

Background: It is unclear how mutations in the coiled-coil tail of β-cardiac myosin cause heart disease.
Results: Effects of disease-causing mutations in the myosin tail were studied in vivo and in vitro.
Conclusion: Mutations that reduce helical content in vitro reduce sarcomere incorporation of myosin in vivo.
Significance: A change in myosin tail structure can lead to heart disease.
Keywords: Adenovirus, Cardiac Hypertrophy, Cardiovascular Disease, Cell Biology, Circular Dichroism (CD), Imaging, Myosin
Abstract
It is unclear why mutations in the filament-forming tail of myosin heavy chain (MHC) cause hypertrophic or dilated cardiomyopathy as these mutations should not directly affect contraction. To investigate this, we first investigated the impact of five hypertrophic cardiomyopathy-causing (N1327K, E1356K, R1382W, E1555K, and R1768K) and one dilated cardiomyopathy-causing (R1500W) tail mutations on their ability to incorporate into muscle sarcomeres in vivo. We used adenoviral delivery to express full-length wild type or mutant enhanced GFP-MHC in isolated adult cardiomyocytes. Three mutations (N1327K, E1356K, and E1555K) reduced enhanced GFP-MHC incorporation into muscle sarcomeres, whereas the remainder had no effect. No mutations significantly affected contraction. Fluorescence recovery after photobleaching showed that fluorescence recovery for the mutation that incorporated least well (N1327K) was significantly faster than that of WT with half-times of 25.1 ± 1.8 and 32.2 ± 2.5 min (mean ± S.E.), respectively. Next, we determined the effects of each mutation on the helical properties of wild type and seven mutant peptides (7, 11, or 15 heptads long) from the myosin tail by circular dichroism. R1382W and E1768K slightly increased the α-helical nature of peptides. The remaining mutations reduced α-helical content, with N1327K showing the greatest reduction. Only peptides containing residues 1301–1329 were highly α-helical suggesting that this region helps in initiation of coiled coil. These results suggest that small effects of mutations on helicity translate into a reduced ability to incorporate into sarcomeres, which may elicit compensatory hypertrophy.
Introduction
β-Cardiac myosin is a conventional class 2 myosin expressed in the heart and in slow muscle. It contains two heavy and four light chains. The heavy chains dimerize by forming a coiled-coil tail in the C-terminal region of the molecule and separate to form two globular heads at their N-terminal region. The globular head contains the motor domain, which binds actin and nucleotide and is responsible for generating contraction. The coiled-coil tail contains two α-helices that interact with each other to bury hydrophobic residues, forming a hydrophobic seam (1). Each contains a distinctive heptad repeat sequence in the form of (abcdefg)n where a and d are commonly hydrophobic. Because the heptad repeat is slightly shorter than two turns of an α-helix, the two helices twist around each other to form a coiled coil with a left-handed superhelix (2). The tail also contains four “skip” residues, nominally at positions Thr-1188, Glu-1385, Glu-1582 and Gly-1807, which interrupt the heptad motif and correlate to bends in the myosin tail in the isolated molecules (3). The distal region of the coiled-coil tail assembles into thick filaments as a result of patterns of alternating charge along the length of the coiled coil (4) and a short assembly competence domain near the C terminus (5).
Mutations in the gene for the heavy chain of β-cardiac myosin (MYH7) (6) cause the disease hypertrophic cardiomyopathy (HCM),3 a global disease that causes heart failure, first described over 50 years ago (7). HCM is the most common cause of sudden death in people under 30 years of age (8) and affects at least 1 in 500 people. Mutations in MYH7 cause ∼40% of cases of inherited HCM (9, 10). The disease is characterized by thickening of the left ventricular wall and myocardial disarray (11). More generally, mutations in genes that encode sarcomeric proteins are responsible for HCM (12) with over 630 mutations in 10 sarcomeric genes described as causing this disease. Mutations in MYH7 also cause dilated cardiomyopathy (DCM), a rarer heart disease that only affects about 1 in 2500 people (13) and is characterized by a thinning of the ventricular walls (ventricular dilatation). Currently, over 200 HCM and 24 DCM mutations have been described for MYH7 (14), many of which result in a highly malignant form of the disease. However, tail mutations are rare, and only limited descriptions of their severity and phenotype are available (supplemental Table S1).
Although a clear link between HCM-causing mutations in the motor domain and HCM can be attributed to direct effects on force output (11), it is unclear why mutations in the distal tail result in HCM. As the main role of the tail is to form filaments, it is unlikely that mutations in the tail directly affect force output. However, effects on coiled-coil structure and/or packing of the coiled coil into the thick filament might affect myosin incorporation into thick filaments in sarcomeres, and thus indirectly affect force output over the longer term.
To test this idea, we investigated five different mutations that result in HCM (15–21) and one (R1500W) that causes DCM (17). Only two of these mutations have been investigated previously (E1356K and R1500W (22, 23)). In those reports, expressed and purified light meromyosin peptides containing the mutation had decreased thermal stability, but the secondary structure was not affected. This suggested to us that any effects on secondary structure could be localized and not observable in the full-length LMM. Thus, in this study, we have used shorter peptides to investigate the effects of the mutations. Neither of these previous reports investigated the effects of these mutations in vivo.
Here, we have investigated the effects of each of these mutations on the incorporation of full-length β-cardiac myosin heavy chain fused to eGFP (eGFP-MHC) in vivo. We used an adenoviral expression system to express eGFP-MHC in adult rat cardiomyocytes, and we determined whether these mutations affect incorporation of the full-length heavy chain into the sarcomere or contraction. To determine whether the effects we observed were correlated to effects on peptide structure in vitro, we analyzed the secondary structure and thermal stability of peptides of different length. We used peptides that were 7, 11, or 15 heptads long from the myosin coiled-coil tail to determine whether any or all of the five different mutations affect molecular structure and to determine whether the effects observed depended on peptide length.
EXPERIMENTAL PROCEDURES
Expression and Characterization of eGFP-MHC in Adult Cardiomyocytes
The full-length cDNA for the heavy chain of MYH7 (24) was cloned into an adenoviral expression vector (pdc315, Addgene) with eGFP fused to the N terminus to generate eGFP-MHC. A second construct, without eGFP, was also constructed. Purified virus (at multiplicities of infection between 50 and 100) was used to infect isolated adult rat cardiomyocytes. These were prepared from isolated rat hearts using a Langendorff retrograde perfusion technique with a collagenase-containing solution (25). After incubation for 24 h at 37 °C, to allow the MHC to be expressed, cells were fixed using 4% formaldehyde in phosphate-buffered saline prior to staining for other proteins of interest. Cells were stained with antibodies against an M-line epitope of titin (Ti51; kind gift of Dieter Furst (26)) or for myosin heavy chain (A4.1025 antibody (27)), and cells were imaged using a Deltavision deconvolution microscope.
Levels of eGFP-MHC expression were compared with endogenous levels of MHC by SDS gel analysis using a protocol that separates MHC isoforms in a minigel system (28). Briefly, the separating gel contained 35% v/v glycerol, 9% total acrylamide of which 1/49 was bisacrylamide, 230 mm Tris-HCl, 115 mm glycine, pH 8.8, and 0.4% w/v SDS; and the stacking gel contained 47% v/v glycerol, 6% w/v acrylamide (1/49 bisacrylamide), 110 mm Tris-HCl, 6 mm EDTA, pH 6.8, and 0.4% w/v SDS. Gels were stained, or proteins were transferred to nitrocellulose paper to perform Western blotting, using an antibody to MHC (A4.1025) or to eGFP (Abcam). These blots confirmed which band was endogenous and which was the eGFP-tagged myosin in the stained gel (Fig. 1).
FIGURE 1.

Expression and incorporation of eGFP-MHC in adult cardiomyocytes. A, fluorescence pattern for eGFP-MHC 24 h after adding adenovirus is similar to that observed for endogenous MHC, using an antibody that recognizes the motor domain of myosin (A4.1025). Co-labeling with an anti-M-line titin antibody (Ti51) confirms that the wider gap in fluorescence observed for eGFP-MHC is at the M-line. Black arrows indicate the positions of Z-discs. B, fluorescence images of part of a cardiomyocyte, corresponding to one myofibril in width, expressing eGFP-MHC, and co-stained for MHC using an antibody. The corresponding line profiles for the fluorescence images are shown below, demonstrating how fluorescence intensity varies along the myofibril for MHC and for eGFP-MHC. Arrows indicate the positions of Z-discs. C, images of a Coomassie-stained SDS gel and corresponding immunoblots probed using antibodies to eGFP or to myosin heavy chain (A4.1025) to identify the bands for eGFP-tagged and endogenous MHC.
As expression levels were ∼10% or less after 24 h, we also attempted to culture MHC expressing cardiomyocytes for longer time periods. However, we found that long term culture of cardiomyocytes resulted in a loss of contractility of the intact cells and in a loss of the characteristic rod shape, as documented previously (29). We also suspect that the large excess of the overexpressed MHC compared with other myosin-binding proteins (light chains, titin, MyBP-C, actin, etc.) that are also needed to incorporate and regulate the MHC into muscle sarcomeres contributed to the loss of contractility as large amounts of unregulated soluble nonincorporated MHC may be toxic to the cells. Thus, 24 h of myosin overexpression is the best compromise between expression of high enough levels of eGFP-MHC to observe the impacts of the mutants within the sarcomere and expression of such high levels that cell viability is lost.
Fluorescence Recovery after Photobleaching (FRAP)
For FRAP experiments, cells were plated onto laminin-coated glass bottomed dishes (35 mm, Iwaki), infected with the adenovirus, cultured overnight, and imaged the following day using an inverted Zeiss 510 confocal microscope. Dishes were placed onto the heated stage (set to 37 °C); 10–15 cells were selected and their positions marked. A Zeiss multitime series macro was programmed to image each cell in turn, bleaching a small rectangular area in each cell, ∼2 sarcomeres in width, using 20 cycles of maximum laser power (at a wavelength of 488 nm) and then imaging each cell every 5–10 min after bleaching for the next 105–125 min at 1% laser power until fluorescence recovery reached a plateau. To analyze the data, the average intensity of the bleached area was measured at each time point using ImageJ. Recordings from cells that contracted or underwent large movements during the experiment were discarded. The average image intensity of the same sized area in an area adjacent to that bleached was also measured, to test that the intensity of nonbleached regions did not change. Data were normalized as described (30). Exponential fits to the data were generated using Prism (GraphPad).
Cardiomyocyte Contraction
Within 18–24 h of infection, live cells were placed in a bath, continuously perfused with physiological saline at room temperature, and stimulated by external platinum electrodes at a frequency of 1 Hz to investigate their ability to contract. Contraction was recorded using video edge detection (24) and analyzed for the times to peak shortening and half-relaxation together with contractility, indexed as the amplitude of shortening as a percent of resting cell length.
Expression and Characterization of Coiled-coil Peptides
Standard PCR-based cloning was used to generate expression vectors for GST fusion proteins, using the MYH7 cDNA as a template. All constructs were sequence-verified after cloning. The resulting expression vectors were used to express peptides in Escherichia coli. During purification, the GST was removed using PreScission Protease. All peptides (supplemental Table S2) contained the N-terminal sequence GPLGS in addition to the peptide sequence. Each peptide expressed was designed to start and end on a residue in the d-position of the heptad repeat. Peptides were 55, 83, or 111 residues long (7, 11, or 15 heptads, respectively). The 7-heptad peptides were designed such that in mutants the mutated residue was in the central heptad with three heptads on either side. The concentrations of the purified proteins were determined by a micro BCA assay (Pierce) and/or by measuring their absorbance at 280 nm in a quartz cuvette on a Cary-50 spectrophotometer and using the extinction coefficient calculated from the sequence (using ProtParam (ExPASy)). The 7-heptad peptides were additionally analyzed by mass spectrometry, to confirm that their molecular mass was correct.
Circular Dichroism
Circular dichroism spectra were measured from 260 to 190 nm at 10 °C using a Jasco J715 spectropolarimeter.
For the 7H peptides, protein concentrations varied from 0.25 to 0.05 mg/ml, and their CD spectra were obtained at 10 °C at pH 8.0 in 150 mm NaCl, 10 mm phosphate buffer. Thermal melt data were obtained either in the same buffer or in 50 mm NaCl, 10 mm phosphate buffer, at pH 7.0. An increase in pH slightly increased the melting temperature (Tm) for all the peptides equally. 1 mm DTT was added to peptides containing a cysteine residue.
The effect of the mutations on α-helical content was estimated by comparing the value of the ellipticity measurements (θ222) at a wavelength of 222 nm, expressed as mean residue ellipticity in degrees cm2 dmol−1 for the peptides at 10 °C for all the constructs. The θ222 values indicate the relative helical content of the peptides. We therefore used θ222 values to compare helical content between the constructs. Repeat measurements for the majority of the 7H constructs gave consistent results, giving confidence that the observed differences between peptides are likely to be significant, especially where differences are large. Where more than two repeats were performed for the 7H constructs, error bars are shown in Fig. 5. To compare helical contents between peptides, the 222 nm value was divided by −36,000 (theoretical value for 100% helical content) (32).
FIGURE 5.

Properties of wild type and mutant peptides. A, diagram of the positions of the tail peptides and mutants investigated in the tail. The black line represents a scale bar of 100 amino acids alongside the coiled-coil tail of the myosin. The positions of the four skip residues are shown by gray dotted lines. The shaded rectangles show the positions, with respect to their positions in the tail of myosin, of each of the peptides investigated, in which overlapping sequence in different peptides is shown in the same color/pattern. For example, heptad H1 is shown in black, and the start of heptad 7H2 is shown as black, as it contains the C-terminal sequence of heptad 7H1. The filled stars denote the relative α-helical content of each peptide (see key). The vertical rectangle shown using a dashed line shows the region of the sequence required for high α-helical content. B, CD spectra for the 7H1 WT and mutant (N1327K) peptides (pH 8.0, 150 mm NaCl, 10 mm phosphate buffer). C, summary plot of the values for θ222 (MRW) (which reports on α-helical content) measured for each of the WT 7H peptides (shown as black bars) and the corresponding mutant (shown as white bars). Conditions used were pH 8.0, 150 mm NaCl, 10 mm phosphate buffer. The mean together with the standard error is shown. The decrease in helical content for N1327K is significant (p < 0.05). D, normalized thermal melt curves for 11H peptides (pH 7.0, 50 mm NaCl, 10 mm phosphate buffer). E, CD spectra for the WT and mutant 11H1 peptides (pH 7.4, 50 mm NaCl, 10 mm phosphate buffer). F, CD spectra for the WT and mutant 15H1 peptides (pH 7.4, 50 mm NaCl, 10 mm phosphate buffer). G, summary plot of the θ222 (MRW) values for the WT 11H1 (black bars) and 15H1 (black bars) and mutant 11H1 and 15H1 peptides (white bars) as shown in E and F, together with the value for the WT 11H2 and 15H2 peptides (gray bars). Significant changes in helical content between WT and mutant peptides are indicated as follows: *, p < 0.05; **, p < 0.01, and ***, p < 0.001. H, normalized thermal melt curves for 15H peptides (pH 7.0, 50 mm NaCl, 10 mm phosphate buffer). MRW, mean residue weight.
For the 11H and 15H peptides, protein concentrations varied from 0.25 to 0.1 mg/ml, and their CD spectra were obtained at 10 °C in 50 mm NaCl, 10 mm phosphate buffer, pH 7.4. Further data obtained in the same buffer at pH 7.0 (data not shown) showed very little change in the spectra at a temperature of 10 °C. Three repeat measurements were made for each of the 11 and 15H peptides, and t tests used to determine whether differences in helical content (estimated from the 222-nm value as described above) were significant.
The effect of temperature on the α-helical content was measured by taking measurements at 222 nm from 5 to 60 °C, for every 1 °C temperature change. The conditions used were 50 mm NaCl, 10 mm phosphate buffer, at pH 7.0. Temperature was changed at a rate of 2 °C/min. The temperature was returned to 10 °C, and the resulting spectrum was close to that measured prior to heating (value at 222 nm, to within a few percent). The θ222 measurements were normalized by setting the value at 5 °C to 0% unfolded and the value at 65 °C to 100% unfolded. The resulting plots of the θ222 values were fit using the Boltzman sigmoidal fit function in GraphPad Prism 6 software (33) to generate values for Tm.
RESULTS
Expression of Full-length MHC in Cardiomyocytes
WT eGFP-MHC was expressed successfully in isolated adult cardiomyocytes, and the eGFP-tagged MHC (in which eGFP is fused to the MHC N terminus (motor domain)) incorporated into the thick filaments in muscle sarcomeres. eGFP-MHC began to be expressed and to incorporate into sarcomeres 6–7 h after viral infection (supplemental Fig. S1). After 24 h, the pattern of fluorescence for eGFP-MHC was similar to the immunostaining pattern for total MHC, using an antibody against an epitope in the motor domain (Fig. 1A).
Both the anti-MHC immunostaining and the eGFP fluorescence show two stripes, separated by an unstained gap, which corresponds to the bare zone in the center of the thick filament. The unstained gap at the M-line was wider (∼0.3 μm) for eGFP-MHC than for endogenous myosin (Fig. 1B), suggesting that there is some limit on incorporation of new myosin in the center of the thick filament, as reported earlier (34). The gap in intensity at the I-band is similar for eGFP-MHC and the immunostained image for MHC (Fig. 1B). Overall, the newly synthesized eGFP-MHC has incorporated generally throughout the thick filaments, and not only at the very edges of the A-bands or at the periphery of the myofibrils, similar to that reported for embryonic chicken MHC in embryonic cardiomyocytes (35), although the fluorescence intensity is higher at the ends of the A-band. This suggests that myosin may exchange more easily at the ends of the thick filaments, because of the reduced number of interactions that myosin there makes with other myosins, as the thick filament tapers at this point. It is also possible that diffusion is faster within the I-band (less congested) than with the A-band, so that exchanging molecules are more likely to encounter thick filament ends.
Impact of Tail Mutations on MHC Uptake into Myofibrils
Using the adenoviral expression system, we expressed and characterized six different tail mutants (N1327K, E1356K, R1382W, R1500W, E1555K, and E1768K) in adult cardiomyocytes. These mutations are all found in the tail of MHC (Fig. 5A and supplemental Table S2). WT and mutant eGFP-MHC were all expressed at a level of 5–10% of the endogenous myosin, following 24 h of infection with the adenovirus as shown by SDS-polyacrylamide gel and Western blotting (Fig. 1B), with the exception of R1500W, which was expressed at slightly lower levels (∼2% of endogenous myosin).
Deconvolution microscopy of fixed cardiomyocytes containing expressed mutant eGFP-MHC isoforms showed that in the case of several mutants, eGFP-MHC did not incorporate into muscle sarcomeres as well as WT eGFP-MHC in substantial numbers of cells (Fig. 2). Instead, eGFP-MHC incorporation was limited to regions at the edges at the A-bands, where it is mainly incorporated into the ends of the thick filament, and there were increased numbers of filamentous aggregates outside the myofibrils (Fig. 2B). These filamentous aggregates are likely to contain eGFP-MHC that is unable to incorporate or exchange into the sarcomeric thick filaments. We repeated these experiments several times and consistently observed this effect.
FIGURE 2.
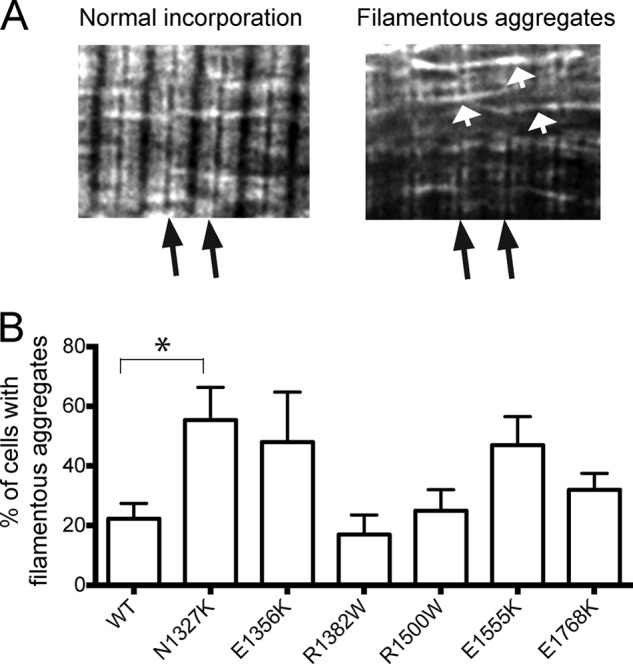
Analysis of eGFP-MHC incorporation in adult cardiomyocytes. A, representative images showing normal incorporation contrasted with poor incorporation with filamentous aggregates. Black arrows show the positions of Z-disks. White arrows show filamentous aggregates. The images shown are for the N1327K mutant. B, quantification of eGFP incorporation for WT and each of the mutants. The results shown are the mean values (± S.E.) for 3–6 experiments. The % of cells with aggregates was significantly higher in N1327K-expressing cells compared with WT cells (p < 0.05) (indicated by the asterisk).
Quantifying the proportions of cells with normal or limited incorporation showed that the mutation N1327K resulted in the highest increase in the proportion of cells with limited incorporation of eGFP-MHC, and this increase was significant (Fig. 2B). Two further mutations, E1356K and E1555K, also showed a trend toward an increase in numbers of cells with limited eGFP MHC incorporation. In the remaining three mutations, R1500W, R138W, and E1768K, eGFP MHC incorporation was similar to wild type. The differences in incorporation are unlikely to be due to different levels of viral infection, as similar multiplicities of infection were used in all the experiments, and the expression levels of the eGFP MHC are low and uncorrelated with these differences (Fig. 1B).
Exchange Rate of MHC into Myofibrils Using FRAP
The expression of full-length eGFP-MHC in living adult cardiomyocytes gave us the opportunity, for the first time, to investigate how quickly eGFP-MHC exchanges into thick filaments using FRAP for WT and mutant isoforms. For WT eGFP-MHC, the fluorescence recovery was slow, with a half-time for recovery of 32.2 ± 2.5 min (mean ± S.E., n = 14) (Fig. 3B). Recovery appeared to occur first in regions close to the Z-discs (arrowed, Fig. 3A) before spreading through the rest of the A-band. Although the rate of recovery was slow, these experiments showed that the myosin molecules are exchanging into and out of the thick filament more dynamically than we might have predicted.
FIGURE 3.

FRAP analysis of eGFP-MHC dynamics in living cardiomyocytes. A, sample images from a FRAP experiment showing a region from a single cardiomyocyte expressing wild type eGFP-MHC, in which the bleached square is in the center. The pre-bleach image, followed by the immediate post-bleach image, and subsequent images at 45 and 125 min are shown. Arrows indicate location of fluorescence recovery near Z-discs. B, time course of FRAP data for WT (n = 14), N1327K (n = 18), and R1382W (n = 18), showing the means ± S.E. at each time point, and the exponential fits to the data.
We extended these FRAP experiments to two different mutants, one that had the most effect on sarcomeric incorporation (N1327K) and one that had the least (R1382W). We found that the half-time for fluorescence recovery was significantly faster (p < 0.05) for N1327K (t½ of 25.1 ± 1.8 min (n = 18)) but not for R1382W (t½ of 31.0 ± 1.6 min (n = 18)) compared with WT eGFP-MHC (Fig. 3B). These results suggest that the poorer incorporation of the N1327K mutation is linked to an increase in the rate of exchange of the MHC into thick filaments. The rate of myosin exchange into the thick filaments is partly determined by the rate at which the incoming myosin incorporates, as well as the rate of myosin dissociation.
Contractile Properties of Transfected Cardiomyocytes
We did not find any significant differences between the contractile properties of untransfected cardiomyocytes and those expressing untagged MHC, WT eGFP-MHC, or mutant isoforms of eGFP-MHC (Fig. 4). This might be thought to be due to the low expression levels of the eGFP MHC in our experiments (5–10% of endogenous myosin). However, modeling has suggested that expressing α-cardiac MHC myosin at similar expression levels on a background of β-MHC has an observable effect on contraction kinetics (36). Therefore, if the mutations studied here were able to exert a strong effect on contraction, we would have observed it in these experiments. Including the eGFP tag did not affect contraction, with no significant difference between any of the parameters measured for cells expressing untagged MHC compared with eGFP-tagged MHC. Moreover, the contractile properties of myocytes expressing WT eGFP-MHC were not significantly different from untransfected cells. These results suggest that none of the tail mutations, expressed at up to 10% of the endogenous level of myosin, directly affect contraction.
FIGURE 4.
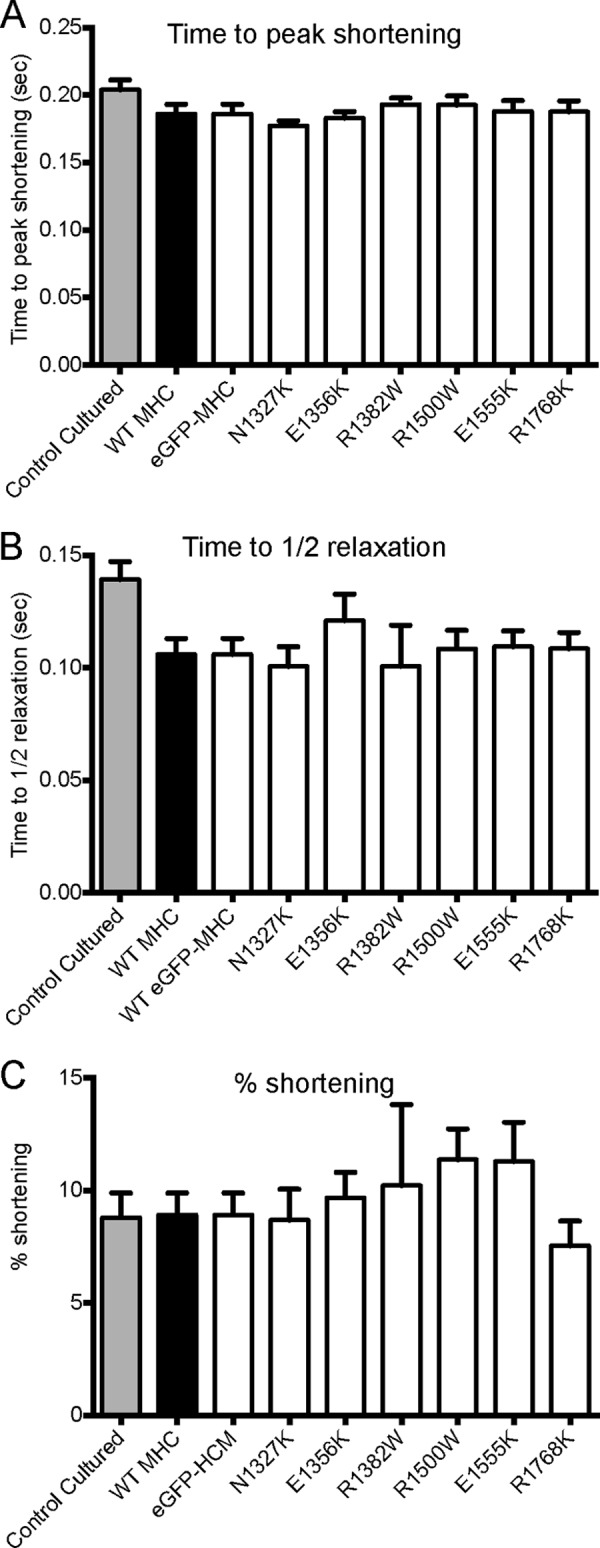
Contraction data for rat ventricular myocytes cultured for 24 h that were untransfected, expressing WT-MHC, WT GFP-MHC, or mutant WT GFP-MHC. Myocytes were stimulated to contract at a frequency of 1 Hz. Data are shown as mean ± S.E. Values for n are as follows: 9 (control untransfected), 14 (WT MHC), 14 (WT GFP-MHC), 12 (N1327K) 20 (E1356K), 11 (R1382W), 14 (R1500W), 11 (E1555K), and 13 (R1768K). A, time from stimulation to peak shortening. B, time of half-relaxation from peak shortening to resting cell length. C, contractility expressed as the amplitude of cell shortening as a % of resting cell length. No significant differences were found between any of the measurements.
Helical Propensity of WT Tail Peptides Containing 7, 11, or 15 Heptads
The reduced incorporation of mutant eGFP-MHC into thick filaments in muscle sarcomeres for three out of the six mutants could be due to alterations in the structure of the coiled-coil tail. To test this, we expressed and purified WT and mutant peptides containing 7, 11, or 15 heptad repeats from the region of coiled coil that contains these mutations (Fig. 5 and supplemental Table S2).
We expected that all of the wild type (WT) peptides would be α-helical and form coiled coils. Unexpectedly, only 7H1, 11H1, and 15H1 WT peptides had a high α-helical content at 10 °C (Fig. 5 and Table 1), with the helical content increasing with length from 57% for 7H1 to 91% for 15H1. The five remaining 7H peptides tested (7H2–7H6, Fig. 5, A and C) as well as 11H2 and 15H2 (Fig. 5F) were all less than 30% α-helical. The low helical content of many peptides indicates that they are unlikely to spend most of their time as a dimer and thus as a coiled coil.
TABLE 1.
Estimates of the % helical contents and melting temperatures (Tm) for each 11H and 15H peptide and associated mutant
Melting curves were only performed once. Tm was estimated as described under “Experimental Procedures.”
| Peptide | Mutation/WT | α-Helical content, pH 7.4, (%) ± S.E. | Tm pH 7.0 (10 °C) | ΔTm compared to WT |
|---|---|---|---|---|
| 11H1 | WT | 79 ± 1 | 26.9 | |
| 11H1 | N1327K | 66 ± 2 | 22.3 | −4.6 |
| 11H1 | E1356K | 62 ± 2 | 26.2 | −0.7 |
| 15H1 | WT | 91 ± 1 | 29.7 | |
| 15H1 | N1327K | 81 ± 3 | 27.8 | −1.9 |
| 15H1 | E1356K | 90 ± 2 | 30.0 | +0.3 |
| 15H1 | R1382W | 98 ± 2 | 33.8 | +4.1 |
The α-helical content of WT 11H1 and 15H1 peptides both showed the sigmoidal dependence on the temperature expected for the cooperative melting of a coiled coil (Fig. 5, G and H). The Tm was 27 °C for 11H1 and increased to 30 °C for 15H1 (Table 1). A melting curve obtained for WT 7H1 (at pH 7.0) was less sigmoidal in shape (supplemental Fig. 2), and the estimated Tm was lower (17 °C). The increasing Tm value with increasing peptide length suggests that the longer peptides are more stable.
These results demonstrate that sequences within the distal myosin tail vary widely in their propensity to form α-helix. Among those that we tested, only those containing residues 1301–1330 had a high helical propensity (Fig. 5A). Plotting the sequence of this region as a heptad net (Fig. 6) (37) shows that there it contains a short leucine zipper surrounded by many oppositely charged residues that can interact to stabilize a helical conformation. This region of sequence is likely to have a high propensity to form a coiled coil and may help longer helical peptides to do so, explaining the high helical nature of the 7, 11, and 15H1 peptides.
FIGURE 6.

Heptad net plots of each of the WT 7H peptides tested. A–F shows peptides 7H1 to 7H6. In each plot, the preferred intra-chain ionic interactions (31, 37) are shown as solid lines. In addition, alternative/other interactions are shown as dotted lines. Every seventh residue is repeated on the right of the plot (in brackets) so that all interactions can be shown. The gray dashed lines indicate the path of the polypeptide backbone; the green dotted line defines the orientation of the α-helix axis. The light gray circles indicate a-positions and the squares indicate d-positions that form the hydrophobic seam. A, boxed region (dashed lines) within this 7H1 peptide that favors coiled coil formation is shown. The black hexagons (dashed lines) indicate the positions of the mutant residues within each sequence.
Impacts of Mutations on Helical Content of Tail Peptides
The mutation that had the most severe effect on sarcomeric incorporation in vivo, N1327K, significantly decreased the α-helical content of 7H1, 11H1 and 15H1 peptides (Fig. 5, B, C, E, and F). The size of this decrease became less marked as the length of the peptide increased (∼27% decrease for 7H1, ∼16% for 11H1, and ∼11% for 15H1 (Fig. 5, C and G, Table 1)). N1327K also reduced the Tm (Fig. 5, D and H, and Table 1), with the effect larger in the 11H1 than in the 15H1 peptide. This mutation could be tested in each of the different length peptides (7H1, 11H1, and 15H1) due to its position in the sequence. A second mutation (E1356K) that reduced sarcomeric incorporation in vivo significantly reduced the helical content of the 11H peptide (by 21%, Fig. 5, E and G), but it had no effect on the 15H peptide (Fig. 5, F and G) or on Tm (Fig. 5, D and H, and Table 1).
A further mutation (R1382W) that did not affect sarcomere incorporation in vivo significantly increased the helical content of the 15H peptide (Fig. 5, F and G). The Tm was also increased for the mutant 15H peptide (Fig. 5H and Table 1). This suggests that R1382W, close to the skip residue (Glu-1385), increases α-helical stability.
The three remaining mutations tested were in regions of the coiled coil that did not appear to form highly helical structures as 7, 11, or 15H peptides. R1500W (in 7H3) and E1555K (in 7H4) both appeared to decrease helical content, and E1768K (in 7H5) was the only mutation in the 7H peptides that appeared to increase helical content. However, the helical contents of the wild type peptides are low, and the differences are not significant.
Overall, these peptide data show that N1327K has the greatest effect on the α-helical nature of the myosin coiled coil, reducing α-helical content and stability, whereas R1382W appears to increase the α-helical content and the stability of the coiled coil.
DISCUSSION
Our data suggest that a reduced incorporation of mutant MHC into muscle sarcomeres is correlated with a large effect on the helical content of isolated peptides. Two mutations (N1327K and E1356K) that reduced the α-helical content of peptides in solution also caused a reduction in sarcomeric incorporation of eGFP-MHC in vivo. In contrast, R1382W increased the α-helical content of a 15-heptad peptide in solution but had little effect on the incorporation of WT eGFP-MHC into sarcomeres in vivo. Local disruptions to the helical nature of the rod portion of MHC may thus contribute to the underlying mechanism by which these mutations result in disease. None of the mutations appeared to have strong effects on contraction at the levels of expression (5–10% of endogenous myosin) obtained in our experiments, which is consistent with the progressive etiology of HCM. Finally, N1327K, the mutation that has the most effect on sarcomeric incorporation and α-helical content, showed a faster rate of exchange into thick filaments in FRAP experiments compared with WT and to R1382W eGFP-MHC.
New Trigger Sequence in the Myosin Tail
Unexpectedly, as part of our research into the effects of mutations in vitro, we discovered that most 50-amino acid residue peptides do not form 100% coiled coil in isolation. We had expected that the α-helical conformation of the seven heptad peptides would be rapidly stabilized by forming a coiled coil, as all of them show a hydrophobic seam in their sequence (Fig. 6, A–F). However, with the exception of 7H1, all the 7H peptides had a low α-helical content, suggesting that they remain largely unfolded and/or form an alternative fold. Moreover, increasing the lengths of such peptides to 11 or even 15 heptads did not necessarily increase the α-helical content. Peptides had to contain the sequence within residues 1301–1329 to contain significant amounts of α-helix/coiled coil, suggesting that this region is important for coiled coil formation, possibly acting as a “trigger sequence” in the tail. Trigger sequences have a high propensity to form α-helix and then assemble to form coiled coil with the trigger sequence region on a second peptide (38). This promotes folding and coiled-coil formation along the whole molecule. Any sequence that favors interchain and intrachain interactions may act as a trigger (39).
Plotting the 7H1 as a heptad net (Fig. 6A) shows that there are many potential attractive intrachain interactions (including Gln-Glu interactions) between successive turns of helix within 1309–1322 (boxed region, Fig. 6A), which will promote formation of α-helix. These interactions surround a short leucine zipper (leucines in the a, d, and subsequent a positions). Together, these may account for the stability and helix propensity of this region of the coiled coil. Fewer interactions and a lack of leucine residues in the hydrophobic seam may explain why downstream sequences (7H2-6, 11H2, and 15H2) have a lower propensity to form coiled coil in isolation. However, when the sequences of 7H2 and 7H3 (Fig. 6, B and C) are incorporated into 11H1 and 15H1, they do become helical presumably due to the influence of the upstream sequence from 7H1.
Molecular Basis of Effects of Tail Mutations
All of the mutations we studied affected the helical content and coiled coil formation of 7H peptides. Three mutations, N1327K, E1356K, and E1555K, which decreased helical content in isolated peptides, also decreased incorporation of full-length myosin into the thick filament in vivo. This suggests that reducing the helical content could contribute to a decreased ability of these mutant isoforms to incorporate normally into thick filaments and an increased chance that the mutant myosin forms aggregates outside the muscle sarcomeres. This may explain why all three of these mutations result in a more severe form of HCM (supplemental Table S1) (18, 20). The nonincorporated aggregated myosin may be proteotoxic, supporting the recently suggested idea that protein misfolding is a major contributor to hypertrophic cardiomyopathy (40).
The reduced helical content that we observed for each of these three mutations may result from the introduction of an intra-chain repulsion. N1327K (b position in the heptad) introduces an intra-chain repulsion between this residue and Lys-1323 in the preceding turn of helix (Fig. 6A). E1356K replaces an intra-chain attraction between this residue and Arg-1359 into a repulsive one (Fig. 6B), which is likely to affect coiled-coil stability. In contrast, E1555K (e position) replaces a potentially repulsive interaction between this residue and Glu-1552 with an attractive one (Fig. 6D). Finally, our observation that the effects of mutations on α-helical content decrease as the lengths of the peptide increase helps to explain why an earlier study, using the full-length filament-forming region of the tail (LMM), found no effect of E1356K on α-helical content (22).
In addition to reducing helical content, each of these mutants, in residues on the outside of the coiled coil, has the potential to affect packing of the myosin heavy chain into the thick filament. Both E1356 (in a “c” position) and E1555K (“e” position) are in patches of glutamates (Fig. 6, B and D). A mutation to lysine decreases the net charge of these patches by two. This has the potential to affect integration of the molecule into the thick filament, which depends on the pattern of alternating negative and positive charges along the coiled coil (41). In addition, E1555K is in a region of LMM implicated in binding to other thick filament proteins such as myosin-binding protein C (thought to lie between residues 1554 and 1581), myomesin, and M-protein (thought to lie between residues 1506 and 1674) (42–44). Although mutation of Glu-1555 to Gln in earlier experiments showed it did not affect myosin-binding protein C binding (42), it has not been determined whether E1555K affects binding of myosin-binding protein C, myomesin, and/or M-line protein.
Two further mutations, R1382W and E1768K, had little effect on sarcomeric incorporation but are associated with increased helical content in the 15H and 7H peptides, respectively. R1382W (a position, Fig. 6F) is close to one of the skip residues (Glu-1385), thought to correlate to the bends in the myosin tail in the extended molecule (3). This mutation may increase α-helical content by stabilizing the sequence close to the skip residue, as tryptophan is more hydrophobic than arginine, and may become more buried (Fig. 6F). In addition, this mutation would abolish any inter-chain attractive forces between Arg-1382 (a position) and both Asp-1378 and Glu-1385 in adjacent d positions. Glu-1768 (g) is partnered with Gln-1773 (e) on the other helix, which cannot form an ionic interaction (Fig. 6E). However, within the chain it can make an ionic interaction with Lys-1771. Its mutation to Lys would result in a repulsive interaction with Lys-1771 (Fig. 6E), but introduce a novel ionic interchain bonding with Glu-1772 (d) emerging from the coiled-coil seam. This might be expected to disfavor helix stability, in contrast to our observation that this mutation increases the helical content of a weakly helical 7H peptide. Thus, the mechanism of disease by these apparently benign mutations remains obscure.
R1500W causes DCM rather than HCM, albeit with a late onset (age 55+ (17)). We found that this mutation abolished the already low helical content of a 7H peptide. R1500W could decrease α-helical content through the loss of a potential attractive force between Arg-1500 (f position) and Glu-1496 (b) (see Fig. 6C). This mutation also replaces a polar residue with a bulky hydrophobic one, in an “f” position in the heptad repeat in which residues are usually polar. In the tail of wild type β-MHC, tryptophan is not found in any of the f positions in the heptad repeats. Introducing a bulky hydrophobic residue out in solution as a result of this mutation might be expected to affect molecular structure. Previous work has shown that this mutation decreases the Tm of LMM (by 2.3 °C) and decreases assembly into filaments in vitro (as observed by light scattering) (23). However, we found that this mutation had little effect on sarcomeric incorporation in vivo.
Exchange of Myosin in Thick Filaments Is Quicker than Turnover
The rate of exchange of eGFP MHC into myofibrils that we observed in FRAP experiments is much faster than the slow rate of turnover of MHC protein in the beating heart. The half-life for myosin turnover is long, recently measured as ∼15 days in hearts from rats (45). Similarly, exchange rates for several Z-disc proteins measured using FRAP were also found to be much faster than the turnover rates for these proteins (46). A rapid exchange rate would enable newly synthesized MHC to be rapidly and efficiently exchanged into the thick filament within the muscle sarcomere, without disassembling the existing myofibrils, thus maintaining force output.
Our experiments with N1327K showed that this mutation increased exchange rates. We think it is likely that the other mutations in the rod domain of myosin that result in HCM could also affect exchange rates. This change in exchange rates could result from the reduction in local helical stability. This suggests the possibility that in patients harboring mutations in the myosin rod, disease results from a defective exchange/turnover process, which has the potential to decrease force output in the heart. Interestingly, this has some analogies with skeletal muscle diseases such as Laing distal myopathy, a disease of skeletal muscle that also involves mutations in the rod portion of myosin, and results in muscle weakness (47). Defective myosin is known to accumulate outside the muscle sarcomeres in this disease. It is also possible that the mutation also affects the 6–10 S equilibrium, although it is unknown whether cardiac muscle myosin adopts a compact conformation in vivo. A partially compact conformer has been observed in vitro (48).
We have reported here, for the first time, that eGFP-MHC can incorporate normally into muscle sarcomeres, without affecting contraction, and that eGFP-MHC can exchange into muscle sarcomeres (half-time of about 30 min). This is the first time that the behavior of a full-length eGFP-MHC has been studied in adult cardiomyocytes and that it has been directly demonstrated the myosin can exchange into thick filaments. Because the thick filament contains 294 myosin molecules (49), the measured FRAP half-time suggests a molecule of myosin exchanges into a thick filament every 10 s on average, making the thick filament a more dynamic assembly than is generally perceived.
Our approach of expressing eGFP-tagged full-length MHC in adult rat cardiomyocytes using an adenoviral delivery system has allowed us to measure the dynamic behavior of myosin in live cells as well as to directly investigate the impact of HCM mutations in vivo. By contrast, previous studies have used a GFP-tagged myosin tail (50, 51), or untagged MHC (52), or expressed the eGFP-MHC in neonatal rather than adult cardiomyocytes (53). This methodology should have wide applicability to gaining understanding of the mechanism of cardiomyopathies derived from any myofibrillar protein.
This work was supported in part by British Heart Foundation Grant PG/07/061/23277 and Biotechnology and Biological Sciences Research Council Grant BB/I007423/1 (to M. P., P. J. K., and M. W.).

This article contains supplemental Figs. 1 and 2, Tables 1 and 2, and additional references.
- HCM
- hypertrophic cardiomyopathy
- FRAP
- fluorescence recovery after photobleaching
- DCM
- dilated cardiomyopathy
- eGFP
- enhanced GFP
- LMM
- light meromyosin
- MHC
- myosin heavy chain.
REFERENCES
- 1. Parry D. A., Fraser R. D., Squire J. M. (2008) Fifty years of coiled-coils and α-helical bundles: a close relationship between sequence and structure. J. Struct. Biol. 163, 258–269 [DOI] [PubMed] [Google Scholar]
- 2. Offer G., Sessions R. (1995) Computer modelling of the α-helical coiled coil: packing of side-chains in the inner core. J. Mol. Biol. 249, 967–987 [DOI] [PubMed] [Google Scholar]
- 3. Offer G. (1990) Skip residues correlate with bends in the myosin tail. J. Mol. Biol. 216, 213–218 [DOI] [PubMed] [Google Scholar]
- 4. Atkinson S. J., Stewart M. (1992) Molecular interactions in myosin assembly. Role of the 28-residue charge repeat in the rod. J. Mol. Biol. 226, 7–13 [DOI] [PubMed] [Google Scholar]
- 5. Cohen C., Parry D. A. (1998) A conserved C-terminal assembly region in paramyosin and myosin rods. J. Struct. Biol. 122, 180–187 [DOI] [PubMed] [Google Scholar]
- 6. Geisterfer-Lowrance A. A., Kass S., Tanigawa G., Vosberg H. P., McKenna W., Seidman C. E., Seidman J. G. (1990) A molecular basis for familial hypertrophic cardiomyopathy: a β cardiac myosin heavy chain gene missense mutation. Cell 62, 999–1006 [DOI] [PubMed] [Google Scholar]
- 7. Maron B. J., Seidman C. E., Ackerman M. J., Towbin J. A., Maron M. S., Ommen S. R., Nishimura R. A., Gersh B. J. (2009) How should hypertrophic cardiomyopathy be classified? What's in a name? Dilemmas in nomenclature characterizing hypertrophic cardiomyopathy and left ventricular hypertrophy. Circ. Cardiovasc. Genet. 2, 81–85 [DOI] [PubMed] [Google Scholar]
- 8. Maron B. J., Chaitman B. R., Ackerman M. J., Bayés de Luna A., Corrado D., Crosson J. E., Deal B. J., Driscoll D. J., Estes N. A., 3rd, Araújo C. G., Liang D. H., Mitten M. J., Myerburg R. J., Pelliccia A., Thompson P. D., Towbin J. A., Van Camp S. P., Working Groups of the American Heart Association Committee on Exercise, Cardiac Rehabilitation, and Prevention, Councils on Clinical Cardiology and Cardiovascular Disease in the Young (2004) Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 109, 2807–2816 [DOI] [PubMed] [Google Scholar]
- 9. Maron B. J. (2010) The 2009 international hypertrophic cardiomyopathy summit. Am. J. Cardiol. 105, 1164–1168 [DOI] [PubMed] [Google Scholar]
- 10. McNally E. M. (2002) β-Myosin heavy chain gene mutations in familial hypertrophic cardiomyopathy: the usual suspect? Circ. Res. 90, 246–247 [PubMed] [Google Scholar]
- 11. Ho C. Y. (2012) Hypertrophic cardiomyopathy in 2012. Circulation 125, 1432–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maron B. J. (2002) Hypertrophic cardiomyopathy: a systematic review. JAMA 287, 1308–1320 [DOI] [PubMed] [Google Scholar]
- 13. Taylor M. R., Carniel E., Mestroni L. (2006) Cardiomyopathy, familial dilated. Orphanet. J. Rare Dis. 1, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu Q., Dewey S., Nguyen S., Gomes A. V. (2010) Malignant and benign mutations in familial cardiomyopathies: insights into mutations linked to complex cardiovascular phenotypes. J. Mol. Cell Cardiol. 48, 899–909 [DOI] [PubMed] [Google Scholar]
- 15. Blair E., Redwood C., de Jesus Oliveira M., Moolman-Smook J. C., Brink P., Corfield V. A., Ostman-Smith I., Watkins H. (2002) Mutations of the light meromyosin domain of the β-myosin heavy chain rod in hypertrophic cardiomyopathy. Circ. Res. 90, 263–269 [DOI] [PubMed] [Google Scholar]
- 16. Hougs L., Havndrup O., Bundgaard H., Køber L., Vuust J., Larsen L. A., Christiansen M., Andersen P. S. (2005) One third of Danish hypertrophic cardiomyopathy patients with MYH7 mutations have mutations (corrected) in MYH7 rod region. Eur. J. Hum. Genet. 13, 161–165 [DOI] [PubMed] [Google Scholar]
- 17. Kärkkäinen S., Heliö T., Jääskeläinen P., Miettinen R., Tuomainen P., Ylitalo K., Kaartinen M., Reissell E., Toivonen L., Nieminen M. S., Kuusisto J., Laakso M., Peuhkurinen K. (2004) Two novel mutations in the β-myosin heavy chain gene associated with dilated cardiomyopathy. Eur. J. Heart Fail. 6, 861–868 [DOI] [PubMed] [Google Scholar]
- 18. Perrot A., Schmidt-Traub H., Hoffmann B., Prager M., Bit-Avragim N., Rudenko R. I., Usupbaeva D. A., Kabaeva Z., Imanov B., Mirrakhimov M. M., Dietz R., Wycisk A., Tendera M., Gessner R., Osterziel K. J. (2005) Prevalence of cardiac β-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J. Mol. Med. 83, 468–477 [DOI] [PubMed] [Google Scholar]
- 19. Richard P., Charron P., Carrier L., Ledeuil C., Cheav T., Pichereau C., Benaiche A., Isnard R., Dubourg O., Burban M., Gueffet J. P., Millaire A., Desnos M., Schwartz K., Hainque B., Komajda M. (2003) Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107, 2227–2232 [DOI] [PubMed] [Google Scholar]
- 20. Van Driest S. L., Jaeger M. A., Ommen S. R., Will M. L., Gersh B. J., Tajik A. J., Ackerman M. J. (2004) Comprehensive analysis of the β-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 44, 602–610 [DOI] [PubMed] [Google Scholar]
- 21. Waldmüller S., Freund P., Mauch S., Toder R., Vosberg H. P. (2002) Low-density DNA microarrays are versatile tools to screen for known mutations in hypertrophic cardiomyopathy. Hum. Mutat. 19, 560–569 [DOI] [PubMed] [Google Scholar]
- 22. Armel T. Z., Leinwand L. A. (2010) A mutation in the β-myosin rod associated with hypertrophic cardiomyopathy has an unexpected molecular phenotype. Biochem. Biophys. Res. Com. 391, 352–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Armel T. Z., Leinwand L. A. (2010) Mutations at the same amino acid in myosin that cause either skeletal or cardiac myopathy have distinct molecular phenotypes. J. Mol. Cell Cardiol. 48, 1007–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miller G., Maycock J., White E., Peckham M., Calaghan S. (2003) Heterologous expression of wild-type and mutant β-cardiac myosin changes the contractile kinetics of cultured mouse myotubes. J. Physiol. 548, 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCrossan Z. A., Billeter R., White E. (2004) Transmural changes in size, contractile and electrical properties of SHR left ventricular myocytes during compensated hypertrophy. Cardiovasc. Res. 63, 283–292 [DOI] [PubMed] [Google Scholar]
- 26. Van der Ven P. F., Ehler E., Perriard J. C., Fürst D. O. (1999) Thick filament assembly occurs after the formation of a cytoskeletal scaffold. J. Muscle Res. Cell Motil. 20, 569–579 [DOI] [PubMed] [Google Scholar]
- 27. Maggs A. M., Taylor-Harris P., Peckham M., Hughes S. M. (2000) Evidence for differential post-translational modifications of slow myosin heavy chain during murine skeletal muscle development. J. Muscle Res. Cell Motil. 21, 101–113 [DOI] [PubMed] [Google Scholar]
- 28. Picard B., Barboiron C., Chadeyron D., Jurie C. (2011) Protocol for high-resolution electrophoresis separation of myosin heavy chain isoforms in bovine skeletal muscle. Electrophoresis 32, 1804–1806 [DOI] [PubMed] [Google Scholar]
- 29. Leach R. N., Desai J. C., Orchard C. H. (2005) Effect of cytoskeleton disruptors on L-type Ca channel distribution in rat ventricular myocytes. Cell Calcium 38, 515–526 [DOI] [PubMed] [Google Scholar]
- 30. Snapp E. L., Altan N., Lippincott-Schwartz J. (2003) Measuring protein mobility by photobleaching GFP chimeras in living cells. Curr. Protoc. Cell Biol. Chapter 21, Unit 21.1 [DOI] [PubMed] [Google Scholar]
- 31. Peckham M., Knight P. J. (2009) When a predicted coiled coil is really a single α-helix, in myosins and other proteins. Soft Matter 5, 2493–2503 [Google Scholar]
- 32. Greenfield N., Fasman G. D. (1969) Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 8, 4108–4116 [DOI] [PubMed] [Google Scholar]
- 33. Greenfield N. J. (2006) Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat. Protoc. 1, 2527–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wenderoth M. P., Eisenberg B. R. (1987) Incorporation of nascent myosin heavy chains into thick filaments of cardiac myocytes in thyroid-treated rabbits. J. Cell Biol. 105, 2771–2780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Q., Moncman C. L., Winkelmann D. A. (2003) Mutations in the motor domain modulate myosin activity and myofibril organization. J. Cell Sci. 116, 4227–4238 [DOI] [PubMed] [Google Scholar]
- 36. Locher M. R., Razumova M. V., Stelzer J. E., Norman H. S., Moss R. L. (2011) Effects of low-level α-myosin heavy chain expression on contractile kinetics in porcine myocardium. Am. J. Physiol. Heart Circ. Physiol. 300, H869–H878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baboolal T. G., Sakamoto T., Forgacs E., White H. D., Jackson S. M., Takagi Y., Farrow R. E., Molloy J. E., Knight P. J., Sellers J. R., Peckham M. (2009) The SAH domain extends the functional length of the myosin lever. Proc. Natl. Acad. Sci. U.S.A. 106, 22193–22198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Steinmetz M. O., Jelesarov I., Matousek W. M., Honnappa S., Jahnke W., Missimer J. H., Frank S., Alexandrescu A. T., Kammerer R. A. (2007) Molecular basis of coiled-coil formation. Proc. Natl. Acad. Sci. U.S.A. 104, 7062–7067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee D. L., Lavigne P., Hodges R. S. (2001) Are trigger sequences essential in the folding of two-stranded α-helical coiled-coils? J. Mol. Biol. 306, 539–553 [DOI] [PubMed] [Google Scholar]
- 40. Willis M. S., Patterson C. (2013) Proteotoxicity and cardiac dysfunction–Alzheimer's disease of the heart? New Engl. J. Med. 368, 455–464 [DOI] [PubMed] [Google Scholar]
- 41. McLachlan A. D., Karn J. (1982) Periodic charge distributions in the myosin rod amino acid sequence match cross-bridge spacings in muscle. Nature 299, 226–231 [DOI] [PubMed] [Google Scholar]
- 42. Flashman E., Watkins H., Redwood C. (2007) Localization of the binding site of the C-terminal domain of cardiac myosin-binding protein-C on the myosin rod. Biochem. J. 401, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Obermann W. M., Gautel M., Weber K., Fürst D. O. (1997) Molecular structure of the sarcomeric M band: mapping of titin and myosin binding domains in myomesin and the identification of a potential regulatory phosphorylation site in myomesin. EMBO J. 16, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Obermann W. M., van der Ven P. F., Steiner F., Weber K., Fürst D. O. (1998) Mapping of a myosin-binding domain and a regulatory phosphorylation site in M-protein, a structural protein of the sarcomeric M band. Mol. Biol. Cell 9, 829–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Papageorgopoulos C., Caldwell K., Schweingrubber H., Neese R. A., Shackleton C. H., Hellerstein M. (2002) Measuring synthesis rates of muscle creatine kinase and myosin with stable isotopes and mass spectrometry. Anal. Biochem. 309, 1–10 [DOI] [PubMed] [Google Scholar]
- 46. Sanger J. W., Wang J., Holloway B., Du A., Sanger J. M. (2009) Myofibrillogenesis in skeletal muscle cells in zebrafish. Cell Motil. Cytoskeleton 66, 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meredith C., Herrmann R., Parry C., Liyanage K., Dye D. E., Durling H. J., Duff R. M., Beckman K., de Visser M., van der Graaff M. M., Hedera P., Fink J. K., Petty E. M., Lamont P., Fabian V., Bridges L., Voit T., Mastaglia F. L., Laing N. G. (2004) Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause Laing early-onset distal myopathy (MPD1). Am. J. Hum. Genet. 75, 703–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jung H. S., Komatsu S., Ikebe M., Craig R. (2008) Head-head and head-tail interaction: a general mechanism for switching off myosin II activity in cells. Mol. Biol. Cell 19, 3234–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Craig R., Offer G. (1976) Axial arrangement of crossbridges in thick filaments of vertebrate skeletal muscle. J. Mol. Biol. 102, 325–332 [DOI] [PubMed] [Google Scholar]
- 50. Buvoli M., Buvoli A., Leinwand L. A. (2012) Effects of pathogenic proline mutations on myosin assembly. J. Mol. Biol. 415, 807–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vandenboom R., Herron T., Favre E., Albayya F. P., Metzger J. M. (2011) Gene transfer, expression, and sarcomeric incorporation of a headless myosin molecule in cardiac myocytes: evidence for a reserve in myofilament motor function. Am. J. Physiol. Heart Circ. Physiol. 300, H574–H582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Herron T. J., Vandenboom R., Fomicheva E., Mundada L., Edwards T., Metzger J. M. (2007) Calcium-independent negative inotropy by β-myosin heavy chain gene transfer in cardiac myocytes. Circ. Res. 100, 1182–1190 [DOI] [PubMed] [Google Scholar]
- 53. Becker K. D., Gottshall K. R., Hickey R., Perriard J. C., Chien K. R. (1997) Point mutations in human β cardiac myosin heavy chain have differential effects on sarcomeric structure and assembly: an ATP binding site change disrupts both thick and thin filaments, whereas hypertrophic cardiomyopathy mutations display normal assembly. J. Cell Biol. 137, 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]