

Background: The β4GalT7 enzyme participates in the synthesis of the linker tetrasaccharide of proteoglycans.
Results: This study for the first time reveals the molecular interactions between a glycosyltransferase and its donor and acceptor substrates bound together in the same crystal structure.
Conclusion: The enzyme follows an SN2 type catalytic mechanism.
Significance: This study offers insight into substrate specificity and enzyme catalytic mechanism.
Keywords: Crystal Structure, Enzyme Mechanisms, Glycosaminoglycan, Glycosyltransferases, Protein Structure, Proteoglycan Synthesis
Abstract
The β-1,4-galactosyltransferase 7 (β4GalT7) enzyme is involved in proteoglycan synthesis. In the presence of a manganese ion, it transfers galactose from UDP-galactose to xylose on a proteoglycan acceptor substrate. We present here the crystal structures of human β4GalT7 in open and closed conformations. A comparison of these crystal structures shows that, upon manganese and UDP or UDP-Gal binding, the enzyme undergoes conformational changes involving a small and a long loop. We also present the crystal structures of Drosophila wild-type β4GalT7 and D211N β4GalT7 mutant enzymes in the closed conformation in the presence of the acceptor substrate xylobiose and the donor substrate UDP-Gal, respectively. To understand the catalytic mechanism, we have crystallized the ternary complex of D211N β4GalT7 mutant enzyme in the presence of manganese with the donor and the acceptor substrates together in the same crystal structure. The galactose moiety of the bound UDP-Gal molecule forms seven hydrogen bonds with the protein molecule. The nonreducing end of the xylose moiety of xylobiose binds to the hydrophobic acceptor sugar binding pocket created by the conformational changes, whereas its extended xylose moiety forms hydrophobic interactions with a Tyr residue. In the ternary complex crystal structure, the nucleophile O4 oxygen atom of the xylose molecule is found in close proximity to the C1 and O5 atoms of the galactose moiety. This is the first time that a Michaelis complex of a glycosyltransferase has been described, and it clearly suggests an SN2 type catalytic mechanism for the β4GalT7 enzyme.
Introduction
Glycosaminoglycans (GAGs)2 are linker chains of monosaccharides, including chondroitin/dermatan sulfate, heparin, and heparan sulfate, that are assembled onto serine residues of proteins known as proteoglycans (PGs) (1). PGs are the major components of the extracellular matrix, and they are involved in various physiological processes, including cellular proliferation and differentiation, inflammatory responses, and wound healing (2–4). Despite their complexity, all GAGs have the same core tetrasaccharide in their stem, GlcAβ1,3Galβ1,3Galβ1,4Xylβ1-O-Ser (1).
Because aberrant PG biosynthesis has been associated with cancer progression and metastasis, using compounds that target enzymes involved in the core tetrasaccharide assembly may be a beneficial method for halting cancer growth (2, 5, 6). For example, hydrophobic xylose analogues, xylopyranosides, were shown to prime GAG synthesis and act as decoy acceptors for human β-1,4-galactosyltransferase 7 (β4GalT7), with an antiproliferative effect (6). In the presence of a manganese ion, β4GalT7 catalyzes the second step of the core tetrasaccharide assembly using UDP-galactose as a donor substrate (7). β4GalT7 resides in the Golgi apparatus and is predicted to be a type II transmembrane glycoprotein comprising a short N-terminal cytoplasmic tail, a transmembrane segment, and a stem region followed by a soluble catalytic domain (1, 7). Several point mutations in the catalytic domain are associated with Ehlers-Danlos syndrome (EDS), which is characterized by frequent joint dislocation, early onset arthritis, and abnormal wound healing (8, 9). The EDS β4GalT7 catalytic domain mutants are either folding-defective or only partially active, and currently there is no effective therapy for treating EDS (8, 9).
An earlier study on the crystal structure of β4GalT7 from Drosophila melanogaster showed that the enzyme undergoes conformational changes upon binding with manganese and UDP, yet the exact nature of the conformational changes is not known (10). The overall amino acid sequence similarity between Drosophila and human β4GalT7 is 58%, with a high sequence similarity of 67% in the catalytic domain (supplemental Fig. S1). Here, we present the crystal structures of human β4GalT7 in two different conformations. Structural comparisons of both conformations reveal residues involved in a conformational change upon binding with UDP and manganese. Furthermore, we present the crystal structures of the Drosophila β4GalT7 in the closed conformation with the donor and acceptor substrates and also with both substrates bound together to the enzyme. These crystal structures show residues that are undergoing conformational changes and those that are important in determining the specificities of the donor and acceptor sugar molecules. These structures also offer a possible molecular mechanism for EDS.
EXPERIMENTAL PROCEDURES
Expression and Purification of Human β4GalT7
A plasmid used in one of our previous studies, pLgals1-Tev-hum-β4-Gal-T7 (11), was modified by deleting the hexa-His (His6) tag from the plasmid and inserting it at the upstream tobacco etch virus (TEV) protease cleavage site. Also, the sequence encoding the N-terminal stem region of human β4GalT7, corresponding to residues 53–80, was deleted using the QuikChange XL II site-directed mutagenesis kit (Agilent Technologies). The resulting construct was used to express and purify the N-terminally truncated human β4GalT7 (β4GalT7Δ81), as described previously (supplemental Fig. S2) (11).
Crystallization and Structure Determination
The purified β4GalT7Δ81 (15 mg/ml) was readily crystallized using the hanging drop vapor diffusion method with 100 mm imidazole (pH 6.5) and 750 mm sodium acetate as a reservoir solution in the presence of 5 mm MnCl2 at 18 °C. The addition of MnCl2 was necessary for growing a larger, 0.1–0.2 mm crystal. The tetragonal crystals appeared during week 4 and grew to full size the following week. The tetragonal crystal was soaked in the same pH 6.5 reservoir solution containing 15% 2-methy-2,4-pentanediol (MPD) at 18 °C for 20 h followed by a brief soak in 100 mm Tris (pH 8.0), 750 mm sodium acetate, 15% MPD, 5 mm MnCl2, and 5 mm UDP-Gal at room temperature. This two-step soaking procedure, combined with the pH change, was necessary for lowering the crystal mosaicity and for improving the overall data quality. The diffraction data from a single tetragonal crystal were collected in-house with a Mar345 area detector and processed using HKL2000 (12). The tetragonal crystal diffracted to a maximum of 2.05 Å resolution, and because of the high similarity (73%) of amino acid sequences between human and Drosophila β4GalT7 (supplemental Fig. S1), the structure was solved by molecular replacement using, the Drosophila β4GalT7 crystal structure (Protein Data Bank (PDB) ID 3LW6) as a search model (10) without the manganese ion, the UDP molecule, and a region encompassing residues 252–272, which includes the long loop and a short α-helical segment. Due to the presence of a disulfide bond in the loop region of Drosophila β4GalT7 (10), the loop deletion from the model structure was necessary for finding the solution by molecular replacement using PHENIX (13). In the tetragonal crystal, there are two β4GalT7 molecules in the asymmetric unit related by a noncrystallographic, two-fold symmetry. Missing residues in the N-terminal region, spanning residues Pro81 to Ser89, and restriction sites containing Gly-Ser-Asp-Ile were built based on the difference electron density map using the Coot program (14). The electron density corresponding to a long loop region, encompassing residues Asp260 to Gln278 in one β4GalT7 molecule and residues His259 to Asp282 in the other molecule, was absent; therefore, those residues were not built. One manganese ion and one UDP molecule are located in the catalytic pocket of each protein molecule. Although we used UDP-Gal in the crystal-soaking solution, only a UDP moiety could be located in the electron density maps. The refinement was carried out using PHENIX with two manganese ions, two UDP molecules, solvent, and water. The data collection and refinement statistics are given in Table 1. The low backbone root mean square deviation (r.m.s.d.) of only 0.27 Å between the two molecules indicates that the overall structure of the two molecules is essentially identical. The most complete model includes the N-terminal restriction site, residues Pro81 to His259, and Phe279 to Ser327. This model was used to assign the secondary structures by DSSP (15), and the figures were made using PyMOL, a molecular graphics program (16).
TABLE 1.
Single crystal x-ray data collection and refinement statistics
The values in the parentheses correspond to their high-resolution shell. The high R-value was noted because 10% of the total residues were missing. This is also due to weak reflection arising from the presence of a strong translational pseudosymmetry. The statistics from the Ramachandran map were calculated using the PROCHECK program, which is a part of the CCP4i package.
| Human β4GalT7 |
Drosophila β4GalT7 (xylobiose complex) |
Drosophila D211N-β4GalT7 |
|||
|---|---|---|---|---|---|
| Open conformation | Closed conformation | UDP-Gal complex | Xylobiose and UDP-Gal complex | ||
| Crystal data | |||||
| Unit cell (Å) | a = b = 125.2, c = 87.3 | a = 67.1, b = 121.5, c = 97.1, β = 95.1 | a = b = 81.8, c = 133.1 | a = b = 82.1, c = 133.4 | a = b = 82.68, c = 131.5 |
| Space group | P41212 | P21 | P43212 | P43212 | P43212 |
| Data collection | |||||
| Resolution (Ä) | 2.1 | 2.3 | 2.4 | 2.0 | 2.2 |
| Unique reflections | 44,450 | 66,815 | 15,814 | 27,652 | 24,123 |
| Data redundancy | 13.1 (11.5) | 4.4 (3.7) | 7.6 (7.0) | 11.2 (6.2) | 9.6 (7.4) |
| Completeness (%) | 99 (94) | 97 (83) | 95 (77) | 97 (85) | 99.9 (99.7) |
| 1/σ(I) | 36.5 (4.6) | 13.8 (2.3) | 21.2 (2.0) | 36.5 (3.2) | 24.7 (2.2) |
| Rsym | 0.063 (0.49) | 0.091 (0.52) | 0.074 (0.65) | 0.057 (0.35) | 0.077 (0.66) |
| Refinement | |||||
| Rfinal | 0.216 (0.24) | 0.193 (0.28) | 0.183 (0.28) | 0.181 (0.22) | 0.188 (0.28) |
| Rfree | 0.27 (0.31) | 0.24 (0.36) | 0.22 (0.33) | 0.20 (0.27) | 0.21 (0.32) |
| r.m.s.d. | |||||
| Bond length (Å) | 0.022 | 0.004 | 0.014 | 0.006 | 0.007 |
| Bond angle (°) | 1.930 | 0.950 | 1.561 | 1.097 | 1.137 |
| Ramachandran map | |||||
| Core region | 89.1 | 87.0 | 86.9 | 84.8 | 83.6 |
| Allowed region | 10.4 | 11.9 | 12.1 | 14.7 | 15.4 |
| Generously allowed | 0.5 | 0.8 | 0.9 | 0.5 | 0.9 |
| PDB entry | 4IRP | 4IRQ | 4LW6 | 4LW3 | 4M4K |
The monoclinic crystals were obtained within 3 days using 100 mm Tris (pH 8.5) and 8% PEG8000 as a reservoir solution by mixing it with 20 mg/ml β4GalT7Δ81 in 10 mm MES (pH 6.5), 150 mm NaCl, 5 mm MnCl2, and 5 mm UDP-galactose at 18 °C. Before data collection, the crystal was briefly soaked in the same reservoir solution, but with 5% PEG8000 and 15% MPD, at room temperature. The crystal diffracted to a maximum resolution of 2.3 Å, and the same Drosophila β4GalT7 search model as that obtained from the tetragonal crystal structure was used for molecular replacement by PHENIX (13). The monoclinic crystal had four β4GalT7 molecules in the asymmetric unit, forming two copies of a dimer. The electron density for the missing residues in the loop region spanning residues 258–284, as well as the eight N-terminal residues Pro81 to Ala88, in the molecular replacement model could be clearly seen in the electron density maps, and therefore the residues were built based on the difference electron density map using the Coot program (14). A Tris buffer molecule, found in all four molecules of the asymmetric unit, was included in the refinement. The refinement was performed as described above. The backbone r.m.s.d. among the four molecules ranged from 0.3 to 0.5 Å, with the most complete model showing clear electron density from the N-terminal residue Pro81 to the C-terminal residue Phe326. Thus, this model was used for the structural comparisons discussed here. The electrostatic potential of the human β4GalT7 structure was obtained using APBS (17) with 0.15 m ion concentrations for +1, +2, −1, and −2 ion species, and the potential was contoured at ± 5 kT/e. The bound UDP, manganese, and Tris were excluded from the potential calculation.
Crystal Structure Determination of Drosophila β4GalT7 with UDP-Gal and Xylobiose
The expression and purification of the catalytic domain of the Drosophila β4GalT7 has been previously described (10). Hanging drop vapor diffusion methods were used to grow crystals with xylobiose from a protein solution containing 10 mg/ml protein, 10 mm MnCl2, 10 mm UDP, and 10 mm xylobiose, with a precipitating solution containing 100 mm HEPES buffer (pH 8.0), 100 mm serinol, 1.0 m NaCl, 5% PEG6000, and 10% MPD. To grow the crystals with UDP-Gal, the D318N β4GalT7 mutant protein was used in the crystallization. The hanging drop vapor diffusion method was use to grow crystals from a protein solution containing 10 mg/ml protein, 10 mm MnCl2, and 10 mm UDP-Gal, with the precipitating solution containing 100 mm HEPES buffer (pH 8.0), 1.0 m NaCl, 5% PEG6000, and 10% MPD. The crystals of Drosophila D211N β4GalT7 protein complex with both the donor and the acceptor substrates were grown using the same condition used to grow the UDP-Gal complex except that 10 mm xylobiose was also added in the crystallization mixture. The single-crystal x-ray data collection statistics are given in Table 1. The crystals were isomorphous to the previously solved Drosophila β4GalT7 crystal structure (10). Therefore, a molecular replacement method without substrates was used as a starting model in the rigid body refinement using REFMAC5 (18). After the initial refinements, the bound substrate molecules were located from difference Fourier maps and included in the refinement using PHENIX (13). The final refinement statistics are given in Table 1.
RESULTS
Structural Comparison of Human β4GalT7 in Its Open and Closed Conformations
The overall structure of human β4GalT7 belongs to a typical GT-A-type glycosyltransferase family (Fig. 1, A and B) (19). In the open conformation crystal structure (Fig. 1A), no electron density was observed for a region encompassing residues 260–278, and only weak backbone electron density could be traced for residues 278–284. In contrast, the same region in the closed conformation had clear electron density corresponding to a long loop, residues 260–285 (Fig. 1B). The appearance of the long loop coincides with the change in the orientation of the Trp224 side chain, from pointing away to pointing toward the binding pocket, forming a hydrogen bond with the β-phosphate oxygen atom of the bound UDP (Fig. 1B). The assignment of these two conformational states is also in accord with the following observations. When the open conformation crystals were grown in the absence of manganese and UDP-Gal and then soaked in a cryoprotecting solution containing manganese and UDP-Gal, clear electron density for manganese and UDP was observed in the structure. This observation suggests that the catalytic pocket is accessible to the solvent; therefore, the structure is named the open conformation. On the other hand, the closed conformation crystal was co-crystallized with MnCl2 and UDP or UDP-Gal. In the crystal structure, the bound manganese and UDP molecule are buried under the long loop of the protein molecule; thus, the structure is designated the closed conformation. In addition to solvent accessibility to the binding pocket, the side chain orientations of catalytic pocket residues Glu227 and Asp228 are different in the open and closed conformations (supplemental Fig. S3). Asp228 is considered to be the catalytic base that binds to the O4 oxygen atom of the acceptor sugar molecule, xylose. Interestingly, in the closed conformation, a Tris molecule is wedged between the UDP and the side chains of Glu227 and Asp228. Due to conformational differences, there is a small difference in the manganese ion coordination observed between these two crystal structures (supplemental Fig. S4, A and B).
FIGURE 1.
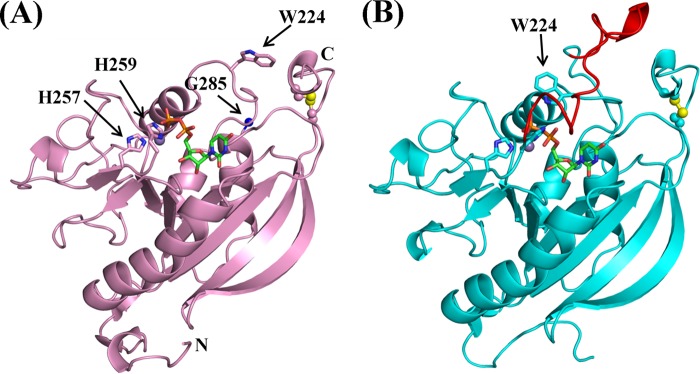
The crystal structures of human β4GalT7 in the open (A) and the closed (B) conformation. A manganese ion and a UDP molecule are shown as a purple sphere and a green-and-orange stick figure, respectively. In the open conformation (A), the electron density was visible up to His259, and the clear electron density appeared again at Gly285 (blue sphere). The His257-X-His259 motif, Trp224, and the N and C termini are indicated. A disulfide bond is shown in a ball-and-stick format, with the yellow spheres indicating sulfur atoms. In B, the long loop region, residues 260–285, invisible in the open conformation, is highlighted in red.
Binding of Xylobiose to Drosophila β4GalT7
The crystal structure of Drosophila β4GalT7 with a xylobiose, (Xylβ1-4Xylβ) molecule, was found in the closed conformation with a manganese ion, UDP molecule, and solvent serinol molecule bound to it. The serinol molecule was wedged between the bound UDP and the xylobiose molecule. The nonreducing end of the xylose moiety of xylobiose was bound to the hydrophobic binding site of the acceptor sugar, created by the Tyr177, Tyr179, Trp207, and Leu209 residues. In the binding site, it forms four hydrogen bonds with the protein molecule. In particular, the O4 hydroxyl group of the bound xylose moiety forms a strong hydrogen bond with the side chain carboxylate oxygen atom of the catalytic base residue Asp211 (Fig. 2B). Only partial electron density for the extended xylose moiety of the bound xylobiose molecule was observed in the crystal structure (supplemental Fig. S5). The extended xylose moiety of the xylobiose forms hydrophobic interactions with Tyr179 (Fig. 2A). A comparison between the xylobiose-bound Drosophila β4GalT7 structure and the open conformation of human β4GalT7 shows a different side chain conformation for the xylose binding Asp228 residue of the human molecule (or Asp211 of Drosophila β4GalT7), suggesting that this difference arises from the presence of the xylose moiety, which forms hydrogen bonds with the side chain carboxyl oxygen atoms of Asp221 residue. Furthermore, a superposition between the xylobiose-bound Drosophila β4GalT7 crystal structure and the closed conformation of human β4GalT7 shows that interactions in the xylobiose binding sites of these two structures are similar. Xylobiose in the Drosophila structure fits into the acceptor binding pocket of human β4GalT7 (supplemental Fig. S5).
FIGURE 2.

Substrate binding to the Drosophila β4GalT7 molecule. The protein molecule is represented by the green graphic, with the interacting residues shown as colored sticks. The substrate molecules UDP-Gal and xylobiose are shown as yellow sticks. The manganese and water molecules are shown as magenta and red spheres. The hydrogen bonds are shown in black dotted lines, and the manganese coordination bond is shown in solid black lines. A, binding of a xylobiose molecule to the Drosophila β4GalT7 molecule in the closed conformation. The acceptor substrate forms four hydrogen bonds with the protein molecule. The C5 and O5 atoms of the acceptor xylose molecule form strong hydrophobic interactions with the aromatic side chain of the Tyr177 residue. Thus, the presence of this residue determines the enzyme acceptor specificity for xylose instead of glucose. The extended xylose moiety forms stacking interactions with the aromatic side chain of the Tyr179 residue. When the PG acceptor substrate binds to the enzyme, with its O-linked β-xylose moiety bound in the acceptor substrate binding site, this extended binding site is likely to facilitate the binding of the protein moiety of the PG acceptor molecule. B, UDP-gal binding to the D211N β4GalT7 mutant protein. The binding of the UDP moiety in the present crystal structure and in the human β4GalT7 closed conformation crystal structure (Fig. 1B) is quite similar (supplemental Fig. S3). The Gal moiety of the bound UDP-Gal forms seven hydrogen bonds with the protein molecule. These hydrogen-bonding interactions are similar to those that occur when UDP-Gal binds to the β4GalT1 molecule. C, the binding of the xylobiose together with UDP-Gal to Drosophila D211N β4GalT7 molecule. The molecular interactions of the substrates with the protein molecule are similar to their individual binding to the enzyme (panels A and B). In the present structure, the O4 oxygen atom of the xylobiose molecule forms a hydrogen bond with the Nϵ2 amino group of the Asn211 residue, which is in contrast to the panel A where the same O4 oxygen atom forms a hydrogen bond with the Oϵ2 atom of the Asp211 residue. The binding of the xylobiose does not seem to affect the binding of the UDP-Gal molecule or vice versa.
Binding of UDP-Gal to Drosophila β4GalT7
The β4GalT7 enzyme exhibits high UDP-Gal hydrolysis activity in the absence of the acceptor substrate. Therefore, to crystallize the β4GalT7 molecule with a UDP-Gal molecule while preventing its hydrolysis, the catalytic base Asp228 of human β4GalT7 or the Asp211 of Drosophila β4GalT7 was mutated to an Asn residue. As expected, the resulting single-mutant enzyme exhibited negligible catalytic activity. The Drosophila D211N β4GalT7 protein readily crystallized in the presence of manganese and UDP-Gal. In the crystal structure, the Drosophila β4GalT7 molecule was found in the closed conformation with a manganese ion and a UDP-Gal molecule bound to the protein molecule (supplemental Fig. S6). The overall protein structure, including the six coordination bonds of the manganese ion and the binding of the UDP moiety of the UDP-Gal, is very similar to the human β4GalT7 crystal structure in the closed conformation (Figs. 1B and 2B). The galactose moiety of the bound UDP-Gal molecule forms seven hydrogen bonds with the catalytic pocket residues Asp145, Glu210, and Asn211 (Fig. 2B). A comparison of the UDP-Gal-bound Drosophila β4GalT7 crystal structure with the human β4GalT7 open conformation crystal structure shows that the galactose binding Glu227 of human β4GalT7 (Glu210 in Drosophila β4GalT7) has a different side chain orientation, suggesting that, in the presence of the Gal moiety, it reorganizes its side chain conformation.
Binding of UDP-Gal and Xylobiose Together to Drosophila β4GalT7
The ternary complex crystals of the Drosophila β4GalT7 were grown using the D211N β4GalT7 mutant enzyme where the catalytic base Asp211 was mutated to Asn residue. The crystals were grown at 4 °C, and the mutant enzyme exhibited no measurable catalytic activity. We tested many crystals and found that all crystals had bound UDP-Gal and xylobiose substrates (supplemental Fig. S7). The best x-ray diffraction data were collected from a single crystal up to 2.2 Å resolution. The binding of both the substrates together to the protein molecule is quite similar to their individually bound crystal structures (Fig. 2C). Thus, it seems that the presence of the acceptor xylose does not seem to influence the binding of the galactose moiety of UDP-Gal or vice versa. The side chain Nϵ2 amino group of Asn211 residue forms a hydrogen bond with the O4 oxygen atom of the nonreducing end xylose molecule. This interaction is quite similar to the xylobiose bound wild-type β4GalT7 crystal structure, where the side chain Oϵ2 atom of Asp211 forms a hydrogen bond with the O4 oxygen atom of the nonreducing end xylose molecule (Fig. 2, B and C). The O4 atom of the xylose molecule is found 3.4 and 3.2 Å away from the C1 and O5 atoms of the galactose moiety of the UDP-Gal molecule, respectively (Fig. 2C). Superposition of the crystal structure of the substrate-bound Drosophila β4GalT7 molecule with the human β4GalT7 molecule in the closed conformation shows a 0.8 Ä r.m.s.d. for the Cα atoms, suggesting that the structures are similar (supplemental Fig. S8). Also, because all the substrate-interacting residues in the Drosophila β4GalT7 molecule are conserved in the human β4GalT7 molecule, the binding of both the substrate molecules to the human β4GalT7 molecule is expected to be similar to their binding to the Drosophila β4GalT7 molecule.
DISCUSSION
β4GalT7 Undergoes Conformational Changes upon Substrate Binding
The present human β4GalT7 crystal structures show that the β4GalT7 enzyme undergoes conformational changes upon binding with manganese and UDP or UDP-Gal. This conformational change involves small and long loops containing residues Gly223-Trp224 and His259 to Gly284, respectively. An earlier isothermal calorimetric study suggested that the binding of manganese and UDP or UDP-Gal molecules induces conformational changes in the β4GalT7 molecule (20). The present study clearly shows the nature of this conformational change at the atomic level. The conformational change also involves reorganization of the Glu227 and Asp228 residues in the catalytic pocket, and it is due to the binding of the galactose and xylose sugar binding (supplemental Fig. S3). Similar conformational changes were observed in other, related, glycosyltransferases, such as β4GalT1 and polypeptidyl-N-acetylgalactosaminy-transferase-2 (ppaGalNAc-T2), in which binding of the donor substrate results in the formation of an acceptor substrate binding site following an ordered, sequential kinetic mechanism (21). Thus, β4GalT7 is also expected to follow the same kinetic mechanism, which involves donor substrate binding followed by the creation of the xylose binding pocket.
The present human β4GalT7 crystal structures also show that the manganese binding site is located at the N-terminal hinge region of the long loop and that the conformational changes also influence the manganese ion coordination, involving the Asp163-X-Asp165 and the His257-X-His259 motifs (supplemental Fig. S4). Upon the conformational changes, those residues are positioned properly to prepare the enzyme for catalysis. A previous mutagenesis study reported that the D165E mutant had a 4-fold increase in the Km for UDP-galactose and a 6-fold increase in the Km for 4-methylumbelliferone-β-d-xylopyranoside as compared with the wild type (22). Although the weaker binding of UDP-galactose observed in the D165E mutant is attributed to an improper coordination of the bound manganese ion, the increase in the Km for the xylose analogue suggests an allosteric communication between the donor and the acceptor binding sites, possibly mediated by the long loop dynamics.
Donor UDP-Gal Substrate Binding
Unlike β4GalT1, β4GalT7 forms an additional hydrogen bond between the side chain hydroxyl group of Tyr177 and the β-phosphate oxygen atom of the bound UDP-Gal molecule (23). A similar hydrogen bond interaction, involving a tyrosine residue and the β-phosphate oxygen atom of UDP-Gal, is observed in the crystal structure of α-1,3-galactosyltransferase. As in the β4GalT7 crystal structure, the galactose moiety of UDP-Gal was not observed in the α-1,3-galactosyltransferase crystal structure unless the catalytic base of the enzyme, Glu317, was mutated to glutamine (24). In contrast to both α-1,3-galactosyltransferase and β4GalT7, the β4GalT1 enzyme is unable to hydrolyze the UDP-Gal molecule in the absence of the acceptor sugar substrate.
The superposition of the UDP-Gal-bound Drosophila D221N β4GalT7 crystal structure with the closed conformation of human β4GalT7 showed that the UDP moieties of these structures overlap, suggesting similar molecular interactions with the bound Gal moiety of the UDP-Gal molecule (supplemental Fig. S6). Thus, in the human enzyme, Glu227 residue is expected to engage in hydrogen bond interactions with the galactose moiety of the UDP-Gal, and the side chain reorientations of this residue, in the open to the closed conformational change, are due to the presence of the Gal moiety of the UDP-Gal molecule (supplemental Figs. S3 and S6).
Acceptor Substrate Xylobiose Binding
The binding of xylobiose to the Drosophila β4GalT7 molecule is comparable with the binding of chitobiose to the β4GalT1 enzyme (25). The nonreducing end of the xylose moiety of xylobiose is the acceptor substrate for β4GalT7, and it is bound to the acceptor binding site, similar to glucose bound to β4GalT1. The O3 and O4 atoms of the acceptor molecule form hydrogen bonds with the side chain of the catalytic base Asp211. Thus, the Asp211 (Asp228 in human) side chain conformational change in the closed conformation is due to the formation of hydrogen bond with the acceptor xylose molecule. Tyr177 determines the acceptor specificity of β4GalT7 as its side chain forms hydrophobic interactions with the C5 and O5 atoms of the acceptor substrate. This is corroborated by a mutagenesis study in which mutating Tyr177 to Ala or Gly alters its acceptor specificity from xylose to glucose, although the specific activity of the mutant enzyme is very poor (data not shown). The hydrophobic environment in the acceptor binding site is formed by two Tyr residues at 177 and 179 (Fig. 2), suggesting that aromatic stacking interaction is a preferred mode for acceptor binding. This is in line with the observation that Km values for the p-nitrophenol-, 4-methylumbelliferone-, and estradiol-β-d-xylosides range from 0.5 to 1 mm as compared with a 16 mm Km value for the xylose monosaccharide (11, 20).
Exploiting the hydrophobic environment in the acceptor binding pocket has been shown to be a promising cancer therapy strategy (6, 26). For instance, a variety of hydrophobic xylosides show antiproliferative activities in human A549 lung carcinoma cells and inhibit proteoglycan synthesis in bovine microvascular endothelial cells (26). GAG synthesis can be primed on these hydrophobic xylosides, and more aromatic rings in xylosides correlate with better GAG priming efficiency (27, 28), low IC50 in A549 cells, and low Km values in in vitro kinetic assays (26). Because similarities exist between the human and Drosophila β4GalT7 xylose binding pocket structures (supplemental Fig. S5), the crystal structures presented in this study provide an opportunity for the development of a structure-based xyloside design approach for cancer therapy.
β4GalT7 binds to Xylβ1-O-Ser on a wide range of protein substrates with various protein surface features, including soluble extracellular matrix, and membrane-spanning glycophosphatidylinositol-anchored proteoglycans (4). Although the xylose binding pocket is hydrophobic, consisting of Tyr194, Tyr196, Tyr199, and Trp224 in the human β4GalT7 enzyme, the region around the xylose binding pocket is highly positively charged (Fig. 3). This charged surface is expected to facilitate the interaction of the enzyme with the acidic residue clusters flanking Ser-Gly repeats found in the xylose attachment sites of various proteoglycan substrates (29). The lack of β-carbon in glycine residues confers structural flexibility to the Ser-Gly repeats in the proteoglycan substrates, thereby facilitating an induced fit between the charged surface of β4GalT7 and the acidic residue clusters of the proteoglycan substrates. Thus, protein shape and charge complementarities are expected to facilitate the interactions between β4GalT7 and proteoglycan substrates.
FIGURE 3.
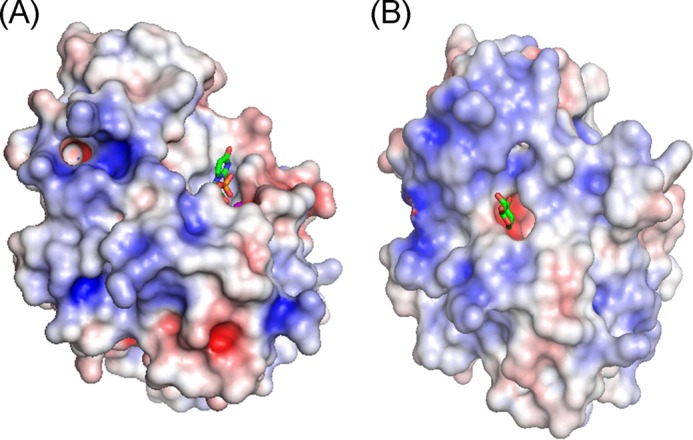
The electrostatic potentials of the open (A) and the closed (B) conformations of human β4GalT7 are shown, with the positively and negatively charged surfaces colored blue and red, respectively. The sticks represent UDP and a modeled xylose molecule in A and B, respectively.
Enzyme Catalytic Mechanism
The present ternary complex between the Drosophila D211N β4GalT7 enzyme with its substrates UDP-Gal and xylobiose and manganese represents a Michaelis complex, and this is the first example of a glycosyltransferase structure in a Michaelis complex. This complex could only be achieved by mutating the catalytic base of the enzyme, Asp211 to Asn211 residue. Because the binding of the xylobiose to the mutant enzyme is quite similar to its binding to the wild-type enzyme, the mutation does not appear to have an effect on the binding of xylobiose molecule. Earlier, the crystal structure of a similar Michaelis complex of β-lactamase mutant enzyme and substrate complex had been reported (30). However, previous crystal structures of glycosyltransferases have been determined either in the presence of substrate analogues or in the presence of a poor donor substrate with the acceptor substrate (31–33). In all these structures, it was not possible to elucidate the actual environment of the nucleophile oxygen atom of the acceptor sugar and the C1 atom of the donor sugar molecules. Although in the present crystal structure the catalytic base Asp residue has been mutated to an Asn residue, the interactions of the Nϵ2 amino group of Asn211 do not seem to alter the binding of the nonreducing end xylose moiety. In β1,4-galactosyltransferase-1, based on the distance between the nucleophile O4 oxygen atom of the acceptor and the O5 ring oxygen atom of the donor substrate, it has been postulated that the O5 ring oxygen may play a role during the catalysis (32, 34). In the present crystal structure, the distance between the nucleophile O4 oxygen atom of the xylose to the galactose ring O5 and C1 atoms is comparable (3.2 and 3.4 Å, respectively); therefore, it is possible that the β4GalT7 enzyme may also follow a very similar SN2-type catalytic mechanism as the β1,4-galactosyltransferase-1 (32, 34).
A Possible Molecular Mechanism for EDS
In the crystal structures of human β4GalT7, the missing and weak electron density from residues 260–284, together with the presence of the Trp224 side chain rotamers in the open conformation (data not shown), suggest structural flexibility in this region. Although the long flexible loop of β4GalT7 and β4GalT1 enzymes have the least protein sequence similarity, they all have high content of basic amino acid residues (35). The fact that the conformational changes of the long flexible loop take place due to the binding of the UDP-Gal to the enzyme, and that the basic amino acid residues on the long flexible loop exhibit molecular interactions with the diphosphate group of the bound UDP-Gal molecule, suggests that the basic amino acid residues in the long flexible loop play an important role in its conformational flexibility and dynamics in these enzymes. One of the EDS mutations, R270C, is located in the middle of the long loop region, and a previous isothermal calorimetric study showed that although the R270C mutation did not affect UDP-galactose binding or the kcat, the Km for a hydrophobic xylose analog, 4-nitrophenyl-β-d-xylose, was increased 6-fold as compared with the wild-type enzyme (36). However, the present crystal structures show that Arg270 engages in neither the donor nor the acceptor sugar binding. Because the acceptor binding site is created by the conformational change on the enzyme, the loss in the acceptor binding in the R270C mutation enzyme might be due to the compromised structural flexibility in the long loop region. In the β4GalT1 enzyme, the conformational flexibility of the enzyme was shown by the limited proteolysis experiments (35, 37). A similar experiment and other biophysical studies are needed with β4GalT7 to prove this hypothesis. Similar to the crystal structure of Drosophila β4GalT7 enzyme, in the human β4GalT7 enzyme, the Ala186 and Leu206 residues are surrounded by hydrophobic residues (supplemental Fig. S9). It has been suggested that the mutations of these residues are expected to disrupt the hydrophobic cores that stabilize the overall fold, thus affecting the catalytic activity of these enzymes (10).
The present study shows that the β4GalT7 enzyme undergoes conformational changes upon binding with the manganese ion and the UDP-Gal molecule, as well as a reorganization of the catalytic pocket residues Glu227 and Asp228. In addition, the study shows the donor and the acceptor substrates binding to the enzyme. It also offers a possible structure-based explanation for EDS. The extended hydrophobic environment created by a cluster of tyrosine residues found at the acceptor binding site could be exploited for the development of effective decoy acceptors for cancer therapy.
Acknowledgments
We thank Dr. Alexander Wlodawer and Mi Li for allowing us to collect data using an in-house x-ray source in the Macromolecular Crystallography Laboratory, Frederick National Laboratory for Cancer Research, Frederick, MD.
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E (to P. K. Q.). This research was supported (in part) by the Intramural Research Program of the National Institutes of Health, NCI, National Institutes of Health.

This article contains supplemental Figs. S1–S10.
- GAG
- glycosaminoglycan
- PG
- proteoglycan
- β4GalT7
- β-1,4-galactosyltransferase 7
- EDS
- Ehlers-Danlos syndrome
- MPD
- 2-methy-2,4-pentanediol
- r.m.s.d.
- root mean square deviation
- UDP-Gal
- α-d-UDP-galactose
- Xyl
- xylobiose.
REFERENCES
- 1. Varki A., Cummings R., Esko J., Freeze H., Hart G., Marth J. (1999) Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 2. Ruoslahti E. (1989) Proteoglycans in cell regulation. J. Biol. Chem. 264, 13369–13372 [PubMed] [Google Scholar]
- 3. Chiodoni C., Colombo M. P., Sangaletti S. (2010) Matricellular proteins: from homeostasis to inflammation, cancer, and metastasis. Cancer Metastasis Rev. 29, 295–307 [DOI] [PubMed] [Google Scholar]
- 4. Bishop J. R., Schuksz M., Esko J. D. (2007) Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 [DOI] [PubMed] [Google Scholar]
- 5. Afratis N., Gialeli C., Nikitovic D., Tsegenidis T., Karousou E., Theocharis A. D., Pavão M. S., Tzanakakis G. N., Karamanos N. K. (2012) Glycosaminoglycans: key players in cancer biology and treatment. FEBS J. 279, 1177–1197 [DOI] [PubMed] [Google Scholar]
- 6. García-García J. F., Corrales G., Casas J., Fernández-Mayoralas A., García-Junceda E. (2011) Synthesis and evaluation of xylopyranoside derivatives as “decoy acceptors” of human β-1,4-galactosyltransferase 7. Mol. Biosyst. 7, 1312–1321 [DOI] [PubMed] [Google Scholar]
- 7. Almeida R., Levery S. B., Mandel U., Kresse H., Schwientek T., Bennett E. P., Clausen H. (1999) Cloning and expression of a proteoglycan UDP-galactose:β-xylose β1,4-galactosyltransferase I. J. Biol. Chem. 274, 26165–26171 [DOI] [PubMed] [Google Scholar]
- 8. Okajima T., Fukumoto S., Furukawa K., Urano T. (1999) Molecular basis for the progeroid variant of Ehlers-Danlos syndrome. J. Biol. Chem. 274, 28841–28844 [DOI] [PubMed] [Google Scholar]
- 9. Seidler D. G., Faiyaz-Ul-Haque M., Hansen U., Yip G. W., Zaidi S. H. E., Teebi A. S., Kiesel L., Götte M. (2006) Defective glycosylation of decorin and biglycan, altered collagen structure, and abnormal phenotype of the skin fibroblasts of an Ehlers-Danlos syndrome patient carrying the novel Arg270Cys substitution in galactosyltransferase I (β4GalT-7). J. Mol. Med. 84, 583–594 [DOI] [PubMed] [Google Scholar]
- 10. Ramakrishnan B., Qasba P. K. (2010) Crystal structure of the catalytic domain of Drosophila β1,4-galactosyltransferase-7. J. Biol. Chem. 285, 15619–15626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pasek M., Boeggeman E., Ramakrishnan B., Qasba P. K. (2010) Galectin-1 as a fusion partner for the production of soluble and folded human β-1,4-galactosyltransferase-T7 in E. coli. Biochem. Biophys. Res. Commun. 394, 679–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 13. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 15. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 16. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.2r3pre, Schrödinger, LLC [Google Scholar]
- 17. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Potterton E., Briggs P., Turkenburg M., Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 19. Lairson L. L., Henrissat B., Davies G. J., Withers S. G. (2008) Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–555 [DOI] [PubMed] [Google Scholar]
- 20. Daligault F., Rahuel-Clermont S., Gulberti S., Cung M. T., Branlant G., Netter P., Magdalou J., Lattard V. (2009) Thermodynamic insights into the structural basis governing the donor substrate recognition by human β1,4-galactosyltransferase 7. Biochem. J. 418, 605–614 [DOI] [PubMed] [Google Scholar]
- 21. Ramakrishnan B., Qasba P. K. (2010) Structure-based evolutionary relationship of glycosyltransferases: a case study of vertebrate β1,4-galactosyltrasferase, invertebrate β1,4-N-acetylgalactosaminyltransferase and α-polypeptidyl-N-acetylgalactosaminyltransferase. Curr. Opin. Struct. Biol. 20, 536–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Talhaoui I., Bui C., Oriol R., Mulliert G., Gulberti S., Netter P., Coughtrie M. W. H., Ouzzine M., Fournel-Gigleux S. (2010) Identification of key functional residues in the active site of human β1,4-galactosyltransferase 7. J. Biol. Chem. 285, 37342–37358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramakrishnan B., Balaji P. V., Qasba P. K. (2002) Crystal structure of β1,4-galactosyltransferase complex with UDP-Gal reveals an oligosaccharide acceptor binding site. J. Mol. Biol. 318, 491–502 [DOI] [PubMed] [Google Scholar]
- 24. Boix E., Swaminathan G. J., Zhang Y., Natesh R., Brew K., Acharya K. R. (2001) Structure of UDP complex of UDP-galactose:β-galactoside-α-1,3-galactosyltransferase at 1.53-Å resolution reveals a conformational change in the catalytically important C terminus. J. Biol. Chem. 276, 48608–48614 [DOI] [PubMed] [Google Scholar]
- 25. Ramakrishnan B., Boeggeman E., Qasba P. K. (2004) Effect of the Met344His mutation on the conformational dynamics of bovine β-1,4-galactosyltransferase: crystal structure of the Met344His mutant in complex with chitobiose. Biochemistry 43, 12513–12522 [DOI] [PubMed] [Google Scholar]
- 26. Raman K., Ninomiya M., Nguyen T. K., Tsuzuki Y., Koketsu M., Kuberan B. (2011) Novel glycosaminoglycan biosynthetic inhibitors affect tumor-associated angiogenesis. Biochem. Biophys. Res. Commun. 404, 86–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fritz T. A., Lugemwa F. N., Sarkar A. K., Esko J. D. (1994) Biosynthesis of heparin sulfate on β-d-xylosides depends on aglycone structure. J. Biol. Chem. 269, 300–307 [PubMed] [Google Scholar]
- 28. Lugemwa F. N., Esko J. D. (1991) Estradiol β-d-xyloside, an efficient primer for heparin sulfate biosynthesis. J. Biol. Chem. 266, 6674–6677 [PubMed] [Google Scholar]
- 29. Esko J. D., Zhang L. (1996) Influence of core protein sequence on glycosaminoglycan assembly. Curr. Opin. Struct. Biol. 6, 663–670 [DOI] [PubMed] [Google Scholar]
- 30. Tremblay L. W., Xu H., Blanchard J. S. (2010) Structures of the Michaelis complex (1.2 Ä) and the covalent acyl intermediate (2.0 Ä) of cefamandole bound in the active sites of the Mycobacterium tuberculosis β-lactamase K73A and E166A mutants. Biochemistry 49, 9685–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Persson K., Ly H. D., Dieckelmann M., Wakarchuk W. W., Withers S. G., Strynadka N. C. J. (2001) Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex. Nat. Struct. Biol. 8, 166–175 [DOI] [PubMed] [Google Scholar]
- 32. Ramakrishnan B., Ramasamy V., Qasba P. K. (2006) Structural snapshots of β-1,4-galactosyltransferase-I along the kinetic pathway. J. Mol. Biol. 357, 1619–1633 [DOI] [PubMed] [Google Scholar]
- 33. Lazarus M. B., Jiang J., Gloster T. M., Zandberg W. F., Whitworth G. E., Vocadlo D. J., Walker S. (2012) Structural snapshots of the reaction coordinate for O-GlcNAc transferase. Nat Chem Biol. 8, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krupicka M., Tvaroska I. (2009) Hybrid Quantum mechanical/molecular mechanical investigation of the β-1,4-galactosyltransferase-I mechanism. J. Phys. Chem. B 113, 11314–11319 [DOI] [PubMed] [Google Scholar]
- 35. Gunasekaran K., Ma B., Ramakrishnan B., Qasba P. K., Nussinov R. (2003) Interdependence of backbone flexibility, residue conservation, and enzyme function: a case study on β1,4-galactosyltransferase-I. Biochemistry 42, 3674–3687 [DOI] [PubMed] [Google Scholar]
- 36. Rahuel-Clermont S., Daligault F., Piet M. H., Gulberti S., Netter P., Branlant G., Magdalou J., Lattard V. (2010) Biochemical and thermodynamic characterization of mutated β1,4-galactosyltransferase 7 involved in the progeroid form of the Ehlers-Danlos syndrome. Biochem. J. 432, 303–311 [DOI] [PubMed] [Google Scholar]
- 37. Ramasamy V., Ramakrishnan B., Boeggeman E., Qasba P. K. (2003) The role of tryptophan 314 in the conformational changes of β1,4-galactosyltransferase-I. J. Mol. Biol. 331, 1065–1076 [DOI] [PubMed] [Google Scholar]