

Abstract
Background. Sepsis and sepsis-associated organ failure are devastating conditions. Understanding the detailed cellular/molecular mechanisms involved in sepsis should lead to the identification of novel therapeutic targets.
Methods. Cecal ligation and puncture (CLP) was used as a polymicrobial sepsis model in vivo to determine mortality and end-organ damage. Macrophages were adopted as the cellular model in vitro for mechanistic studies.
Results. PTRF+/− mice survived longer and suffered less organ damage after CLP. Reductions in nitric oxide (NO) and iNOS biosynthesis were observed in plasma, macrophages, and vital organs in the PTRF+/− mice. Using an acute sepsis model after CLP, we found that iNOS−/− mice had a comparable level of survival as the PTRF+/− mice. Similarly, polymerase I transcript release factor (PTRF) deficiency resulted in decreased iNOS and NO/ROS production in macrophages in vitro. Mechanistically, lipopolysaccharide (LPS) enhanced the co-localization and interaction between PTRF and TLR4 in lipid rafts. Deletion of PTRF blocked formation of the TLR4/Myd88 complex after LPS. Consistent with this, lack of PTRF impaired the TLR4 signaling, as shown by the decreased p-JNK, p-ERK, and p-p38, which are upstream factors involved in iNOS transcription.
Conclusion. PTRF is a crucial regulator of TLR4 signaling in the development of sepsis.
Keywords: PTRF, sepsis, macrophage, CLP, nitric oxide, ROS, TLR4
Sepsis and sepsis-associated end organ failure, commonly resulting from bacterial infections, are leading causes of both mortality and morbidity worldwide. An initial infection frequently triggers a devastating systemic inflammatory response syndrome (SIRS), which results in multiple organ failure (MOF) and intense immunological abnormalities [1, 2]. An imbalance between bactericidal effects and over-response of host defense has been thought to be the mechanism underlying sepsis-derived SIRS and MOF [1–3].
Among all of the parenchymal and inflammatory cells that participate in the development of sepsis and MOF, macrophages are first-line innate immune cells, which are both efficient microorganism scavengers and major source of proinflammatory factors [4, 5]. However, macrophage activation has also been shown to be associated with severe SIRS and death [4–6]. During the classic activation of macrophages, besides their role in proinflammatory cytokines and chemokines, macrophages also up-regulate iNOS and promote nitric oxide (NO) release, as well as the generation of reactive oxygen species (ROS). In addition, other parenchymal cells also release NO/ROS during the acute phase of sepsis [7, 8]. This exaggerated production of NO and ROS is effectively a double-edged sword. Both NO and ROS induce cell apoptosis and result in tissue damage [7–10]. Thus, NO and ROS not only mediate host defense but also lead to significant SIRS, tissue destruction, MOF, and mortality. Macrophages robustly produce NO/ROS after LPS in cell culture in vitro [11, 12]. Therefore, macrophages were used as the cellular model in this study to explore the detailed molecular mechanisms.
Before macrophages initiate the immune response, they have to detect and recognize the invading microbes. Most of these “detection” and “recognition” events rely on a group of microbial-sensing proteins called Toll-like receptors (TLRs) [13]. Among the TLRs, TLR4 is well known as the receptor for lipopolysaccharide (LPS). TLR4 receptors are located primarily on the plasma membrane, where they interact with pathogens. Upon activation, MyD88 interacts with interleukin 1 (IL-1) receptor-associated kinase-4 (IRAK-4), and TLR4 via the death domain in each of these molecules. This complex induces the phosphorylation of IRAK-1. Phosphorylated IRAK-1 (activated) then interacts with TRAF6, which subsequently leads to the activation of the MAP kinases (JNK and p38 MAPK), as well as NF-κB. The activation of these MAP kinases ultimately results in the up-regulation of iNOS, an increased production of NO, ROS, and matrix metalloproteinases (MMPs), as well as cytokine and chemokine release [13].
Previous studies have shown that lipid rafts and their protein components, such as caveolin-1 (cav-1) and caveolin-2 (cav-2), are involved in regulating immune function [14, 15]. Disruption of lipid rafts using pharmacological approaches has been shown to significantly affect the normal immune cell response to bacterial infection. Accumulating evidence further suggests that lipid rafts are used by bacterial pathogens as ports of entry to enter into host cells [14]. Interestingly, various roles for the caveolins in sepsis have been reported [16, 17], suggesting a homeostasis is at work among the lipid raft proteins. Thus, it is important to explore the function and regulation of other lipid raft components, in addition to cav-1, in the pathogenesis of bacterial infection-associated inflammation.
Polymerase I transcript release factor (PTRF) is a soluble protein containing a putative leucine zipper, a nuclear localization signal and a PEST domain [18–20]. Recently, this relatively unstudied protein has been suggested to be a potential caveolar coat protein that controls and stabilizes both caveolar structure and function [18]. Although ubiquitously expressed in all the vital organs and tissues, the exact activities of PTRF in sepsis and macrophage function have not been reported. We hypothesized that PTRF plays a crucial role in regulating the function of macrophages after polymicrobial infection. We further hypothesized that PTRF is required for macrophages to generate NO and ROS by providing support for TLR4 signaling. To the best of our knowledge, the current study is the first report on the cellular function of PTRF in sepsis, and it provides a novel molecular target for therapeutic development.
MATERIAL AND METHODS
More experimental details are provided in the Supplementary Data.
Animal Studies
PTRF+/− mice, iNOS and matched wild-type C57BL/6 mice (male, 8–10 weeks of age), were obtained from Jackson Lab (Maine, USA). All mice were maintained under specific pathogen-free conditions. Animal experiments were carried out in compliance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and the guidelines of the Harvard Medical Laboratory Animal Care and Use Committee.
Mice and Cecal Ligation and Puncture
Cecal ligation and puncture (CLP) was performed as described elsewhere [21, 22]. Detailed methods can be found in the Supplementary Data.
Cells and Cell Culture
Bone-marrow derived macrophages (BMDMs) and peritoneal macrophages were isolated as described elsewhere [23]. Raw 264.7 cell lines were purchased from ATCC (American Type Culture Collection Manassas, USA). Detailed information can be found in the Supplementary Data.
Western Blot Analysis and Co-immunoprecipitation Assay
Co-immunoprecipitation assay (Co-IP) assay and Western blot was performed as reported elsewhere [24]. Signals were detected using the SuperSignal West Pico system (Pierce, IL) and exposed to ChemiDoc XRS imaging system (Bio-Rad, Hercules, CA).
Measurement of NO Production
NO production in supernatant was assessed as described elsewhere [25]; please refer to the Supplementary Data for details.
Lipid Raft Isolation
Lipid raft fractions were isolated by sucrose gradient ultracentrifugation as described, in the absence of detergent [24]; details can be found in the Supplementary Data.
Confocal Microscopy
Confocal microscopy was performed as reported elsewhere [24]; details can be found in the Supplementary Data.
Statistical Analysis
Data were expressed as mean ± SD, from 3 independent experiments. For all statistical tests, PRISM 5.0 (GraphPad Software Inc, San Diego, CA) was used. P values <.05 were considered statistically significant.
RESULTS
PTRF+/− Mice Exhibited a Reduction in Both Mortality and Organ Damage in a CLP-induced Acute Sepsis Model, and Also had Significantly Reduced NO and iNOS Production After CLP
To evaluate the effect of PTRF knocking down on mortality after acute sepsis in vivo, we used the CLP-induced acute sepsis model in mice. WT and PTRF+/− mice underwent CLP (21 G, 2 holes). Survival (percentage) was much higher in the PTRF+/− mice than WT mice (Figure 1A). Liver injury is one of the prominent features in CLP-induced sepsis and SIRS [26, 27]. We evaluated liver injury using PAS staining for cytoplasmic glycogen deposition and hepatocyte damage [27]. Tissues and plasma were collected 24 hours after CLP. As shown in Figure 1B, robust glycogen deposition (PAS positive, green arrow) was found in the liver tissue of WT mice 24 hours after CLP, whereas much less staining was found PTRF+/− mice. Furthermore, lymphocyte apoptosis in the spleen and epithelial cell apoptosis in the colon were markedly reduced in PTRF+/− mice after CLP compared to WT mice, detected by TUNEL staining (Supplementary Figure 1B). Interestingly, NO production in the plasma from the PTRF+/− mice was also significantly lower compared with the WT mice (Figure 1C). This observation was further confirmed by reduced iNOS expression in PTRF+/− mice using IHC staining on liver tissue sections (Figure 1D, red arrow).
Figure 1.

Deletion of PTRF decreased iNOS expression in vivo, reduced mortality and organ damage in CLP-induced sepsis. A, PTRF+/− mice (open circles; n = 10) and WT mice (closed circles; n = 10) were subjected to CLP and monitored for survival. B–E, Tissues and plasma were collected 24 hours after CLP. B, PAS staining was performed on liver tissue to evaluate acute hepatocyte injury. Light arrow: positive for PAS staining (C) NO level in plasma was measured using Griess reagent. D, Paraffin-embedded sections of liver tissue were obtained from WT mice (left panel) and PTRF+/− mice (right panel) after CLP. Liver tissue was stained for iNOS expression (dark arrow) using polyclonal anti-iNOS. E, iNOS positive cells were quantified in the liver sections obtained from WT mice or PTRF+/− mice. All figures above represented 3 independent experiments with similar results. *P < .05. Abbreviations: CLP, cecal ligation and puncture; PTRF, polymerase I transcript release factor; WT, wild type.
Deletion of iNOS (iNOS−/−) Resulted in Reduced Mortality and Less Liver Damage After CLP, Similar to the PTRF+/− Mice
To determine whether the survival benefit in PTRF+/− mice after sepsis is potentially associated with the PTRF+/− induced iNOS suppression, we performed CLP in iNOS−/− mice. In this acute sepsis model, we found that iNOS−/− mice also survived better after CLP (21G, 2 puncture holes) compared with the WT mice (Figure 2A), similar to the results observed in the PTRF+/− mice. Again, the liver damage was also robustly reduced in the iNOS−/− mice (Figure 2B).
Figure 2.
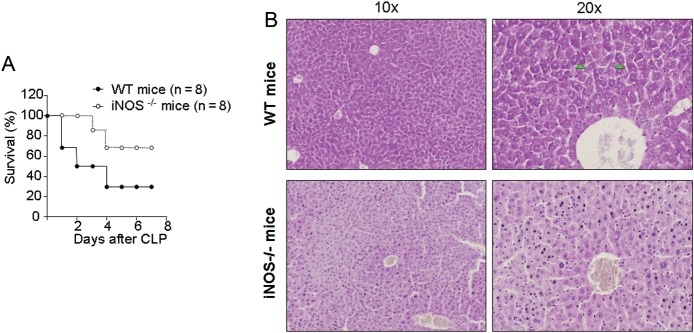
Deletion of iNOS reduced mortality and organ damage in CLP-induced sepsis. A, iNOS−/− mice (open circles; n = 8) and WT mice (closed circles; n = 8) were subjected to CLP and monitored for survival. B, Liver tissue was collected from WT and iNOS−/− mice 24 hours after CLP, and PAS stain was obtained. Arrow: positive for PAS staining. All figures represented 3 independent experiments with similar results. Abbreviations: CLP, cecal ligation and puncture; WT, wild type.
PTRF Is Ubiquitously Expressed in Vital Organs and Macrophages
PTRF is a relatively underexplored protein that may play a crucial function in both lipid rafts and the nucleus [19]. To further investigate the role of PTRF in sepsis, we next explored whether this protein is expressed in the major vital organs. Indeed, we found that PTRF was ubiquitously expressed in multiple organs and tissues, including the lung, liver, kidney, and spleen (Supplementary Figure 2A). To determine whether PTRF is localized to leukocytes, we co-stained the tissue slides using anti-PTRF and anti-leukocyte common antigen (CD45, LCA). We found a strong co-localization pattern for PTRF and CD45 (Supplementary Figure 2B). During the pathogenesis of sepsis and MOF, many types of tissue-resident parenchymal cells and inflammatory cells generate NO. Macrophages are first-line defenders in the innate immunity involved in bacteria-associated sepsis [12]. Therefore, we determined the expression pattern of PTRF in Raw 264.7 macrophages, mouse peritoneal macrophages, and macrophages derived from bone marrow (BMDM) , at the mRNA level using RT-PCR (Supplementary Figure 2C). PTRF was highly expressed in all of these macrophages. The expression of PTRF in these cells was further confirmed at the protein level using Western blot analysis (Supplementary Figure 2D).
PTRF Was Required for LPS-mediated iNOS Induction and NO Production in Macrophages
To further investigate the mechanisms by which PTRF regulates sepsis and MOF, we took macrophages as our cellular models in vitro. Macrophages are known to produce NO after LPS stimulation in vitro [11, 12]. Initially, we deleted PTRF in Raw264.7 macrophages by means of PTRF shRNA transfection. More than 60%–80% efficiency on PTRF deletion (PTRF−/−) was achieved (Figure 3A and Supplementary Figure 1C). The LPS-induced iNOS mRNA transcription in these PTRF−/− macrophages was significantly decreased compared with the control shRNA-transfected cells, particularly within the first 12 hours after LPS stimulation (Figure 3B). This observation of an iNOS reduction was confirmed at the protein level by Western blot analysis (Figure 3C). Furthermore, NO, the product of iNOS, was significantly decreased in response to LPS in PTRF−/− cells (Figure 3D). Next, we obtained BMDMs from wild-type (WT) or heterozygous mice (PTRF+/−). Similarly, iNOS mRNA, iNOS protein, and NO production were markedly reduced in PTRF+/− BMDM in response to LPS compared with WT BMDM (Figure 3E, 3F and 3G, respectively). Additionally, we confirmed the above observations in peritoneal macrophages isolated from WT or PTRF+/− mice. As shown in Figure 1H and 1I, LPS-induced iNOS and NO upregulation were highly suppressed in PTRF+/− macrophages compared with the WT macrophages (Figure 3H and 3I).
Figure 3.

Deletion of PTRF inhibited LPS-induced iNOS expression and NO synthesis in macrophages. A, Raw264.7 cells were transfected with PTRF shRNA plasmid (ShRNA) and negative control plasmid (CTL). After 48 hours, the efficiency of deleting PTRF was assessed by western blot analysis. Densitometry was used to quantify the density of bands. B–D, The transfected Raw264.7 cells were stimulated with LPS (200 ng/mL) for indicated time. B, iNOS mRNA was determined by real-time PCR and standardized to the endogenous control GAPDH. C, iNOS protein expression was determined using western blot analysis and standardized to the endogenous control GAPDH. D, LPS induced NO production was measured using the Greiss reagent in Raw 264.7 cells transfected with shRNA or control plasmid. E–H, Bone marrow derived macrophages (BMDM) were obtained from PTRF+/− mice. Cells were then stimulated with LPS (200 ng/mL) for indicated time. E, iNOS mRNA transcription was measured by real-time PCR and standardized to the endogenous control GAPDH. F, iNOS protein expression was determined using western blot analysis and standardized to the endogenous control GAPDH. G, NO production was measured using the Griess reagent. H and I, PTRF+/− peritoneal macrophages were derived from PTRF+/− mice. Cells were then stimulated with LPS (200 ng/mL) for indicated time. H, iNOS mRNA was measured by real-time PCR and standardized to the endogenous control GAPDH. I, NO production was detected using the Griess reagent. All figures above represented three independent experiments with similar results. *P < .05; **P < .01. Abbreviations: LPS, lipopolysaccharide; mRNA, messenger RNA; NO, nitric oxide; PCR, polymerase chain reaction; PTRF, polymerase I transcript release factor.
PTRF Was Required for LPS-induced ROS Generation in Macrophages
Along with NO, ROS is also a factor that is involved in end-organ damage [28–30]. We next determined the extent of LPS-derived ROS generation using PTRF−/− macrophages (Raw264.7 transfected with PTRF siRNA). As shown in Figure 4A, LPS-induced ROS generation was dramatically reduced in the PTRF−/− cells (green line) compared with the WT cells (red line). Previous reports have suggested that ROS is partially derived from NO [29, 30]. In Figure 4B, we confirmed that reduced ROS generation was found in iNOS knockout macrophages (iNOS−/−, isolated from iNOS knockout mice; green line) with WT macrophages (red line) in the presence of LPS (Figure 4B). Additionally, we confirmed via immunofluorescence staining that the deletion of PTRF prevented ROS generation after LPS (Supplementary Figure 1D).
Figure 4.

Deletion of PTRF or iNOS reduced the LPS-induced ROS generation. A, Raw264.7 cells were transfected with PTRF siRNA or control siRNA (CTL). After 36 hours, cells were stimulated with LPS (200 ng/mL). After additional 6 hours, the production of total ROS was determined using flow cytometry. Mean fluorescence intensity (MFI) was analyzed by 3 independent experiments. B, BMDM was obtained from iNOS−/− mice. Cells were then stimulated with LPS (200 ng/mL). After 6 hours, the total ROS was measured as above. MFI was analyzed by three independent experiments. **P < .01. Abbreviations: BMDM, bone-marrow derived macrophages; LPS, lipopolysaccharide; PTRF, polymerase I transcript release factor; ROS, reactive oxygen species.
PTRF Interacts With TLR4 Complex Components in Lipid Rafts After LPS Stimulation
Cluster of differentiation 14 (CD14), TLR4, and Myd88 are components of the pattern recognition receptor complex that is activated in response to LPS stimulation [13]. Shortly after being exposed to LPS (30 minutes), CD14, TLR4, and Myd88 trafficked to the lipid raft portion (Figure 5). As expected, lipid raft proteins (cav-1, Flot-1, and PTRF) all were present in the lipid rafts at this time point (Figure 5), thus potentially allowing physical interaction with the TLR4 complex components to occur in lipid rafts. We next determined the co-localization of PTRF and TLR4 in the absence and presence of LPS stimulation. As early as 30 minutes of LPS exposure, PTRF and TLR4 co-localization increased significantly on the cell surface (Figure 6A, red arrow). This observation was confirmed using co-IP assays (Figure 6B). In addition to TLR4, the interactions between PTRF, Myd88, and CD14 were also evaluated. Similar to TLR4, the interaction between PTRF and Myd88 was decreased after LPS, whereas no changes between PTRF and CD14 were found (Figure 6C).
Figure 5.
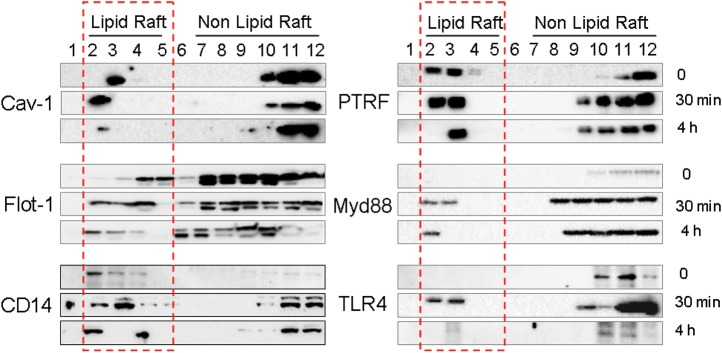
TLR4 signaling components trafficked into lipid rafts after LPS stimulation. Raw264.7 cells were treated with LPS (200 ng/mL). After 30 minutes and 4 hours respectively, lipid raft and non raft fractions were isolated as described in Material and Method. Fractions were then analyzed for cav-1, PTRF, Flot-1, Myd88, CD14, and TLR4 using Western blot analysis. Red frame: lipid raft portion. All figures represented 3 independent experiments with similar results. Abbreviations: LPS, lipopolysaccharide; PTRF, polymerase I transcript release factor.
Figure 6.
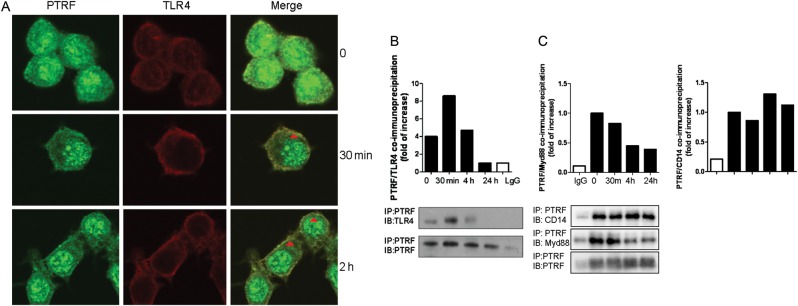
PTRF co-localized and interacted with TLR4 complex. A, Raw264.7 cells were treated with LPS (200 ng/mL). After 30 minutes or 4 hours, cells were stained with anti-PTRF, anti-TLR4, and DAPI (not shown). Co-localization of PTRF and TLR4 was observed using confocal microscopy. Light cells: PTRF; Dark cells: TLR4; Areas indicated by arrows: merge, co-localization of PTRF and TLR4; (B) Raw264.7 cells were treated with LPS (200 ng/mL). After 30 minutes, 4 hours, and 24 hours, respectively, cell lysate was collected for co-IP assays. PTRF was precipitated with rabbit polyclonal anti-PTRF antibody. TLR4 and PTRF (input) levels were determined using western blot analysis. Density of the bands was quantified by densitometry. All figures above represented at least 3 independent experiments with similar results. C, PTRF interacted with Myd88 and CD14. Raw264.7 cells were treated with LPS (200 ng/mL). After 30 minutes, 4 hours, and 24 hours, respectively, cell lysate was collected for co-IP assays. Myd88, CD14, and PTRF (input) levels were determined using Western blot analysis. Density of the bands was quantified by densitometry. Abbreviations: co-IP, co-immunoprecipitation assay; LPS, lipopolysaccharide; PTRF, polymerase I transcript release factor.
PTRF Is Required for LPS-induced TLR4/Myd88 Interaction in Macrophages
Again, using PTRF shRNA to knock down PTRF in Raw264.7 cells, we determined the effect of PTRF on TLR4/Myd88 interaction in the absence or presence of LPS. Figure 7A shows the “knock down” efficiency. TLR4/Myd88 interaction was decreased when PTRF was suppressed (Figure 7B). Consequently, the activation of TLR4 down-stream signaling, including the p38, ERK, and JNK pathways, was also significantly suppressed in PTRF+/− cells compared with control cells. As shown in Figure 7C, LPS-induced phosphorylation of p38, ERK, and JNK was markedly diminished in PTRF+/− cells compared with control cells (Figure 7C).
Figure 7.

Deletion of PTRF inhibited the activation of TLR4 signaling. A, Raw264.7 cells were transfected with PTRF ShRNA plasmid (ShRNA) and control plasmid (CTL). The transfection efficiency was assessed by Western blot analysis. B, Deletion of PTRF affected TLR4/Myd88 complex formation. Transfected cells were treated with LPS (200 ng/mL) for indicated time. Interactions between TLR4 and Myd88 in control cells and PTRF shRNA transfected cells were determined using co-IP assays. Cell lysates were subjected to immunoprecipitation (IP) with anti-TLR4 and blotted with anti-Myd88 or anti-TLR4 (input). C, Deficiency of PTRF affected TLR4 down-stream signaling. BMDMs were obtained from WT or PTRF+/− mice. Cells were then stimulated with LPS for indicated time. Cell lysates were blotted with anti-phospho-p38 (p-p38), anti-phospho-ERK (p-ERK), anti-phospho-JNK (p-JNK). The relative expressions were standardized to the endogenous control GAPDH. Density of bands was quantified using densitometry. All figures above represented at least three independent experiments with similar results. D, Schemata of proposed mechanisms. PTRF interacts with TLR4 and assists the TLR4/Myd88 complex formation in the presence of LPS stimulation thus, promotes LPS induced TLR4/Myd88 signaling. Subsequently, this complex activates the down-stream MARK pathways and MARK pathway associated iNOS transcription. These augmented iNOS expression and NO production, along with fluxed ROS release lead to cellular injury and tissue damage in CLP induced sepsis. When PTRF is deleted, the LPS induced TLR4/Myd88 interaction decreases. The activation of MARK pathway and its down-stream iNOS/NO production are impaired. Thus, deletion of PTRF confers a protective role in CLP/LPS induced cellular injury and tissue damage. Abbreviations: co-IP, co-immunoprecipitation assay; LPS, lipopolysaccharide; PTRF, polymerase I transcript release factor; ROS, reactive oxygen species.
DISCUSSION
Sepsis, SIRS, and sepsis-associated MOF are common causes of morbidity and mortality in critical care units. An understanding of the detailed mechanisms involved in sepsis will potentially lead to novel therapeutic targets. In the current study, we found that PTRF (cavin-1) plays a crucial role in TLR4 signaling in the pathogenesis of sepsis. PTRF regulates the mortality and end-organ damage after CLP.
TLRs are involved in the pathophysiology of sepsis and MOF [13]. As shown in the schema in Figure 7D, PTRF is required for TLR4 signaling assembly, especially after LPS stimulation. TLR4 physically binds with PTRF in the lipid raft portion of the plasma membrane at a basal level. Upon LPS or pathogen-associated molecular patterns (PAMPs) being recognized, TLR4 and its down-stream components traffic from the non-raft portion to the lipid raft portion on the plasma membrane. At this point, PTRF helps to “hold” the TLR4 pathway components together in the rafts. Consequently, the tight interaction between TLR4 and Myd88 trigger the down-stream signal cascades, including ERK, p38, and JNK. In fact, deletion of PTRF affects all of these 3 pathways (Figure 7). This result shows that the key step of PTRF regulation falls on the initial TLR4 signaling. Thus, given that PTRF targets the TLR4/Myd88 complex, it is easily understood that deletion of PTRF attenuates TLR4 down-stream products, including NO production, iNOS synthesis, and ROS release. TLR4-mediated cytokine release presumably is affected by PTRF as well. In fact, our preliminary data indeed suggested an attenuated tumor necrosis factor α release after the deletion of PTRF in RAW 264.7 cells (data not shown). Interestingly, deletion of PTRF also augmented macrophage-mediated phagocytosis (Supplementary Figure 1E). This observation indicates that PTRF mediates a broad spectrum of cellular function in addition to the regulation of caveolae formation [18]. For example, our preliminary data showed a potential role of PTRF on mitochondria-mediated apoptosis, probably related to NF-keppa B signaling (Supplementary Figure 3A and not shown). Future investigation of the role of PTRF in sepsis and MOF should include its effect on cell death, such as the lymphocyte and colon epithelial cell apoptosis that occurs after ischemic injury.
This CLP model is clearly an acute to subacute model in which wild-type mice die at a significant rate within the first 3 days. In this model, the initial cytokine flux is robust and results in significant hemodynamic instability [31–33]. NO production and its associated ROS generation further induce profound organ damage [34, 35]. In our model, the deletion of PTRF delays death and protects against vital organ damage by eliminating NO production. NO is known to be a potent vasodilatory agent and a free radical generator [31–33]. A reduction in NO production, particularly in the initial stage of acute sepsis, not only reduces the hemodynamic instability but also free radical generation. However, it is also well established that NO exerts a bactericidal effect that is crucial in limiting the spread of both local infection and systemic inflammation [36–38]. We think that the beneficial effects of NO likely occur in chronic sepsis models but not an acute model such as ours. Indeed, there is controversy on the role of NO in sepsis. Cobb et al reported that iNOS deficiency leads to early mortality [31]. On the other hand, deletion of iNOS protects mice from death after CLP [32]. In support of the theory that hemodynamic compromise takes place in early sepsis, in studies conducted by Hollenberg et al, microvascular catecholamine responsiveness was shown to be significantly improved in iNOS-deficient mice, as was mortality [32]. Other studies have also reported the role of NO in sepsis-associated organ injury, and a lack of NO has been shown to result in less damage in the lung and brain, as well as a reduction in the systemic inflammatory response [33–35, 39]. Clearly, the controversies regarding NO in sepsis depend on differences in the models and mice used.
Currently, the CLP model is considered a favorable one for sepsis research because it reproduces the hemodynamic changes in cardiovascular function seen in septic patients [40, 41]. Further, the CLP model can induce the progressive release of proinflammatory mediators, which is considered highly important in human sepsis [40, 41]. In the study by Cobb et al, female mice were used, and the CLP model was a chronic one, based on the death curve of the control mice [31]. In contrast, Hollenberg et al used an acute to subacute model with male mice [32] and their death curve was highly similar to the one in our study. The discrepancy on the effect of iNOS/NO deletion between the acute and chronic models would seem to be straightforwardly understood. NO exerts a vasodilatory function [39] and thus may induce hemodynamic instability in the acute models, in which overwhelming cytokine release and inflammation occur. However, in the chronic model, the slow release of inflammatory factors makes NO more tolerable and does not induce immediate hemodynamic compromise. Thus, NO may function as a bactericidal agent [37, 38] and delay mortality in this type of model. Additionally, the age and gender of the mice certainly play a significant role in the outcome of sepsis [42–45]. In fact, female mice, like those used in the study of Cobb et al [31], are more resistant than male mice to CLP-induced mortality, as shown in the reports by Dienstknecht et al and Zellweger et al [44, 45].
This study has a number of limitations. First, the results can not be extrapolated to all other lipid raft proteins, for instance, cav-1. Previous studies on cav-1, like the case of nitric oxide, also have produced divergent results and competing hypotheses on their roles in sepsis and SIRS [15–17]. These discrepancies partially reflect the importance of the acuity of the CLP model, the level of hemodynamic compromise, the technical details of CLP, the gender, age, and strain of the animals used, and so forth. Thus, all of the CLP-related models and studies, including the current one, require careful interpretation of the results, with careful consideration of all of the above mentioned factors.
Furthermore, besides PTRF (cavin-1), serum deprivation response factor (SDPR, cavin-2) also has crucial regulatory functions in caveolae [46, 47]. SDPR is a phosphatidylserine-binding protein and is a homolog of PRTF. It interacts with PTRF and recruits PTRF to the caveolar membrane. Overexpression of SDPR induces deformation of caveolae [47]. Interestingly, our data indicated a transient interaction between PTRF and SDPR after LPS in macrophages (Supplementary Figure 3B), suggesting an effect of SDPR on TLR4 signaling, in addition to the effect of PTRF. However, an elucidation of the relationship between PTRF and the other lipid raft proteins is not a main focus of this investigation, even though it is certainly a subject we intend to address in the future.
The second limitation is that we were not able to use homozygous PTRF−/− mice in this study because these homozygous mice exhibit a reduced metabolic capacity and lean body mass [48]. The reduced metabolic capacity and lean body mass result in a bias in the CLP-induced sepsis in vivo, given that the severity of intra-abdominal infection is closely associated with the amount of fat tissue. Further, CLP-induced sepsis involves multiple cell types, including not only inflammatory cells but also parenchymal cells in the vital organs. Cell-specific conditional knock-outs would likely cause bias too. Therefore, in our study, we simply used heterozygous mice to avoid this problem.
Third, we did not explore the effect of PTRF on other immunomodulatory cells, such as lymphocytes or neutrophils. Our results can not be extrapolated to infectious processes largely dependent on these cells.
Finally, the sepsis that mice developed in response to polymicrobial challenge after CLP seems to differ from the clinical response in humans. Our initial observations on the role of PTRF in sepsis require additional studies using human cells.
In summary, we identified a novel role of PTRF in the regulation of TLR4 signaling in sepsis. PTRF is required for TLR4 complex assembly upon LPS stimulation. Deletion of PTRF results in a lack of TLR4-mediated signaling and subsequently a decrease in iNOS biosynthesis and NO production, as well as ROS generation. In this acute/subacute CLP-induced sepsis model, the suppression of PTRF and subsequently decreased NO production confer a protective role on sepsis-associated mortality and vital organ injury.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Financial support. This work was supported by National Institutes of Health R01 HL102076 (Y. J.), the Ministry of Education, Science and Technology (2012R1A1A2005472)/KRIBB Research Initiative Program (S. J. L.)
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Levy MM, Fink MP, Marshall JC, et al. 2001 Sccm/Esicm/Accp/Ats/Sis international sepsis definitions conference. Crit Care Med. 2003;31:1250–6. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- 2.Munford RS. Severe sepsis and septic shock: the role of gram-negative bacteremia. Annu Rev Pathol. 2006;1:467–96. doi: 10.1146/annurev.pathol.1.110304.100200. [DOI] [PubMed] [Google Scholar]
- 3.Annane D, Bellissant E, Cavaillon JM. Septic shock. Lancet. 2005;365:63–78. doi: 10.1016/S0140-6736(04)17667-8. [DOI] [PubMed] [Google Scholar]
- 4.Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage polarization and plasticity in health and disease. Immunol Res. 2012;53:11–24. doi: 10.1007/s12026-012-8291-9. [DOI] [PubMed] [Google Scholar]
- 5.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Jong HK, Van der Poll T, Wiersinga WJ. The systemic proinflammatory response in sepsis. J Innate Immun. 2010;2:422–30. doi: 10.1159/000316286. [DOI] [PubMed] [Google Scholar]
- 7.Heemskerk S, Masereeuw R, Russel FG, Pickkers P. Selective iNOS inhibition for the treatment of sepsis-induced acute kidney injury. Nat Rev Nephrol. 2009;5:629–40. doi: 10.1038/nrneph.2009.155. [DOI] [PubMed] [Google Scholar]
- 8.Aasen AO, Wang JE. Mediator responses in surgical infections. Surg Infect (Larchmt) 2006;7:3–4. doi: 10.1089/sur.2006.7.s2-3. [DOI] [PubMed] [Google Scholar]
- 9.Simkova V, Baumgart K, Radermacher P, Barth E, Calzia E. Year in review 2006: critical care—multiple organ failure, sepsis, and shock. Crit Care. 2007;11:221. doi: 10.1186/cc5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bae YS, Lee JH, Choi SH, Kim S, Almazan F, Witztum JL, Miller YI. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: Toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ Res. 2009;104:210–8. doi: 10.1161/CIRCRESAHA.108.181040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogawa R, Pacelli R, Espey MG, et al. Comparison of control of listeria by nitric oxide redox chemistry from murine macrophages and no donors: insights into listeriocidal activity of oxidative and nitrosative stress. Free Radic Biol Med. 2001;30:268–76. doi: 10.1016/s0891-5849(00)00470-6. [DOI] [PubMed] [Google Scholar]
- 12.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salomao R, Brunialti MK, Rapozo MM, Baggio-Zappia GL, Galanos C, Freudenberg M. Bacterial sensing, cell signaling, and modulation of the immune response during sepsis. Shock. 2012;38:227–42. doi: 10.1097/SHK.0b013e318262c4b0. [DOI] [PubMed] [Google Scholar]
- 14.Fessler MB, Parks JS. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J Immunol. 2011;187:1529–35. doi: 10.4049/jimmunol.1100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Almeida CJ, Witkiewicz AK, Jasmin JF, Tanowitz HB, Sotgia F, Frank PG, Lisanti MP. Caveolin-2-deficient mice show increased sensitivity to endotoxemia. Cell Cycle. 2011;10:2151–61. doi: 10.4161/cc.10.13.16234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai TH, Chen SF, Huang TY, et al. Impaired CD14 and CD36 expression, bacterial clearance, and Toll-like receptor 4-Myd88 signaling in caveolin-1-deleted macrophages and mice. Shock. 2011;35:92–9. doi: 10.1097/SHK.0b013e3181ea45ca. [DOI] [PubMed] [Google Scholar]
- 17.Feng H, Guo L, Song Z, et al. Caveolin-1 protects against sepsis by modulating inflammatory response, alleviating bacterial burden, and suppressing thymocyte apoptosis. J Biol Chem. 2010;285:25154–60. doi: 10.1074/jbc.M110.116897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hill MM, Bastiani M, Luetterforst R, et al. PTRF-cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell. 2008;132:113–24. doi: 10.1016/j.cell.2007.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mason SW, Sander EE, Grummt I. Identification of a transcript release activity acting on ternary transcription complexes containing murine RNA polymerase I. Embo J. 1997;16:163–72. doi: 10.1093/emboj/16.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jansa P, Mason SW, Hoffmann-Rohrer U, Grummt I. Cloning and functional characterization of PTRF, a novel protein which induces dissociation of paused ternary transcription complexes. Embo J. 1998;17:2855–64. doi: 10.1093/emboj/17.10.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker CC, Chaudry IH, Gaines HO, Baue AE. Evaluation of factors affecting mortality rate after sepsis in a murine cecal ligation and puncture model. Surgery. 1983;94:331–5. [PubMed] [Google Scholar]
- 22.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–6. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett B. Isolation and cultivation in vitro of macrophages from various sources in the mouse. Am J Pathol. 1966;48:165–81. [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang M, Lee SJ, An C, et al. Caveolin-1 mediates fas-bid signaling in hyperoxia-induced apoptosis. Free Radic Biol Med. 2011;50:1252–62. doi: 10.1016/j.freeradbiomed.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin Y, Heck DE, DeGeorge G, Tian Y, Laskin JD. 5-fluorouracil suppresses nitric oxide biosynthesis in colon carcinoma cells. Cancer Res. 1996;56:1978–82. [PubMed] [Google Scholar]
- 26.Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LR, Bland KI, Chaudry IH. Cecal ligation and puncture. Shock. 2005;24:52–7. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 27.Banks JG, Foulis AK, Ledingham IM, Macsween RN. Liver function in septic shock. J Clin Pathol. 1982;35:1249–52. doi: 10.1136/jcp.35.11.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stuehr DJ, Gross SS, Sakuma I, Levi R, Nathan CF. Activated murine macrophages secrete a metabolite of arginine with the bioactivity of endothelium-derived relaxing factor and the chemical reactivity of nitric oxide. J Exp Med. 1989;169:1011–20. doi: 10.1084/jem.169.3.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Victor VM, Rocha M, Esplugues JV, De la Fuente M. Role of free radicals in sepsis: antioxidant therapy. Curr Pharm Des. 2005;11:3141–58. doi: 10.2174/1381612054864894. [DOI] [PubMed] [Google Scholar]
- 30.Fubini B, Hubbard A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic Biol Med. 2003;34:1507–16. doi: 10.1016/s0891-5849(03)00149-7. [DOI] [PubMed] [Google Scholar]
- 31.Cobb JP, Hotchkiss RS, Swanson PE, et al. Inducible nitric oxide synthase (iNOS) gene deficiency increases the mortality of sepsis in mice. Surgery. 1999;126:438–42. [PubMed] [Google Scholar]
- 32.Hollenberg SM, Broussard M, Osman J, Parrillo JE. Increased microvascular reactivity and improved mortality in septic mice lacking inducible nitric oxide synthase. Circ Res. 2000;86:774–8. doi: 10.1161/01.res.86.7.774. [DOI] [PubMed] [Google Scholar]
- 33.Lidington D, Li F, Tyml K. Deletion of neuronal NOS prevents impaired vasodilation in septic mouse skeletal muscle. Cardiovasc Res. 2007;74:151–8. doi: 10.1016/j.cardiores.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 34.Weberpals M, Hermes M, Hermann S, et al. NOS2 gene deficiency protects from sepsis-induced long-term cognitive deficits. J Neurosci. 2009;29:14177–84. doi: 10.1523/JNEUROSCI.3238-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duma D, Fernandes D, Bonini MG, Stadler K, Mason RP, Assreuy J. NOS-1-derived NO is an essential triggering signal for the development of systemic inflammatory responses. Eur J Pharmacol. 2011;668:285–92. doi: 10.1016/j.ejphar.2011.05.065. [DOI] [PubMed] [Google Scholar]
- 36.Gross A, Spiesser S, Terraza A, Rouot B, Caron E, Dornand J. Expression and bactericidal activity of nitric oxide synthase in Brucella suis-infected murine macrophages. Infect Immun. 1998;66:1309–16. doi: 10.1128/iai.66.4.1309-1316.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alam MS, Akaike T, Okamoto S, et al. Role of nitric oxide in host defense in murine salmonellosis as a function of its antibacterial and antiapoptotic activities. Infect Immun. 2002;70:3130–42. doi: 10.1128/IAI.70.6.3130-3142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eriksson S, Chambers BJ, Rhen M. Nitric oxide produced by murine dendritic cells is cytotoxic for intracellular Salmonella enterica sv. Typhimurium. Scand J Immunol. 2003;58:493–502. doi: 10.1046/j.1365-3083.2003.01330.x. [DOI] [PubMed] [Google Scholar]
- 39.Ketteler M, Cetto C, Kirdorf M, Jeschke GS, Schafer JH, Distler A. Nitric oxide in sepsis-syndrome: potential treatment of septic shock by nitric oxide synthase antagonists. Kidney Int. 1998;64:27–30. [PubMed] [Google Scholar]
- 40.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov. 2005;4:854–65. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- 41.Ebong S, Call D, Nemzek J, Bolgos G, Newcomb D, Remick D. Immunopathologic alterations in murine models of sepsis of increasing severity. Infect Immun. 1999;67:6603–10. doi: 10.1128/iai.67.12.6603-6610.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turnbull IR, Buchman TG, Javadi P, Woolsey CA, Hotchkiss RS, Karl IE, Coopersmith CM. Age disproportionately increases sepsis-induced apoptosis in the spleen and gut epithelium. Shock. 2004;22:364–8. doi: 10.1097/01.shk.0000142552.77473.7d. [DOI] [PubMed] [Google Scholar]
- 43.De Maio A, Torres MB, Reeves RH. Genetic determinants influencing the response to injury, inflammation, and sepsis. Shock. 2005;23:11–7. doi: 10.1097/01.shk.0000144134.03598.c5. [DOI] [PubMed] [Google Scholar]
- 44.Dienstknecht T, Schwacha MG, Kang SC, Rue LW, Bland KI, Chaudry IH. Sex steroid-mediated regulation of macrophage/monocyte function in a two-hit model of trauma-hemorrhage and sepsis. Cytokine. 2004;25:110–8. doi: 10.1016/j.cyto.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 45.Zellweger R, Zhu XH, Wichmann MW, Ayala A, DeMaso CM, Chaudry IH. Prolactin administration following hemorrhagic shock improves macrophage cytokine release capacity and decreases mortality from subsequent sepsis. J Immunol. 1996;157:5748–54. [PubMed] [Google Scholar]
- 46.Nabi IR. Cavin fever: regulating caveolae. Nat Cell Biol. 2009;11:789–91. doi: 10.1038/ncb0709-789. [DOI] [PubMed] [Google Scholar]
- 47.Hansen CG, Bright NA, Howard G, Nichols BJ. SDPR induces membrane curvature and functions in the formation of caveolae. Nat Cell Biol. 2009;11:807–14. doi: 10.1038/ncb1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu L, Brown D, McKee M, et al. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 2008;8:310–7. doi: 10.1016/j.cmet.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.