basal colonic inflammation reflected by increased levels of pro-inflammatory cytokines IL-6 and IL-17A in colon along with increased colonic myeloperoxidase activity. The inflammations in KO mice were associated with shortening of colon length, reduced cecum length, decreased crypt heights, and increased severity toward dextran sulfate sodium-induced colitis. In differentiated human colonic epithelial cells, Jak3 redistributed to basolateral surfaces and interacted with adherens junction (AJ) protein β-catenin. Jak3 expression in these cells was essential for AJ localization of β-catenin and maintenance of epithelial barrier functions. Collectively, these results demonstrate the essential role of Jak3 in the colon where it facilitated mucosal differentiation by promoting the expression of differentiation markers and enhanced colonic barrier functions through AJ localization of β-catenin.
Introduction
Differentiation in the gastrointestinal tract is a highly organized process by which proliferative compartments of the intestinal epithelium (known as crypt) contribute to the renewal of intestinal epithelial mucosa. A small number of cells at the bottom of the crypt (known as stem cells) slowly proliferate to give a transient population of progenitor cells that rapidly divide at the same time migrating toward the lumen of the intestine. Depending on the extracellular cues and cytokine profiles, progenitor cells become committed toward one of the three different lineages as follows: absorptive cells displaying a brush border, goblet cells containing mucous granules, and enteroendocrine cells containing dense secretary granules (1). The integrity of epithelial barriers comprising these differentiated cells was impaired and compromised in the course of various intestinal disorders such as inflammatory bowel disease (IBD),2 celiac disease, and intestinal infection (2). Intestinal differentiation is essential for the protection of the mucosal surfaces. Ulcerative colitis is associated with defects in differentiation with reduced numbers of differentiated cells and reduced thickness of the mucous layer overlaying the colonic mucosa (3, 4). Although different genes have been implicated in IBD (5), the role of epithelial Jak3 in ulcerative colitis is not known.
Janus kinases (Jaks) are a family of nonreceptor tyrosine kinase with four members as follows: Jak1, Jak2, Jak3, and Tyk2. Like other members, Jak3 mediates signals initiated by cytokines through interactions with receptors for IL-2, IL-5, IL-7, IL-9, and IL-15 via the common γ chain of these receptors (6). Different studies have shown that Jak3 is widely expressed in different organs, including intestine and kidney of both humans and mice (6, 7). Previous studies with IL-2Rγ-null mice showed that it developed spontaneous IBD symptoms (8). Moreover, abnormal activation of Jak3 was associated with human hematological and epithelial malignancies (9–11), indicating that a tight balance in its activity was not only essential for normal hematopoietic development but also for epithelial functions. Recently, we reported the essential functions of Jak3 in cytoskeletal remodeling and epithelial wound repair (12), through its interactions with cytoskeletal proteins (13). Although we also reported that Jak3 played an important role in mucosal homeostasis through its interactions with adapter protein p52ShcA (14), the in vivo mucosal functions of Jak3 are less understood. In this study, we characterize the role of Jak3 on mucosal differentiation, colonic inflammation, and structural changes in mucosal surfaces of colon, and we demonstrate the mechanism of Jak3-mediated mucosal differentiation, where Jak3 regulated the expression of differentiation markers, formation of mucus in mice, and facilitated barrier functions through its interactions and adherens junction (AJ) localization of β-catenin in human intestinal epithelial cells (IEC).
EXPERIMENTAL PROCEDURES
Materials
Materials were obtained from the following sources: HT-29 CL-19A, a permanently differentiated homogeneous clone derived from human colonic epithelial cells HT-29, was a gift from Dr. A. P. Naren, University of Tennessee Health Science Center at Memphis, TN (15); HEK293T cells, Jak3-shRNA, and Shc-shRNA (Open Biosystems); doxycycline (Sigma); dextran sulfate sodium (DSS) 40,000 kDa (United States Biochemical); hematoxylin, eosin, and xylene (VWR Scientific); Alcian blue and tetramethylbenzidine substrate kit (Thermo Scientific). Antibodies and kits were obtained from the following sources: pY20 (MP Biomedicals); p(Y142) β-catenin (Abcam®); Jak3, villin, IgG, β-catenin, β-actin, and carbonic anhydrase (Santa Cruz Biotechnology); muc2 (Novus Biologicals); α1-antitrypsin (Sigma); MultiAnalyte cytokine assay kit (Qiagen); 3,3′-diaminobenzidine substrate kit (Vector Laboratories); and Pierce® BCA protein assay kit (Thermo Scientific).
Animals
Six- to 8-week-old C57BL/6 mice (WT) or C57BL/6-background jak3−/− mice (KO) were purchased from The Jackson Laboratory. This study adhered to the institutional guidelines of Texas A&M University Institutional Animal Care and Use Committee.
Induction of Colitis by DSS
Male WT and KO mice weighing 20–25 g were used for the experiments. The animals were housed in a specific pathogen-free facility in individually ventilated cages. Acute colitis was induced by feeding 2.5% DSS in drinking water over a period of 9 days. This procedure is known to induce colonic epithelial injury (16, 17). Animals were provided unlimited access to food and water throughout the experiment (ad libitum). Body weights of mice were measured daily. The mice that lost ≥20% of initial body weight were euthanized, and all the data shown are based on this end point. Following treatment, the distal colon and serum were sampled for analysis. Images were taken by a Nikon camera. Quantitation of colon length, colon to body weight ratio, length of cecum, and crypt lengths were calculated using NIS element imaging software (Nikon®).
Disease Score in Colitis
Progression of colitis was evaluated daily by measuring the body weight loss, stool consistency, and presence of fecal blood. An earlier validated clinical disease activity index was calculated according to the following parameters: stool consistency (0–4), the presence or absence of fecal blood (0–4), and body weight loss (0–4). The maximum possible score taken was 12 (18, 19).
Tissue Staining and Histological Damage Scoring for Colitis
The distal colons of WT and KO mice with or without treatment were dissected out and cut into small pieces (3–4 mm long). The fresh tissues were placed in a mold containing OCT (Sakura Inc.) solution and frozen. Tissue cross-sections (5–10 μm thick) were stained with hematoxylin and eosin (H&E) using standard protocol. The mean degree of acute colitis in the colon was calculated from observation of 25 different fields of H&E-stained sections of colon from each animal (20). The degree of colitis was evaluated by the following histopathological score grading system. (a) Areas of activity (erosions and inflammation), possible dysplasia, crypt distortion, and plasmatic infiltrates were considered indicative of colonic inflammation. Two independent parameters were measured as reported by others (21) as follows: the extent of inflammation (0, none; 1, slight; 2, moderate; 3, severe; 4, massive) and the extent of crypt damage (0, none; 1, the basal one-third portion damaged; 2, the basal two-thirds portion damaged; 3, the entire crypt damaged but the surface epithelium intact; 4, the entire crypt and epithelium lost). The maximum possible score was 6. H&E-stained slides were visualized using a Nikon microscope, and the images were processed with NIS element software (Nikon®) to calculate crypt lengths and cecum lengths.
Myeloperoxidase (MPO) Assay
MPO activities were measured by colorimetric assay and used as an index of inflammation (22). Briefly, tissue samples from colon were homogenized in ice-cold buffer (50 mm K2HPO4 and 50 mm KH2PO4, pH 6.0) containing 0.5% hexadecyltrimethyl ammonium bromide (Sigma). The homogenates were sonicated, freeze-thawed three times, and centrifuged. Supernatants (25 μl) were used for MPO activities. The enzymatic reactions were carried out by adding 25 μl of 1.6 mm tetramethylbenzidine in 80 mm sodium phosphate (NaPO4). The reactions were stopped by adding 100 μl of 0.3 mm H2O2. MPO activity was measured spectrophotometrically at 650 nm, and results were expressed as optical density per mg of tissue.
Immunofluorescence Microscopy (IFM) and Immunohistochemistry
For IFM, OCT mold-embedded tissue sections were air-dried for 20 min at RT, fixed using 4% paraformaldehyde, and blocked using 5% BSA in PBS for 30 min. Sections were than incubated with primary antibodies for Jak3, villin, carbonic anhydrase, and β-catenin (Santa Cruz Biotechnology) at 1:100 dilutions in 1% BSA/PBS at 4 °C followed by incubation with cy3 or Alexa-Fluor 488-conjugated secondary antibodies. The sections were then rinsed twice with PBS and mounted using Vectashield (Vector Laboratories). For all negative controls, primary antibodies were replaced with a control nonimmune IgG at the same concentrations. IFM for HT-29 Cl-19A cells transduced with RFP-tagged Jak3 shRNA were done as reported previously (14). The immunostained slides were visualized using C1-plus Nikon laser scanning confocal microscope, and the images were processed using NIS element software (Nikon®). All experiments were conducted at least in triplicate, and representative images were shown. For immunohistochemistry, the aforementioned sections from frozen tissue block were fixed with pre-cooled acetone at −20 °C. Endogenous peroxidase activity was inactivated by incubating the tissue with 0.3% H2O2 for 10 min. Nonspecific binding was blocked by 45 min of incubation with 10% fetal bovine serum prior to incubation with primary antibodies. The sections were then incubated with biotinylated secondary antibody followed by staining with Vectastain ABC kit (Vector Laboratories). The reaction was visualized using the 3,3′-diaminobenzidine substrate.
Alcian Blue Staining
The aforementioned tissues sections were air-dried and fixed with anhydrous alcohol followed by staining with Alcian blue for 30 min and counter-staining with the diluted eosin. Tissue sections were dehydrated in anhydrous alcohol and then immersed in clearing reagent followed by mounting for visualization.
Cytokine and α1-Antitrypsin Assays
IL-6 and IL-17a were determined in the sera/colonic tissues lysates from WT and KO mice using Multi-Analyte ELISA array kit (SA Biosciences) as per the manufacturer's protocol. Stool α1-antitrypsin from WT and KO mice were determined by ELISA using α1-antitrypsin antibody and peroxidase-conjugated anti-chicken secondary antibody. A negative control (without stool sample) was performed for each stool sample tested. The absorbance at 450 nm was measured using a microplate reader (Awareness Technology Inc.).
Serum Total Protein and Albumin Analysis
Total serum protein from WT and KO mice was measured from equal volumes of blood serum using BCA reagent. Serum albumin was determined by densitometry and by the bromcresol purple method as reported (23, 24). Densitometric analysis of PAGE-separated proteins were done using Molecular Imager® (ChemiDoc LabTM XRS, Bio-Rad) equipped with Image LabTM software.
Immunoprecipitation (IP), Western Blot (IB), and Dot Blot Analysis
The frozen colon tissue samples from WT and KO mice were washed and resuspended (2 ml/g of tissue) in ice-cold lysis buffer (50 mmol/liter Tris, pH 7.4, containing 0.1 mmol/liter egtazic acid (EGTA), 0.1 mmol/liter EDTA, 2 mmol/liter β-mercaptoethanol, 5 μmol/liter leupeptin, and 4 μmol/liter pepstatin). The suspended tissues were cut into small pieces using sterilized surgical scissors and homogenized using a homogenizer (Wheaton Overhead Stirrer, CT) with 20 brief pulses. The resulting homogenates were centrifuged at 14,000 rpm for 10 min at 4 °C. Protein concentrations in the postnuclear supernatants (total protein extract) were determined using BCA® reagent. Samples were aliquoted and stored at −80 °C. SDS-PAGE was separated, and electroblotted samples were analyzed for protein expression using the indicated antibodies. IP and IB using cell lysates from HT-29 CL-19A cells grown under the indicated experimental conditions were done as reported previously (14). For dot blot analysis, a grid was drawn on a nitrocellulose membrane, and 3 μl of stool samples were loaded at the center of the grid followed by air-drying the membrane. Nonspecific sites on the membrane were blocked by soaking in 5% BSA in TBST followed by incubating with α1-antitrypsin primary antibody and peroxidase-conjugated anti-chicken secondary antibody. The secondary antibodies were detected by IB.
Cell Culture and WHI-P131 Treatment
HT-29 Cl-19A cells were cultured and treated with Jak3 inhibitor WHI-P131 as reported before (12–14). For differentiation studies, cells were grown to subconfluence, confluence, and 2- and 4-week postconfluence. The expression of differentiation markers was determined using Western analysis of the cell lysates from these cells.
Measurement of Trans-epithelial Electrical Resistance (TEER)
TEER was measured using Millicell electrical resistance system (Millipore) as reported (14, 26) and expressed as ohms/cm2, where ohm is the observed value of resistance, and cm2 is the surface area of the membrane. The background resistance of the trans-well membrane (∼30 ohms/cm2) was subtracted from the observed resistance values.
Data Analysis
Differences in the survival rates were evaluated by the log rank test. All data are presented as mean ± S.E. and analyzed with the univariant analysis of variance using Origin® software version 9.2. Differences in the parametric data were evaluated by the Student's t test. Significance in all tests was set at a 95% or greater confidence level.
RESULTS
Knock-out of jak3 Gene Leads to Increased Severity toward DSS-induced Colitis
The DSS-induced colitis in the murine model is commonly used to address the pathogenesis of IBD (27). Previously, we reported the essential roles of Jak3 in mucosal wound healing and homeostasis (12–14); here, we determined the effects of Jak3 KO in DSS-induced colitis. Eight-week-old WT and c57/bl jak3−/− mice were given 2.5% (w/v) DSS in drinking water for 9 days using the previously described model (28, 29), and the time course of body weight change and parameters for inflammation were determined to measure the induction and severity of colitis. Although the body weight was slightly increased in untreated Jak3 KO mice compared with WT, the body weight decreased progressively in both WT and KO mice following DSS treatment, and the KO mice started showing the signs of illness and severity much earlier than the WT. There was significant weight loss and mortality following DSS treatment in Jak3 KO group compared with the wild type (Fig. 1A). The decreased survival in Jak3 KO mice started on day 8 (32% versus 8% in WT p < 0.08) and worsened on day 9 (74% versus 35% in WT p < 0.05). The effect of DSS treatment on the time course of weight loss showed that although WT mice lost about 10–14% of initial weight following 9 days of DSS treatment, Jak3 KO mice started losing weight as early as 5 days post-treatment, and about 20–26% of initial weight was lost at 9 days of DSS treatment, which precluded further observation of the mice. The time course of disease activity index as calculated by weight loss, stool consistency, and presence of fecal blood showed that it was significantly higher in KO mice compared with WT from days 6 to 9 (Fig. 1B). During this treatment period, inflammation was enhanced in the KO mouse group, as indicated by rises in the extent of diarrhea and rectal bleeding. Because of the substantial mortality after 9 days of DSS treatment in Jak3 KO group, we used this time point to further confirm colonic inflammation by MPO activity and histology. Measurement of MPO activity as a measure of inflammation showed that it increased by 1.5-fold in KO mice compared with the WT (Fig. 1C). H&E-stained day 9 colon sections of DSS-treated mice showed severe lesions throughout the mucosa, alteration of epithelial structure, high levels of neutrophil and lymphocyte infiltration into the mucosal and submucosal areas, and loss of crypts, whereas all of these were higher in KO mice (Fig. 1D). These observed alterations were combined to obtain histological scores (Fig. 1E), which were 3-fold higher in KO mice compared with WT. To confirm further, we measured the colon to body weight ratios in these two groups. These ratios were significantly decreased in KO mice, again indicating a higher colonic inflammation in Jak3 KO mice (Fig. 1F). Taken together, these results indicated that KO of Jak3 resulted in increased severity toward DSS-induced colitis in mice.
FIGURE 1.

Knock-out of jak3 gene leads to increased severity toward DSS-induced colitis. A, time course of body weight change during DSS-induced colitis. WT and jak3−/− (KO) mice (n = 6, each group) were treated with water or 2.5% DSS for the indicated periods, and the percent change in body weight was plotted versus days of treatment. B, time course of the disease activity index during DSS treatment. The disease activity index was determined in WT and KO mice as described under “Experimental Procedures” and plotted versus days of treatment. C, MPO in colonic mucosal scrapings. MPO activity was determined as described under “Experimental Procedures.” D and E, histological analysis of DSS-induced colitis. Fresh frozen colonic sections from WT and KO mice were stained with H&E; representative sections are shown with indicated magnifications from each group (n = 6) (D). E, colonic H&E sections from each animal under D were scored in a blinded fashion using protocol described under “Experimental Procedures.” F, colon to body weight ratios of DSS-treated WT (control) and Jak3 KO mice were calculated, and values are shown. A–C, E, and F, values are mean ± S.E. Asterisks indicate statistically significant differences between WT and KO groups (p < 0.05 in at least n = 3 independent experiments).
Jak3 KO Leads to Increased Basal Colonic Inflammation
Because Jak3 KO mice showed increased severity toward DSS-induced colitis and previously we reported that Jak3 plays a role in different mucosal functions (12–14), we determined whether the KO of Jak3 had an impact on colon and colonic mucosa. As shown in Fig. 2, A and B, KO of Jak3 resulted in significant shortening of average colon lengths that were about 26% shorter than their WT counterpart. Additionally, the KO mice also had significantly shorter ceca (Fig. 2C). Because Jak3 KO mice had shorter colons, we determined whether colonic mucosae of these mice were affected. As shown in representative H&E sections in Fig. 2D (×40 and ×100), the epithelial architecture toward the colonic lumen was perturbed in KO mice with interruptions in epithelial lining compared with the WT controls. Further analysis at higher magnifications showed that the mucous layer above the epithelium was discontinuous (Fig. 2D, red arrows in ×100 and ×200), and the arrangement of cells from the base of the crypt to the surface was disturbed in KO mice (Fig. 2D, black arrow, ×400). In addition, both crypt lengths (Fig. 2D, ×200, yellow arrows) and widths (×400, blue lines) were decreased in KO mice. Further analysis showed that there was a 2-fold decrease in crypt length of KO mice (Fig. 2E). Increased body weight is associated with IL-17a- and IL-6-mediated chronic low grade inflammation (30). Because the epithelial architectures were disturbed in untreated Jak3 KO mice, where these mice also showed increased body weight (Fig. 1A) compared with WT, we determined whether KO of Jak3 had an effect on basal inflammation in these mice. Indeed, the KO mice not only showed more than a 6-fold increase in colonic MPO activity. there was also a substantial increase in proinflammatory cytokines IL-6 and IL-17a in these tissues (Fig. 2, F–H). To determine whether the basal inflammation was restricted to the colon, we also determined mucosal structure, Jak3 expression, and MPO activities in small intestine (supplemental Fig. S1, A–C), which indicated that the perturbation of epithelial architecture and basal inflammation was not restricted to the colon only but extended to the small intestine as well. These results showed that KO of Jak3 led to disturbed colonic mucosal architecture that was associated with increased basal inflammation of the colon and small intestine. Although separate studies are underway (data not shown here) to determine the reason for weight gain in KO mice, this study focused on the mechanism of mucosal defects in these mice.
FIGURE 2.

Increased basal colonic inflammation in Jak3 KO mice. A–C, shortening of colon and cecum lengths in Jak3 KO mice. A, representative (n = 5) macroscopic images of the colons with cecum from WT and KO mice are shown. Colon (B) and cecum (C) lengths of WT and KO mice were calculated using NIS Element software (Nikon®), and mean values (n = 5) were plotted for each group. D, histological analysis of colon sections from mice treated with water. Fresh frozen colonic sections from WT and KO mice were stained with H&E; D, representative sections are shown with the indicated magnifications from each group (n = 6). Black arrows (×40) show comparison of colonic mucosa indicating perturbed epithelial architecture with interruptions in epithelial lining in KO mice. Red arrows (×100 and ×200) indicate discontinuous mucous layer overlaying epithelial mucosa. Yellow double arrows (×200) with oval lining around indicates reduced crypt lengths in KO mice. Black arrows and blue lines (×400) indicate reduced crypt width with irregular arrangement of cells from the base of the crypt to the luminal surface in KO mice. E, epidermal thickness (crypt length) from H&E-stained colonic sections (from D) of WT and KO was quantified using the NIS Element imaging software (Nikon®), and mean values from each group (n = 6 mice per group) are plotted. F, increased colonic MPO activity in untreated KO mice. MPO activities in colonic tissue lysates from untreated mice were determined, and mean values from each group (n = 6 mice per group) were plotted. G and H, increased pro-inflammatory cytokines in colon of KO mice. Cytokine level in the colonic tissue lysates from WT and KO mice were measured using a mouse MultiAnalyte cytokine assay kit (Qiagen) as per the manufacturer's protocol, and mean values from each group (n = 6 mice per group) are shown. B, C, and E–H, values are mean ± S.E. Asterisks indicate statistically significant differences between WT and KO groups (p < 0.05 from at least n = 3 independent experiments).
Mucosal Differentiations Were Affected in Jak3 KO Mice
Because Jak3 KO mice showed increased severity toward DSS-induced colitis and Jak3 is known to regulate differentiation of different cells (31, 32), we determined whether KO of Jak3 had effects on the differentiation of colonic mucosae. Our results demonstrate that this was indeed the case where reduced expressions of differentiation markers were evident in the mucosal cells of Jak3 KO mice. These data show that Jak3 was not only expressed in colonic mucosae of mice (Fig. 3A), but the absence of Jak3 led to decreased expression of the differentiation markers for enterocytes, villin, and carbonic anhydrase (33, 34). Western analysis using colonic tissue lysates from these mice further confirmed the loss of Jak3 expression and reduced expression of villin and carbonic anhydrase in colon of KO mice (Fig. 3B). Because Jak3 KO mice showed a discontinuous mucous layer, we determined whether the number of goblet cells and the expression of secretory mucin (muc2) were affected in these mice. As shown in Fig. 3C, there was a nonsignificant decrease in the number of goblet cells per crypt in KO mice, but the surface localizations of these goblet cells in crypts were severely affected in colonic mucosae (Fig. 3D, top and middle panels) of these mice. Additionally, the expression of secretory mucin muc2 was also decreased in KO mice (Fig. 3D, bottom panel). Because decreased goblet cells and decreased muc2 expression are associated with increased incidence of ulcerative colitis in humans (3), these results establish a potential mechanistic link between goblet cell differentiation and increased severity toward DSS-induced colitis in Jak3 KO mice.
FIGURE 3.

Compromised colonic mucosal differentiations in Jak3 KO mice. A, immunofluorescence staining of colonic mucosa. Colonic tissue sections from WT and KO mice were stained using indicated primary antibodies. Images were acquired using Nikon C1-plus laser confocal microscope, and representative images are shown from each group (n = 6). Note that Jak3 is localized to both crypt and villus of colonic mucosa in WT but is completely absent in KO mice. Bar, 550 μm. B, colonic tissue lysates were analyzed using IB for indicated proteins using β-actin as control. Representative blots (n = 3) are shown. C and D, mucin-secreting goblets cells were affected in Jak3 KO mice. Periodic acid-Schiff staining for goblet cells (D) in colonic tissue sections of WT and KO mice were performed, and average numbers of goblet cells per crypt were determined using NIS Element imaging software for each group (n = 6 mice per group with 5 crypts per mice) using protocol described under “Experimental Procedures.”. The mean values (C) of goblet cells per crypt are shown. Black arrows (D, middle panels) show the comparison of luminal surface-localized goblet cells. Notice that KO mice had dispersed goblet cells with less localization toward luminal surfaces. D, bottom panels, immunohistochemical staining for muc2 expression in colonic mucosa of WT and KO mice was performed using the protocol described under “Experimental Procedures.” The data shown are representative (n = 6 per group) of positive-stained areas for muc-2. Black arrows (D, bottom panels) show the absence of muc2-positive cells in KO mice.
Jak3 Expression Facilitates Barrier Functions in Human IEC
Because loss of Jak3 expression led to a compromise in colonic differentiation and increased inflammatory state of colonic mucosa, by using an in vitro cell culture system we determined whether Jak3 expression was changed during the differentiation of IEC, and whether Jak3 expression influenced the epithelial barrier functions. Fig. 4A (left panel) shows that the expression of Jak3 increased with the differentiation of IEC with a maximum expression at 2 weeks post-confluence. This was associated with increased expression of enterocytic differentiation markers villin and carbonic anhydrase. To determine whether Jak3 expression had a role in IEC differentiation, HT-29 cells were stably transfected with HA-tagged Jak3, and expressions of villin and carbonic anhydrase were monitored in Jak3-overexpressing cells. As shown in Fig. 4A (right panel), Jak3 overexpression led to increased expression of enterocytic differentiation markers in subconfluent, confluent, and postconfluent cells. To determine whether Jak3 expression facilitated epithelial barrier function, we determined TEER in HT-29 (control), Jak3-overexpressing (HT-29-HA-Jak3), and doxycycline-regulated Jak3 shRNA-expressing (HT-29-HA-Jak3) cells. Previously, we showed that doxycycline-regulated expression of RFP-tagged Jak3-shRNA using the lentivirus system led to substantial loss of Jak3 protein expression (14). Fig. 4B shows that overexpression of Jak3 increased the TEER of IEC, although its knockdown in these cells by shRNA led to a decrease in TEER. Increased gut permeability is associated with protein-losing enteropathy (PLE) (35). Because Jak3 influenced the barrier functions, we determined whether Jak3 KO mice had PLE symptoms. Fig. 4C shows that both serum total protein (upper panel) and albumin (lower panel) (supplemental Fig. S2) were significantly decreased in KO mice compared with WT. We also determined the fecal α1-antitrypsin as a marker for PLE (36), using ELISA (Fig. 4D, upper panel) and dot blot (corresponding lower panel), both of which indicated an increased level in the fecal extracts of Jak3 KO mice. Together, these results demonstrate that Jak3 expression facilitates the expression of differentiation markers thereby promoting epithelial barrier functions in both differentiated IEC and in mice.
FIGURE 4.

Jak3 promotes expression of enterocytic differentiation markers in human IEC. A, left panel, human-derived colonic epithelial cells HT-29 Cl-19A were grown until subconfluence (Sub), confluence (Conf), and 2 and 4 weeks (wks) postconfluence. Cells lysates were analyzed by IB for the indicated proteins using β-actin as control. Representative blots (n = 3) are shown. Right panel, similar experiments were performed as under the left panel except using HT-29 Cl-19a cells that were stably transfected with wild type HA-tagged Jak3. B, Jak3 expression promotes TEER. Control, HA-tagged Jak3-expressing (Jak3-HT-29) and doxycycline-regulated RFP-tagged Jak3-shRNA (shRNA-HT-29) expressing in Jak3-HT-29 cells were cultured in a 24-transwell plate to confluence, and TEER was measured. Mean values (n = 6 independent experiments) of TEER are shown. For Jak3 shRNA-HT-29, the expression of RFP-tagged Jak3-shRNA was induced by supplementing doxycycline in the growth media. Previously, we reported that this leads to knock down of Jak3 expression in HT-29 CL-19A cells where treatment with doxycyline alone in untransduced cells had no effect on Jak3 expression (25). C and D, Jak3 KO mice show symptoms of PLE. Serum total protein and albumin (C) and fecal α1-antitrypsin (D) were determined in serum and fecal extracts, respectively, from WT and KO mice using methods described under “Experimental Procedures.” Lower panel in C and D show representative images (n = 3). B–D, values are means ± S.E. (n = 6). Asterisks indicate statistically significant differences (p < 0.05) from the control cells.
Jak3 Regulates β-Catenin Localization to AJ
Because Jak3 expression facilitated the epithelial barrier functions, we determined the effects of Jak3 knockdown on AJ in differentiated IEC. Fig. 5A shows that although expression of Jak3 was mostly restricted to apical surfaces in the confluent (bright field image in upper left panel and corresponding XZ images on the right panel) IEC, there was substantial increase in Jak3 expression at the basolateral surfaces of post-confluent differentiated IECs (bright field image in lower left panel and corresponding XZ images on right panels). For these experiments, AJ protein β-catenin was taken as control, the expression of which was also increased at the basolateral surfaces in the differentiated IEC (Fig. 5A, right, 2nd and 4th panels from the top). Because β-catenin localization to AJ is essential for the assembly of the junctions and maintenance of epithelial barrier functions (37), we determined whether the expression of Jak3 played a role in β-catenin localization to AJ. Fig. 5B, left panel, shows a continuous expression of β-catenin around the cellular periphery indicating an intact AJ in control differentiated IEC that was transduced with lentiviruses containing RFP-tagged Jak3 shRNA but was not supplemented with doxycycline in the growth media. However, when doxycycline was added to the growth media of these cells, it led to the expression of RFP-tagged Jak3 shRNA (indicated by red fluorescence, Fig. 5B, right panel) and substantial loss of β-catenin expression at AJ as indicated by relatively discontinuous and punctate staining for β-catenin at the cell periphery in these differentiated IEC. Doxycycline alone had no effect on the expression or localization of β-catenin, which showed similar results as the control (data not shown). Next, we determined whether Jak3 directly interacted with β-catenin and the conditions for their interactions. Fig. 5C (left panel, 1st and 2nd from the top) shows that Jak3 antibody co-immunoprecipitated β-catenin and vice versa from the cell lysates of differentiated IEC grown in the presence of serum. β-Catenin is known to be phosphorylated at tyrosine residues 142 and 645, where tyrosine 142-phosphorylated β-catenin is targeted to the nucleus to participate as a transcription factor for the expression of different genes (38). We determined whether Jak3-associated β-catenin was tyrosine-phosphorylated and whether the transcription factor pool (Tyr(P)-142) of β-catenin was present in this complex. Fig. 5C (left panel, 3rd from the top) shows that Jak3-associated β-catenin was tyrosine-phosphorylated but not at tyrosine 142, the transcription factor pool of β-catenin (left panel 4th from the top). Fig. 5C (5th panel from the top) shows that Jak3-associated β-catenin was phosphorylated mostly at tyrosine 654. We determined whether Jak3 activation was necessary for its interaction with β-catenin. Fig. 5C (top right panel) shows that β-catenin antibody immunoprecipitated β-catenin from the cell lysates of differentiated IEC that was treated with Jak3 inhibitor WHI-P131. However, Jak3-associated β-catenin was substantially reduced (Fig. 5C, compare with left panel, 2nd from the top) in the presence of the Jak3 inhibitor as indicated by Jak3 antibody co-immunoprecipitated β-catenin in the presence of WHI-P131. These results show that Jak3 activation was necessary for the interactions between Jak3 and β-catenin where tyrosine residue 654 rather than 142 of β-catenin was phosphorylated through these interactions. Because Jak3 interacted with β-catenin and the expression of Jak3 was necessary for AJ localization of β-catenin, we determined the effect of loss of Jak3 on the localization of β-catenin in the colonic mucosa of mice. As shown in Fig. 5D, β-catenin was localized at the AJ toward the luminal surfaces of the differentiated colonic epithelial mucosa of WT mice (white arrow). In these mice, the undifferentiated crypt cells situated toward the bottom of the columnar crypt axis also showed β-catenin expression, which did not appear to be at the AJ (Fig. 5D, yellow arrow). In contrast, loss of Jak3 expression resulted in crypts that were not restricted to the bottom of the crypt axis but also migrated toward the luminal side of the mucosa (Fig. 5D, yellow arrow) and the columnar structures of the crypt axis were lost. Moreover, β-catenin was localized exclusively to these noncolumnar crypts only. Taken together, these results show that Jak3 not only interacted with β-catenin but also facilitated the localization of β-catenin to AJ in both differentiated IEC and in differentiated colonic mucosa of mice.
FIGURE 5.

Jak3 facilitates AJ formation through interactions with β-catenin. A, Jak3 redistributes to basolateral surfaces in differentiated human IEC. Cellular redistribution of Jak3 was determined using IFM in confluent (nondifferentiated) and 2-week post-confluent (as these had maximum expression of differentiation markers, Fig. 4A) HT-29 Cl-19a cells. Bright field images (left panels) and corresponding XZ section (right panels) of Jak3 (green)-stained cells were taken to determine the basolateral redistribution using AJ protein β-catenin as a positive control. Representative images are shown (n = 5 experiments). B, Jak3 regulates β-catenin localization to AJ. AJ localization of β-catenin was determined in 2-week post-confluent HT-29 CL-19a cells transduced with lentiviruses mentioned in Fig. 4B and grown in the presence (+Dox) or absence (−Dox) of doxycycline, and IFM was performed using antibody for β-catenin. Representative images are shown (n = 6 experiments), where the green color indicates β-catenin and the red color confirms RFP-tagged Jak3-shRNA expression-mediated knockdown of Jak3 expression (25). Note that knockdown of Jak3 expression disrupts the AJ localization of β-catenin (white arrows) as denoted by punctate green staining in Jak3-shRNA-expressing (+Dox) cells. Bar 14 μm. C, Jak3 activation facilitates its interactions with tyrosine 654-phosphorylated pool of β-catenin. Left panel, co-IP followed by IB studies were done using cell lysates from HT-29 CL-19a cells and indicated antibodies. Representative blots are shown (n = 3 experiments). C, right panel, effect of inhibition of Jak3 on the interactions between Jak3 and β-catenin was studied using co-IP followed by IB studies from lysates of HT-29 CL-19A cells treated with Jak3 inhibitor WHI-P131 (25 μm). Previously, we reported that treatment with WHI-P131 leads to inhibition of Jak3 in HT-29 CL-19A cells (12, 13, 25). D, knock-out of Jak3 affects localization of β-catenin in colonic mucosa of mice. Localization of β-catenin was studied in colonic tissue sections from WT and KO mice using IFM as described in Fig. 3A. Representative images are shown (n = 5 experiments with three tissue sections per experiment). Profuse β-catenin staining in WT colon is evident in the crypt and villus regions (yellow and white arrows, respectively), which were absent in KO mice. Scale bar, 550 μm.
DISCUSSION
The gastrointestinal tract is a highly organized tissue that maintains homeostasis on three fronts as follows: (a) cells of microbiome situated over the mucosal surfaces; (b) the cells of mucosal surfaces itself, and (c) the cells of the immune system situated below the mucosal surface. Recently, we showed that Jak3 plays an essential role during mucosal wound repair and homeostasis (12, 14). Interestingly, although Jak3 interactions with cytoskeletal and adapter proteins were essential for mucosal wound repair function (12) and intestinal epithelial homeostasis, respectively (14), the in vivo significance of these functions was lacking. To achieve this, we first determined whether the absence of Jak3 expression influenced the severity of DSS-induced colitis in mice. Administration of synthetic DSS in drinking water leads to acute and chronic experimental ulcerative colitis in mice and is a good model to assess mucosal integrity (16). Our data suggested that although untreated Jak3 KO mice slightly gained weight over time compared with WT, treatment with DSS led to a substantial loss of body weight in KO mice that was associated with loss of colonic mucosal integrity. These resulted in an increased severity of colitis in KO mice compared with WT. Although reduced expression of Jak3 has been reported in human ulcerative colitis patients (39), it was not known if the reduced expression of Jak3 was the cause or the effect of colitis. To find out if the absence of Jak3 was a reason for increased severity of colitis, we determined the basal colonic inflammation in Jak3 KO mice. Our data indicated that absence of Jak3 expression resulted in shortened colon length, reduced colon to body weight ratio, and reduced cecum length in untreated mice. Although several studies indicated reduced colonic length post-DSS-induced colitis, it was not known if reduced colon length predisposes toward colitis (40, 41). We showed that Jak3-deficient mice had reduced colon length, and when these mice were subjected to DSS-induced colitis, they developed more severe colitis symptoms than their WT counterparts suggesting that Jak3 played a protective role during DSS-induced colitis (Fig. 1). Increased body weight and obesity are considered to be inflammatory predispositions, and these conditions predispose toward different inflammatory disorders, including inflammatory bowel disease. These in turn are mediated by increased expression of proinflammatory cytokines such as IL-6 and IL-17A (42, 43). Interestingly, our data suggested that although KO of Jak3 led to a marginal increase in body weight, the colonic levels of IL-6 and IL17A were substantially increased. This led to a low grade basal chronic inflammation as reflected by shortening of colon and cecum lengths with disturbed colonic and intestinal mucosal architecture and increased MPO activities (Fig. 2 and supplemental Fig. S1). Thus, it appears that KO of Jak3 was responsible for inflammatory predisposition of not only the colon but also the small intestine. Moreover, these conditions were aggravated when KO mice were subjected to colitis as reflected by increased severity of colonic inflammation during DSS-induced colitis.
To find out how the absence of Jak3 contributed to inflammation of the colon, we determined mucosal differentiation in the colon of Jak3 KO mice. Jak3 is widely expressed in different immune cells where, among other functions, it helps in their differentiation (44). Although Jak3 is also known to regulate differentiation of other cells types (31, 32), and our previous studies showed a role of Jak3 in mucosal wound repair and homeostasis (12–14), the role of Jak3 in mucosal differentiation was not known. Our data suggested that Jak3 not only promoted expression of colonocyte differentiation markers in human IEC but the lack of Jak3 led to compromise in the differentiation of colonic mucosae as reflected by diminished expression of villin and carbonic anhydrase, the differentiation markers for enterocytic lineages (Figs. 3 and 4). Colonic mucosae of both human and mice are overlaid with a thick mucous layer that prevents the direct access of microbe to mucosal epithelia. The decreased mucous layer increases the severity of colitis in both humans and mice (45). Goblet cells are the major mucin-producing cells in colon where they produce secretory glycoproteins mucin (muc2), which is the major constituent of the protective mucus layer. Decreased muc2 expression is associated with the increased incidence of ulcerative colitis in humans (3, 46). In contrast, mice lacking intestinal mucin (muc-2 KO) or mice with single missense mutations in the muc2 gene develop colitis with mucosal friability and spontaneous wound formation of the mucosa (47). Our data suggested that loss of Jak3 not only resulted in decreased surface localization of goblet cells toward the colonic lumen, it also affected the expression of secretory mucin, muc-2, that could be the reason for the discontinuous mucous layer and increased predisposition to colitis in mice (Fig. 3).
An intrinsic defect in the mucosal barrier has been implicated to be one of several factors that contribute to the exaggerated immune response to otherwise normal commensal microbiota. This leads to chronic inflammation during IBD (48). We showed that Jak3 promoted barrier functions in human IEC and its KO in mice predisposed toward colitis. Thus, it is safe to speculate that mucosal barrier in Jak3 KO mice was compromised as reflected by chronic low grade basal inflammation in colon, PLE symptoms, and increased severity during DSS-induced colitis. To understand mechanistically how Jak3 might be facilitating barrier functions, our data showed that Jak3 redistributed to basolateral surfaces in differentiated human IECs, Jak3 interacted with β-catenin, Jak3 activations were necessary for these interactions, and knockdown of Jak3 decreased TEER and AJ localizations of β-catenin. Expression of β-catenin at AJ is essential for the differentiation and barrier functions of colonic mucosa (49, 50). Altogether, these point to an essential role of Jak3 not only in mucosal differentiation but also in facilitating barrier functions through its interactions with β-catenin (Fig. 5). Although phosphorylation of tyrosine 142 of β-catenin facilitates its participation in transcription factor activity, phosphorylation of tyrosine 654 decreases intramolecular association between the C-terminal tail and the armadillo repeat domain, where armadillo repeat domain facilitates β-catenin interactions with E-cadherin (38, 51). Moreover, the phosphomimetic mutant Y654E of β-catenin that mimics constitutive phosphorylation at tyrosine 654 forms a functional cadherin-catenin complex to mediate strong cell adhesion (52). Our data suggest that phosphorylation of 654 and not of 142 was important for β-catenin interactions with Jak3 upon Jak3 activation. These indicate the physiological significance of these interactions where Jak3 activation led to β-catenin phosphorylation at Tyr-654, which promoted β-catenin interactions with E-cadherin, thereby facilitating AJ formation and enhancing the IEC barrier functions (Fig. 4). Because we showed that Jak3 was located at the apical surfaces in nondifferentiated/differentiated cells and previously we reported that the less phosphorylated form of Jak3 translocates to the nucleus in mucosal epithelial cells (14), it is possible that Jak3 interactions with β-catenin may facilitate nuclear translocation of these where they can influence the expression of differentiation markers through transcription factor activity in those cells that are not fully differentiated. Under in vivo conditions, because Jak3 played a key role in expression of differentiation markers, this role of Jak3 could be through its activation by different cytokine receptors at the apical surfaces in nondifferentiated/differentiated cells. However, Jak3 translocated to basolateral surfaces in fully differentiated cells where it facilitated cellular junction formation. Together, these data indicate that Jak3 expression was essential for differentiation, mucin expression, and barrier functions of colonic mucosa (Fig. 6). Additionally, these could have direct implications for patients with immunological disorders, including IBD where using immunosuppressive drugs having Jak3 inhibition activities may complicate the therapeutic outcome through compromised mucosal barrier functions (25, 53). Alternatively, using a delivery route with the least accumulation of Jak3 inhibitor at the intestinal mucosa may improve the outcome.
FIGURE 6.
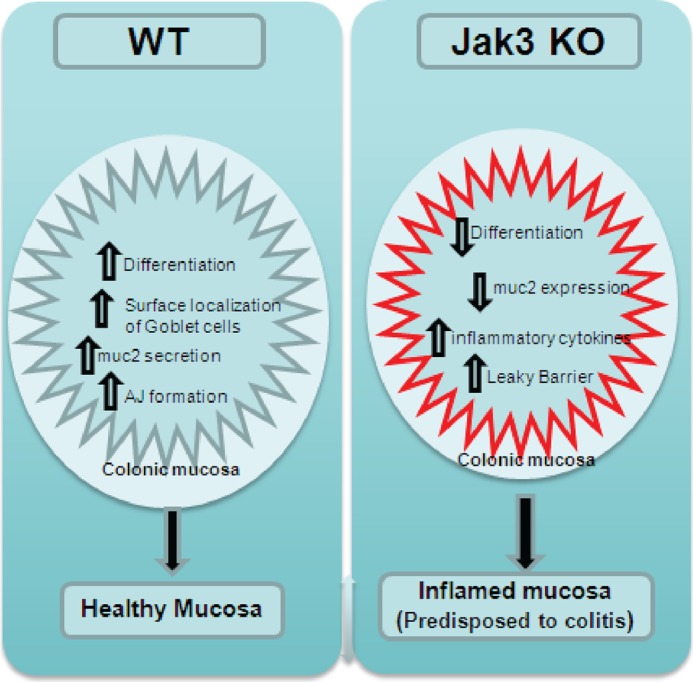
Proposed model for the role of Jak3 in colonic mucosal health and predisposition to colitis.
In summary, we showed that Jak3 played a critical role in the pathogenesis of colitis induced by DSS, where colonic expression of Jak3 was essential for a healthy mucosal barrier (Fig. 6), and knock-out of Jak3 resulted in a low grade chronic inflammation in colon and PLE symptoms, which predisposed the mice toward increased severity of colitis. In IEC, we demonstrated that Jak3 facilitated enhanced barrier functions through interactions and AJ localization of β-catenin. Thus, these results showed for the first time that Jak3 was essential for mucosal differentiation and barrier functions where it contributed to the expression of differentiation markers and reinforced barrier functions through its interactions with β-catenin.
Acknowledgment
We thank Dr. Sohel Quazi for technical help.
This work was supported, in whole or in part, by National Institutes of Health Grant DK081661 (to N. K.). This work was also supported by Crohn's and Colitis Foundation of America Grant CCFA 2188 and the Dr. Nicholas C. Hightower Centennial Chair of Gastroenterology Endowment from the Scott and White Hospital (to G. A.).

This article contains supplemental Figs. S1 and S2.
- IBD
- inflammatory bowel disease
- AJ
- adherens junction
- TEER
- trans-epithelial electrical resistance
- IEC
- intestinal epithelial cells
- IP
- immunoprecipitation
- IB
- immunoblotting
- Jak3
- Janus kinase 3
- PLE
- protein-losing enteropathy
- MPO
- myeloperoxidase
- DSS
- dextran sulfate sodium
- RFP
- red fluorescence protein
- IFM
- immunofluorescence microscopy.
REFERENCES
- 1. Sancho E., Batlle E., Clevers H. (2003) Live and let die in the intestinal epithelium. Curr. Opin. Cell Biol. 15, 763–770 [DOI] [PubMed] [Google Scholar]
- 2. Dignass A. U. (2001) Mechanisms and modulation of intestinal epithelial repair. Inflamm. Bowel. Dis. 7, 68–77 [DOI] [PubMed] [Google Scholar]
- 3. Gersemann M., Becker S., Kübler I., Koslowski M., Wang G., Herrlinger K. R., Griger J., Fritz P., Fellermann K., Schwab M., Wehkamp J., Stange E. F. (2009) Differences in goblet cell differentiation between Crohn's disease and ulcerative colitis. Differentiation 77, 84–94 [DOI] [PubMed] [Google Scholar]
- 4. Pullan R. D., Thomas G. A., Rhodes M., Newcombe R. G., Williams G. T., Allen A., Rhodes J. (1994) Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut 35, 353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Christodoulou K., Wiskin A. E., Gibson J., Tapper W., Willis C., Afzal N. A., Upstill-Goddard R., Holloway J. W., Simpson M. A., Beattie R. M., Collins A., Ennis S. (2013) Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes. Gut 62, 977–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Safford M. G., Levenstein M., Tsifrina E., Amin S., Hawkins A. L., Griffin C. A., Civin C. I., Small D. (1997) JAK3: expression and mapping to chromosome 19p12–13.1. Exp. Hematol. 25, 374–386 [PubMed] [Google Scholar]
- 7. Takahashi T., Shirasawa T. (1994) Molecular cloning of rat JAK3, a novel member of the JAK family of protein-tyrosine kinases. FEBS Lett. 342, 124–128 [DOI] [PubMed] [Google Scholar]
- 8. Murata Y., Yamashita A., Saito T., Sugamura K., Hamuro J. (2002) The conversion of redox status of peritoneal macrophages during pathological progression of spontaneous inflammatory bowel disease in Janus family tyrosine kinase 3−/− and IL-2 receptor γ−/− mice. Int. Immunol. 14, 627–636 [DOI] [PubMed] [Google Scholar]
- 9. Cornejo M. G., Boggon T. J., Mercher T. (2009) JAK3: a two-faced player in hematological disorders. Int. J. Biochem. Cell Biol. 41, 2376–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin Q., Lai R., Chirieac L. R., Li C., Thomazy V. A., Grammatikakis I., Rassidakis G. Z., Zhang W., Fujio Y., Kunisada K., Hamilton S. R., Amin H. M. (2005) Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and cell lines: inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. Am. J. Pathol. 167, 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mishra J., Drummond J., Quazi S. H., Karanki S. S., Shaw J. J., Chen B., Kumar N. (2013) Prospective of colon cancer treatments and scope for combinatorial approach to enhanced cancer cell apoptosis. Crit. Rev. Oncol. Hematol. 86, 232–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kumar N., Mishra J., Narang V. S., Waters C. M. (2007) Janus kinase 3 regulates interleukin 2-induced mucosal wound repair through tyrosine phosphorylation of villin. J. Biol. Chem. 282, 30341–30345 [DOI] [PubMed] [Google Scholar]
- 13. Mishra J., Karanki S. S., Kumar N. (2012) Identification of molecular switch regulating interactions of Janus kinase 3 with cytoskeletal proteins. J. Biol. Chem. 287, 41386–41391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mishra J., Waters C. M., Kumar N. (2012) Molecular mechanism of interleukin-2-induced mucosal homeostasis. Am. J. Physiol. Cell Physiol. 302, C735–C747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cormet-Boyaka E., Di A., Chang S. Y., Naren A. P., Tousson A., Nelson D. J., Kirk K. L. (2002) CFTR chloride channels are regulated by a SNAP-23/syntaxin 1A complex. Proc. Natl. Acad. Sci. U.S.A. 99, 12477–12482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Okayasu I., Hatakeyama S., Yamada M., Ohkusa T., Inagaki Y., Nakaya R. (1990) A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98, 694–702 [DOI] [PubMed] [Google Scholar]
- 17. Mashimo H., Wu D. C., Podolsky D. K., Fishman M. C. (1996) Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science 274, 262–265 [DOI] [PubMed] [Google Scholar]
- 18. Cooper H. S., Murthy S. N., Shah R. S., Sedergran D. J. (1993) Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Invest. 69, 238–249 [PubMed] [Google Scholar]
- 19. Xu Y., Hunt N. H., Bao S. (2008) The role of granulocyte macrophage-colony-stimulating factor in acute intestinal inflammation. Cell Res. 18, 1220–1229 [DOI] [PubMed] [Google Scholar]
- 20. Mackay F., Browning J. L., Lawton P., Shah S. A., Comiskey M., Bhan A. K., Mizoguchi E., Terhorst C., Simpson S. J. (1998) Both the lymphotoxin and tumor necrosis factor pathways are involved in experimental murine models of colitis. Gastroenterology 115, 1464–1475 [DOI] [PubMed] [Google Scholar]
- 21. Kabashima K., Saji T., Murata T., Nagamachi M., Matsuoka T., Segi E., Tsuboi K., Sugimoto Y., Kobayashi T., Miyachi Y., Ichikawa A., Narumiya S. (2002) The prostaglandin receptor EP4 suppresses colitis, mucosal damage, and CD4 cell activation in the gut. J. Clin. Invest. 109, 883–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elrod J. W., Laroux F. S., Houghton J., Carpenter A., Ando T., Jennings M. H., Grisham M., Walker N., Alexander J. S. (2005) DSS-induced colitis is exacerbated in STAT-6 knockout mice. Inflamm. Bowel Dis. 11, 883–889 [DOI] [PubMed] [Google Scholar]
- 23. Ganta V. C., Cromer W., Mills G. L., Traylor J., Jennings M., Daley S., Clark B., Mathis J. M., Bernas M., Boktor M., Jordan P., Witte M., Alexander J. S. (2010) Angiopoietin-2 in experimental colitis. Inflamm. Bowel Dis. 16, 1029–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lo S. F., Miller W. G., Doumas B. T. (2013) Laboratory performance in albumin and total protein measurement using a commutable specimen: results of a College of American Pathologists study. Arch. Pathol. Lab. Med. 137, 912–920 [DOI] [PubMed] [Google Scholar]
- 25. Papageorgiou A. C., Wikman L. E. (2004) Is JAK3 a new drug target for immunomodulation-based therapies? Trends Pharmacol. Sci. 25, 558–562 [DOI] [PubMed] [Google Scholar]
- 26. Sheth P., Delos Santos N., Seth A., LaRusso N. F., Rao R. K. (2007) Lipopolysaccharide disrupts tight junctions in cholangiocyte monolayers by a c-Src-, TLR4-, and LBP-dependent mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G308–G318 [DOI] [PubMed] [Google Scholar]
- 27. Sann H., Erichsen Jv, Hessmann M., Pahl A., Hoffmeyer A. (2013) Efficacy of drugs used in the treatment of IBD and combinations thereof in acute DSS-induced colitis in mice. Life Sci. 92, 708–718 [DOI] [PubMed] [Google Scholar]
- 28. Zhang H., Kuai X. Y., Yu P., Lin L., Shi R. (2012) Protective role of uncoupling protein-2 against dextran sodium sulfate-induced colitis. J. Gastroenterol. Hepatol. 27, 603–608 [DOI] [PubMed] [Google Scholar]
- 29. Beck P. L., Ihara E., Hirota S. A., MacDonald J. A., Meng D., Nanthakumar N. N., Podolsky D. K., Xavier R. J. (2010) Exploring the interplay of barrier function and leukocyte recruitment in intestinal inflammation by targeting fucosyltransferase VII and trefoil factor 3. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G43–G53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ahmed M., Gaffen S. L. (2010) IL-17 in obesity and adipogenesis. Cytokine Growth Factor Rev. 21, 449–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jang Y. N., Lee I. J., Park M. C., Baik E. J. (2012) Role of JAK3 in myogenic differentiation. Cell. Signal. 24, 742–749 [DOI] [PubMed] [Google Scholar]
- 32. Murphy K. M. (2008) Permission to proceed: Jak3 and STAT5 signaling molecules give the green light for T helper 1 cell differentiation. Immunity 28, 725–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chantret I., Barbat A., Dussaulx E., Brattain M. G., Zweibaum A. (1988) Epithelial polarity, villin expression, and enterocytic differentiation of cultured human colon carcinoma cells: a survey of twenty cell lines. Cancer Res. 48, 1936–1942 [PubMed] [Google Scholar]
- 34. Bekku S., Mochizuki H., Takayama E., Shinomiya N., Fukamachi H., Ichinose M., Tadakuma T., Yamamoto T. (1998) Carbonic anhydrase I and II as a differentiation marker of human and rat colonic enterocytes. Res. Exp. Med. 198, 175–185 [DOI] [PubMed] [Google Scholar]
- 35. Shin J. K., Jung Y. H., Bae M. N., Baek I. W., Kim K. J., Cho C. S. (2013) Successful treatment of protein-losing enteropathy due to AA amyloidosis with octreotide in a patient with rheumatoid arthritis. Mod. Rheumatol. 23, 406–411 [DOI] [PubMed] [Google Scholar]
- 36. Brimnes J., Reimann J., Claesson M. H. (2000) Immunoglobulin leakiness in scid mice with CD4+ T-cell-induced chronic colitis. Clin. Immunol. 96, 222–229 [DOI] [PubMed] [Google Scholar]
- 37. You H., Padmashali R. M., Ranganathan A., Lei P., Girnius N., Davis R. J., Andreadis S. T. (2013) JNK regulates compliance-induced adherens junctions formation in epithelial cells and tissues. J. Cell Sci. 126, 2718–2729 [DOI] [PubMed] [Google Scholar]
- 38. Piedra J., Miravet S., Castaño J., Pálmer H. G., Heisterkamp N., García de Herreros A., Duñach M. (2003) p120 Catenin-associated Fer and Fyn tyrosine kinases regulate β-catenin Tyr-142 phosphorylation and β-catenin-α-catenin interaction. Mol. Cell. Biol. 23, 2287–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dai S. X., Wu G., Zou Y., Feng Y. L., Liu H. B., Feng J. S., Chi H. G., Lv R. X., Zheng X. B. (2013) Balance of CD8+ CD28+/CD8+ CD28− T lymphocytes is vital for patients with ulcerative colitis. Dig. Dis. Sci. 58, 88–96 [DOI] [PubMed] [Google Scholar]
- 40. Assi K., Mills J., Owen D., Ong C., St Arnaud R., Dedhar S., Salh B. (2008) Integrin-linked kinase regulates cell proliferation and tumour growth in murine colitis-associated carcinogenesis. Gut 57, 931–940 [DOI] [PubMed] [Google Scholar]
- 41. Banner K. H., Cattaneo C., Le Net J. L., Popovic A., Collins D., Gale J. D. (2004) Macroscopic, microscopic and biochemical characterisation of spontaneous colitis in a transgenic mouse, deficient in the multiple drug resistance 1a gene. Br. J. Pharmacol. 143, 590–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hass D. J., Brensinger C. M., Lewis J. D., Lichtenstein G. R. (2006) The impact of increased body mass index on the clinical course of Crohn's disease. Clin. Gastroenterol. Hepatol. 4, 482–488 [DOI] [PubMed] [Google Scholar]
- 43. Zúñiga L. A., Shen W. J., Joyce-Shaikh B., Pyatnova E. A., Richards A. G., Thom C., Andrade S. M., Cua D. J., Kraemer F. B., Butcher E. C. (2010) IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J. Immunol. 185, 6947–6959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi M., Lin T. H., Appell K. C., Berg L. J. (2008) Janus-kinase-3-dependent signals induce chromatin remodeling at the Ifng locus during T helper 1 cell differentiation. Immunity 28, 763–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Johansson M. E., Gustafsson J. K., Holmén-Larsson J., Jabbar K. S., Xia L., Xu H., Ghishan F. K., Carvalho F. A., Gewirtz A. T., Sjövall H., Hansson G. C. (2013) Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut, 10.1136/gutjnl-2012-303207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dorofeyev A. E., Vasilenko I. V., Rassokhina O. A., Kondratiuk R. B. (2013) Mucosal barrier in ulcerative colitis and Crohn's disease. Gastroenterol. Res. Pract. 2013, 431231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heazlewood C. K., Cook M. C., Eri R., Price G. R., Tauro S. B., Taupin D., Thornton D. J., Png C. W., Crockford T. L., Cornall R. J., Adams R., Kato M., Nelms K. A., Hong N. A., Florin T. H., Goodnow C. C., McGuckin M. A. (2008) Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 5, e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kosa P., Szabo R., Molinolo A. A., Bugge T. H. (2012) Suppression of Tumorigenicity-14, encoding matriptase, is a critical suppressor of colitis and colitis-associated colon carcinogenesis. Oncogene 31, 3679–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Joo M., Shahsafaei A., Odze R. D. (2009) Paneth cell differentiation in colonic epithelial neoplasms: evidence for the role of the Apc/β-catenin/Tcf pathway. Hum. Pathol. 40, 872–880 [DOI] [PubMed] [Google Scholar]
- 50. Mariadason J. M., Bordonaro M., Aslam F., Shi L., Kuraguchi M., Velcich A., Augenlicht L. H. (2001) Down-regulation of β-catenin TCF signaling is linked to colonic epithelial cell differentiation. Cancer Res. 61, 3465–3471 [PubMed] [Google Scholar]
- 51. Piedra J., Martinez D., Castano J., Miravet S., Dunach M., de Herreros A. G. (2001) Regulation of β-catenin structure and activity by tyrosine phosphorylation. J. Biol. Chem. 276, 20436–20443 [DOI] [PubMed] [Google Scholar]
- 52. Tominaga J., Fukunaga Y., Abelardo E., Nagafuchi A. (2008) Defining the function of β-catenin tyrosine phosphorylation in cadherin-mediated cell-cell adhesion. Genes Cells 13, 67–77 [DOI] [PubMed] [Google Scholar]
- 53. Vuitton L., Koch S., Peyrin-Biroulet L. (2013) Janus kinase inhibition with Tofacitinib: Changing the face of inflammatory bowel disease treatment. Curr. Drug Targets, in press [DOI] [PubMed] [Google Scholar]