

Background: Aberrant mRNPs are targeted and degraded by an Rrp6p-dependent nuclear quality control system.
Results: The exosome cofactors Rrp47p, Mpp6p, and two TRAMP complexes are cotranscriptionally recruited like Rrp6p and contribute to the elimination process.
Conclusion: The exosome cofactors assist Rrp6p in the targeting of aberrant mRNPs.
Significance: We provide an integrated view of how cofactors of the RNA degradation machinery cooperate to target aberrant mRNPs.
Keywords: mRNA, mRNA Decay, RNA, RNA Metabolism, RNA Processing
Abstract
The cotranscriptional mRNA processing and packaging reactions that lead to the formation of export-competent messenger ribonucleoprotein particles (mRNPs) are under the surveillance of quality control steps. Aberrant mRNPs resulting from faulty events are retained in the nucleus with ensuing elimination of their mRNA component. The molecular mechanisms by which the surveillance system recognizes defective mRNPs and stimulates their destruction by the RNA degradation machinery are still not completely elucidated. Using an experimental approach in which mRNP formation in yeast is disturbed by the action of the bacterial Rho helicase, we have shown previously that the targeting of Rho-induced aberrant mRNPs is mediated by Rrp6p, which is recruited cotranscriptionally in association with Nrd1p following Rho action. Here we investigated the specific involvement in this quality control process of different cofactors associated with the nuclear RNA degradation machinery. We show that, in addition to the main hydrolytic action of the exonuclease Rrp6p, the cofactors Rrp47p, Mpp6p as well as the Trf-Air-Mtr4 polyadenylation (TRAMP) components Trf4p, Trf5p, and Air2p contribute significantly by stimulating the degradation process upon their cotranscriptional recruitment. Trf4p and Trf5p are apparently recruited in two distinct TRAMP complexes that both contain Air2p as component. Surprisingly, Rrp47p appears to play an important role in mutual protein stabilization with Rrp6p, which highlights a close association between the two partners. Together, our results provide an integrated view of how different cofactors of the RNA degradation machinery cooperate to target and eliminate aberrant mRNPs.
Introduction
The expression of protein coding genes in eukaryotic cells is a multistep process in which the genetic message is transcribed from DNA into a pre-mRNA molecule that undergoes numerous modifications such as 5′ end capping, splicing, 3′ end cleavage, and polyadenylation while being assembled into a messenger ribonucleoprotein particle (mRNP)4 that will be exported to the cytoplasm for translation (1, 2). These events, coupled to transcript elongation by RNA polymerase II, are error-prone, and, thus, the cells have developed surveillance mechanisms that detect malformed mRNPs resulting from inappropriate or inefficient processing and packaging reactions. Aberrant mRNPs that fail to pass the quality control steps are retained in the nucleus with ensuing degradation of their mRNA component by ribonuclease activities associated with the RNA exosome (3–7).
Most studies regarding nuclear mRNP surveillance have relied on the use of Saccharomyces cerevisiae mutant strains with defects in the THO-Sub2p complex, which mediates mRNP assembly and export of a subset of transcripts. It has been shown that deletion or mutation of any component of the complex leads to a large reduction in the steady-state level of some THO-Sub2p-dependent transcripts, with the heat shock-inducible HSP104 mRNA as the prominent example. The level of these transcripts can be recovered when the RNA degradation machinery is compromised by removal or inactivation of some components of the RNA exosome (8–12). The precise nature of the defects inflicted to the mRNPs in this particular system and how they are detected by the quality control apparatus and directed to the RNA degradation machinery is still not completely delineated.
The RNA exosome is a highly conserved multisubunit complex that plays a central role in whole cellular RNA metabolism, including processing, maturation, and surveillance of non-coding RNAs (such as rRNA, tRNA, snRNA, small nucleolar RNA, and cryptic unstable transcripts) as well as surveillance and turnover of protein-coding mRNAs. A special feature of the RNA exosome is that it requires several cofactors to promote its catalytic activity and determine its cellular localization (13, 14). In yeast, a ring-shaped core complex formed by nine subunits is associated with two additional subunits, Rrp6p and Dis3p (Rrp44p), which provide the RNA degradation activities of the exosome. Rrp6p is confined in the nucleus and possesses a 3′-to-5′ exonuclease activity, whereas Dis3p is associated with both the nuclear and the cytoplasmic forms of the exosome and displays both endonuclease and 3′-to-5′ exonuclease activities (15–17). So far, it is still unclear whether the two ribonucleases always require the core exosome scaffold to perform their functions in the cell or whether they can carry out hydrolytic activities independently or within other protein complexes, depending on the functional tasks. However, several reports suggest that at least Rrp6p appears to possess some functional activities that are distinct from those performed by the exosome in the cell (18–20).
Two additional exosome cofactors restricted to the nucleus (Rrp47p and Mpp6p) were identified as components of Rrp6p-containing exosome complexes (21–23). The two cofactors seem to have distinct RNA-binding preferences, suggesting that their role could be to direct the exosome complex to specific RNA substrates. The catalytic functions of the exosome are also stimulated by another cofactor, named the TRAMP complex, which is formed by one of two non-conventional poly(A) polymerases Trf4p or Trf5p, one of two homologous RNA-binding proteins Air1p or Air2p, and the RNA helicase Mtr4p. The TRAMP complex has been shown to participate in the nuclear quality control of several types of RNAs (24–26). However, the mechanisms by which it stimulates the RNA degradation activity of the exosome and/or promotes the targeting of specific RNA substrates remain poorly understood.
Recently, we reported the implementation of a new assay to study nuclear mRNP surveillance in S. cerevisiae based on perturbation of mRNP biogenesis by the RNA-dependent helicase/translocase activity of the bacterial Rho factor. In this model system, the heterologous expression of Rho in the yeast nucleus induces the production of full-length but aberrant mRNPs that are targeted and eliminated by the Rrp6p-dependent RNA degradation machinery, leading to a growth defect phenotype (27). We have shown previously that the recognition and removal of Rho-induced aberrant mRNPs is mediated by a large increase in cotranscriptional recruitment of the RNA-binding protein Nrd1p, with a concomitant association of the exonuclease Rrp6p along the transcription unit (28).
In this work, we asked the question whether, following its cotranscriptional recruitment in association with Nrd1p, Rrp6p proceeds independently to degrade the Rho-induced aberrant mRNPs or whether it acts in concert with the other exosome cofactors to accomplish this task. Therefore, we conducted a systematic analysis to evaluate the individual contribution of each exosome cofactor and TRAMP component in the degradation process as well as their potential cotranscriptional recruitment with Rrp6p along the transcription unit. By focusing on the PMA1 transcript, which is highly affected by Rho activity (27, 28), we show that, in addition to the main catalytic action of Rrp6p, the cofactors Rrp47p, Mpp6p as well as the TRAMP components Trf4p, Trf5p, and Air2p contribute significantly by stimulating the degradation process upon their cotranscriptional recruitment. Surprisingly, Rrp47p appears to intervene primarily by stabilizing Rrp6p, which is consistent with the reported close association between the two proteins (22).
EXPERIMENTAL PROCEDURES
Yeast Strains and Plasmids
MPP6 genomic deletion was generated by one-step gene replacement (29) after PCR amplification on plasmid pFA6a-kanMX4 with MPP6 primers. The other deletion strains were generated by crossing the appropriate BY4741 MATa genomic deletion strain (Euroscarf) with BMA41 MATα (30) and selection for the KanR progeny after sporulation. All gene disruptions were confirmed by PCR analyses. A PCR-based one-step tagging technique was used to create strains expressing the various C-terminally tagged proteins with appropriate cassette plasmids and oligonucleotides (31, 32). For the RRP6-TAP strain, the PCR amplification was performed on template genomic DNA from the strain RRP6-TAP::URA obtained from Euroscarf (strain Sc1480). The strains were eventually screened by PCR and immunoblotting. To construct the RRP6-Y361A strain, the PCR amplicon made on genomic DNA from strain RRP6-MYC (with the forward primer harboring the mutated codon) was used to transform BMA41 cells. The selected transformants were analyzed by PCR amplification and sequencing of the PCR products to confirm the presence of the mutated codon, and then immunoblotting was used to verify the presence of the tag. The strain MPP6-TAP in the rrp47Δ background was constructed in two steps. First, the kanamycin marker in the MPP6-TAP strain was replaced by a HIS3 marker by PCR amplification using the plasmid pYM19 (31). The genomic DNA from the created strain (Mpp6-TAP::HIS) was used as template to make an amplicon that was used to transform the rrp47Δ strain.
The pRrp6 plasmid derived from pAG425GPD (Addgene) was generated by Gateway cloning (Invitrogen) of PCR products made on genomic DNA from BMA41. To create the plasmid pY361A, two amplicons made on pRrp6 with two pairs of primers were used as templates in overlapping PCR reactions. Sequencing and immunoblotting were used to confirm the integrity of both expression plasmids. Plasmids expressing Rho under the control of the TetO7 promoter, pCM185-Rho-NLS and pCM189-Rho-NLS, derived from pCM185 and pCM189 (Euroscarf), respectively, were described previously (27, 28). Tables containing the lists of yeast strains, plasmids, and oligonucleotide primers used in this study can be obtained upon request.
Cell Growth and Rho Induction
S. cerevisiae cells were grown according to standard procedures at 25 °C in synthetic complete medium with bases and amino acids omitted as necessary for selection and with glucose (2%) as a carbon source. The cell growth was monitored by measuring the optical density at 600 nm. The expression of Rho was repressed by growing the cells in the presence of doxycycline (Doxy) at a final concentration of 1 μg/ml, whereas the omission of the antibiotic in the growth medium allowed maximum expression. To induce Rho, the cells harboring the expression plasmid were first pregrown with 0.5 μg/ml of Doxy (moderate expression) for 7 h. The cells were then washed extensively (three to four times) with medium lacking Doxy, diluted, and allowed to grow overnight in the selective medium lacking Doxy to achieve maximum Rho expression. After the overnight growth (14–16 h), the Rho-induced cells were typically at an optical density of 0.25–0.3, whereas the non-induced cells were at an optical density of around 0.8. To analyze the samples, the non-induced cultures were diluted with fresh medium to adjust the optical density to the induced cultures before harvesting the cells (protein and RNA extractions) or cross-linking (ChIP).
RNA Isolation and Northern Blotting
Yeast cells were grown with or without Rho induction, and total RNAs were extracted by the hot phenol method (33) and quantified with a Nanodrop spectrophotometer. Northern blot analysis and hybridization were done as described (27). Briefly, 12 μg of RNA was fractionated on a 1.2% agarose-formaldehyde-MOPS gel, transferred onto a Nylon membrane by capillary blotting in 10× SSC for 12–15 h, and then the RNA was cross-linked to the membrane with a UV cross-linker. RNAs were detected by hybridization with a 5′ end-labeled probe. Hybridization signals were quantified with a Storm 860 PhosphorImager (Molecular Dynamics), and the data were processed with ImageQuant software version 2005.
RT-PCR and RT-qPCR
RNAs were treated by DNase using an RTS DNaseTM kit (Mo Bio Laboratories, Inc.) according to the instructions of the manufacturer. One microgram of total RNA was used in a Superscript II RT reaction (Invitrogen) with the two gene-specific oligonucleotides in the same 25-μl reaction mixture (18 S and PMA1). The cDNA (1 μl) was PCR-amplified with specific oligonucleotides. To achieve linear range amplification, 20 cycles (PMA1 and 18 S cDNA) or 21 cycles (RRP6 and RRP47) were applied. The PCR products were analyzed by agarose gel electrophoresis. For real-time PCR quantifications, 2 μl of 100- to 1000-fold diluted cDNA was amplified in a Roche LightCycler 480 with the Maxima SYBR Green qPCR Master Mix detection kit from Fermentas as recommended by the supplier. The qPCR data sets were analyzed using the ΔΔCt method, and the results were normalized to 18 S rRNA RT-qPCR amplification, which was used as internal control. The level of PMA1 mRNA for each sample was expressed relative to the PMA1 mRNA abundance in wild-type BMA41 cells and in the absence of Rho, which was set as 1. Amplifications were done in duplicate for each sample, and three independent RNA extractions were analyzed.
Western Blotting
Total proteins in whole-cell extracts were obtained as described (34), resolved on SDS-10% polyacrylamide electrophoresis gels, and analyzed by Western blotting according to standard procedures. The Rho protein was detected with rabbit anti-Rho antibodies (a custom preparation from Eurogentec). Rrp6p, Rrp47p, and Tfs1p proteins were detected with specific polyclonal antibodies (provided by T. H. Jensen, P. Mitchell, and H. Benedetti, respectively). Tagged proteins were detected with anti-myc-HRP (Santa Cruz Biotechnology) or anti-HA (Invitrogen) antibodies or with peroxidase-anti-peroxidase (Sigma) for the detection of protein A. Secondary antibodies (goat anti-rabbit-HRP IgG, Promega) were used whenever necessary. Visualization on x-ray films was performed following enhanced chemiluminescence (Pierce ECL Western blotting substrate).
ChIP
Strains harboring appropriate C-terminal-tagged proteins and the “no tag” control strain (BMA41) were grown in selective media with Doxy (1 μg/ml final concentration) or without Doxy as described above. Preparations of chromatin were performed essentially as described (35). Forty milliliters of grown cells were fixed with 1% formaldehyde for 20 min. After glycine addition to stop the reaction, the cells were washed and lysed with glass beads to isolate chromatin. The cross-linked chromatin was sheared by sonication to reduce average fragment size to ∼500 bp. Chromatin fractions of 400 μl were taken for each immunoprecipitation reaction made with 40 μl of IgG-Sepharose (GE Healthcare), protein G-Plus-agarose premixed with anti-Myc antibodies (sc-40 X, Santa Cruz Biotechnology), or protein G-Plus-agarose premixed with anti-RNA polymerase II antibodies (8WG16, Covance). After overnight incubation at 4 °C, the beads were washed extensively, and the chromatin was eluted. Eluted supernatants (output) and the input controls were hydrolyzed with Pronase (0.8 mg/ml final concentration, Sigma) for 2 h at 42 °C, followed by 5 h incubation at 65 °C to reverse cross-linked DNA complexes. DNA was extracted using the Qiagen PCR cleanup columns. The immunoprecipitated DNAs (output) were quantitated by real-time PCR (LightCycler 480, Roche, with the products and instructions recommended by the supplier) using six sets (Fig. 1) or three sets (in the other figures) of primers located along the PMA1 gene and normalized to a 1:200 dilution of input DNA. Amplifications were done in duplicate for each sample, and averages and S.D. were calculated on the basis of three independent experiments.
FIGURE 1.
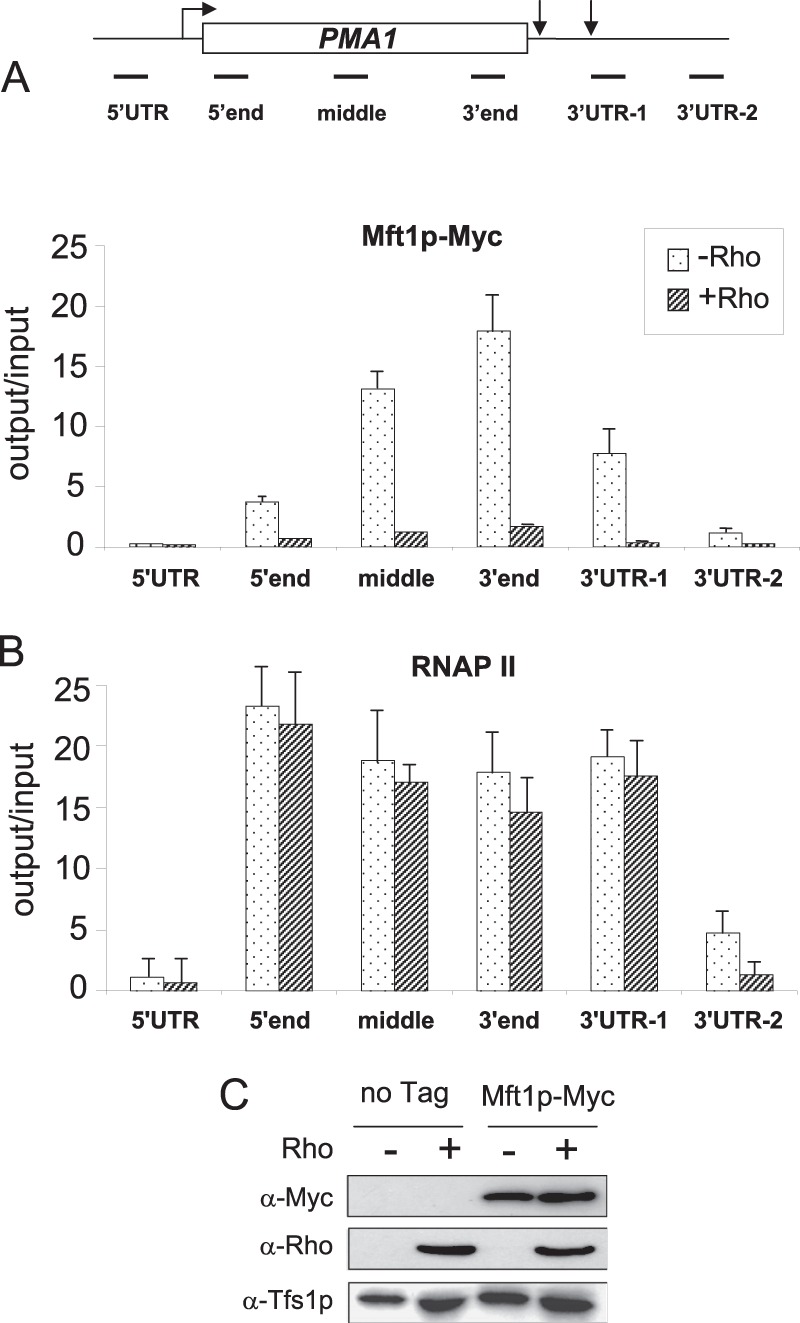
Rho action interferes in the normal cotranscriptional deposition of the mRNA packaging factor Mft1p. A, quantification of the fold enrichment for PMA1 DNA in immunoprecipitates from cells harboring Myc-tagged THO subunit Mft1p and grown under repressing (−Rho) or inducing (+Rho) conditions. Immunoprecipitated samples (output) were normalized to input following quantification by real-time PCR. The average of three independent experiments is shown, with error bars representing S.D. The top panel illustrates the positions of the six sets of oligonucleotides (5′ end, middle, and 3′ end regions of the ORF (28) as well as the 5′ UTR and 3′ UTR regions (48)) that were used in the qPCR quantifications. Arrows indicate the positions of transcription start site and poly(A) sites. B, quantification by ChIP of RNA polymerase II distribution along the PMA1 gene in the absence and presence of Rho using specific antibodies 8WG16. The analyses were performed as in A. C, Western blot analyses of whole protein extracts isolated from wild-type cells (no Tag) or cells harboring Myc-tagged Mft1p and grown under Rho-inducing or repressing conditions. The extracts were fractionated by SDS-PAGE, transferred onto a membrane, and then the detections were performed with anti-Myc antibodies (α-Myc) and specific polyclonal anti-Rho antibodies (α-Rho). As a loading control, the yeast protein Tfs1p, whose function is unrelated to RNA metabolism, was also detected by specific antibodies (α-Tfs1p).
RESULTS
The Removal of Rho-induced Aberrant mRNPs Requires the Catalytic Activity of the Recruited Rrp6p
Recently, we implemented a Rho-based approach to study mRNP quality control in yeast. In this experimental system, sufficient amounts of defective mRNPs can be produced by Rho action within the cell nucleus to serve as substrates for the quality control process. Rho protein is directed to the nucleus by the fusion of a nuclear localization signal at its C terminus (27). Apparently, Rho action along nascent transcripts interferes with normal deposition of mRNA processing and packaging factors, yielding mRNPs that are recognized as defective and eliminated by the nuclear quality control apparatus. This Rho effect on mRNP protein composition is exemplified by the displacement of the THO subunit Mft1p along the PMA1 gene, as revealed by ChIP experiments shown in Fig. 1A. A systematic analysis of the effect of Rho on the deposition of numerous mRNA processing and packaging factors will be described elsewhere.5 The Rho-induced loss of Mft1p enrichment along the PMA1 gene did not result from a transcriptional defect that would cause premature transcription termination, as revealed by ChIP analyses of RNA polymerase II distribution in the presence and absence of Rho (Fig. 1B). Also, protein measurements shown in Fig. 1C ruled out the possibility of an indirect effect in which Rho would decrease the cellular level of Mft1p.
As shown in Fig. 2A, the growth of yeast cells expressing the bacterial Rho factor under the control of a doxycycline-regulated TetO7 promoter within a centromeric plasmid is severely reduced under inducing conditions (−Doxy). The Rho-induced growth defect phenotype is associated with a large drop (around 70% reduction) in the steady-state level of the model transcript PMA1, as shown by Northern blot analysis and RT-PCR quantifications in Fig. 2, B and C. Previous mRNA 3′ end mapping analyses have shown that Rho helicase/translocase action leads to the production of transcripts having normal lengths and poly(A) tails but that are recognized as aberrant and eliminated by the Rrp6p-mediated nuclear mRNA quality control system (27). Indeed, the absence of Rrp6p in the rrp6Δ strain relieves the Rho-induced growth defect and restores the PMA1 mRNA to a nearly normal level (Fig. 2). A similar suppression of Rho-induced growth defects and rescue of PMA1 mRNA is observed in the strain rrp6-Y361A, which harbors a catalytically inactive version of Rrp6p as the sole source of the protein expressed from its chromosomal locus. The Y361A mutation is within the catalytic site of the exonuclease, which leads to its inactivation in vitro and in vivo (8, 36). Thus, the exonuclease function of Rrp6p appears to be the essential hydrolytic activity for the degradation of Rho-induced defective mRNPs. This conclusion was substantiated by applying a competition approach in which the action of endogenous wild-type Rrp6p was challenged by the catalytically dead variant Rrp6p-Y361A overproduced from a 2μ plasmid. This was seen by the relief of growth defects and restoration of PMA1 mRNA in the wild-type strain when Rrp6p-Y361A was overexpressed from the plasmid. As controls, the overexpression of the active form of Rrp6p did not relieve the Rho-induced defects in the wild-type strain and, as expected, renders the strains rrp6Δ or rrp6-Y361A sensitive to Rho action with the accompanying drop in the level of PMA1 mRNA (results not shown).
FIGURE 2.

The catalytic activity of the recruited Rrp6p is required for the removal of Rho-induced aberrant mRNPs. A, the conditional expression of Rho in yeast induces a growth defect phenotype. The rrp6Δ mutant, the corresponding wild-type isogenic BMA41 strain (RRP6), and a strain that harbors a catalytically inactive version of Rrp6p (rrp6-Y361A) were transformed with the Rho expression plasmid (pCM185-Rho-NLS). 10-fold serial dilutions of the transformants grown selectively in liquid medium with 1 μg/ml Doxy (non-induced conditions) were spotted on Rho-repressing (+Doxy) or Rho-inducing (−Doxy) plates. The plates were photographed after 3 days at 25 °C. B, Northern blot analyses of total RNAs extracted from cultures of cells transformed with the Rho expression plasmid and grown with Doxy at a concentration of 1 μg/ml (−Rho) or without Doxy (+Rho). Samples of total RNAs (12 μg) were fractionated on agarose gel, transferred to a membrane, and then the steady-state levels of PMA1 were detected using a 5′ end-labeled probe. Ethidium bromide staining of the gel before membrane transfer was performed to visualize the 18 S rRNA as a loading control. C, agarose gel showing semiquantitative RT-PCR analysis performed on total RNAs (1 μg) using specific primers to PMA1 mRNA and 18 S rRNA designed to give PCR products with different sizes. The histogram shows the level of PMA1 mRNA relative to 18 S rRNA as determined by quantitative RT-PCR and by setting the value of PMA1 signal to 1 for the wild-type BMA41 strain in the absence of Rho expression. The average of three independent experiments is shown, with error bars representing S.D. D, quantification of the fold enrichment for PMA1 DNA in immunoprecipitates from cells harboring Myc-tagged Rrp6p variants or Rrp6p without a tag (no Tag) and grown under repressing (−Rho) or inducing (+Rho) conditions. Immunoprecipitated samples (output) were normalized to input following quantification by real-time PCR of three regions of the PMA1 gene (5′, middle, and 3′ regions) as represented schematically. The average of three independent experiments is shown, with error bars representing S.D.
The involvement of Rrp6p in the targeting and elimination of Rho-induced aberrant mRNPs was also revealed by ChIP experiments where Rho action brought about cotranscriptional recruitment of Rrp6p in association with a large increase in Nrd1p enrichment along the PMA1 gene (28). To verify that the absence of the Rho effect in the strain rrp6-Y361A is linked to the lack of catalytic activity of the mutant protein rather than a deficiency in its recruitment, we conducted parallel ChIP analyses using wild-type and mutant strains harboring Myc-tagged versions of the Rrp6p variants expressed from the chromosomal locus under the control of the endogenous promoter. The results show clearly that Rho action triggers cotranscriptional recruitment of the inactive exonuclease Rrp6p-Y631A over the whole PMA1 gene to a similar extent than for the wild-type Rrp6p (Fig. 2D). Because the Rho-dependent disappearance of the PMA1 mRNA is fully alleviated in the mutant strain, this indicates that the elimination of the defective mRNPs cannot proceed if the recruited Rrp6p is catalytically inactive.
Dis3p Plays Only a Minor Role in the Degradation of Rho-induced Aberrant mRNPs
To show further that the exonuclease function of Rrp6p is the main degradation activity required for the elimination of the aberrant mRNPs, we tested the possible involvement of the other exosome-associated factor having hydrolytic activity, Dis3p. The Rho expression system was introduced into two mutant strains having single mutations in Dis3p that abolish either the exonuclease (exo−, dis3-D551N allele) or the endonuclease (endo−, dis3-D171N allele) activities of the exosome cofactor (15, 16). Growth tests conducted in parallel with the isogenic wild-type strain show clearly that the Dis3p mutations did not relieve the growth defect induced by Rho action (Fig. 3A). RT-PCR quantifications of the PMA1 mRNA are consistent with the growth results because both the exo and the endo mutations were unable to significantly rescue the transcript under Rho-inducing conditions, although a slight recovery (about 10% increase relative to the wild-type) is observed when the endonuclease activity of Dis3p is abolished (Fig. 3B). Hence, the catalytic activities of Dis3p are obviously not the main hydrolytic function involved in the elimination of Rho-induced aberrant mRNPs.
FIGURE 3.
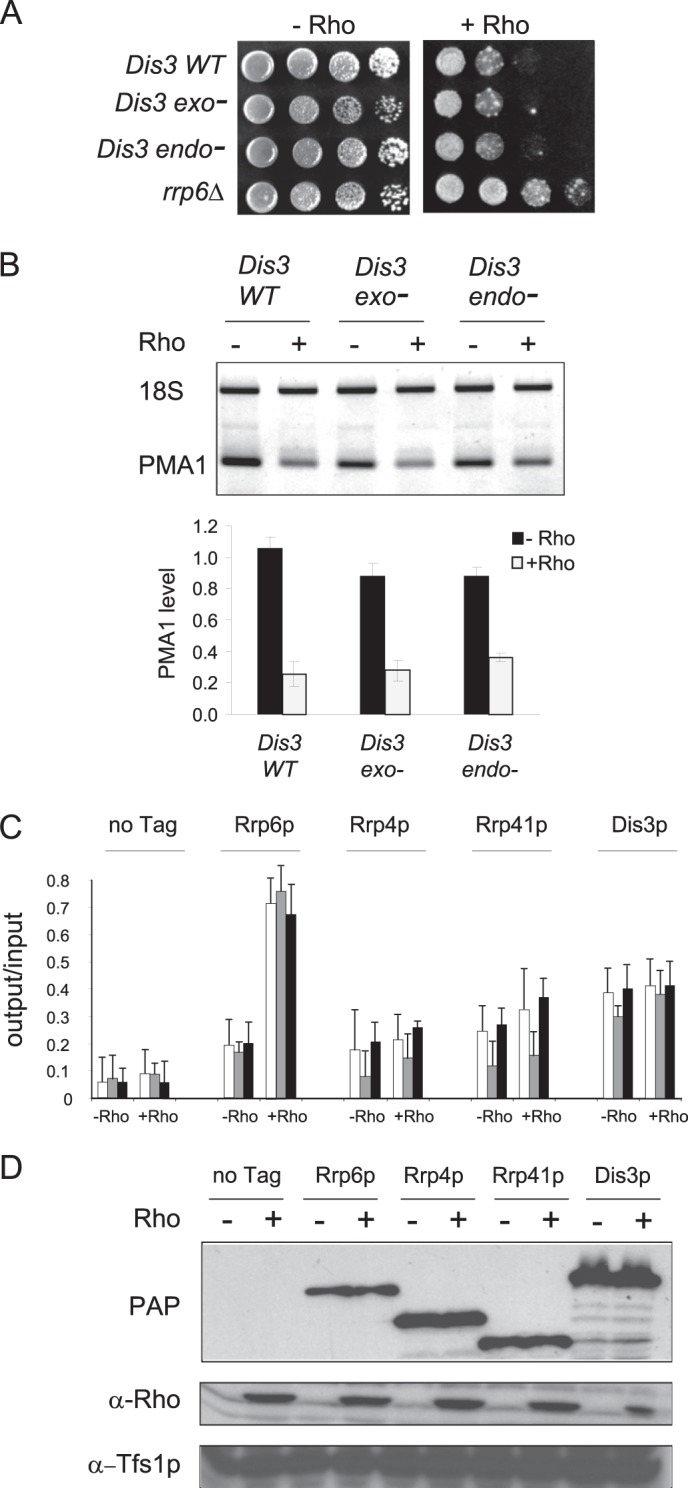
The core exosome and Dis3p play only a minor role in the elimination of Rho-induced aberrant mRNPs. A, the strains with mutations in Dis3p that abrogate either exonuclease (exo-) or endonuclease (endo-) activities of the exosome cofactor as well as the isogenic wild-type strain were transformed with the plasmid expressing Rho (pCM185-Rho-NLS). Strains grown selectively under repressing conditions were spotted on agar plates for growth under inducing or repressing conditions along with the control strain (rrp6Δ) as described in Fig. 2A. B, agarose gel showing semiquantitative RT-PCR analysis of the steady-state level of PMA1 mRNA. Real-time RT-PCR measurements were conducted to calculate PMA1 abundance, which is relative to the level in wild-type BMA41 strain in the absence of Rho expression as described in Fig. 2C. C, quantification of the fold enrichment for PMA1 DNA (5′, middle, and 3′ regions) in immunoprecipitates from cells harboring TAP-tagged core exosome subunits (Rrp4p and Rrp41p) or exosome cofactors (Rrp6p and Dis3p) or cells having no tag (no Tag) as a control and grown under repressing (−Rho) or inducing (+Rho) conditions. Immunoprecipitated samples (output) were normalized to input as in Fig. 2D. D, Western blot analyses of whole protein extracts isolated from wild-type cells (no Tag) or cells harboring TAP-tagged versions of the proteins analyzed by ChIP in C and grown under Rho-inducing or Rho-repressing conditions. The detection of the tagged proteins was performed using peroxidase-anti-peroxidase (PAP), whereas the detection of Rho and Tfs1p was made as in Fig. 1C.
We next tested the potential Rho-dependent, cotranscriptional recruitment of Dis3p using a yeast strain harboring the protein A-tagged version of the cofactor. Surprisingly, moderate ChIP signals of Dis3p were detected along the PMA1 gene. However, the intensity of the signals was similar in the absence or the presence of Rho, indicating that the enrichment is independent of Rho action (Fig. 3C). These results contrast with the profiles obtained with TAP-tagged Rrp6p, which was run in parallel as positive control. Because Dis3p is known to be ubiquitously associated with the core exosome (13, 14, 37), we examined the recruitment of two core exosomal subunits (Rrp4p and Rrp41p) tagged with a TAP epitope at the C terminus. Again, modest ChIP signals above the background were detected along the PMA1 gene for the two core exosome subunits, but no Rho-dependent enhancement of the recruitment could be observed (Fig. 3C). Protein measurements indicated that, as for Rrp6p, the levels of Dis3p, Rrp4p and Rrp41p were not affected by Rho expression (Fig. 3D). Thus, these results suggest that Dis3p and the core exosome may reside within the chromatin but that they are not specifically recruited to target the Rho-induced defective mRNPs. However, their possible participation as part of a basal surveillance mechanism cannot be excluded (see “Discussion”).
The Removal of Rho-induced Aberrant mRNPs Requires Rrp47p and Mpp6p, Which Are Cotranscriptionally Recruited Like Rrp6p
The Rrp47p and Mpp6p cofactors have been shown to promote the catalytic activity of Rrp6p-containing exosome complexes, and it has been suggested that their RNA-binding ability may help to direct the exosome complex to specific RNA substrates (21–23). To address whether the two cofactors contribute to the targeting and elimination of Rho-induced aberrant mRNPs, we generated deletion mutants (rrp47Δ and mpp6Δ) and evaluated their sensitivity to Rho action. The absence of either Rrp47p or Mpp6p in these mutant strains fully suppressed the Rho-induced growth defect and restored the PMA1 mRNA to nearly normal level (Figs. 4, A–C). The extent by which the absence of either cofactor alleviates the Rho effects indicates that they both play an essential role in the Rrp6p-mediated targeting and/or degradation of the defective mRNPs. The reported close association of the two cofactors with Rrp6p prompted us to assess their potential Rho-dependent cotranscriptional recruitment, as observed for Rrp6p. ChIP analyses using yeast strains harboring at their chromosomal locations Myc-tagged or TAP-tagged versions of Rrp47p and Mpp6p, respectively, revealed that Rho action leads to a stimulation of the recruitment of both proteins along the PMA1 gene (Fig. 4D). The Rho-induced enrichment did not result from an increase in the cellular levels of the two proteins, as indicated by the Western blot measurements shown in Fig. 4E. Interestingly, the Rho-induced recruitment of TAP-tagged Mpp6p was suppressed in the rrp47Δ background, suggesting that the two cofactors are involved in a common functional pathway, presumably with Rrp6p (see below).
FIGURE 4.

The exosome cofactors Rrp47p and Mpp6p contribute to the targeting and elimination of Rho-induced aberrant mRNPs upon cotranscriptional recruitment. A, the wild-type strain and single deletion mutants rrp47Δ and mpp6Δ were transformed with the plasmid expressing Rho. Strains grown selectively under repressing conditions were spotted for growth as described in Fig. 2A. B, Northern blot analysis was made as in Fig. 2B. C, semiquantitative RT-PCR analysis and RT-qPCR quantifications to measure the level of PMA1 mRNA were done as in Fig. 2C. D, quantification of the fold enrichment for PMA1 DNA (5′, middle, and 3′ regions) in immunoprecipitates from cells harboring Myc-tagged Rrp47p or TAP-tagged Mpp6p. Immunoprecipitated samples (output) were normalized to input as in Fig. 2D. E, Western blot analysis of whole protein extracts isolated from cells harboring Myc-tagged Rrp47p or TAP-tagged Mpp6p was conducted as in Fig. 1C.
Previous studies have shown that the cellular level of Rrp47p can be affected in strains lacking Rrp6p or bearing N-terminal deletions of Rrp6p. This was ascribed to a close physical interaction found in vitro between Rrp47p and the N-terminal part of Rrp6p, which may stabilize the Rrp47p protein (22, 38, 39). Indeed, a Western blot analysis using specific anti-Rrp47p antibodies (Fig. 5A) confirms these previous observations by revealing an even more drastic effect of Rrp6p depletion on the accumulation of Rrp47p, with a complete disappearance of the protein in the rrp6Δ strain. Quantification of the RRP47 mRNA by RT-PCR (Fig. 5B) indicated that the disappearance of Rrp47p in the rrp6Δ strain results from a down-regulation at the protein level. This stronger effect observed in our experiments could be explained by the fact that Rrp47p is not tagged as compared with the previous study, where the protein was fused to a protein A tag at its C terminus, which probably increases its stability (22).
FIGURE 5.
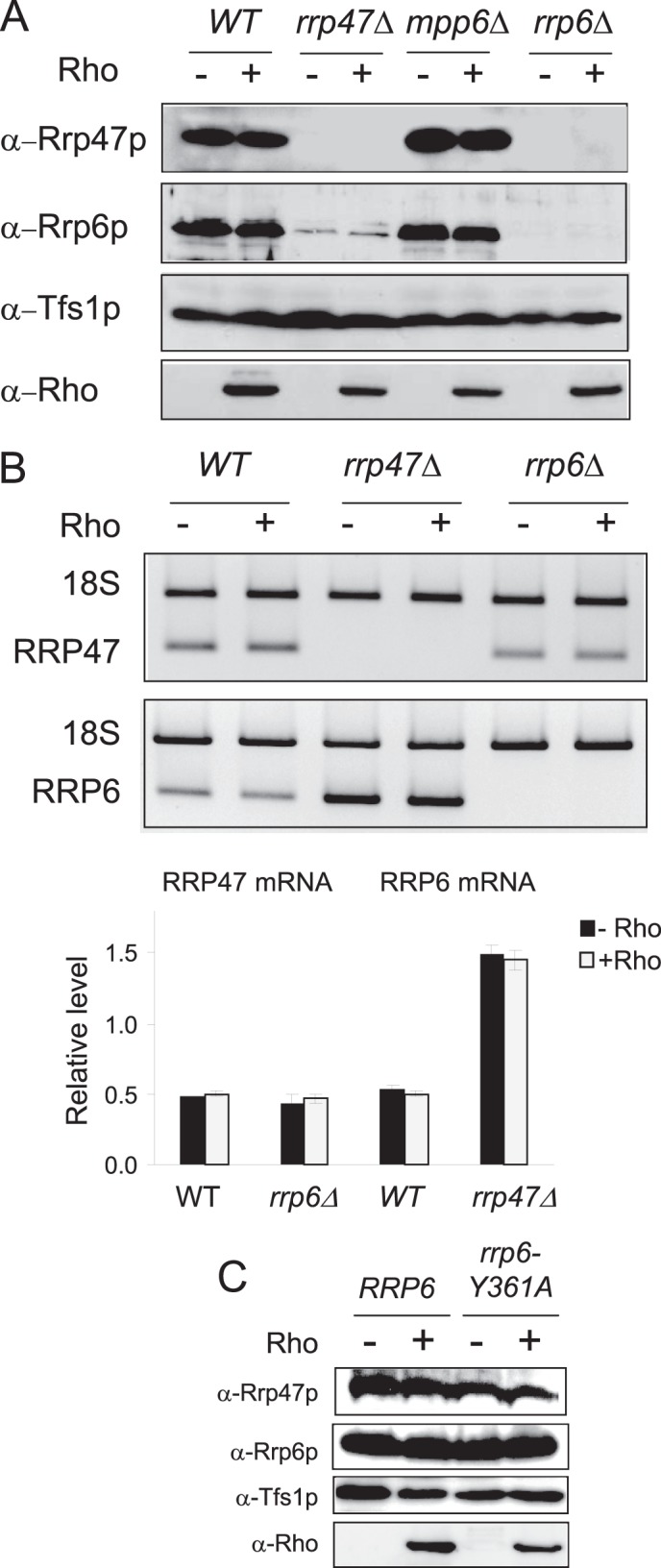
Mutual stabilization of Rrp6p and Rrp47p proteins in vivo. A, Western blot analyses of whole protein extracts isolated from wild-type, rrp47Δ, mpp6Δ, and rrp6Δ cells grown under Rho-repressing or Rho-inducing conditions. The extracts were analyzed as described in Fig. 1C, and the detections were performed with the indicated specific polyclonal antibodies. B, quantification by RT-PCR of steady-state levels of RRP47 and RRP6 mRNAs in wild-type, rrp47Δ, and rrp6Δ cells grown under Rho-repressing or Rho-inducing conditions. Reverse transcriptions were carried out with specific primers for the mRNAs and a primer for 18 S rRNA. Then, 20 cycles of PCR were performed using sets of primers. As loading control, 20 cycles of PCR were performed on the same cDNA using a set of primers for 18 S rRNA. The histogram shows the relative levels of the mRNAs as determined by quantitative RT-PCR. C, comparative analysis of the levels of Rrp6p and Rrp47p in wild-type and rrp6-Y361A strains. The protein extracts were analyzed as in A.
To find out whether a similar impact on the accumulation of Rrp6p could be observed in the rrp47Δ or mpp6Δ backgrounds, we measured its level in the deletion strains using specific anti-Rrp6p antibodies. Surprisingly, the analyses revealed that, in the absence of Rrp47p, the level of Rrp6p dropped by more than 90%, whereas the removal of Mpp6p did not have any effect on the accumulation of Rrp6p (Fig. 5A). RT-PCR quantifications of the steady-state level of RRP6 mRNA indicate that the drop in the protein amount does not result from a down-regulation at the transcriptional level or a more rapid mRNA decay because the RRP6 mRNA accumulation is even higher in the rrp47Δ strain as compared with the wild-type strain (Fig. 5B). These results, along with previous findings (22), indicate that, by interacting, Rrp6p and Rrp47p protect each other from degradation. As shown in Fig. 5C, the protection of Rrp47p from degradation is also provided by the catalytically inactive version of Rrp6p (rrp6-Y361A allele). Remarkably, these data extend our ChIP results described above by suggesting that Rrp47p is presumably recruited within the same complex than Rrp6p following Rho action. However, the catalytic function of Rrp6p within the complex is required for the elimination of the Rho-induced defective mRNPs.
The TRAMP Components Trf4p and Trf5p Are Both Involved in the Targeting of Rho-induced Aberrant mRNPs
The TRAMP complex is an essential cofactor that was shown to stimulate the targeting and degradation of a large variety of RNA substrates by the nuclear exosome. Moreover, in vitro experiments have shown that the TRAMP complex can stimulate the exonuclease activity of Rrp6p independently from the exosome (40). The two paralogous non-canonical poly(A) polymerases Trf4p and Trf5p are alternative subunits of the TRAMP complex, and simultaneous loss of the two proteins is lethal in S. cerevisiae. However, it has been suggested that the two components may not substitute for each other in a single TRAMP complex but, rather, that they are part of alternative complexes (TRAMP4 and TRAMP5) having distinct functional implications (24–26, 41, 42). To investigate the possible role of the TRAMP complex in the elimination of Rho-induced aberrant mRNPs, we constructed single deletion mutant strains of the TRF4 and TRF5 genes and tested their sensitivity to Rho action. As shown in Fig. 6A, the deletion of the TRF4 gene mitigates the growth inhibition induced by Rho expression, whereas the deletion of TRF5 did not show any relief of the growth defect. We next analyzed the steady-state level of the model transcript PMA1. The RT-PCR quantifications shown in Fig. 6B revealed that, under Rho expression conditions, a large part of the transcript is recovered in the trf4Δ strain (a ratio of 0.9 as compared with 0.3 in the wild-type strain), whereas only a moderate fraction is rescued in the case of the trf5Δ mutant (a ratio of 0.5 as compared with 0.3 in the wild-type strain). These results are consistent with the growth tests because the substantial rescue in the absence of Trf4p is sufficient to mitigate the growth defect, whereas the minor transcript recovery observed in the absence of Trf5p is not sufficient to improve the relative growth. The possibility of an indirect effect through differential depletion of Rrp6p, as in the case of the rrp47Δ strain, was excluded by Western blot analyses (Fig. 6C). Together, these data suggest that the two TRAMP components Trf4p and Trf5p are involved in the surveillance process that confers the Rho-mediated phenotype. However, the fact that the absence of either component allows the restoration of only a fraction of the model transcript and does not confer a full growth recovery raises the possibility that they may intervene within distinct TRAMP complexes. Thus, a full recovery should require the removal of both components. We tested this hypothesis by applying a metabolic depletion approach in which a double mutant strain trf4Δ trf5Δ was complemented by Trf4p or Trf5p expressed under a Gal1 promoter from a plasmid (42). However, despite many attempts, the growth results in glucose versus galactose did not provide clear conclusions, probably because of the leakiness of the expression system.
FIGURE 6.

The removal of Rho-induced aberrant mRNPs requires TRAMP components Trf4p and Trf5p. A, the wild-type strain and single deletion mutants trf4Δ and trf5Δ were transformed with the plasmid expressing Rho. Cells grown selectively under repressing conditions were spotted for growth as described in Fig. 2A, with the strain rrp6Δ as control. B, semiquantitative RT-PCR analyses and RT-qPCR measurements to evaluate the level of PMA1 mRNA were made as in Fig. 2C. C, Western blot analysis to evaluate the presence of Rrp6p in the different strains was performed as in Fig. 5A. D, quantification of the fold enrichment for PMA1 DNA (3′, middle, and 5′ regions) in immunoprecipitates from cells harboring Myc-tagged Trf4p or Trf5p in the wild-type background or Myc-tagged Trf5p in the trf4Δ background. Immunoprecipitated samples (output) were normalized to input as in Fig. 2D. E, Western blot analysis to evaluate the level of Trf4p and Trf5p in the strains analyzed by ChIP.
Strains carrying Myc-tagged versions of Trf4p or Trf5p were constructed to assess the potential cotranscriptional recruitment of the two TRAMP components following Rho action. The ChIP analyses presented in Fig. 6D show clearly that Rho action triggers a large recruitment of both proteins to the PMA1 gene. This Rho-induced enrichment did not result from an increase in the cellular level of the two proteins, as verified by Western blot (Fig. 6E). We reasoned that if the two components are functionally redundant and, thus, can replace each other within a unique TRAMP complex, a depletion of one component should increase the recruitment level of the other. To explore this possibility, we generated a trf4Δ strain harboring Myc-tagged Trf5p. Comparative ChIP analyses indicated that Trf5p is recruited to the PMA1 gene at the same level whether Trf4p is present or not (Fig. 6D). These results led us to conclude that Trf4p and Trf5p are both involved in the targeting and degradation of Rho-induced aberrant mRNPs but, presumably, as part of two different forms of the TRAMP complex that are recruited cotranscriptionally.
The Removal of Rho-induced Aberrant mRNPs Requires Air2p but Not Air1p
The TRAMP complex also contains one of two homologous RNA-binding proteins, Air1p or Air2p, which are about 50% identical (24, 26, 42, 43). Because most previous studies have focused only on Air2p as the partner of Trf4p in the TRAMP complex, it is still unclear whether there is functional redundancy between Air1p and Air2p. However, previous genetic screens have found that mpp6Δ is synthetic lethal in combination with air1Δ but not with air2Δ, which suggests that there are presumably functional distinctions between the two TRAMP components (23). Moreover, a more recent study on the basis of phenotypic analyses and RNA deep sequencing revealed differential functions of the two Air proteins in RNA metabolism (43). We have shown previously that the double deletion mutant air1Δ air2Δ is a robust suppressor of the Rho-induced effects, with a complete relief of the growth defect and a restoration of the model PMA1 mRNA to a nearly normal level (Ref. 27 and Fig. 7). Guided by our results on Trf4p and Trf5p, we wanted to evaluate the specific contributions of Air1p and Air2p in the removal of Rho-induced aberrant mRNPs. Single deletion mutant strains were generated, and their sensitivity to Rho action was tested. Surprisingly, the elimination of Air1p alone (air1Δ) did not relieve the sensitivity to Rho action, whereas the elimination of Air2p (air2Δ) showed a strong suppressing phenotype similar to the one observed with the double mutant (Fig. 7, A–C). Western blot analyses indicated that the removal of Air1p and/or Air2p did not affect the cellular accumulation of Rrp6p or the expression of Rho, which excludes a possible indirect effect (Fig. 7D). Thus, these results show that, for the removal of Rho-induced aberrant mRNPs, there are indeed functional distinctions between Air1p and Air2p because Air1p is not participating in the process. These findings were substantiated by ChIP analyses using strains harboring TAP-tagged versions of Air1p or Air2p. Significant ChIP signals over the PMA1 gene were observed for Air2p under Rho inducing conditions, whereas only background levels were repeatedly detected for Air1p (Fig. 7E). The Rho-induced stimulation of ChIP signals for Air2p did not result from an increase in the cellular level of the protein (Fig. 7F).
FIGURE 7.

The removal of Rho-induced aberrant mRNPs requires TRAMP component Air2p but not Air1p. A, the wild-type strain, single deletion mutants air1Δ and air2Δ, as well as a double mutant, air1Δair2Δ, were transformed with the Rho-expressing plasmid. Strains grown selectively under repressing conditions were spotted for growth as described in Fig. 2A. B, Northern blot analysis was made as in Fig. 2B. C, semiquantitative RT-PCR analyses and PMA1 mRNA level determinations by RT-qPCR were performed as in Fig. 2C. D, Western blot analysis to evaluate the presence of Rrp6p in the different strains was performed as in Fig. 5A. E, ChIP analyses of the recruitment of TAP-tagged Air1p or Air2p to the PMA1 gene were performed as in Fig. 2D. F, Western blot analysis to evaluate the level of Air1p and Air2p in the strains analyzed by ChIP. PAP, peroxidase-anti-peroxidase. G, comparison of the recruitment to the PMA1 gene of Myc-tagged Trf4p and Trf5p in wild-type or air2Δ backgrounds. The ChIP analyses were performed as in Fig. 2D. H, evaluation of Trf4p and Trf5p levels in the strains analyzed by ChIP.
The Absence of Air2p Impedes the Recruitment of Trf4p and Trf5p
The results described above suggest that Trf4p and Trf5p are recruited to the Rho-induced aberrant mRNPs as part of two distinct TRAMP complexes that, presumably, both contain Air2p as a component. This led us to ask the question whether the lack of Air2p could have any impact on the recruitment of Trf4p and Trf5p. We thus constructed air2Δ strains harboring Myc-tagged Trf4p or Trf5p and compared the recruitment of the two tagged proteins in the absence and presence of Air2p. The ChIP analyses shown in Fig. 7G revealed that the Rho-dependent recruitment of Trf4p and Trf5p to the PMA1 gene is reduced by more than 60% in the absence of Air2p. This reduction did not result from loss of the two proteins in the air2Δ background (Fig. 7H). These results support our conclusion that the TRAMP complex formed by Trf5p, which targets Rho-induced aberrant mRNPs, contains Air2p instead of Air1p. The results also suggest that the TRAMP complexes containing Air2p and either Trf4p or Trf5p are, presumably, formed prior to their cotranscriptional recruitment.
The Removal of Rho-induced Aberrant mRNPs Does Not Require Mtr4p Helicase Activity
The ATP-dependent RNA helicase Mtr4p is the third component forming the TRAMP complex. The physiological function of Mtr4p in the degradation of RNA substrates has been correlated with its ability to hydrolyze ATP and unwind RNA duplexes in vitro. Because the deletion of the MTR4 gene is lethal in S. cerevisiae, we took advantage of the mutant mtr4–20 allele, which was shown to be functionally inactive because of a mutation in a conserved helicase motif of the protein (44). The sensitivity to Rho action of the mutant strain mtr4–20 was tested in parallel with the isogenic wild-type strain. As shown in Fig. 8, the mutation of Mtr4p did not relieve the growth defect induced by Rho, nor did it rescue the model PMA1 mRNA. These results most likely indicate that the ATPase/helicase functions of Mtr4p do not play an essential role in the ability of the TRAMP complex to stimulate the removal of Rho-induced aberrant mRNPs. This is in agreement with previous in vitro findings showing that the TRAMP complex is able to stimulate the exonuclease activity of Rrp6p in the absence of ATP or without Mtr4p (40).
FIGURE 8.
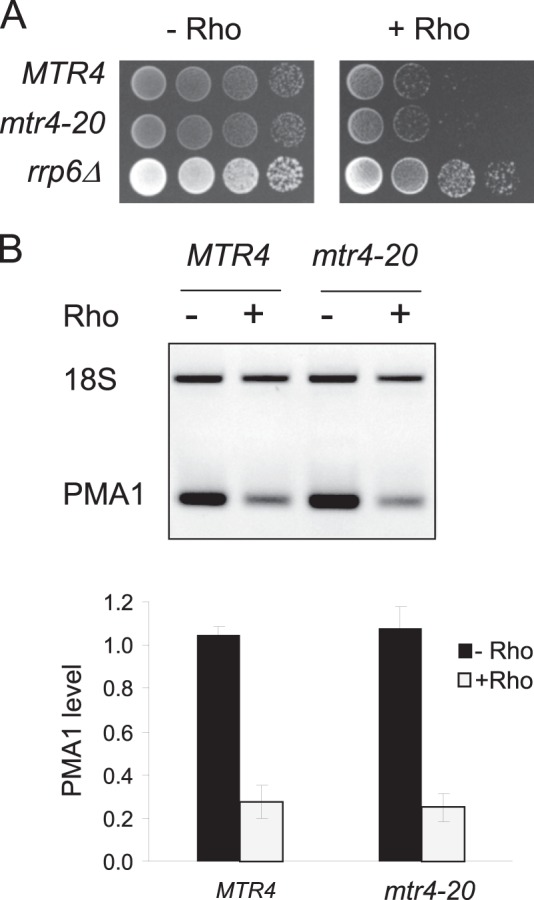
The removal of Rho-induced aberrant mRNPs does not require helicase activity of the TRAMP component Mtr4p. A, the strain harboring a mutation in a helicase motif of Mtr4p (mtr4-20) and the corresponding wild-type isogenic strain (MTR4) were transformed with the plasmid expressing Rho (pCM189-Rho-NLS). Cells grown selectively under repressing conditions were spotted for growth as described in Fig. 2A, with the strain rrp6Δ as a control. B, semiquantitative RT-PCR analysis and RT-qPCR quantifications of the level of PMA1 mRNA were done as in Fig. 2C.
DISCUSSION
Recently we implemented an experimental system in which the activity of the bacterial Rho helicase in the yeast nucleus impedes the mRNA processing and packaging reactions, thus producing sufficient amounts of aberrant mRNPs that can serve as substrates for the surveillance apparatus (27). Using this system, we have shown that the targeting of these defective mRNPs is mediated by Rrp6p, which is recruited cotranscriptionally in association with Nrd1p following Rho action (28).
Here we examined the involvement of different cofactors associated with the nuclear RNA degradation machinery in the elimination of Rho-induced aberrant mRNPs. To this end, we focused our analyses on a model transcript (PMA1) that is highly affected by Rho action, probably because of the presence of an efficient loading site for the RNA-dependent bacterial helicase. Our results show clearly that the exonuclease function of Rrp6p is the main hydrolytic activity responsible for the degradation of malformed mRNPs. Indeed, Dis3p appears to play a rather minor role in the process through its endonuclease activity because only a very small fraction (about 10%) of the PMA1 transcript was rescued in the endo− mutant strain. However, our results do not exclude a more significant participation of Dis3p in the elimination of other malformed mRNPs not analyzed here that may be targeted by alternative quality control pathways. For instance, by monitoring the fate of HSP104 mRNA in cells having an impaired THO complex (mft1Δ strain), it has been shown previously that the exonuclease activity of Dis3p contributes partly to mRNP quality control in this particular system (8). Moreover, recent transcriptome-wide analyses revealed the existence of a widespread nuclear surveillance system that constitutively targets most RNAs, including mRNAs, and in which Rrp6p and Dis3p have both specific and overlapping roles (45, 46). This basal surveillance activity could explain the detection of moderate signals in our ChIP experiments indicating the presence of Dis3p and the core exosomal subunits Rrp4p and Rrp41p along the PMA1 gene independently from Rho action.
The Rrp47p cofactor has been shown to interact with the N-terminal region of Rrp6p and to promote the catalytic activity of the exonuclease by facilitating its binding to structural elements within the RNA targets (22, 38). Previous observations in THO mutant strains suggested that the physical interaction between Rrp6p and Rrp47p may play an important role in mRNP quality control. In effect, deletion of the RRP47 gene was found to somewhat mimic the rrp6Δ phenotype (12). Alternatively, truncation of the N-terminal part of Rrp6p, which impacts the normal accumulation of Rrp47p, has been shown to affect the quality control of HSP104 mRNA (8). Our results elucidate these previous observations by showing a strong interdependence in the stability of the two protein partners where the absence of Rrp6p leads to the complete disappearance of Rrp47p, whereas the absence of Rrp47p induces a large depletion of Rrp6p. The requirement for a mutual stabilization between Rrp6p and Rrp47p implies that they probably associate in a complex prior to their cotranscriptional recruitment.
Remarkably, the recent transcriptome-wide analysis on the basis of RNA-protein cross-linking revealed that Rrp6p is preferentially associated with structured RNA targets that do not frequently associate with the core exosome (46). The first important information that can be taken from these findings is that they probably highlight the key contribution of Rrp47p, which should tether Rrp6p to the structural motifs within the RNA targets. In this regard, we expect that the displacement of RNA processing and packaging factors by Rho action along the transcript (as in the case of Mft1p, shown in Fig. 1) should favor the formation of RNA structures and, thus, enhance the tethering of the Rrp6p-Rrp47p complex. The second clue from these findings relates to the infrequent association of core exosome to the Rrp6p-preferred sites. This could be an indication for exosome-independent function of Rrp6p, which is in agreement with previous biochemical and genetic studies suggesting that Rrp6p performs some of its essential functions without physical association with the core exosome (20). The absence of Rho-stimulated recruitment of Rrp4p and Rrp41p in our ChIP experiments (Fig. 3C) supports this core-independent functioning of Rrp6p in the targeting of defective mRNPs, at least in the early steps of the process when the defects are detected cotranscriptionally.
Our results reveal for the first time an important contribution to mRNP quality control of the Mpp6p cofactor, which is also recruited cotranscriptionally in a Rho-dependent manner and at a similar stimulation level than Rrp6p or Rrp47p. Obviously, the participation of Mpp6p is essential for the targeting and elimination of Rho-induced aberrant mRNPs because its absence completely abolishes the growth defect induced by Rho and restores the model transcript to a nearly normal level. Mpp6p is a nuclear RNA-binding protein that was discovered recently in a screen for synthetic lethal interactions with loss of Rrp47p, and it has been shown further that the lack of Mpp6p was also lethal in the absence of Rrp6p. These genetic interactions were corroborated by coimmunoprecipitation experiments showing the presence of the three exosome cofactors in a large RNA degradation complex (23). Our results do not provide direct evidence that Mpp6p is physically associated with Rrp6p and Rrp47p upon its cotranscriptional recruitment. Nevertheless, the prominent suppressing effect conferred by Mpp6p removal strongly suggests that this cofactor is actively involved in a common functional pathway with Rrp6p and Rrp47p. Thus, possible roles for Mpp6p in mRNP quality control could be to participate with Rrp47p in securing the recruited complex tethered to the aberrant transcript or to directly stimulate the catalytic activity of Rrp6p. Given that Mpp6p has been poorly studied since its discovery, it remains to be investigated further precisely how this new cofactor operates within the RNA degradation machinery.
Previous studies investigating the role of the TRAMP complex in mRNP quality control using the impaired THO system have focused on TRAMP4 containing Trf4p and Air2p as components with the assumption that the other respective paralogues, Trf5p and Air1p, are functionally redundant (12). Thus, the most intriguing result in this work is the finding that the TRAMP complex participates in the targeting and elimination of Rho-induced aberrant mRNPs as two distinct complexes containing either Trf4p or Trf5p that both harbor Air2p as the RNA binding component. The paralogous Air1p component is clearly not involved in the process (Fig. 7). This interpretation is made on the basis of several lines of evidence that emerged from our data. First, the ChIP experiments show robust Rho-dependent recruitment of Trf4p and Trf5p along the PMA1 gene. The recruitment is stimulated at a similar level for both components, and it does not increase when one component is absent (Fig. 6D), indicating that the two proteins do not replace each other. Second, although Trf4p seems to play the major role in the process because its removal (trf4Δ strain) restores a large part of the PMA1 transcript, a moderate but significant fraction of the model transcript is readily recovered when Trf4p is present and Trf5p is absent (trf5Δ strain). Again, this indicates that Trf4p cannot functionally replace Trf5p because otherwise no transcript restoration should be observed in a trf5Δ background. Third, the deletion of the TRF4 gene does not lead to a full suppression of the Rho-induced growth defect but only improves the relative growth as compared with TRF5 deletion. This is in line with the differential restoration of the PMA1 transcript induced by the absence of either component and, thus, reflects their respective participation in the targeting and elimination process. Finally, the participation of the two components in two distinct TRAMP complexes is also supported by the comparative ChIP analyses between wild-type and air2Δ cells where the Rho-induced recruitment of both Trf4p and Trf5p is severely reduced when Air2p is absent (Fig. 7G). In addition, these results indicate clearly that the two TRAMP complexes formed by Trf4p or Trf5p are assembled with Air2p prior to their cotranscriptional recruitment, which is consistent with the reported critical role of this protein in the assembly of the TRAMP complex (47). This also confirms the importance of Air2p, whose absence fully suppresses the Rho-induced defects (Fig. 7), a phenotype that mimics conditions where the contributions of Trf4p and Trf5p in the Rho-mediated defect would be absent because they both require Air2p for this specific activity. As a reminder, such a double deletion is lethal in yeast.
In summary, our study provides an integrated view of how different cofactors of the RNA degradation machinery assist the exonuclease Rrp6p in the targeting and elimination of aberrant mRNPs upon their cotranscriptional recruitment. The loading of the cofactors during transcript elongation most likely facilitates a rapid degradation of the malformed mRNP as soon as the RNA 3′ end becomes available upon transcription completion. The results are consistent with previous reports showing that Rrp6p can carry out hydrolytic activities in the absence of the core exosome scaffold and that these activities are specifically stimulated by the TRAMP complex (20, 40). The implication of two distinct TRAMP complexes is also supported by a previous large-scale analysis using DNA microarrays that suggested that TRAMP4 and TRAMP5 have distinct RNA selectivities with functional implications in RNA surveillance (42). The precise mechanism by which the TRAMP complexes stimulate Rrp6p activity is so far unknown and will require further work to be elucidated.
Acknowledgments
We thank Bertrand Seraphin, Domenico Libri, Mathieu Rougemaille, and James Anderson for strains. We also thank Torben Heick Jensen (antiRrp6), Phil Mitchell (antiRrp47) and Helene Benedetti (antiTfs1) for the gift of antibodies and Claudine Kieda for unlimited access to the Roche LightCycler 480 within her research group.
This work was supported in part by la Ligue contre le Cancer Grand Ouest (Comité du Loiret) and by recurrent funding from the CNRS. This work was also supported by a postdoctoral fellowship from the CNRS (to I. S.) and by a doctoral fellowship from the Ecole Doctorale Santé, Sciences Biologiques et Chimie du Vivant of the University of Orléans (to M. R.).
I. Stuparevic, C. Mosrin-Huaman, N. Hervouet-Coste, M. Remenaric, and A. Rachid Rahmouni, manuscript in preparation.
- mRNP
- messenger ribonucleoprotein particle
- TRAMP
- Trf-Air-Mtr4 polyadenylation complex
- Doxy
- doxycycline
- qPCR
- quantitative PCR
- TAP
- tandem affinity purification.
REFERENCES
- 1. Aguilera A. (2005) Cotranscriptional mRNP assembly: from the DNA to the nuclear pore. Curr. Opin. Cell Biol. 17, 242–250 [DOI] [PubMed] [Google Scholar]
- 2. Luna R., Gaillard H., González-Aguilera C., Aguilera A. (2008) Biogenesis of mRNPs. Integrating different processes in the eukaryotic nucleus. Chromosoma 117, 319–331 [DOI] [PubMed] [Google Scholar]
- 3. Doma M. K., Parker R. (2007) RNA quality control in eukaryotes. Cell 131, 660–668 [DOI] [PubMed] [Google Scholar]
- 4. Fasken M. B., Corbett A. H. (2005) Process or perish. Quality control in mRNA biogenesis. Nat. Struct. Mol. Biol. 12, 482–488 [DOI] [PubMed] [Google Scholar]
- 5. Saguez C., Olesen J. R., Jensen T. H. (2005) Formation of export-competent mRNP. Escaping nuclear destruction. Curr. Opin. Cell Biol. 17, 287–293 [DOI] [PubMed] [Google Scholar]
- 6. Villa T., Rougemaille M., Libri D. (2008) Nuclear quality control of RNA polymerase II ribonucleoproteins in yeast. Tilting the balance to shape the transcriptome. Biochim. Biophys. Acta 1779, 524–531 [DOI] [PubMed] [Google Scholar]
- 7. Schmid M., Jensen T. H. (2008) Quality control of mRNP in the nucleus. Chromosoma 117, 419–429 [DOI] [PubMed] [Google Scholar]
- 8. Assenholt J., Mouaikel J., Andersen K. R., Brodersen D. E., Libri D., Jensen T. H. (2008) Exonucleolysis is required for nuclear mRNA quality control in yeast THO mutants. RNA 14, 2305–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jensen T. H., Patricio K., McCarthy T., Rosbash M. (2001) A block to mRNA nuclear export in S. cerevisiae leads to hyperadenylation of transcripts that accumulate at the site of transcription. Mol. Cell 7, 887–898 [DOI] [PubMed] [Google Scholar]
- 10. Libri D., Dower K., Boulay J., Thomsen R., Rosbash M., Jensen T. H. (2002) Interactions between mRNA export commitment, 3′-end quality control, and nuclear degradation. Mol. Cell. Biol. 22, 8254–8266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rougemaille M., Dieppois G., Kisseleva-Romanova E., Gudipati R. K., Lemoine S., Blugeon C., Boulay J., Jensen T. H., Stutz F., Devaux F., Libri D. (2008) THO/Sub2p functions to coordinate 3′-end processing with gene-nuclear pore association. Cell 135, 308–321 [DOI] [PubMed] [Google Scholar]
- 12. Rougemaille M., Gudipati R. K., Olesen J. R., Thomsen R., Seraphin B., Libri D., Jensen T. H. (2007) Dissecting mechanisms of nuclear mRNA surveillance in THO/sub2 complex mutants. EMBO J. 26, 2317–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lykke-Andersen S., Brodersen D. E., Jensen T. H. (2009) Origins and activities of the eukaryotic exosome. J. Cell Sci. 122, 1487–1494 [DOI] [PubMed] [Google Scholar]
- 14. Schmid M., Jensen T. H. (2008) The exosome: a multipurpose RNA-decay machine. Trends Biochem. Sci. 33, 501–510 [DOI] [PubMed] [Google Scholar]
- 15. Dziembowski A., Lorentzen E., Conti E., Séraphin B. (2007) A single subunit, Dis3, is essentially responsible for yeast exosome core activity. Nat. Struct. Mol. Biol. 14, 15–22 [DOI] [PubMed] [Google Scholar]
- 16. Lebreton A., Tomecki R., Dziembowski A., Séraphin B. (2008) Endonucleolytic RNA cleavage by a eukaryotic exosome. Nature 456, 993–996 [DOI] [PubMed] [Google Scholar]
- 17. Schneider C., Leung E., Brown J., Tollervey D. (2009) The N-terminal PIN domain of the exosome subunit Rrp44 harbors endonuclease activity and tethers Rrp44 to the yeast core exosome. Nucleic Acids Res. 37, 1127–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Houseley J., Tollervey D. (2008) The nuclear RNA surveillance machinery. The link between ncRNAs and genome structure in budding yeast? Biochim. Biophys. Acta 1779, 239–246 [DOI] [PubMed] [Google Scholar]
- 19. Kiss D.L., Andrulis E.D. (2011) The exozyme model. A continuum of functionally distinct complexes. RNA 17, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Callahan K.P., Butler J.S. (2008) Evidence for core exosome independent function of the nuclear exoribonuclease Rrp6p. Nucleic Acids Res. 36, 6645–6655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mitchell P., Petfalski E., Houalla R., Podtelejnikov A., Mann M., Tollervey D. (2003) Rrp47p is an exosome-associated protein required for the 3′ processing of stable RNAs. Mol. Cell. Biol. 23, 6982–6992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stead J.A., Costello J.L., Livingstone M.J., Mitchell P. (2007) The PMC2NT domain of the catalytic exosome subunit Rrp6p provides the interface for binding with its cofactor Rrp47p, a nucleic acid-binding protein. Nucleic Acids Res. 35, 5556–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Milligan L., Decourty L., Saveanu C., Rappsilber J., Ceulemans H., Jacquier A., Tollervey D. (2008) A yeast exosome cofactor, Mpp6, functions in RNA surveillance and in the degradation of noncoding RNA transcripts. Mol. Cell. Biol. 28, 5446–5457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. LaCava J., Houseley J., Saveanu C., Petfalski E., Thompson E., Jacquier A., Tollervey D. (2005) RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121, 713–724 [DOI] [PubMed] [Google Scholar]
- 25. Vanácová S., Wolf J., Martin G., Blank D., Dettwiler S., Friedlein A., Langen H., Keith G., Keller W. (2005) A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol. 3, e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wyers F., Rougemaille M., Badis G., Rousselle J. C., Dufour M. E., Boulay J., Régnault B., Devaux F., Namane A., Séraphin B., Libri D., Jacquier A. (2005) Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121, 725–737 [DOI] [PubMed] [Google Scholar]
- 27. Mosrin-Huaman C., Honorine R., Rahmouni A. R. (2009) Expression of bacterial Rho factor in yeast identifies new factors involved in the functional interplay between transcription and mRNP biogenesis. Mol. Cell. Biol. 29, 4033–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Honorine R., Mosrin-Huaman C., Hervouet-Coste N., Libri D., Rahmouni A. R. (2011) Nuclear mRNA quality control in yeast is mediated by Nrd1 co-transcriptional recruitment, as revealed by the targeting of Rho-induced aberrant transcripts. Nucleic Acids Res. 39, 2809–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wach A., Brachat A., Pöhlmann R., Philippsen P. (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10, 1793–1808 [DOI] [PubMed] [Google Scholar]
- 30. Baudin-Baillieu A., Guillemet E., Cullin C., Lacroute F. (1997) Construction of a yeast strain deleted for the TRP1 promoter and coding region that enhances the efficiency of the polymerase chain reaction-disruption method. Yeast 13, 353–356 [DOI] [PubMed] [Google Scholar]
- 31. Janke C., Magiera M. M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., Knop M. (2004) A versatile toolbox for PCR-based tagging of yeast genes. New fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962 [DOI] [PubMed] [Google Scholar]
- 32. Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Séraphin B. (2001). The tandem affinity purification (TAP) method. A general procedure of protein complex purification. Methods 24, 218–229 [DOI] [PubMed] [Google Scholar]
- 33. Schmitt M. E., Brown T. A., Trumpower B. L. (1990) A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 18, 3091–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kushnirov V. V. (2000) Rapid and reliable protein extraction from yeast. Yeast 16, 857–860 [DOI] [PubMed] [Google Scholar]
- 35. Kuras L., Struhl K. (1999) Binding of TBP to promoters in vivo is stimulated by activators and requires Pol II holoenzyme. Nature 399, 609–613 [DOI] [PubMed] [Google Scholar]
- 36. Phillips S., Butler J. S. (2003) Contribution of domain structure to the RNA 3′ end processing and degradation functions of the nuclear exosome subunit Rrp6p. RNA 9, 1098–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lorentzen E., Basquin J., Tomecki R., Dziembowski A., Conti E. (2008) Structure of the active subunit of the yeast exosome core, Rrp44. Diverse modes of substrate recruitment in the RNase II nuclease family. Mol. Cell 29, 717–728 [DOI] [PubMed] [Google Scholar]
- 38. Costello J. L., Stead J. A., Feigenbutz M., Jones R. M., Mitchell P. (2011) The C-terminal region of the exosome-associated protein Rrp47 is specifically required for box C/D small nucleolar RNA 3′-maturation. J. Biol. Chem. 286, 4535–4543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feigenbutz M., Jones R., Besong T. M., Harding S. E., Mitchell P. (2013) Assembly of the yeast exoribonuclease Rrp6 with its associated cofactor Rrp47 occurs in the nucleus and is critical for the controlled expression of Rrp47. J. Biol. Chem. 288, 15959–15970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Callahan K. P., Butler J. S. (2010) TRAMP complex enhances RNA degradation by the nuclear exosome component Rrp6. J. Biol. Chem. 285, 3540–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Houseley J., Tollervey D. (2006) Yeast Trf5p is a nuclear poly(A) polymerase. EMBO Rep. 7, 205–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. San Paolo S., Vanacova S., Schenk L., Scherrer T., Blank D., Keller W., Gerber A.P. (2009) Distinct roles of non-canonical poly(A) polymerases in RNA metabolism. PLoS Genet. 5, e1000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmidt K., Xu Z., Mathews D.H., Butler J. S. (2012) Air proteins control differential TRAMP substrate specificity for nuclear RNA surveillance. RNA 18, 1934–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang X., Jia H., Jankowsky E., Anderson J. T. (2008) Degradation of hypomodified tRNA(iMet) in vivo involves RNA-dependent ATPase activity of the DExH helicase Mtr4p. RNA 14, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gudipati R. K., Xu Z., Lebreton A., Séraphin B., Steinmetz L. M., Jacquier A., Libri D. (2012) Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol. Cell 48, 409–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schneider C., Kudla G., Wlotzka W., Tuck A., Tollervey D. (2012) Transcriptome-wide analysis of exosome targets. Mol. Cell 48, 422–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Holub P., Lalakova J., Cerna H., Pasulka J., Sarazova M., Hrazdilova K., Arce M. S., Hobor F., Stefl R., Vanacova S. (2012) Air2p is critical for the assembly and RNA-binding of the TRAMP complex and the KOW domain of Mtr4p is crucial for exosome activation. Nucleic Acids Res. 40, 5679–5693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Johnson S.A., Cubberley G., Bentley D.L. (2009) Cotranscriptional recruitment of the mRNA export factor Yra1 by direct interaction with the 3′ end processing factor Pcf11. Mol. Cell 33, 215–226 [DOI] [PMC free article] [PubMed] [Google Scholar]