

Abstract
The female genital epithelium plays a protective role against invading pathogens; however, sexual transmission of human immunodeficiency virus type 1 (HIV-1) still occurs in healthy women. To model virus–cell interactions in this barrier during sexual transmission, we studied the uptake and infection of ectocervical and endocervical cell lines with cell-free fluorescent protein-expressing recombinant HIV-1 carrying primary transmitted/founder envelope genes. We observed that a subset of both the ectocervical and endocervical epithelial cells become productively infected with cell-free HIV-1 in a CD4-independent manner. In addition, the ability of the semen-derived enhancer of virus infection (SEVI) to enhance virus-epithelial cell interactions was studied. This infection is increased approximately 2–5 fold when inoculation occurs in the presence of SEVI fibrils. Once infected, the epithelial cells are capable of transmitting the virus to target CD4 T cells in coculture in a contact-dependent manner that uses conventional CD4- and coreceptor-dependent entry. The infection of target CD4 T cells only occurs when de novo HIV-1 is produced within the epithelial cells. These findings suggest that a subset of cervical epithelial cells may be actively involved in establishing a systemic HIV infection and should be a target when designing prevention strategies to protect against HIV-1 sexual transmission.
Keywords: cervical epithelium infection, SEVI, transmitted and founder HIV envelope, coculture
Women aged 15–24 years represent the most at-risk demographic for human immunodeficiency virus type 1 (HIV-1), accounting for approximately 22% of all new infections worldwide [1]. Sexual transmission remains the most common mode of HIV-1 transmission [2], with 1 in every 1000 to 10 000 coital acts resulting in infection in the absence of facilitating factors [3]. Without an effective HIV-1 vaccine, prevention strategies for women to efficiently protect themselves against the sexual transmission of HIV-1 are of vital importance in controlling dissemination of HIV-1. The weak and variable efficacy of recent prophylactic strategies [4–8] underlines the need for a greater understanding of the interaction between HIV-1 and the female genital epithelium during sexual transmission, as well as the factors that affect this interaction.
During sexual transmission of HIV-1, the female genital mucosa is exposed to HIV-1 and endogenous factors found in semen that enhance virus infection [9–11]. One such factor has been identified as semen-derived enhancer of virus (SEVI), a peptide fragment of the abundant seminal protein prostatic acid phosphatase that corresponds to amino acids 248–286 [9]. This peptide fragment can be efficiently assembled into amyloid fibrils after agitation in polar solvents [9]. In vivo, seminal plasma accelerates the formation of these fibrils, and they can be readily detected from human semen samples [11, 12]. SEVI fibrils are proposed to be responsible for the donor-dependent, HIV-enhancing ability of semen by serving as a polycationic bridge that neutralizes negative charges between the HIV-1 virion and target cells and facilitates virion attachment and fusion [9–11, 13–19].
The cervix is composed of stratified squamous (ectocervical) and simple columnar (endocervical) epithelium. It is frequently compromised by sexually transmitted infections (STIs) and has been suggested to be the site of initial HIV-1 infection during sexual transmission [20–22]. Studies in the rhesus macaque model found SIV-infected cells within the lamina propria of the endocervix as early as 3 days following SIV inoculation. In comparison, SIV-positive cells were not detected in the vaginal epithelium until approximately 12 days following virus inoculation [23], suggesting that viral infection may initiate at the cervix and spread to the vagina. Additionally, the endocervix has been implicated in an “outside-in” signaling system that involves the chemokine MIP-3α, plasmacytoid dendritic cells, and C-C chemokine receptor 5 (CCR5)-activating chemokines that recruit CD4+ target cells in close proximity to the epithelial cells during initial SIV infection [24]. While epithelial cells primarily serve a barrier function that excludes the entry of pathogens, some studies suggest that HIV-1 may exploit these cells to gain entry into the body. Cervical epithelial cells have been reported to support HIV-1 transcytosis [25] and the “transinfection” of target CD4+ T cells [26, 27]. While these researchers suggest that the cervical epithelial cells remain refractory to infection, others have demonstrated the susceptibility of tissue and cells from the female reproductive tract to HIV-1 infection [28]. In the current study, ectocervical (Ect1) and endocervical (End1) epithelial cell lines [29] were used to model the interaction of cell-free HIV-1 with the cervical epithelium. These cell lines exhibit the same in vitro phenotype and cytokeratin expression as their primary cell counterparts [29]. They have been used to characterize the immunological response of the genital epithelium, in vitro, to pathogens and genital irritants and reflect in vivo responses [30–32]. Using novel fluorescent protein-expressing HIV-1 constructs that carry lab isolate or transmitted-founder HIV-1 Env, we characterized de novo gene expression and infection following viral challenge of the cervical epithelial cells. We found surprising levels of viral gene expression in the epithelial cells that could be transmitted to CD4+ T cells following cell–cell contact.
METHODS
Cell Culture
Ectocervical (Ect1/E6E7) and endocervical (End1/E6E7) cell lines were obtained from the American Type Culture Collection and grown in cell-specific media as indicated by the manufacturer.
Primary CD4+ T cells were isolated from blood using CD4+ T-cell isolation kit II (Miltenyi Biotec Inc.) and activated with 4 µg/mL phytohemagglutinin and 50 U/mL interleukin-2 for 72 hours in Roswell Park Memorial Institute complete media with penicillin streptomycin and 10% fetal bovine serum.
Virus Description and Preparation
Fluorescent virus particles were derived from recombinant molecular clones, HIV Gag-internal green fluorescent protein (iGFP), HIV Gag-iCherry, and HIV Gag-iGFP-ΔEnv. Plasmid design was described previously [33]. These recombinant clones were used in capture and internalization experiments.
HIV-1 molecular clones designated NL-GI or HIV NL-CI are viruses based on molecular clone NL4-3, which express GFP or mCherry, respectively, in place of the early-transcribed gene, nef, with nef expression restored by the insertion of an internal ribosomal entry site (IRES). These recombinant clones were used in infection experiments. Primary envelope sequences were obtained from the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program (ARP) from Drs B. H. Hahn and J. F. Salazar-Gonzalez: pWITO4160 clone 33 (SVPB18), RHPA4259 clone 36 (SVPB14), Qh0692.42 clone 42 (SVPB6), and PVO.4 clone 4 (SVPB11). Primary envelopes were cloned into HIV NL-CI construct to give rise to variants (NL-CIenvWITO4160 and NL-CIenvRHPA4259) that express envelope genes representing transmitted founder viruses [34]. The full-length, nonfluorescent, transmitted/founder HIV-1 infectious molecular clone that corresponds to envWITO4160 (HIVWITO4160) was obtained through the NIH ARP, Panel of Infectious Molecular Clones, from Dr John Kappes [35].
Transfection of EndoFree DNA into 293 T cells was performed using PolyJet (SignaGen Laboratories). HIV-p24 was measured using enzyme-linked immunosorbent assay following protocol 2 (D7320) by Aalto Bio Reagents Ltd.
TZM Assay
The TZM-bl infectivity assay was performed as described previously [36].
Immunofluorescence and Microscopy
Cells were incubated with HIV Gag-iCherry and stained with wheat germ agglutinin-fluorescein isothiocyanate (FITC) at 1 µg/mL (Invitrogen). Cells were washed, fixed in 4% paraformaldehyde, and mounted using 4,6 diamidino-2-phenylindole-Vectashield mounting medium (Vector Laboratories Inc.). Confocal microscopy was done on a Leica SP5 DMI microscope (MSSM Microscopy SRF). Images were enhanced using AutoQuant X2 Deconvolution software (Media Cybernetics). Post analysis was performed using Volocity 3D image analysis software (PerkinElmer).
Flow cytometry was performed on BD LSR II and BD LSRFortessa.
Reagents
The following inhibitors were used at the following nontoxic concentrations: Leu3A, 0.5 µg/mL; isotype control, 0.5 µg/mL (BD Pharmingen); Tak779, 60 µM; integrase inhibitor 118-D-24, 50 µM; azidothymidine (AZT), 50 µM; Pro20000 (Pro2K), 20 µg/mL; indinavir, 4 µM (NIH ARP); chondroitinase ABC, 2 U/mL; dextran sulfate, 25 µg/mL; iota carrageenan, 100 µg/mL; and heparinase III, 25 U/mL (Sigma-Aldrich). SEVI fibrils were kindly provided by Frank Kirchhoff [9] and used at 10 µg/mL, and anti-CD8 and anti-CD3 antibodies were obtained from Biolegend.
RNA Isolation and Quantitative Polymerase Chain Reaction
RNA was isolated using the RNeasy Mini kit (Qiagen). cDNA synthesis from approximately 1 ng of DNase-treated RNA (Promega) was measured using a high-capacity RNA-to-cDNA kit (Applied Biosystems). Viral RNA was detected using a Taqman gene expression assay and Taqman universal polymerase chain reaction (PCR) master mix on the ABI 7300 Prism real-time PCR instrument (Applied Biosystems). Glyceraldehyde 3-phosphate dehydrogenase and S18 were used as housekeeping genes, and the geometric mean was calculated. HIV-Gag PCR primers were specifically designed for this study (Applied Biosystems): forward-5′-TCTGGCTAACTAGGGAACCCA-3′, reverse-5′-GTGCCCGTCTGTTGTGTGAC-3′. Quantitative PCR (qPCR) plates were run at the Mount Sinai School of Medicine's qPCR Shared Resource Facility.
DNA Isolation and HIV Integration Assay
Genomic DNA was isolated using DNeasy blood and tissue kit (Qiagen) protocol. The integration assay was performed using 70 ng of DNA, following the protocol described elsewhere [37]. Pre-amp PCR was done using AmpliTaq Gold Fast PCR Master Mix (Applied Biosystems) according to the manufacturer's protocol. PCR product was then subjected to nested kinetic PCR (qPCR) using the Gag RNA primer, resulting in the exponential amplification of only the integrated DNA.
Statistics
All experiments were repeated a minimum of 3 times and analyzed using a 2-tailed, unpaired t-test performed using Prism 5 for Mac OS X (GraphPad software, Inc.).
RESULTS
Internalization of Cell-Free HIV-1
To examine the interaction between cervical epithelial cells and cell-free HIV-1 particles, we exposed the cells to fluorescent recombinant cell-free virus. HIV-1 Gag-iGFP and HIV Gag-iCherry are labeled virus particles where the structural protein Gag is fused to a fluorescent protein [33]. Ect1 and End1 cells were treated with R5-tropic cell-free HIV Gag-iGFPenvJRFL or HIV Gag-iCherryenvJRFL at either 4 or 37°C for 2 hours, trypsinized to remove surface-adsorbed virus, and analyzed by flow cytometry. An increase in GFP fluorescence was observed in the HIV-1 Gag-iGFP–incubated samples at 37°C, but not at 4°C, indicating temperature-dependent HIV-1 internalization (Figure 1A). To visualize the distribution of virus in the cervical epithelial cells, confocal microscopy was performed (Figure 1B and 1C). When exposed to cells at 37°C, the HIV Gag-iCherry particles were located in an internal location within the boundaries of the cervical epithelial cell plasma membrane, as defined by wheat germ agglutinin staining (Figure 1C). When incubated with cells at 4°C, virus particles were only observed on the plasma membrane and not internalized (Figure 1B). To determine whether the internalization of virus was a receptor-mediated process, we measured uptake of an Env-deficient HIV-1 mutant, HIV Gag-iGFP-ΔEnv. This mutant virus was not internalized, suggesting that the engagement of Env is required for this process (Figure 1D, End1 not shown). Similar results were seen with End1 cells, but to a lesser extent.
Figure 1.

Cervical epithelial cells internalize cell-free human immunodeficiency virus type 1 (HIV-1) Gag-iGFPJRFL. Ectocervical (Ect1) and endocervical (End1) cells (approximately 150 000 cells/well, 24-well plate) were examined for the ability to internalize cell-free HIV (30 ng/mL p24) in Dulbecco's modified Eagle's medium without serum for duration of virus incubation. A, Internalization of HIV Gag-iGFPJRFL occurs at 37°C but not at 4°C. Ect1 (top) and End1 (bottom) were incubated for 2 hours with HIV Gag-iGFPJRFL at 37°C (blue histogram) or 4°C (green histogram), washed vigorously with phosphate-buffered saline, and trypsinized for 10 minutes at 37°C to remove surface-bound virus. Cells are assessed via flow cytometry for internalization of fluorescent HIV. Internalization of HIV Gag-iGFPJRFL at 37°C was significant, Ect1 P = .005 and End1 P = .003. B, Confocal microscopy of HIV Gag-iCherryJRFL (red) adsorbed to the surface of epithelial cells at 4°C. Plasma membrane was stained with wheat germ agglutinin (green); yellow arrows indicate virus particles. Perpendicular confocal slices through the cell representing xy, xz, and zy planes are shown. Cells were not treated with trypsin prior to analysis. C, Confocal images of internalized HIV Gag-iCherryJRFL particles incubated with cells as in (B), but at 37°C. D, Internalization of fluorescent HIV-1 requires Env. Ect1 cells were incubated for 2 hours with either HIV Gag-iGFPJRFL or HIV Gag-iGFPΔEnv, and trypsinized cells were subjected to flow cytometry. End1 not shown. E, Kinetics of nonfluorescent HIV-1 internalization measured by viral RNA accumulation in cells; quantitative polymerase chain reaction (qPCR) for HIV-Gag RNA within Ect1 or End1 cells after incubation with HIV-pNL4.3 for the indicated time. qPCR values were normalized to the housekeeping genes Glyceraldehyde 3-phosphate dehydrogenase and S18 (geometric mean) and represent log-fold change compared with untreated samples. Internalization of virus by both Ect1 and End1 cells after 5 hours at 4°C are also depicted on graph and show minimal internalization.
The kinetics of internalization of the nonfluorescent HIV-1NL4.3 variant by both Ect1 and End1 cells was determined by measuring the acquisition of viral RNA within the cells by qPCR. The cervical epithelial cells were trypsinized after being incubated for various times with HIVNL4.3. Internalization of HIV-1NL4.3 was observed at the earliest observed time point (30 minutes) and increased steadily for 4–5 hours until the levels of internalized virus stabilized (Figure 1E). Internalization of viral RNA did not occur when cells were incubated at 4°C.
Cervical Epithelial Cells Become Infected With HIV
Prior studies have shown that epithelial cells internalize HIV-1 but do not complete a full infectious cycle [25–27]. We used fluorescent protein-expressing HIV-1 clones that contain the envelope sequence from transmitted/founder viral sequences [34, 35] to examine epithelial cell infection. Fluorescent protein expression in cells challenged with these viruses is indicative of early gene expression [38]. This allowed us to examine virus-epithelial cell infection on a single-cell level using flow cytometry.
Ect1 and End1 cells were incubated with NL-GIenvNL4.3 (X4-tropic) or NL-CIenvQH0692.42, NL-CIenvPVO.4, NL-CIenvRHPA4259, NL-CIenvWITO4160 (R5-tropic) viruses for 2 hours, trypsinized, and replated for an additional 72 hours. Both Ect1 and End1 cells became infected with X4-tropic and all 4 R5-tropic viruses at comparable levels (Figure 2A). This infection was inhibited by the pretreatment of epithelial cells with either reverse transcriptase inhibitor, AZT, or the integrase inhibitor, 118-D-24, indicating that viral gene expression in these cells requires reverse transcription and integration of provirus (Figure 2B). Since all of the R5-tropic viruses infected the cervical epithelial cell lines to comparable levels, we used the NL-CIenvWITO4160 for subsequent experiments. The duration of NL-CIenvWITO4160 infection was assessed in Ect1 cells (Figure 2C). At 72 hours following virus incubation, cells were either collected for analysis or replated and cultured until day 7 and day 14. The observed decrease in infection at day 7 and day 14 was due to dilution of the initially infected population over time and the inability of the virus to spread. Cell proliferation and viability were also assessed (Supplementary Figure 1) and demonstrated no difference in overall proliferation of HIV-exposed or unexposed cells.
Figure 2.

Cell-free human immunodeficiency virus type 1 (HIV-1) infection of cervical epithelial cells. Ect1 and End1 (approximately 150 000 cells/well, 24-well plate) were examined for infection 72 hours after exposure to recombinant fluorescent protein-expressing HIV NL-CIenv (30 ng/mL). Cells were incubated with virus for 2 hours in Dulbecco's modified Eagle's medium. Cells were washed vigorously with phosphate-buffered saline prior to trypsinization for 10 minutes at 37°C or until adherent cells were visibly removed from the well. Cells were then replated and allowed to grow in culture for an additional 72 hours (A–C, flow cytometry) or for the indicated time (D and E, quantitative real-time polymerase chain reaction [qRTPCR]). A, Ect1 (top) and End1 (bottom) cells were infected with cell-free NL-GIenvNL4.3 (X4-tropic) or NL-CIenvQH0692.42, NL-CIenvPVO.4, NL-CIenvRHPA4259, NL-CIenvWITO4160 (R5-tropic) viruses and examined 72 hours post infection via flow cytometry. FL1 denotes empty fluorescent channel. B, Ect1 and End1 NL-CIenvRHPA4259 infection was inhibited by both reverse transcription inhibitor, AZT (50 µM), and integrase inhibitor (50 µM). Cells were preincubated for 15 minutes with either inhibitor prior to NL-CIenvRHPA4259 incubation, and inhibitors were maintained after cells were replated. Graph shows the mean and standard deviations of 4 independent experiments; inhibition of infection was significant using a 2-tailed, unpaired t-test comparing infected cells to inhibitor-treated cells (Ect1-AZT, P = .0005; Ect1-integrase, P = .0013; End1-AZT, P = .007; End1-integrase, P = .009). C, NL-CIenvWITO4160 infection of Ect1 cells persists for up to 14 days following initial infection. Cells were passaged as necessary to avoid overconfluency. D, Kinetics of viral Gag-encoding RNA measured by qRTPCR following exposure to HIV. Ect1 and End1 were incubated with cell-free HIVenvWITO4160 for 2 hours, trypsinized, and replated for the indicated times. Control cells treated with 50 µM AZT show no rebound of HIV-Gag RNA. E, Integration of HIV-DNA using the Alu-Gag integration assay, fold change over untreated samples. Ect1 and End1 integration correlates with Gag RNA levels observed in (D). Samples showing integration Signal Below Background (S.B.B.) are shown on the graph. (*, **, *** indicate increasing degree of significance).
To confirm observations using fluorescent NL-CIenvWITO4160, we measured the abundance of viral RNA and synthesis of proviral DNA with qPCR-based methods. Epithelial cells were incubated with HIV-1WITO4160 for 2 hours (either in the presence or absence of AZT), trypsinized, and replated for the indicated times. Examination of the unspliced Gag viral mRNA revealed high levels of HIV-Gag RNA within cells immediately following 2-hour incubation with HIV (Figure 2D). A 100- to 1000-fold decrease in Gag RNA was observed until 15 hours after HIV-1 incubation, followed by a rebound in HIV-Gag RNA in untreated samples. This rebound in Gag RNA was not observed in the AZT-treated samples, indicating that new RNA production required reverse transcription of the incoming viral RNA. This observation is consistent with new viral transcription within the epithelial cells and indicative of HIV-1 infection. To determine whether integration was taking place, genomic DNA samples were analyzed for integrated virus (Figure 2E). In both Ect1 and End1 cells, integrated HIV DNA with kinetics similar to the increase in viral RNA levels was detected (Figure 2D and 2E). AZT treatment effectively blocked viral integration in the cells.
Consistent with previous reports, we found that cervical epithelial cells do not express CD4, CXCR4, and CCR5 (Supplementary Figure 2) [25, 26], which implies that HIV internalization does not occur via standard HIV-1 receptor and coreceptor interactions. Neither the anti-CD4 monoclonal antibody, Leu3A, nor TAK779, a CCR5 agonist, blocked infection of cervical epithelial cells (Figure 3A). Next, we tested the role of charged surface proteoglycans in HIV-1 infection of cervical epithelial cells, which have been implicated in nonclassical HIV-1 entry pathways. Chrondroitinase ABC had no effect on epithelial cell infection, while dextran sulfate, iota carageenan, and heparinase III all decreased cervical cell HIV-1 infection significantly; the polyanion (Pro2K) completely inhibited epithelial cell infection consistent with charge-dependent interactions between HIV-1 and cervical epithelial cells (Figure 3B).
Figure 3.

Inhibition and enhancement of cell-free human immunodeficiency virus type 1 (HIV-1) infection of cervical epithelial cells. A, CD4 and coreceptor antagonists did not inhibit infection of Ect1 (top) and End1 (bottom). Cells (approximately 150 000 cells/well, 24-well plate) were pretreated with HIV-1 inhibitors that target CD4: Leu3A (0.5 µg/mL); target CCR5: TAK779 (60 µM); or serve as an antibody isotype control Isotype control (Iso CT; 0.5 µg/mL). Cells were then incubated with NL-CIenvWITO4160 for 2 hours, replated, and analyzed for infection by flow cytometry at 72 hours. Leu3A and TAK779 did inhibit CD4+ T cell infection. Graphs indicate the mean and standard deviation of 3 independent experiments. B, Inhibition of HIV infection of cervical epithelial cells by modulation of polyanionic/heparan sulfate–mediated interactions. Cells were incubated with NL-CIenvWITO4160 in the absence (−) of inhibitors or in the presence of 2 U/mL chondroitinase ABC (CH ABC; not significant), 25 µg/mL dextran sulfate (Dex Sul; Ect1 P = .003, End1 P = .02), 100 µg/mL iota carrageenan (IC; Ect1 P = .003, End1 P = .03), 25 U/mL heparinase III (Hep III; Ect1 P = .008, End1 P = .02), or 20 µg/mL Pro2000 (Pro2K; Ect1 P = .001, End1 P = .01). The graph represents the mean of at least 3 independent experiments. C, Semen-derived enhancer of virus (SEVI) fibrils enhanced HIV infection of cervical epithelial cells. NL-CIenvWITO4160 (10 ng/mL) was incubated with SEVI fibrils (10 µg/mL) before adding virus to the epithelial cells. A lower concentration of cell-free NL-CIenvWITO4160 was used because the enhancing effect of SEVI fibrils is often observed at lower concentrations of virus [9]. Cells were replated following 2-hour virus inoculation at which point SEVI fibrils were removed (Ect1, P = .03; End1, P = .04). P values were determined using an unpaired, 2-tailed T test comparing infected epithelial cells to inhibitor treated–infected cells. (*, **, *** indicate increasing degree of significance).
After examination of the effect of polyanion-blocking molecules on the infection of cervical epithelial cells, we examined the effect of SEVI fibrils on epithelial infection. SEVI fibrils have been shown to enhance HIV infection up to 5-fold in T cells in a charge-dependent manner [9, 10]. We observed a 2- to 3-fold increase in cervical epithelial cell infection when SEVI fibrils were incubated with NL-CIenvWITO4160 (10 ng/mL) before epithelial cell inoculation (Figure 3C).
HIV Released From Infected Epithelial Cells
To determine if cell-free HIV-1 was released from infected epithelial cells, supernatants were collected from NL-CIenvWITO4160–infected Ect1 cells 3 and 6 days following infection. There was no detectable HIV-1 p24 in the supernatant of virus-infected epithelial cells (data not shown). Supernatants from infected Ect1 cells did not infect the TZM-bl indicator cell line (Figure 4A) or the primary CD4+ T cells when spinoculation was used to promote infection (Figure 4B), confirming that no infectious cell-free NL-CIenvWITO4160 was released from the epithelial cells.
Figure 4.
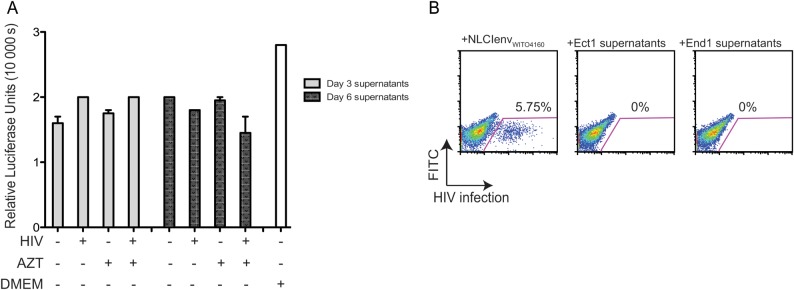
Cell-free human immunodeficiency virus type 1 (HIV-1) release from infected cervical epithelial cells. A, Supernatants from + azidothymidine (AZT) or −AZT, NL-CIenvWITO4160–infected Ect1 cells were collected at day 3 and day 6. Supernatants were applied to the TZMbl indicator cell line for 24 hours. Cells were washed after supernatant incubation; 24 hours later, the luciferase assay was performed. Results display no luciferase induction beyond background (Dulbecco's modified Eagle's medium alone), indicating the absence of infectious HIV in supernatants. B, Supernatants from polybrene-enhanced (4 µg/mL) NL-CIenvWITO4160–infected (30 ng) Ect1 and End1 cells were collected 3 days after initial infection. Supernatants were used to spinoculate primary CD4+ T cells. Cells and supernatants were spun in a flat-bottom 96-well plate at 1200 × g for 2 hours and then incubated for 48 hours. Results show no HIV infection when compared with NL-CIenveWITO4160 spinoculated CD4+ T cells.
HIV-Infected Cervical Epithelial Cells Transmit Virus to CD4+ T Cells in Contact-Dependent Manner
Next, we examined the ability of infected cervical epithelial cells to infect target CD4+ T cells in a coculture assay where cervical epithelial cells were exposed to HIV-1, replated, and incubated for 3 days prior to the addition of CD4+ T cells (Figure 5A). Infection of the cocultured T cells was assessed by flow cytometry. The Cell Proliferation Dye e670 marked CD4+ T cells, and Cherry fluorescence from NL-CIenvWITO4160 measured new infection of these cells. Gating on e670-positive cells in the small lymphocyte gate readily discriminated T cells from epithelial cells (Supplementary Figure 3A). When epithelial cells were infected with NL-CIenvWITO4160 and cocultured with CD4+ T cells, infection of target CD4+ T cells occurred (Figure 5B). The percentage of infected CD4+ T cells with NL-CIenvWITO4160 was increased when the epithelial cells were infected in the presence of SEVI fibrils or polybrene. However, only SEVI fibrils used to enhance initial epithelial cell infection significantly increased infection of CD4+ T cells in coculture (Figure 5B). The purity of the T-cell population was confirmed by surface CD3 and CD8 staining (Supplementary Figure 3B).
Figure 5.

Infected epithelial cells are capable of transmitting the infection to cocultured target CD4+ cells. Infected Ect1 and End1 cells (approximately 300 000 cells/well, 12-well plate) were cocultured with approximately 2 million activated CD4+ T cells per well in Roswell Park Memorial Institute complete media following coculture assay. A, Outline of experimental design that indicate when inhibitors or enhancing factors were added. B, CD4+ T-cell human immunodeficiency virus type 1 (HIV-1) infection (x-axis) vs CD4+ T-cell proliferation (y-axis). Infection of CD4+ T cells was enhanced when epithelial cells were incubated with HIV and semen-derived enhancer of virus (SEVI) fibrils at day 0 (Ect1, P = .041; End1, P = .02), or polybrene (PB; Ect1, P = .1; End1, P = .3) based on 3 separate experiments. Noninfected epithelial cells cocultured with CD4+ T cells acted as a negative control. C, Infection of CD4+ T cells was sensitive to inhibition by the viral protease inhibitor indinavir, indicating that new viral maturation was required to transmit the infection to CD4+ T cells. Ect1 (top, black graph) or End1 (bottom, gray graph) were treated with indinavir on day 2 prior to and during coculture (Ect1, P = .0074; End1, P = .005). D, Coculture infection of CD4+ T cells was inhibited by Leu3A (Ect1, P = .03; End1, P = .04) and TAK779 (Ect1, P = .03; End1, P = .04), indicating a CD4- and coreceptor-dependent infection. Inhibitors were added on day 3 prior to addition of CD4+ T cells. E, Addition of SEVI fibrils on day 3 prior to coculture with CD4+ T cells had no effect on coculture infection of CD4+ T cells. P values were determined using an unpaired, 2-tailed T test comparing infected epithelial cell coculture with inhibitor treated–infected coculture. Graphs show mean and standard deviation of 3 separate experiments. (*, **, *** indicate increasing degree of significance).
We determined whether de novo virus production within the epithelial cells was necessary for infection of cocultured CD4+ T cells. The HIV-1 protease inhibitor, indinavir, will inhibit mature cell-free virus infection, but inhibition of virus infection is dependent on a mature, fully cleaved virion. Infection of CD4+ T cells was significantly inhibited when indinavir was added to the coculture, suggesting that mature virus production from the epithelium was necessary for infection of CD4+ T cells (Figure 5B). Addition of indinavir at the same time as incubation with NL-CIenvWITO4160 had no effect on cell-free epithelial or CD4+ T-cell infection (data not shown).
Next, we determined whether the epithelial cell–mediated infection of CD4+ T cells required CD4 and CCR5 receptors for viral entry by pretreating the CD4+ T cells with Leu3A and TAK779 prior to coculture with infected epithelial cells. Leu3A and TAK779 completely blocked this infection (Figure 5D). Last, we determined whether the addition of SEVI fibrils after epithelial cell infection would enhance infection of cocultured CD4+ T cells. SEVI fibrils did not enhance infection of CD4+ T cells mediated by contact with the infected epithelial cells (Figure 5E).
DISCUSSION
Previous studies have suggested that cervical epithelial cells are capable of internalizing, transcytosing, and transinfecting CD4+ T cells with HIV-1 without becoming infected [25–27]. Here we demonstrate that cervical epithelial cells can be infected by HIV-1, suggesting a potential role for viral production from the cervical epithelium during heterosexual transmission. Both R5- and X4-tropic HIV-1 can infect cervical epithelial cells despite the absence of CD4 and coreceptors. Cervical cell infection was inhibited by reverse transcriptase and integrase inhibitors, confirming that infection required enzymatic steps associated with an HIV-1 infectious life cycle (Figure 2). These data could relate to the recently reported effectiveness of intravaginally placed 1% tenofovir gel for the prevention of sexually transmitted HIV-1 [39]. Earlier reports failed to demonstrate active virus replication [25–27]; however, these studies used methods that may be less sensitive. Our use of fluorescent HIV-1 clones allowed for the detection of viral gene expression from individual infected cells. In addition, analysis of RNA and DNA isolated from cervical epithelial cells incubated with a primary isolate (HIV-1WITO4160) confirms the findings with engineered NL-CIenvWITO4160 virus (Figure 2).
Uptake of HIV by epithelial cells does not involve classic envelope interactions with CD4 and coreceptors, as neither Leu3A nor Tak779, which block CD4-gp120 interaction and CCR5-gp120 interaction, respectively, blocked the infection of cervical epithelial cells. However, inhibition of cervical cell infection was observed with polyanion antagonists of heparan sulfate–mediated interactions (Figure 3C). This heparan sulfate proteoglycan-dependent pathway resembles other cells that have been found to use glipican or syndecan-1 to internalize HIV-1 in the absence of CD4 and CCR5 [25, 26, 40, 41]. Additional experiments are required to determine the specific receptor necessary for cervical epithelial cell infection. Just as polyanions block infection, the addition of molecules that induce polycationic bridges, such as SEVI fibrils, can promote infection [9]. SEVI fibrils enhanced epithelial cell infection and the subsequent contact-dependent infection of CD4+ T cells when cocultured with these cells (Figures 3C and 5B). However, the latter effect can be accounted for by the enhanced epithelial infection and not by direct enhancement of infection of CD4+ T cells, as SEVI fibrils did not enhance infection when present after epithelial cell uptake (Figure 5E).
After an early peak of HIV-1 gene expression in epithelial cells, the infection gradually decreased over 14 days in these cells. Since there was no cell-free virus released from the epithelial cells and no effect on proliferation and viability of cells, this gradual decline in infection was most likely due to dilution of the infected cells (Supplementary Figure 1). At a minimum, it shows that viral expression cannot be sustained in these nonlymphoid cells. However, the infection was not a dead-end infection when rescued by exposure to CD4+ cells in coculture. Infection of target CD4+ T cells required de novo virus production from the epithelial cells, and infection was inhibited when CD4 or CCR5 was blocked (Figure 5C and 5D). These data support an epithelial cell–T-cell infection model that is analogous to the virological synapse between an infected and uninfected T cell wherein cell contact is required for cell-to-cell transmission [42–44]. In this model system, R5 and X4-tropic viruses infect the cervical epithelial cells at comparable levels. However, the lamina propria of the cervix and vagina are populated with CCR5 CD4+ T cells [45, 46], which are increased in the setting of genital exposure to HIV-1, providing an opportunity for epithelial cells to infect the predominant resident T cell. In vitro, this observed infection is small and inefficient on its own (Figure 5B) but can be amplified by contact with T cells. Current studies of acute transmission support an inefficient initial infection process that propagates locally [23], leading to acute infection by a single viral sequence in most cases [35, 47–50]. These data may suggest that virus–epithelial interactions that transmit small amounts of virus to T cells are highly relevant.
Thus, we underscore the importance of considering virus interaction with nonlymphoid cells during transmission and point to the cervical epithelium as a possible infection-promoting cell type during HIV-1 sexual transmission. Future studies might consider the productive potential of the cervical epithelium when developing preventive strategies against the sexual transmission of HIV-1.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We are grateful to Frank Kirchoff and Jan Münch who supplied SEVI and helped design SEVI experiments. The Mount Sinai Microscopy Shared Resource Facility assisted in acquiring the confocal images.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID; R21 AI79776–01). This work was also partly funded by a grant to BKC from the National Institute on Drug Abuse (NIDA; DA028866).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.WHO. Global summary of the AIDS epidemic 2010. http://www.who.int/hiv/data/2011_epi_core_en.png . Accessed 12 October 2012. [Google Scholar]
- 2.Haase AT. Perils at mucosal front lines for HIV and SIV and their hosts. Nat Rev Immunol. 2005;5:783–92. doi: 10.1038/nri1706. [DOI] [PubMed] [Google Scholar]
- 3.Gray RH, Wawer MJ, Brookmeyer R, et al. Probability of HIV-1 transmission per coital act in monogamous, heterosexual, HIV-1-discordant couples in Rakai, Uganda. Lancet. 2001;357:1149–53. doi: 10.1016/S0140-6736(00)04331-2. [DOI] [PubMed] [Google Scholar]
- 4.McCormack S, Ramjee G, Kamali A, et al. PRO2000 vaginal gel for prevention of HIV-1 infection (Microbicides Development Programme 301): a phase 3, randomised, double-blind, parallel-group trial. Lancet. 2010;376:1329–37. doi: 10.1016/S0140-6736(10)61086-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdool Karim SS, Richardson BA, Ramjee G, et al. Safety and effectiveness of BufferGel and 0.5% PRO2000 gel for the prevention of HIV infection in women. AIDS. 2011;25:957–66. doi: 10.1097/QAD.0b013e32834541d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skoler-Karpoff S, Ramjee G, Ahmed K, et al. Efficacy of Carraguard for prevention of HIV infection in women in South Africa: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:1977–87. doi: 10.1016/S0140-6736(08)61842-5. [DOI] [PubMed] [Google Scholar]
- 7.Van Damme L, Govinden R, Mirembe FM, et al. Lack of effectiveness of cellulose sulfate gel for the prevention of vaginal HIV transmission. N Engl J Med. 2008;359:463–72. doi: 10.1056/NEJMoa0707957. [DOI] [PubMed] [Google Scholar]
- 8.Feldblum PJ, Adeiga A, Bakare R, et al. SAVVY vaginal gel (C31G) for prevention of HIV infection: a randomized controlled trial in Nigeria. PLoS One. 2008;3:e1474. doi: 10.1371/journal.pone.0001474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munch J, Rucker E, Standker L, et al. Semen-derived amyloid fibrils drastically enhance HIV infection. Cell. 2007;131:1059–71. doi: 10.1016/j.cell.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 10.Roan NR, Munch J, Arhel N, et al. The cationic properties of SEVI underlie its ability to enhance human immunodeficiency virus infection. J Virol. 2009;83:73–80. doi: 10.1128/JVI.01366-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim KA, Yolamanova M, Zirafi O, et al. Semen-mediated enhancement of HIV infection is donor-dependent and correlates with the levels of SEVI. Retrovirology. 2010;7:55. doi: 10.1186/1742-4690-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olsen JS, DiMaio JT, Doran TM, Brown C, Nilsson BL, Dewhurst S. Seminal plasma accelerates semen-derived enhancer of viral infection (SEVI) fibril formation by the prostatic acid phosphatase (PAP248–286) peptide. J Biol Chem. 2012;287:11842–9. doi: 10.1074/jbc.M111.314336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tovanabutra S, Robison V, Wongtrakul J, et al. Male viral load and heterosexual transmission of HIV-1 subtype E in northern Thailand. J Acquir Immune Defic Syndr. 2002;29:275–83. doi: 10.1097/00126334-200203010-00008. [DOI] [PubMed] [Google Scholar]
- 14.Chakraborty H, Sen PK, Helms RW, et al. Viral burden in genital secretions determines male-to-female sexual transmission of HIV-1: a probabilistic empiric model. AIDS. 2001;15:621–7. doi: 10.1097/00002030-200103300-00012. [DOI] [PubMed] [Google Scholar]
- 15.Kaushic C, Ferreira VH, Kafka JK, Nazli A. HIV infection in the female genital tract: discrete influence of the local mucosal microenvironment. Am J Reprod Immunol. 2010;63:566–75. doi: 10.1111/j.1600-0897.2010.00843.x. [DOI] [PubMed] [Google Scholar]
- 16.Gupta P, Mellors J, Kingsley L, et al. High viral load in semen of human immunodeficiency virus type 1-infected men at all stages of disease and its reduction by therapy with protease and nonnucleoside reverse transcriptase inhibitors. J Virol. 1997;71:6271–5. doi: 10.1128/jvi.71.8.6271-6275.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dyer JR, Gilliam BL, Eron JJ, Jr, Grosso L, Cohen MS, Fiscus SA. Quantitation of human immunodeficiency virus type 1 RNA in cell free seminal plasma: comparison of NASBA with Amplicor reverse transcription-PCR amplification and correlation with quantitative culture. J Virol Methods. 1996;60:161–70. doi: 10.1016/0166-0934(96)02063-0. [DOI] [PubMed] [Google Scholar]
- 18.Butler DM, Delport W, Kosakovsky Pond SL, et al. The origins of sexually transmitted HIV among men who have sex with men. Sci Transl Med. 2010;2:18re1. doi: 10.1126/scitranslmed.3000447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bagasra O, Farzadegan H, Seshamma T, Oakes JW, Saah A, Pomerantz RJ. Detection of HIV-1 proviral DNA in sperm from HIV-1-infected men. AIDS. 1994;8:1669–74. doi: 10.1097/00002030-199412000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Moench TR, Chipato T, Padian NS. Preventing disease by protecting the cervix: the unexplored promise of internal vaginal barrier devices. AIDS. 2001;15:1595–602. doi: 10.1097/00002030-200109070-00001. [DOI] [PubMed] [Google Scholar]
- 21.Moss GB, Clemetson D, D'Costa L, et al. Association of cervical ectopy with heterosexual transmission of human immunodeficiency virus: results of a study of couples in Nairobi, Kenya. J Infect Dis. 1991;164:588–91. doi: 10.1093/infdis/164.3.588. [DOI] [PubMed] [Google Scholar]
- 22.Tan X, Pearce-Pratt R, Phillips DM. Productive infection of a cervical epithelial cell line with human immunodeficiency virus: implications for sexual transmission. J Virol. 1993;67:6447–52. doi: 10.1128/jvi.67.11.6447-6452.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z, Schuler T, Zupancic M, et al. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science. 1999;286:1353–7. doi: 10.1126/science.286.5443.1353. [DOI] [PubMed] [Google Scholar]
- 24.Li Q, Estes JD, Schlievert PM, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature. 2009;458:1034–8. doi: 10.1038/nature07831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bobardt MD, Chatterji U, Selvarajah S, et al. Cell-free human immunodeficiency virus type 1 transcytosis through primary genital epithelial cells. J Virol. 2007;81:395–405. doi: 10.1128/JVI.01303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Z, Chen Z, Phillips DM. Human genital epithelial cells capture cell-free human immunodeficiency virus type 1 and transmit the virus to CD4+ cells: implications for mechanisms of sexual transmission. J Infect Dis. 2003;188:1473–82. doi: 10.1086/379248. [DOI] [PubMed] [Google Scholar]
- 27.Dezzutti CS, Guenthner PC, Cummins JE, Jr, et al. Cervical and prostate primary epithelial cells are not productively infected but sequester human immunodeficiency virus type 1. J Infect Dis. 2001;183:1204–13. doi: 10.1086/319676. [DOI] [PubMed] [Google Scholar]
- 28.Howell AL, Edkins RD, Rier SE, et al. Human immunodeficiency virus type 1 infection of cells and tissues from the upper and lower human female reproductive tract. J Virol. 1997;71:3498–506. doi: 10.1128/jvi.71.5.3498-3506.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fichorova RN, Rheinwald JG, Anderson DJ. Generation of papillomavirus-immortalized cell lines from normal human ectocervical, endocervical, and vaginal epithelium that maintain expression of tissue-specific differentiation proteins. Biol Reprod. 1997;57:847–55. doi: 10.1095/biolreprod57.4.847. [DOI] [PubMed] [Google Scholar]
- 30.Fichorova RN, Anderson DJ. Differential expression of immunobiological mediators by immortalized human cervical and vaginal epithelial cells. Biol Reprod. 1999;60:508–14. doi: 10.1095/biolreprod60.2.508. [DOI] [PubMed] [Google Scholar]
- 31.Fichorova RN, Cronin AO, Lien E, Anderson DJ, Ingalls RR. Response to Neisseria gonorrhoeae by cervicovaginal epithelial cells occurs in the absence of toll-like receptor 4-mediated signaling. J Immunol. 2002;168:2424–32. doi: 10.4049/jimmunol.168.5.2424. [DOI] [PubMed] [Google Scholar]
- 32.Herbst-Kralovetz MM, Quayle AJ, Ficarra M, et al. Quantification and comparison of toll-like receptor expression and responsiveness in primary and immortalized human female lower genital tract epithelia. Am J Reprod Immunol. 2008;59:212–24. doi: 10.1111/j.1600-0897.2007.00566.x. [DOI] [PubMed] [Google Scholar]
- 33.Hubner W, Chen P, Del Portillo A, Liu Y, Gordon RE, Chen BK. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J Virol. 2007;81:12596–607. doi: 10.1128/JVI.01088-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li M, Gao F, Mascola JR, et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79:10108–25. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salazar-Gonzalez JF, Salazar MG, Keele BF, et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med. 2009;206:1273–89. doi: 10.1084/jem.20090378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abela IA, Berlinger L, Schanz M, et al. Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathog. 2012;8:e1002634. doi: 10.1371/journal.ppat.1002634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liszewski MK, Yu JJ, O'Doherty U. Detecting HIV-1 integration by repetitive-sampling Alu-gag PCR. Methods. 2009;47:254–60. doi: 10.1016/j.ymeth.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen BK, Gandhi RT, Baltimore D. CD4 down-modulation during infection of human T cells with human immunodeficiency virus type 1 involves independent activities of vpu, env, and nef. J Virol. 1996;70:6044–53. doi: 10.1128/jvi.70.9.6044-6053.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klasse PJ, Shattock R, Moore JP. Antiretroviral drug-based microbicides to prevent HIV-1 sexual transmission. Annu Rev Med. 2008;59:455–71. doi: 10.1146/annurev.med.59.061206.112737. [DOI] [PubMed] [Google Scholar]
- 40.Bobardt MD, Saphire AC, Hung HC, et al. Syndecan captures, protects, and transmits HIV to T lymphocytes. Immunity. 2003;18:27–39. doi: 10.1016/s1074-7613(02)00504-6. [DOI] [PubMed] [Google Scholar]
- 41.de Parseval A, Bobardt MD, Chatterji A, et al. A highly conserved arginine in gp120 governs HIV-1 binding to both syndecans and CCR5 via sulfated motifs. J Biol Chem. 2005;280:39493–504. doi: 10.1074/jbc.M504233200. [DOI] [PubMed] [Google Scholar]
- 42.Jolly C, Sattentau QJ. Retroviral spread by induction of virological synapses. Traffic. 2004;5:643–50. doi: 10.1111/j.1600-0854.2004.00209.x. [DOI] [PubMed] [Google Scholar]
- 43.Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J Exp Med. 2004;199:283–93. doi: 10.1084/jem.20030648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen P, Hubner W, Spinelli MA, Chen BK. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J Virol. 2007;81:12582–95. doi: 10.1128/JVI.00381-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller CJ, McChesney M, Moore PF. Langerhans cells, macrophages and lymphocyte subsets in the cervix and vagina of rhesus macaques. Lab Invest. 1992;67:628–34. [PubMed] [Google Scholar]
- 46.Veazey RS, Marx PA, Lackner AA. Vaginal CD4+ T cells express high levels of CCR5 and are rapidly depleted in simian immunodeficiency virus infection. J Infect Dis. 2003;187:769–76. doi: 10.1086/368386. [DOI] [PubMed] [Google Scholar]
- 47.Fischer W, Ganusov VV, Giorgi EE, et al. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One. 2010;5:e12303. doi: 10.1371/journal.pone.0012303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee HY, Giorgi EE, Keele BF, et al. Modeling sequence evolution in acute HIV-1 infection. J Theor Biol. 2009;261:341–60. doi: 10.1016/j.jtbi.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keele BF, Giorgi EE, Salazar-Gonzalez JF, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A. 2008;105:7552–7. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goonetilleke N, Liu MK, Salazar-Gonzalez JF, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009;206:1253–72. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.