

Abstract
We have devised a method for isolating virtually pure and comprehensive libraries of restriction fragments that contained replication initiation sites (bubbles) in vivo. We have now sequenced and mapped the bubble-containing fragments from GM06990, a near-normal EBV-transformed lymphoblastoid cell line, and have compared origin distributions with a comprehensive replication timing study recently published for this cell line. We find that early-firing origins, which represent ∼32% of all origins, overwhelmingly represent zones, associate only marginally with active transcription units, are localized within large domains of open chromatin, and are significantly associated with DNase I hypersensitivity. Origin “density” falls from early- to mid-S-phase, but rises again in late S-phase to levels only 17% lower than in early S-phase. Unexpectedly, late origin density calculated on the 1-Mb scale increases as a function of increasing chromatin compaction. Furthermore, the median efficiency of origins in late-replicating, heterochromatic domains is only 25% lower than in early-replicating euchromatic loci. Thus, the activation of early- and late-firing origins must be regulated by quintessentially different mechanisms. The aggregate data can be unified into a model in which initiation site selection is driven almost entirely by epigenetic factors that fashion both the long-range and local chromatin environments, with underlying DNA sequence and local transcriptional activity playing only minor roles. Importantly, the comprehensive origin map we have prepared for GM06990 overlaps moderately well with origin maps recently reported for the genomes of four different human cell lines based on the distributions of small nascent strands.
There are estimated to be at least 50,000 origins of replication embedded in the six billion base pairs that constitute the typical mammalian somatic genome. A few dozen origins have been localized and analyzed by molecular biological approaches after enormous effort and expense, and fall into two distinct classes: (1) zones of clustered, inefficient, initiation sites; and (2) very circumscribed sites analogous to the classic genetic replicators of bacteria and the simple yeasts (for reviews, see Aladjem 2007; Hamlin et al. 2008). These data raise the question whether true replicators exist in mammalian genomes. This question has been difficult to address by the standard autonomously replicating sequence (ARS) assay used to identify replicators in bacteria and yeast, since virtually any DNA fragment from any source replicates to some extent when transfected into a suitable mammalian host cell (Krysan et al. 1989; Lin et al. 2005). Thus, any shared, relevant sequence elements have been difficult to identify by standard approaches. Whether the two origin types distribute differently relative to the nature of the local genes or other aspects of chromatin architecture is largely unknown, owing to the small number of origins that have been characterized within a given cell type or species. Furthermore, most of the known mammalian origins are early-firing, as they were identified near active genes for which mapping data and relevant reagents were already available (Aladjem and Fanning 2004; Aladjem 2007; Hamlin et al. 2008). Therefore, little is known about the distributions and natures of origins in mid- and late-replicating regions.
Because physical access to these important elements/regions is paramount to successful orchestration of the DNA synthetic program, identifying origins and their epigenetic characteristics is extremely important in understanding the nature of the information embedded in the genome. To paint a reliable, unbiased, picture of mammalian origin structure and function on a genome-wide scale, methods were clearly needed for isolating all of the origins in any given mammalian cell line—preferably one for which there are large publicly available data sets relating transcription patterns and chromatin characteristics to the underlying DNA sequences.
In a genome-wide study, the challenge is to identify origins no matter when they fire in the S-period, and then to map them onto the genome. Since replication forks diverge from origins, the various identification schemes depend in some way on monitoring fork direction or detecting the smallest nascent strands (NSs) surrounding initiation sites. By and large, attention has been focused on origins that fire at the beginning of S-phase in cell populations obtained either after synchronizing regimens or by cell sorting; this approach eliminates the complication of read-through replication of inefficient origins, which assigns them to both early and later S-phase timing windows. The latter problem is exemplified by recent whole-genome studies that have been only partially successful at localizing origin positions based on the dynamics of replication timing (for review, see Gilbert 2010).
Theoretically, the only successful genome-wide approach prepares saturating collections of validated origins and identifies their genomic positions either by hybridization to genomic tiling microarrays or by sequencing. In both cases, the origin identification scheme has to end up with a physical product for analysis. Only two approaches actually yield such a product. In the first of these, purified genomic DNA preparations are heated above the average genomic melting temperature to release the small NSs centered over replication start sites (Vassilev and Johnson 1989). Since the melting procedure itself generates overwhelming amounts of fragmented, non-origin DNA, an important additional step uses lambda exonuclease to attempt to digest contaminating non-replicating DNA strands that are not primed by RNA (Bielinsky and Gerbi 1998).
Several recent reports have prepared human origin maps based on the distributions of these small NS preparations (Cadoret et al. 2008; Karnani et al. 2010; Martin et al. 2011). Disappointingly, overlaps among these NS preparations are marginal—even within the same cell line (Cadoret et al. 2008; Karnani et al. 2010) and, in some cases, even within the same laboratory (Cadoret et al. 2008) .Unfortunately, since methodological details are necessarily brief in these reports, it is difficult to uncover the possible reasons for discordance. As we suggested previously (Mesner et al. 2011), a lack of concordance may possibly result from a lack of saturation resulting from the small number of starting cells (∼108), as well as the very short time period in vivo between RNA primer synthesis and its removal during synthesis of the next Okazaki fragment on the retrograde arm of the replication fork (Balakrishnan and Bambara 2011). However, in none of these studies has the product actually been validated by an independent origin identification scheme, although in one study, the presence of the origin recognition complex (ORC) (Bell and Stillman 1992) at several of the identified sites was verified (Karnani et al. 2010). Importantly, stringent validation schemes have since been published by other laboratories studying murine and other higher eukaryotic replication programs utilizing the NS origin isolation method (e.g., Cayrou et al. 2012b).
In a different approach, we have taken advantage of the circular nature of fragments containing replication bubbles to trap them in gelling agarose (Mesner et al. 2006). We showed in a pilot study on Chinese hamster ovary cells that the method yields almost pure (∼90%) populations of restriction fragments that sustained initiation events in vivo. In a subsequent study, more than 106 independent fragments were isolated and cloned from both synchronized and log-phase human HeLa cells, as well as from log-phase human GM06990 lymphoblastoid cells. These samples were then hybridized to microarrays representing the 1% of the human genome selected for study by the human ENCODE Project (The ENCODE Project Consortium 2007; Mesner et al. 2011). The excellent reproducibility of biological replicates suggested that these origin preparations might be close to saturating (77% of the smaller library’s clones were also found in the larger library). Importantly, the very low background levels concurred with 2D gel replicon mapping studies, suggesting that these human origin libraries were very pure.
We were able to reliably map origins within the 44 ENCODE pilot regions and to relate their positions to local transcriptional activity as well as to the few chromatin features then available through publicly available databases (Mesner et al. 2011). The results supported a model in which most origins correspond to zones of clustered, inefficient initiation sites that are localized to open chromatin. Surprisingly, however, origins were positioned not only within intergenic regions, but many overlapped or resided within both active and inactive genes. This suggested that the underlying sequence of initiation sites might be less important than the epigenetic features that characterize their environments.
Because of the artificial boundaries that define the 44 ENCODE regions, as well as the largely arbitrary nature of the majority of the selected genomic loci, the origin maps we and others have prepared are necessarily biased and, furthermore, cannot be related in a meaningful way to the larger domain structure of chromatin modifications and transcriptional activity. In addition, since our previous study, an enormous amount of genome-wide epigenetic information has accrued via the ENCODE Consortium (The ENCODE Project Consortium 2012).
The success of the pilot studies, access to high-throughput sequencing technology, and the availability of large, validated libraries has allowed us to prepare comprehensive genome-wide origin maps. Of special interest are the GM06990 origin libraries, which derive from an EBV-immortalized lymphoblastoid cell line with an essentially normal karyotype (http://www.genome.gov/12513455). The recent construction of high-quality replication timing profiles for GM06990 obtained by cell sorting (Hansen et al. 2010) allowed us for the first time to classify origins of replication according to firing times on a genome-wide scale. We relate these origin maps to a companion transcriptome for GM06990, as well as to DNase I hypersensitivity, relevant chromatin marks, and Hi-C data.
The genome-wide picture that emerges validates most of the studies that have been carried out on individual origins. Importantly, this global view provides information on mid- and late-firing origins, for which almost nothing was known previously owing to the inability to study their activities in log-phase cell populations. This has allowed us to uncover an important new paradigm for epigenetic differences between early- and late-firing origins. Finally, we have compared the bubble map to recently published small nascent strand (NS) maps for the genomes of four different human cell lines (Besnard et al. 2012). Although there is good concordance between bubbles and NS distributions in many regions of the genome, there is serious discordance in many others. Our results will be discussed in light of these recent contradictions.
Results
Origin purification, validation, and definition
We previously described the preparation of origin libraries from log-phase GM06990 lymphoblastoid cells (Mesner et al. 2011). Two independent trapping experiments (B1 and B2) were performed on replication intermediates (RIs) prepared from ∼1010 cells each. A sensitive 2D gel analysis of the trapped material suggested that 85%–90% of trapped fragments contained replication bubbles in vivo. After cloning, pooled DNAs from each library were sequenced in parallel lanes of the Illumina Genome Analyzer II (two samples from the B1 library and one from B2). We were able to map 10.0 M, 17.7 M, and 14.9 M reads to the human genome (hg18), respectively, using the read-mapping tool, BWA (Li and Durbin 2009).
To establish statistically significant read levels for library fragments, we first calculated a robust estimate of the read depth (RD), defined as the number of reads in each EcoRI fragment divided by the fragment length in base pairs. We then fit the low end of the RD distribution (background or noise) to a negative binomial distribution, and defined bona fide bubble-containing fragments at a false discovery rate (FDR) of 0.1% (Fig. 1A; Methods). Note that the negative binomial distribution has been shown to successfully model the noisy end of ChIP-seq data (Ji et al. 2008), and fits the low end of our RD data much better than a Poisson or Gaussian distribution (not shown). The validity of this cutoff value also is supported by the observation that fragments narrowly exceeding it display reads throughout the majority of their lengths, which terminate abruptly at the EcoRI sites (Fig. 1C). Furthermore, signals from the integrated EBV genome, which serves as an internal control, are consistent with replicon mapping studies showing that every fragment within the viral genome initiates replication at a low level (Supplemental Table S1; Norio and Schildkraut 2004). Finally, we applied filters to remove artifacts such as EcoRI fragments <500 bp, most of which contained alpha-satellite and other low complexity DNA sequences. These small inserts are greatly overrepresented in the cloned libraries undoubtedly because of a bias in cloning efficiency, as the uncloned material was greatly depleted of fragments <500 bp in length (LD Mesner, unpubl.). Any larger fragments containing ≥80% alpha-satellite also were excluded.
Figure 1.

Establishing and validating the read-depth (RD) cutoff value and the negative fragment exclusion rule for zones. (A) Distribution of read depths multiplied by 10,000 and rounded to the nearest integer (gray bins), and the negative binomial distribution (red) fitted to the lowest RDs in the sequencing data. The fitted negative binomial distribution was used to calculate P-values and false discovery rates (FDRs) for every EcoRI fragment in the genome. (B) Scatter plot of the fraction of each EcoRI fragment that is covered by reads (y-axis) versus EcoRI RD for all EcoRI fragments. The red line represents a moving average calculated using the loess package in R (Becker et al. 1988). As can be seen, the 0.0144 RD cutoff applied to the combined sequencing data sets corresponds to ∼60% coverage of EcoRI fragments on average. (C) IGV screen shot of EcoRI fragment boundaries, sequencing read profiles (blue), RDs calculated within EcoRI fragments (fuchsia), and fragments that pass the 0.1% FDR cutoff value of 0.0144 (red). This 30-kb region highlights a typical fragment (left of center) whose RD just surpassed the cutoff, as well as the constrained RD pile-ups within EcoRI fragment boundaries. (D) Histogram of the number of negative EcoRI fragments between any two bubble-containing fragments in the genome (gray bins), and the discrete distribution of the null model 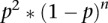 after scaling (red dots). p is the probability (0.1599365) of finding a bubble-containing fragment in the genome and n is the number of negative fragments between two bubble-containing fragments (See Supplemental Methods for details). Based on the ∼3.6-fold enrichment of actual data over that predicted by the null model for n = 1 (i.e., one negative fragment between two positive ones), a one-fragment joining rule was adopted wherein a “zone” was defined as a cluster of adjacent bubble-containing EcoRI fragments, no two of which are separated by more than one negative fragment.
after scaling (red dots). p is the probability (0.1599365) of finding a bubble-containing fragment in the genome and n is the number of negative fragments between two bubble-containing fragments (See Supplemental Methods for details). Based on the ∼3.6-fold enrichment of actual data over that predicted by the null model for n = 1 (i.e., one negative fragment between two positive ones), a one-fragment joining rule was adopted wherein a “zone” was defined as a cluster of adjacent bubble-containing EcoRI fragments, no two of which are separated by more than one negative fragment.
We identified 34,184, 39,971, and 36,225 bubble-containing fragments in the B1A, B1B, and B2 GM06990 samples at a 0.1% FDR, which corresponded to RD cutoffs of 0.0081, 0.0139, and 0.0123, respectively. To assess the reproducibility of the trapping and sequencing procedures, we made pairwise comparisons among the three data sets. In this analysis, 72% of B1A calls were also scored in the B1B sequence replicate, while 62% of B1B calls were found in B1A. For the biological replicates, 22.4% of the combined B1 calls (76,891) also were called in the B2 library and 47.6% of B2 calls coincided with those in B1, for a total of 59,655 unique fragments. Given that only 7%–8% of the genome is covered by bubble-containing fragments in each data set, these overlaps are highly significant. However, even though the GM06990 bubble libraries are of similar purities as the HeLa libraries we reported on previously (∼90%; Mesner et al. 2011), these overlaps are less impressive than replicates in that study in which the libraries were hybridized to ENCODE microarrays (89.9% and 84.0% for technical replicates and 77.1% and 42.9% for biological replicates). The apparent reasons for these discrepancies will be addressed below.
Reads from the three samples were combined and remapped to the genome, and a 0.1% FDR cutoff was applied together with the filters described above, which yielded 123,297 bubble-containing fragments. This cutoff corresponds to an RD value of 0.0144, which represents ∼60% coverage of each fragment on average (Fig. 1B). This makes intuitive sense, since an EcoRI fragment sequenced to 1× coverage with ∼64-bp reads corresponds to a slightly larger RD value of 0.0156 (1/64). Within the 1% of the genome covered by the ENCODE pilot regions, 1362 bubble-containing fragments were identified by sequencing in the present study, while 988 were identified in the previous microarray study (Mesner et al. 2011) using the same 0.1% FDR cutoff. Thus, the depth of sequencing we have performed here is clearly more sensitive than the previous microarray analysis. The two methods detected 631 shared fragments, which corresponded to 46% of those identified by sequencing and 64% of those identified with microarrays. Given that the percentage of bubble-containing fragments identified by bubble-chip and bubble-seq analysis represent 16% and 22%, respectively, of all genomic EcoRI fragments in the ENCODE regions, these overlaps are highly significant.
Note that large numbers of fragments in each of the sequencing data sets had RDs just below the respective library’s cutoff, but when the three data sets were combined, their RDs were elevated above the newly established cutoff. As we will show below, the lower concordance among replicates in the present study results primarily from an important difference in the genomic distributions of early- and late-firing origins, as well as a dramatic difference in their epigenetic characteristics.
Painting the origin landscape of a near-normal human genome
The 123,297 origin-containing EcoRI fragments that we mapped in GM06990 cells comprise 24.35% of the genome. Given the lower sensitivity of the microarray analysis, this value is consistent with the coverage we reported for origin fragments in GM06990 in the ENCODE pilot regions (21.8%; Mesner et al. 2011). A representative distribution of bubble-containing fragments for a 90-Mb region comprising most of chr14 is shown in Figure 2A (labeled “Comb”). Even in this low-resolution view, it is clear that the RDs of fragments vary widely along the chromosome, with some of the more efficient origins clustering amid long stretches of less efficient ones.
Figure 2.

Origin distributions differ widely in both read depths (RDs) and reproducibility in different segments of the human genome. Integrative Genomics Viewer (IGV) screen shots of bubble-containing fragment RDs within a ∼90-Mb region of chr14. (A) TR50: time at which 50% of the DNA is replicated (scale: 0 to 6, representing the early-to-late-replication time windows defined in the text; Jeon et al. 2005). Comb: origin calls for the aggregate B1A, B1B, and B2 data sets (see text), where B1 and B2 correspond to biological replicate libraries, and B1A and B1B to sequencing replicates of the B1 library. Calls: bubble-containing EcoRI fragments with RDs greater than the respective cutoffs (0.0081 for B1A, 0.0139 for B1B, 0.0123 for B2, and 0.0144 for the combined set; Supplemental Table SI). (B) A 600-kb generally early-replicating region of chr14 corresponding to the region delimited by the green box in A. The scales of the IGV graphs were adjusted to aid comparison so that the highest RD in each data set corresponded to 100%, with the absolute range of values as follows: B1A, 0–0.273; B1B, 0–0.145; B2, 0–0.198; and Comb, 0–0.477. (C) A 600-kb late-replicating region of chr14 corresponding to the region outlined with the red rectangle in A. This IGV view has been expanded vertically by 2.5-fold to aid comparison of the four different data sets. The absolute scales remain the same except for this correction.
Of the bubble-containing fragments identified in the present genome-wide study, 61.9% are contained within zones of two or more adjacent fragments and 38.1% are isolated. Note that a single negative fragment was considered to be part of a zone if it was adjacent to two positive fragments, based on a statistically significant 3.6-fold enrichment of single fragment interruptions over that predicted by a null model (Fig. 1D). Such fragments could represent part of a zone that does not support initiation (as with the Chinese hamster dihydrofolate reductase origin; Dijkwel et al. 2002), may result from a lack of saturation in the biological or sequencing samples, or, less likely, may represent bona fide false negatives.
The mean and median “isolated” fragment sizes are comparable to those for “zonal” bubble-containing fragments (Supplemental Table SIIC). As discussed previously (Mesner et al. 2011), these fragments are larger on average than the mean genomic EcoRI fragment (Supplemental Table SII) because the isolation scheme selects somewhat against smaller fragments in which the dwell times of bubbles are shorter, and because branch migration of NSs during isolation is more likely. The mean number of fragments in zones was found to be 3.63 (including any single negative fragments), while the mean and median zone sizes are 20 kb and 16 kb, respectively. The largest zone is located on chromosome 16, contains 38 EcoRI fragments, and is 373 kb in length.
Summarizing GM06990 DNA replication timing data for assigning origin firing times
Most of the few dozen mammalian origins that have been studied at the molecular level are early-firing, since they were initially identified by labeling with DNA precursors as cells entered S-phase (for reviews, see Aladjem and Fanning 2004; Hamlin et al. 2008). However, there are a few examples of mid- and late-firing origins (Ma et al. 1990; Aladjem 2004), and little is known about their relative numbers genome-wide, their regulation, or their distributions vis-à-vis other features of the genome. Although DNA synthesized at the very beginning of S-phase is sure to contain early-firing origins, DNA synthesized at later times represents a mixture of later-firing origins and DNA passively replicated by forks from distant earlier-firing ones. Thus, identifying mid- and late-firing origins based on timing data alone has proved difficult in mammalian cells (for review, see Gilbert 2010), and even in the simple genomes of yeast (Czajkowsky et al. 2008; Yang et al. 2010).
To classify origins according to firing times, we compared the GM06990 origin map to a careful replication timing study recently published for this same cell line (Hansen et al. 2010). DNA was isolated from log-phase cells flow-sorted into six successive cell cycle intervals according to DNA content (designated G1b, S1–S4, and G2; Hansen et al. 2010). The sequences of the labeled DNA were then mapped onto the reference human genome. To summarize this timing data for comparison to the origin map, we calculated the time during S-phase by which 50% of the DNA is replicated for each EcoRI fragment, as well as for 50-kb and 1-Mb sliding windows (termed the TR50; Karnani et al. 2007).
The TR50 values were estimated as follows: (1) The enriched read profiles for each time interval were normalized as described in Supplemental Methods; (2) the normalized number of reads for the six S-phase intervals were then summed in each EcoRI fragment or sliding window to arrive at the 100% replication value; and (3) the time by which 50% of that EcoRI fragment or sliding window is replicated was determined by interpolation (see Supplemental Methods). Note that if the replication time is localized to a relatively narrow window during S-phase and the distribution of replicated DNA is symmetric about that time, the TR50 corresponds to the mode, mean, and median of that EcoRI fragment or sliding window. TR50s ranged from 0 (earliest) to 6 (latest), where we simply indexed them to the six cell cycle intervals from which the replicated DNA fractions were recovered (i.e., 0–1, 1–2, etc.). DNA replication times were classified as “early” if the TR50 was less than or equal to 2, “mid” if it was greater than 2 but less than or equal to 4, and “late” if the TR50 was greater than 4.
We additionally characterized the extent to which the replication of a given genomic segment is either confined to a particular part of S-phase or is more broadly distributed, by estimating the “variance” of replication times about the TR50 value (see Supplemental Methods). Timing variances ranged from 0 (all the reads occur in a single interval) to 12 (equal numbers of reads occur in the first and last intervals but none in between). A variance of 3.17 corresponds to equal amounts of DNA replicated in each one of the six time intervals (a so-called pan-S replication pattern) (Jeon et al. 2005; Karnani et al. 2007).
Raw replication timing data for the six S-phase intervals (G1b, S1–4, and G2) are shown in Supplemental Figure S2. Also shown are the smoothed TR50s (orange plot) and the variances (purple plot) calculated for overlapping 50-kb windows. Four illustrative ∼1-Mb genomic segments are presented: (1) a region with a low TR50 (early-replicating) and low variance (Supplemental Fig. S2A); (2) a region with a high TR50 (late-replicating) and low variance (Supplemental Fig. S2B); (3) a region with a generally low TR50 but high variance (Supplemental Fig. S2C); and (4) a region with a high TR50 and relatively high variance (Supplemental Fig. S2D).
It is clear that selected segments of the genome can replicate in relatively narrow timing windows. For early- and late-replicating DNA segments with low variances (as in Supplemental Fig. S2A,B), a probable explanation is that their origins are reasonably efficient and the sequences replicated from these origins are reasonably close together, so that a relatively inactive initiation site does not have to wait long to be replicated from a neighboring origin. Regions that display high variances (as in Supplemental Fig. S2C,D) are the regions that are the most difficult to classify vis-à-vis origin distributions based solely on timing data (see Discussion).
Relating origin distributions and activities to replication timing
With TR50s in hand for the GM06990 genome, we were able to categorize members of the origin collection according to replication times (note that 123,076 of 123,297 bubble-containing fragments mapped to regions for which timing sequence data were available) (Supplemental Table SIIB). In Figure 2A, the origin map and TR50s are compared for a large part of chr14. The region outlined with the green rectangle in Figure 2A is expanded horizontally ∼60-fold in Figure 2B, and now includes the three individual data sets used to compile the combined RDs. The red box in Panel A outlines an adjacent, late-replicating region also expanded ∼60-fold in the horizontal direction in Figure 2C, but also about 2.5-fold in the vertical direction to aid comparison among data sets. Clearly, there are fundamental differences between early- and late-replicating DNA in origin distributions and activities.
“Early-firing” bubble-containing fragments appear to be focused within relatively narrow domains (i.e., clusters are surrounded on each side by relatively vacant origin landscapes), and the highest RDs are considerably higher than those that fire in late S-phase. Furthermore, the B1A, B1B, and B2 samples overall appear to detect the same spectrum of fragments (compare the RD values to one another and to the combined data set represented by the dark blue map in Fig. 2B).
On the other hand, bubble-containing fragments in the “late-firing” domain outlined with the red box in Figure 2A and expanded in Figure 2C are less efficient than early-firing ones judging from their RDs, and the individual sequence reads for the three replicates are far less reproducible (in Fig. 2B,C, yellow ovals encompass a few examples of calls made in fewer than all three samples). Indeed, for late-firing origins (but not early-firing ones), combining the three data sets clearly increases the number of detectable bubble-containing fragments two- to threefold. This is visually apparent in the lower panel in Figure 2C. Thus, the distribution of late-firing origins vis-à-vis the template could possibly be stochastic.
Substantiating these visual impressions are the statistics themselves. For “early-firing” fragments, correlation coefficients are quite reasonable for both technical and biological replicate RDs (0.62 for B1A versus B1B, 0.55 for B1A versus B2, and 0.59 for B1B versus B2) (Supplemental Fig. S2A–C). For “late-firing” fragments, however, only the comparison of sequencing replicate RDs yielded a reasonable correlation coefficient (0.40 for B1A versus B1B, 0.20 for B1A versus B2, and 0.08 for B1B versus B2) (Supplemental Fig. S2G–I). The coefficients for mid-firing bubbles were similarly unimpressive (Supplemental Fig. S2D–F). Furthermore, based on replication timing data, only 23.7% of the genome sorted into the early-replicating G1b and S1 channels, compared with 35.3% and 32.3% for mid- (S2 + S3) and late (S4 + G2) S-phase, respectively (Supplemental Table SIVA; note that contigs were not mapped for ∼7.3% of the genome and timing data were not available for an additional 1.4%). Yet, 31.9%, 31.9%, and 36.2% of the 123,076 bubble-containing fragments for which timing data were available reside within early-, mid-, and late-replicating chromosomal domains (Supplemental Table SIVB).
Using simple ratios for each timing window, we determined the normalized average “density” of origins in early-, mid-, and late-replicating regions (i.e., the percentage of bubble-containing fragments in each timing sector divided by the percentage of the genome that partitions to that sector). The densities are, respectively, 1.34, 0.90, and 1.12. Surprisingly, therefore, the density of late-firing origins is only ∼17% less than early-firing origins. Furthermore, the typical “efficiency” (median RD) for origins in late-replicating, heterochromatic environments defined by a negative Hi-C eigenvector (HCE) value is only 25% less than that of origins in early-replicating euchromatic environments (Supplemental Table SIIE).
We also found that origins in early-replicating regions are skewed toward initiation zones: Applying the one-fragment rule, 78.7% of early-firing bubble-containing fragments reside in zones, while 61.4% and 60.3% of those that fire in mid- and late-S-phase reside in zones, respectively (Supplemental Table SIID; Fig. 2B,C). Early-firing initiation zones also contain larger numbers of fragments on average than do those in mid- and late-replicating regions (Supplemental Table SIID).
Again, it is surprising that the actual number of bubble-containing fragments (whether isolated or zonal) is larger in late- relative to early-replicating domains (44,527 versus 39,236; Supplemental Table SIIB). This is because late-firing origins are comprised mainly of isolated fragments or small zones and are more densely and uniformly spaced (compare “Comb” data in Fig. 2B,C). In fact, the distributions of end-to-end distances between origins (isolated fragments and zones treated as units) are narrower and peak at shorter intervals in late-replicating DNA (15.4 kb on average; Supplemental Fig. S4D) compared with those in early-replicating regions (22.7 kb; Supplemental Fig. S4A). To further characterize the relationships between replication timing and origin distributions, we generated boxplots of the standard deviation of replication timing about the TR50 value (i.e., the square root of the variance described above) for bubble-containing fragments and for all genomic EcoRI fragments in the same timing window. Except for very late-replicating regions (TR50 > 5), the distribution of timing variations is somewhat narrower for bubble-containing fragments than for all genomic fragments in the same window (Fig. 3 cf. A and B). Furthermore, there is a dramatic monotonic decrease in TR50s toward earlier replication times as the number of fragments in zones increases (Fig. 3C), as well as a monotonic narrowing of the distribution of variation in timing, as specifically applied here to initiation zones (Fig. 3D). These data are consistent with stochastic origin-firing models that were fit to Saccharomyces cerevisiae replication timing data (see Discussion and Yang et al. 2010).
Figure 3.

Larger initiation zones replicate earlier on average and with less variable replication times than smaller zones. (A,B) Boxplots of SDs about TR50s as a function of S-phase window for all genomic EcoRI fragments (A), and for all bubble-containing fragments (B). (C) Boxplot of TR50s as a function of fragment number for isolated fragments (green) and zones (blue). (D) Boxplots of SDs about TR50s for all genomic isolated fragments (green) and for zones (blue) of increasing fragment numbers.
Associations between active transcription units and origins
In our previous studies on the ENCODE pilot regions, origin distributions were compared to the published transcriptomes for HeLa and GM06990 cell lines (Mesner et al. 2011). For GM06990, we found that, in the ENCODE regions, initiation zones were significantly associated with the 5′ ends of transcribed genes, as well as with transcribed genes that were completely encompassed by a zone. Importantly, however, these categories together accounted for only 20% of all origin positions in the GM06990 ENCODE regions in log-phase cells, suggesting that there are significant drivers of origin activity other than transcription per se. To extend this comparison to the entire genome in a more pristine way, we elaborated a companion transcriptome with replicate cDNA libraries constructed from log-phase GM06990 cells propagated in our own laboratory under the same conditions used to isolate origins, and compared bubble distributions to transcribed and non-transcribed genes (see Methods for RNA preparation and sequencing procedures).
To estimate the significance of overlaps between origins and genes (active or inactive), we adopted a random permutation null model that we developed for the earlier analysis of bubble distributions in the ENCODE pilot regions (Mesner et al. 2011). Specifically, the genome was divided into 1-Mb windows within which all genomic EcoRI fragments were randomly permuted 10,000 times (see Methods). The bubble-containing fragments in each iteration were then tracked for further comparisons (in this case, to transcription features). This approach minimizes local biases related to GC content, gene density, and chromatin state. For overlap estimates within particular timing windows, the TR50 value then was determined for each bubble-containing fragment (see Supplemental Methods).
As in the previous study (Mesner et al. 2011), bubble-containing EcoRI fragments were sorted into nine different gene-related categories (Fig. 4E), and the significance of their associations with each category was calculated for the genome as a whole and for fragments residing in each of the timing windows (Methods; Supplemental Table SV). In agreement with earlier studies, for the genome as a whole, bubble-containing fragments containing active genes or overlapping their 5′ ends (categories 2 and 3) displayed the lowest P-values and the highest fold enrichments over mean random. However, this analysis was less informative for distributions within each timing window, because the actual bubble density in early-replicating DNA is ∼1.5-fold higher than in the genome as a whole, with the result that all but category 5 fragments are significantly enriched over mean random (Supplemental Table SV).
Figure 4.

The majority of origins reside in non-transcribed chromosomal regions regardless of their firing times, but are significantly associated with transcribed genes in early-replicating regions. (Left) Pie charts of origin distributions across the genome (A), early- (B), mid- (C), and late-replicating (D) regions among the nine annotation categories indicated by the key in E. (Right) Percentages of the genome represented by the nine annotation categories. See Supplemental Tables SIV and SV for numerical distributions and Methods for details of the statistical analysis.
Therefore, we calculated the percentage that each category represents of the total fragment numbers (actual and mean random) within each timing window (“normalized to fragment number” in Supplemental Table SV). When presented as pie charts (Fig. 4B–D), it is apparent that origin distributions are not strikingly different between actual and mean random values. However, the categories that display the highest fold-enrichment values for the genome as a whole and for early-firing origins are those that completely encompass a transcribed gene (category 2) and those that overlap the 5′ end of a transcribed gene (category 3): For the genome as a whole, these values are 1.77 and 1.80, respectively, and for early-firing origins, 1.43 and 1.49 (Supplemental Table SV; Fig. 4A,B). For mid-firing origins, only the overlaps with the 5′ ends of transcribed and non-transcribed genes were particularly enriched (categories 3 and 7) (Supplemental Table SVC). The only remarkable associations of bubbles in late-replicating regions are those containing a non-transcribed gene or overlapping its 5′ end (categories 6 and 7) (Supplemental Table SVD). We also found that origins are significantly depleted in three transcribed categories (1, 2, and 4), based on the ratios of the normalized actual to mean random values.
Associations of epigenetic factors with origins
Even though the overlap between bubble-containing fragments and the 5′ ends of transcribed genes is statistically significant in the genome as a whole, it is clear from Figure 4 that the overwhelming majority of origin activity in the GM06990 genome (regardless of S-phase interval) occurs on non-transcribed inter- and intragenic templates (i.e., all the blue and green sectors). These findings suggest that factors in addition to, or other than, transcription per se constitute the major drivers of origin activation and perhaps in selective ways. Therefore, we compared the origin map for GM06990 to distributions of epigenetic attributes available for this cell line via the ENCODE project (The ENCODE Project Consortium 2012). These included broad- and narrow-peak data for CTCF binding sites, DNase I hypersensitive sites (DNase I HSS), and H3K4me3, H3K27me3, and H3K36me3 histone modifications (Supplemental Table SVI). Broad-peak algorithms search for regions in which even modest positive signals persist over a statistically significant contiguous stretch of the chromosome (Landt et al. 2012). Narrow-peak algorithms determine significant peak heights irrespective of neighboring signal levels, based on a background null model constructed either from low-level signals in the data or in a control data set (e.g., DNA input; Landt et al. 2012).
The origin and epigenetic maps were compared across the entire genome, as well as for early-, mid- and late-replicating regions. The significance of each association was initially assessed with the random permutation null model (Methods). Results of these analyses are summarized in Supplemental Table SVI. For the “entire genomic” origin collection, all associations between origins and these epigenetic attributes appeared to be significant (P-values < 10−4), with the exception of two of the H3k27me3 comparisons (Supplemental Table SVIA). As a group, DNase I HSS signatures were modestly enriched in the actual bubble-containing fragments relative to the mean random values, as were H3K4me3 histone modifications (Supplemental Table SVI). When the analysis focused only on “early-replicating” regions, all associations again appeared to be significant, but now every category of interaction was highly enriched relative to mean random (Supplemental Table SVIB).
As with origin–gene interactions, this outcome is predicted by the very high density of actual origins in early-replicating DNA (the numerator) relative to mean random values (the denominator), and somewhat obscures the relative enrichments among the query categories. This, in turn, drives results for origin–factor overlaps in the genome as a whole. We therefore expressed the actual and mean random values as percentages of total bubble-containing fragments in each timing window, as we did for origin–gene interactions. This procedure resulted in more modest but informative fold enrichments among the various categories (Supplemental Table SVI). The highest overlaps for all genomic origins appear to be with narrow-peak DNase I HSS (Supplemental Table SVIA; Fig. 5), and with activating H3K4me3 narrow-peak sites (Supplemental Table SVIA). An average of 18% of bubbles overlap narrow-peak HSS, while 50% of HSS narrow-peak sites overlap bubble-containing fragments (calculated from Supplemental Table SVIIA). Likewise, 10% of bubbles overlap H3K4me3 narrow-peak sites, while 58% of the latter sites overlap bubble-containing fragments. Thus, a fraction of origins seems to require a DNA-accessible, activating chromatin environment.
Figure 5.

DNase I accessibility is strongly associated with origin activity in early-replicating chromatin. An IGV screen shot of a 25-Mb window of chromosome 22 displaying (from top to bottom) smoothed TR50 estimates over 50-kb windows in sliding steps of 1 kb, origin fragment RDs (bubbles), initiation zone calls (i.e., two or more adjacent bubble-containing fragments obeying the one-negative-fragment rule; see text), DNase I HSS read profiles at narrow-peak calls (Rep1), density of DNase I narrow-peak calls in 1-Mb windows (i.e., genomic coverage of the sites in base pairs divided by 1 Mb), DNase I HSS read profiles at broad-peak calls (Rep1), density of DNase I broad-peak calls in 1-Mb windows, transcript read profiles, and RefSeq gene annotations. (B,C) Boxplots of average DNase I HSS densities per bubble-containing fragment (i.e., genomic coverage of the sites across each EcoRI fragment divided by its length) as a function of origin fragment number, either in early-firing origins (B) or in all origins (C).
Data for early-firing bubbles are shown in Figure 5A and tabulated in Supplemental Table SVIB. Overlaps with DNase I and H3K4me3 narrow-peak sites again were the most substantial. In this case, an average of 36% of bubble-containing fragments overlap narrow-peak DNase I HSS, while an average of 56% of narrow-peak DNase I HSS overlap bubbles (Supplemental Table SVIID). For H3K4me3 interactions, 23% of bubbles overlap the narrow-peak sites, while 62% of narrow-peak sites overlap bubbles. Taken together, these data show that origins in early-replicating genomic regions manifest significant associations with activating characteristics. Importantly, no epigenetic factors were found to be significantly associated with origins in “mid-” or “late-replicating” regions.
As shown for the 90-Mb region in Figure 2A, origin distributions, replication timing, and gene densities and activities appear to be correlated on a megabase scale. These associations are likely to be driven by higher order chromatin structure. To attempt to quantify this phenomenon, we calculated the densities of origins and epigenetic marks in non-overlapping, contiguous 1-Mb windows over the entire genome. Note that density is defined here as the base pair coverage of bubbles or epigenetic sites divided by the base pair coverage of the window. We also recalculated TR50s and reclassified regions as early, mid-, or late-replicating on the same scale. Correlation coefficients for these associations and their P-values are summarized in Supplemental Table SVIII.
Not surprisingly, the highest correlations between origin fragments and calls for chromatin marks again were found in early-replicating regions. With one exception (H3K27me3), all Pearson correlation coefficients were between 0.74 and 0.87 (Supplemental Table SVIIIB-F). In contrast, the Pearson correlation between factor densities and origins was much more modest in “mid-replicating” regions (0.02 to 0.21), and was strongly anti-correlated in “late-replicating” regions (−0.42 to −0.61; see below and Supplemental Table SVIIIB–F). The “genome-wide” Pearson correlation coefficients between origin and epigenetic factor densities are somewhat lower (0.53–0.61), owing to the admixture of values for the three timing windows.
To address the question whether the predominant origin type (zonal) might rely on a particular epigenetic signature, we asked whether such associations were manifested in zonal origins with increasing fragment numbers. In Figure 5B,C are shown boxplots of DNase I HSS densities for early-firing bubble-containing fragments and for all genomic bubble-containing fragments as a function of fragment number. HSS densities clearly increase with increasing numbers of contiguous fragments in zones, and this trend is more dramatic in early-replicating regions than in the genome as a whole. While no epigenetic factors appear to be significantly associated with late-firing origins at the site level, for all epigenetic factors studied here, origin and factor densities calculated in 1-Mb intervals in late-replicating regions are strongly anti-correlated with such factors, ranging from −0.42 to −0.61 (Supplemental Table SVIII). Together, these data imply that origins in late-replicating regions tend to avoid not only activating epigenetic marks, but also repressive ones.
We also compared origin distributions to domains defined by the Hi-C eigenvector (HCE), which measures the physical interactions between genomic sequences and, among other things, reflects the relative degree of chromatin compaction in various regions: Positive values are associated with open chromatin and negative values with closed chromatin (Lieberman-Aiden et al. 2009; see Supplemental Methods for details of the HCE analysis). While the Pearson correlation coefficient between origin densities and the HCE values (positive and negative) was modest (0.26) (Supplemental Table SVIIIA), the correlation between the absolute values of HCE and origin densities was twofold higher (0.5) (Supplemental Table SVIIIA). The simplest explanation is that origin densities are highest in highly accessible (early-replicating) and, surprisingly, in highly compact (late-replicating) chromatin. Thus, unexpectedly, the density of late-firing origins increases in increasingly heterochromatic environments.
Comparison to genome-wide distributions of small NSs
A recent report describes genome-wide maps of replication origins for four different human cell lines (Besnard et al. 2012). These origin maps were elaborated with small nascent strands (NSs) ∼1.5 knt in length isolated by extrusion, sizing, and digestion of irrelevant non-RNA-primed DNA with lambda exonuclease. Importantly, these preparations were isolated from about the same number of cells as used in similar NS isolation schemes in the past (∼108), and whose maps displayed only modest concordance with one another (Cadoret et al. 2008; Karnani et al. 2010). The NS preparations in this latest study were sequenced to demonstrable saturation, and 200,000–250,000 origin sequences with a median length of ∼450 nt were identified in each cell line. As shown in Figure 6, the NS locations were remarkably concordant among the four cell lines (65%–84% pairwise overlaps; data from Besnard et al. 2012). This suggested to the authors that origin selection at the sequence level is only marginally affected by epigenetic differences resulting from developmental or tumorigenic status.
Figure 6.

Concordance among bubble-containing fragments, small nascent strands, active transcripts, and G-quadruplexes. IGV screen shots of demonstrative segments of chr 14 at increasing magnifications, comparing TR50, bubble-containing fragment calls, NS calls, GM06990 cDNAs, and the G-quadruplex sequence identified as shared by >90% of all NSs (Besnard et al. 2012). (A) An ∼8-Mb global view of the left end of chr 14 suggesting relatively good concordance between NS calls and bubble-containing fragments identified in the present report, particularly in early-replicating regions (i.e., low TR50s). (B) An ∼1-Mb region from the right end of chr 14 displaying two different subregions that exhibit both high and low concordance between NSs and bubble-containing fragments, as well as a clear association between the G-quadruplex sequence and NS (but not bubbles). (C) An ∼70-kb window of good concordance between NSs and bubble-containing fragments, illustrating the presence of NSs throughout the length of the larger bubble-containing fragments. IGV data scales or settings are as follows: TR50/0–5.4; GM06990 bubble distributions/0–0.3; G-quadruplex distributions/collapsed setting; NS distributions/0–400; and GM06990 cDNA distributions/0–0.5.
Because the GM06990 cell line was not among those examined in this NS study, and because we did not observe a high degree of overlap between bubble-containing fragments from two different cell lines (HeLa and GM06990) in our previous ENCODE pilot study (Mesner et al. 2011), we did not perform a detailed statistical analysis of overlaps between their NS calls and the present GM06990 origin map. However, as shown in Supplemental Table SIX, there is relatively good concordance with the NS assignments on a global scale (45%–46% of the NS calls fall within bubble-containing EcoRI fragments, while 33%–37% of bubble-containing fragments overlap NS calls, with P-values < 1 × 104 and fold increases over random ranging from 1.17 to 1.23). Furthermore, visual appraisal of the overlap between the two origin assignments in the birds-eye view of a typical 8-Mb region of the genome on the left end of chr 14 indicates that both approaches generally detect origin activity in the early-replicating regions of the genome, as might be predicted (Fig. 6A). At a more local level, however, there are many regions of marked discordance between the two data sets, as illustrated for an ∼1-Mb region at the right end of chr 14 (Fig. 6B). Nevertheless, in regions of high concordance (e.g., an early-replicating ∼70-kb region on the left end of chr 14; Fig. 6C), each of the bubble-containing EcoRI fragments in this zone of initiation sites overlaps several NSs, as we would predict from many earlier studies (e.g., Kalejta and Hamlin 1996; Dijkwel et al. 2002).
The study of Besnard et al. (2012) also identified a common repetitive sequence element that resides in 91%–95% of NSs that were isolated by the melting, sizing, and lambda exonuclease protocol from the four cell lines under study (the so-called G-quadruplex: G3 + N1–15 G3 + N1–15 G3 + N1–15 G3+; Bochman et al. 2012). This same sequence was uncovered in a large percentage of NSs isolated by a similar protocol from mouse and Drosophila melanogaster (Cayrou et al. 2011, 2012a). This particular quadruplex contains a minimum of three Gs, with loops of 1–15 nt. In fact, when we measured the distribution of these motifs among the entire ensemble of bubble-containing EcoRI fragments in the GM06990 genome, we found very little enrichment over that predicted by the random null model (1.05-fold) (Supplemental Table SIX). It is also interesting to note that in regions of discordance (e.g., as shown in Fig. 6B), the high concentration of NSs in the relatively depleted portion of the bubble landscape coincides rather closely with a very high concentration of G-quadruplexes, while the region that is more densely populated with bubbles occurs in a region which is relatively depleted of these G-rich sequences.
Discussion
Mammalian origins are organized into zones whose firing times are consistent with a stochastic firing-time model
The two GM06990 origin libraries we analyzed in the present study were obtained in two independent bubble-trapping experiments (Mesner et al. 2011). We demonstrated previously that ∼90% of the cloned EcoRI fragments in these libraries correspond to fragments that initiated replication in vivo, as demonstrated on 2D gels (Brewer and Fangman 1987; Mesner et al. 2006, 2011). Thus, these are the only reported human origin preparations that have been independently validated by a universally accepted origin mapping procedure. The GM06990 cell line originally was selected by the ENCODE Project for analyses of several epigenetic features (including Hi-C), and these data are publicly available (The ENCODE Project Consortium 2012). Here, we present a genome-wide origin map for GM06990, as well as origin distributions vis-à-vis a companion transcription map and other published epigenetic characteristics. Most importantly, a careful, comprehensive genome-wide replication timing study was recently published for GM06990 (Hansen et al. 2010), allowing us to assign firing times to the more than 120,000 origins that we have identified.
In our previous study (Mesner et al. 2011), two replicate HeLa cell libraries and one of the GM06990 libraries were analyzed by hybridization to microarrays representing the 44 ENCODE genomic regions (The ENCODE Project Consortium 2007). With some important exceptions, that circumscribed and somewhat biased view of origin distributions vis-à-vis other genomic elements proved to be a reasonably faithful predictor of the genome-wide view reported here. For example, the previous study suggested that 22% of the GM06990 genome is a template for replication initiation, while the more comprehensive whole-genome view suggests a value of 24%. In addition, the ENCODE hybridization study suggested that 46% of bubble-containing fragments reside within initiation zones in GM06990, while in the present study, 62% or 67% of all bubble-containing fragments (excluding or including single negative fragments) reside in zones.
The fact that so many bubble-containing fragments are arrayed contiguously in zones suggests that these two GM06990 libraries are close to comprehensive, particularly in early-replicating genomic regions. Clearly, a small number of start sites are lost from analysis because they lie too close to an EcoRI site or within a very small fragment. In fact, all of the mammalian libraries we have prepared are somewhat depleted of fragments less than ∼3 kb in length (Mesner et al. 2011; LD Mesner, unpubl.). This problem could be partially ameliorated by additionally using a different restriction enzyme to digest the DNA prior to trapping. In our view, however, the limited additional information gained does not justify the enormous effort and expense this would entail.
Our studies suggest that early-firing origins are more efficient, are usually organized into zones of initiation compressed epigenetically into permissive chromatin milieus, and inter-origin distances are more in line with expectations from earlier studies (Huberman and Riggs 1968). Conversely, there are 13% more late-firing origins, most of which correspond to isolated fragments or very small zones, and their inter-origin distances are shorter. A potentially major finding is that the distribution of late-firing origins may be stochastic on the scale of ≤100 kb. The fact that half of the 44 ENCODE regions were somewhat biased toward biologically interesting, gene-dense, and likely early-replicating loci undoubtedly explains the greater reproducibility of the biological and technical replicates in that study (Mesner et al. 2011).
The finding that larger zones replicate earlier in general than smaller zones and isolated fragments suggests that the larger the available template, the greater chance that it will attract an initiation complex (i.e., ORC). Thus, firing times might not be strictly tethered to some biological clock, but may result in part from the greater chance of activating a potential site in a large zone early in S-phase as opposed to a small zone or isolated fragment.
Our results are consistent with analytical stochastic origin firing models that were fit to S. cerevisiae replication timing data (Yang et al. 2010). These timing data were fit well by a multiple-initiator model (MIM) in which origin firing depended only on the number of loaded initiators per origin. Fitting the MIM to the replication timing data suggested that increasing numbers of initiators would result in earlier, more narrowly distributed, origin firing times. The essential insight is that in loci with multiple initiators, there is a higher probability of one of the initiators firing early in S-phase when compared with loci with only one initiator. For the case of S. cerevisiae, Yang and colleagues propose that the minichromosome maintenance (MCM) complex functions as the initiator. In contrast, human origins are clustered into zones. Yet they display the stochastic firing-time behavior of the MIM model developed for yeast, perhaps because of this zonal characteristic.
Significant associations are evident between early-firing origins and actively transcribed genes, but transcription is clearly not the major driver of most origins regardless of firing times
A potential mechanistic cause-and-effect relationship has often been suggested between origin locations and local transcriptional activity (Heintz 1992; van der Vliet 1996; Karnani et al. 2010; Martin et al. 2011). In the several attempts to identify a larger, less-biased, spectrum of origins in mammalian cells—either in the ENCODE pilot regions or in the genome as a whole—a modest association with active genes, including their transcription start sites, has been noted, and in most cases has been shown to be statistically significant (Cadoret et al. 2008; Karnani et al. 2010; Martin et al. 2011; Mesner et al. 2011; Aladjem 2012; Besnard et al. 2012). However, the overwhelming majority of origins in the more global studies, as well as in the present report, are not within nor even near transcribed genes (see Fig. 4). These data are somewhat in agreement with a recent genome-wide study of two different human cell lines in which small NSs were localized vis-à-vis active transcription units (Martin et al. 2011). In this study, small NSs were actually excluded from transcription start sites, as well as from the bodies of either actively or weakly transcribed genes. However, regions containing genes with moderate transcription levels were characterized by significant origin activity. Whether this latter observation might be explained by allelic exclusion is not yet clear.
In sum, it is difficult to argue that transcription is a major driver of origin activity. It is more likely that a permissive chromatin architecture that facilitates transcription also facilitates initiation at potential origins, probably by exposing the template for loading of important components such as ORC, MCMs, and the multiple other proteins required to activate an initiation site. Alternatively (or in addition), some factors required by both activities may be ambidextrous, preparing the template for the melting reactions required by both processes.
DNase I hypersensitivity appears to be a reliable marker of a chromatin structure required for some early-firing origins, but there appears to be no reliable signature for late-firing origins
Clearly, additional features of chromosomal environment must play an important role in both the efficiency and timing of origin activation. This is exemplified by the positive and highly significant association between early-firing origins and DNase I HSS, which report on modifications to the canonical nucleosomal substructure of chromatin. However, many late-firing origins correspond to isolated fragments or small zones, and there is little transcription late in S-phase; thus, it is not surprising that the association between late origins and HSS is negligible. These findings also suggest that the structural arrangements that DNase I is sensing in early origins are minor enough or absent in late origins to be undetectable in the log-phase cell populations on which the hypersensitivity experiments were performed (Thurman et al. 2012). The fact that these sites also track significantly with active transcription units again makes it difficult to determine whether there is a causal relationship between the two activities or merely a mutually accommodating structural alteration. However, in early-replicating regions, 58% of bubble-containing fragments contain or are associated with DNase I HSS, while only 43% are associated with actively transcribed genes, suggesting the possibility in many early-firing origins of an origin-specific local chromatin environment.
Early-firing, but not late-firing, origins appear to have been sequenced to saturation
The fact that such a large fraction of bubble-containing fragments are arrayed contiguously in zones suggests that these two GM06990 libraries are very close to comprehensive in early-replicating regions. However, the biological and sequencing replicates for the whole libraries analyzed herein do not attain the precision of the earlier ENCODE microarray study. What was lacking in the ENCODE study was the more global view of origin distributions vis-à-vis architectural characteristics and replication timing. Indeed, prior to the present study, very little was known about the actual numbers or natures of mid- or late-firing origins in mammalian genomes.
Despite the fact that both early- and late-firing origins were called in the present study with the same highly stringent 0.1% FDR cutoff, the concordance of late-firing origin calls between biological replicate libraries is much lower than for early-firing ones. The near-random association of origins with transcription characteristics and epigenetic factors at the site level in late-replicating regions also leaves the impression that the distribution of late-firing origins could possibly be stochastic. Indeed, the fact that half of the 44 ENCODE regions were somewhat biased toward biologically interesting, gene-dense, and likely early-replicating loci may explain the greater reproducibility of the biological and technical replicates in that study (Mesner et al. 2011). Moreover, based on concordance values of the sequencing replicates, it is likely that neither biological replicate library has been sequenced sufficiently to comprehensively map all of the relatively inefficient origins, whether late- or early-firing. In hindsight, it is also likely that the two biological replicate libraries did not trap all inefficient origins—regardless of firing times.
These considerations raise the interesting possibility that late-replicating regions might be extremely densely populated with a large number of relatively inefficient origins. To address this issue, it would be necessary to prepare several biological replicate origin libraries from large numbers of cells (e.g., 1010), sequence each to demonstrable saturation, and then apply the IDR framework developed by the ENCODE Consortium (Li et al. 2011; Landt et al. 2012) to identify bona fide sites based on biological replicate concordance. Clearly, this would be an enormous undertaking, but could possibly suggest the real densities of less-efficient origins and whether the late-firing ones are distributed randomly or are focused to more specific regions.
Nevertheless, our current study has sampled a relatively large number of late-firing origins, and their distributions display a significant and surprising signature with respect to HCE and epigenetic factor densities. These unexpected associations may have surfaced even though we sampled only a fraction of late-firing origins. Thus, we were able to capture robust trends with respect to their densities. It is also noteworthy that, while origin efficiency does monotonically fall with replication time, the median RD level—a measure of typical origin efficiency—is only ∼25% less for late-firing origins than the median RD for early-firing bubbles housed in euchromatic regions. Therefore, we may not be overlooking a major component of the late-firing origin population.
The density of late-firing origins increases with increasing degree of chromatin compaction
Only in early-firing origins did we uncover significant associations with activating chromatin marks at the site level (i.e., 10–100 kb). In contrast, no significant associations were identified with mid- or late-firing origins at the site level. On the megabase scale, the density of origins in late-replicating DNA is actually strongly “anti-correlated” with the densities of all the epigenetic factors (either active or repressive) that have been mapped by the ENCODE Consortium. In addition, the correlation between origin density and the absolute value of HCE, whose positive and negative values correspond to open and closed chromatin, respectively, is twofold higher than the correlation between origin density and HCE values (retaining their sign). This is consistent with the density of origins in late-replicating regions being significantly anti-correlated with the density of all the epigenetic factors we studied. These surprising results suggest that the density of late-firing origins increases with increasing chromatin compaction.
Unfortunately, the data for H3K9me3, which is a modulator of “constitutive” heterochromatin, is not presently available for GM06990. The H3K9me3 modification is bound by HP1, which has been shown to interact with both Drosophila and human ORC (Pak et al. 1997; Shareef et al. 2001; Prasanth et al. 2010). This surprising finding could form the molecular basis for a novel and important activation mechanism unique to late-firing origins in the heterochromatic domains of complex genomes.
The distribution of bubble-containing fragments in the GM06990 genome is moderately concordant with the recent genome-wide maps of small NSs prepared for four other human cell lines
The question arises how well our data compare with those obtained by analyses of small NS distributions. In two earlier studies (Cadoret et al. 2008; Karnani et al. 2010), NS preparations were hybridized to microarrays representing the 44 ENCODE regions. In fact, fewer than ∼35% of calls in either independent NS preparation overlapped the bubble map that we had constructed for HeLa cells, while <14% of the bubble-containing fragments overlapped calls in either NS preparation. In addition, only 13%–14% of calls for the two NS preparations overlapped one another (Karnani et al. 2010). Since each preparation was isolated from fewer than 108 cells (contrasted with ∼1010 cells for each bubble library), we suggested that neither was likely to be saturating either in the biological sampling or in the hybridization step (see arguments in Mesner et al. 2011).
A recent report describes the isolation of small (1.0–1.5 knt) NSs, again from ∼108 cells, from four different human cell lines. Importantly, each preparation then was exhaustively sequenced to near-saturation, and 200,000–250,000 fragments containing potential initiation sites were identified. Many were clustered into zones, generally agreeing with the organization of origins that we have suggested previously (e.g., Mesner et al. 2011) and further illuminated by the present study. The larger number of NSs relative to the bubble collection is consistent with our previous studies showing that fragments in the ∼6-kb range host multiple initiation events throughout their lengths (Fig. 6C; Kalejta and Hamlin 1996; Kalejta et al. 1996; Dijkwel et al. 2002). Importantly, although GM06990 cells were not included in this recent NS analysis, origin locations were surprisingly similar among the four human cell lines they examined, ranging from 65% to 84% pairwise overlaps. This finding led the authors to conclude that origin locations are relatively fixed regardless of the developmental lineages or tumorigenic states of the host cell lines (Besnard et al. 2012).
However, the GM06990 origin map we have elaborated here does not concord with the NS assignments for any of these four human cell lines nearly as well as the four NS maps concord with one another (Supplemental Table SIX): Only 45%–46% of NSs overlap or fall within bubble-containing EcoRI fragments. Conversely, only 33%–37% of bubble-containing fragments overlap NS calls. Given that Besnard and colleagues convincingly sequenced NS libraries to saturation, revisiting the comparison between NS and sequence-saturated bubble-seq origin maps could improve the concordance somewhat in cases where an origin is detected by NS and not by bubble-seq in this study, especially in late-replicating regions. Nevertheless, based on our findings and those of Besnard and colleagues, neither differences among cell lines nor a lack of better sampling or sequencing saturation is likely to adequately explain the lack of better concordance between the bubble and NS maps.
These unlikely considerations notwithstanding, this recent NS whole-genome origin mapping study has uncovered a common repetitive sequence element (a G-quadruplex sequence; Huppert and Balasubramanian 2005) that resides in 91%–95% of the NSs isolated from each of the four human cell lines by the melting, sizing, and lambda exonuclease protocol. This same sequence resides in a large percentage of NSs prepared in the same way and mapped to the murine and D. melanogaster genomes (Cayrou et al. 2011, 2012a; Besnard et al. 2012).
This potentially important finding would seem to represent the “Holy Grail” that has been sought by the eukaryotic replication field for almost 45 yr, i.e., a common “replicator” element that attracts initiation complexes. The G-quadruplex occurs ∼106 times in the haploid human genome (every 3.4 kb on average). Therefore, its presence in almost all NSs is impressive. Indeed, among the 123,297 bubble-containing fragments in the GM06990 genome, we found a 1.65-fold enrichment of G-quartets compared with that of all EcoRI fragments. However, 36% of bubble-containing fragments do not contain G-quadruplexes, and a statistical analysis suggests that the presence of the G-quadruplex sequence in bubble-containing fragments is only 1.05-fold enriched over mean random (Supplemental Table SIX).
It is also worth noting that G-quadruplexes do occur in a few of the well-studied mammalian origins that have been sequenced (e.g., Chinese hamster ori-beta, human MYC, and human beta-globin), but not in several others (e.g., Chinese hamster ori-beta′, human lamin B2) (LD Mesner, data not shown). Additionally, several laboratories have attempted to identify mammalian sequences that preferentially bind the ORC complex with little, if any, success. Whether the unusual quadruplex structure itself could actually aid melting of NSs in the initial step of their isolation, could stall local fork movement in a way that would enrich for non-origin-centered NSs, and/or could impede lambda exonuclease activity needs to be considered. It is also worth pointing out that in NS-rich regions, such as that depicted in Figure 6B wherein bubbles appear to be relatively depleted, there is a high density of G–Q sequences. This could mean either that our bubble-trapping procedure selects against bona fide bubble-containing fragments that contain such sequences or that the NS isolation procedure selects for some sequences that are not actually nascent strands. One way to resolve this issue would be to analyze NS-rich but bubble-poor regions such as that shown in Figure 6B by the very stringent neutral/neutral two-dimensional gel or fiber FISH replicon mapping procedures.
Therefore, there is once again a serious disconnect between the nature of sequences isolated as small NSs by melting and lambda exonuclease treatment, and those isolated by trapping via their internal bubble structures, which are supposed to be formed from the same NSs. On several grounds, we conclude that there are important and selective methodological differences between the two kinds of “origin” preparations. Thus, rather than unifying the two sides of an ongoing argument about the accuracy and relevance of origin isolation schemes, our studies and this recent NS origin map again bring this issue front and center. Importantly, however, only the bubble-trapping procedure has been validated by an independent origin mapping protocol (i.e., two-dimensional gel analysis).
Methods
Cell lines and culture conditions
The lymphoblastoid cell line, GM06990, was obtained from Coriell Institute for Medical Research and was propagated in RPMI 1640 supplemented with 50μg/mL Gentamicin and 15% serum (FetalClone II, HyClone). Cells were passaged every 2–3 d to maintain a cell density of 3 − 8 × 105/mL, and were cultured for no longer than 3 wk after sampling from frozen stocks. Under these conditions, the population doubling time was ∼40 h. For the preparation of replication intermediates and RNA, log-phase cultures were best harvested at 5 − 6 × 105 cells/mL 36–48 h after the last passage.
Preparation of origin and cDNA libraries for sequencing
Each duplicate “origin” library was prepared from 10 independent replication intermediate (RI) isolations from 109 cells each (1010 cells per library). RI isolation and bubble trapping procedures were exactly as described previously (Dijkwel et al. 1991; Mesner and Hamlin 2009; Mesner et al. 2009, 2011). The resulting preparations were assessed by a sensitive 2D gel replicon mapping technique (Brewer and Fangman 1987) as described previously (Mesner et al. 2011), and were judged to be 85%–90% pure for each library. For the duplicate cDNA libraries, total RNA was isolated from 108 log-phase cells by standard methods (Chomczynski and Sacchi 1987), and poly(A)+ RNA was purified on oligo-dT cellulose. This material was then fragmented, converted to its cDNA, and libraries were constructed according to the recommendations of the supplier (Illumina Genome Analyzer IIx manual).
Identifying statistically significant “origin” calls in the sequencing data
Sequence reads for the three log-phase GM06990 libraries (B1A, B1B, and B2) were mapped to NCBI human genome assembly v36 (hg18) using BWA version 0.5.7 (Li and Durbin 2009). Since the GM06990 cell line was derived from a female, chrY data were excluded. The output of the Illumina analytic pipeline was converted to individual enriched-read “wig” files for the B1A, B1B, B1 (i.e., B1A and B1B combined), and B2 data sets, as well as for a combination of B1A, B1B, and B2. Since the bubble libraries are constructed of EcoRI fragments, comparisons to other data sets first required a definition of an EcoRI-based measure. The read depth (RD) of each EcoRI fragment was defined as the density of coverage of enriched reads over the EcoRI fragment (i.e., the number of reads divided by the length of the EcoRI fragment in base pairs):
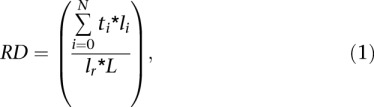 |
where N equals the total number of genomic segments over which the number of overlapping reads is a constant (i.e., wig-file start-stop segments), ti equals the number of overlapping reads for each genomic segment i (i.e., the height of a given wig-file segment), li equals the length of genomic segment i, lr equals sequence read length, and L signifies the EcoRI fragment length. Outliers were excluded in the read depth calculation for each EcoRI fragment by excluding genomic segments whose overlapping read depths were not in the range defined by (Q1 − 1.5*[Q3 − Q1], Q3 + 1.5*[Q3 − Q1]), where Q1 is the lower quartile and Q3 is the upper quartile of a given genomic segment’s overlapping read depths. RDs were calculated for B1A, B1B, B2, and the combination of the three data sets. As described in the main text, we excluded cloned EcoRI fragments <500 bp in length, as well as any fragments containing >80% alpha-satellite DNA.
A negative binomial distribution was used to model background or noisy RD levels and identify EcoRI fragments with RDs enriched over background. RD values were first multiplied by a scaling factor of 10,000, and then rounded to the nearest integer. EcoRI fragments with RDs of zero were removed as a preprocessing step. Given that a single lane of sequencing does not sequence each library to saturation, combining technical and/or biological replicate data yielded a higher proportion of reads (signal) in each bubble-containing fragment. B1A, B1B, B1, B2, and the combination of B1A, B1B, and B2 (“Comb”) yielded 10 M, 17.7 M, 27.7 M, 14.9 M, and 42.6 M mapped reads, respectively. To avoid biasing the background null model by the signal contribution of the extended right tail of the scaled RD distributions, the scaled, positive RDs were randomly sampled from B1A, B1B, B1, B2, and Comb fragments with the 60%, 60%, 50%, 60%, and 40% lowest RD values, respectively. These values produced the best fit of the negative binomial distribution to the scaled background RD distributions. A P-value cutoff corresponding to a very conservative false discovery rate of 0.1% was applied to determine a high confidence level for origin calls, leading to RD cutoffs of 0.0081, 0.0139, 0.0136, 0.0123, and 0.0144 for B1A, B1B, B1, B2, and the combination of B1A, B1B, and B2, respectively. Bubble-containing fragments with <20% read coverage across their lengths were filtered out. After these steps, 34,184 (B1A), 39,971 (B1B), 76,891 (B1), 36,225 (B2), and 123,297 (B1A, B1B, and B2 combined) EcoRI fragments in each data set were judged to be significantly enriched over the whole genome and therefore should represent bona fide bubble-containing fragments.
EcoRI permutation null model
The significance of the number of bubble-containing fragments in each timing window (early, mid, late) (Supplemental Table SIII), the overlaps between bubble-containing fragments and transcription units in the nine annotation categories (Supplemental Table SV), the overlaps between bubbles and epigenetic factor sites (Supplemental Table SVI), and the overlaps between bubble-containing fragments and G-quadruplex sequences were assessed using a random permutation model. All chromosomes were first divided into ∼1-Mb non-overlapping windows. When an EcoRI fragment straddled two 1-Mb windows, the boundary of the window with >50% of the EcoRI fragment was adjusted to entirely encompass that fragment. All EcoRI fragments (bubbles and non-bubbles) within each ∼1-Mb window then were randomly permuted 10,000 times. This approach preserved the local composition bias, and also the genomic coverage of the bubbles within each window. With each individual permutation, the new location of each bubble-containing EcoRI fragment within that window was tracked. TR50 values were computed as described below in the TR50 analysis (using timing data for G1b, S1–4, and G2 channels), corresponding to the new location of each bubble-containing EcoRI fragment for all 10,000 iterations. The permuted bubble-containing fragments for all 10,000 instances were then reclassified as early-, mid-, or late-firing, if their newly calculated TR50 values fell in the intervals [0, 2), [2, 4), or [4, 6), respectively. Using the 10,000 instances, we derived the “mean random” bubble distributions or number of overlaps between bubbles and annotations or epigenetic sites expected by chance alone, as well as the SDs about the means. Fold changes of actual over mean random and empirical, one-sided, upper tail P-values (associated with positive enrichment) were also calculated for each data set, as well as the Z-score, which is the actual minus the mean of the random divided by the SD of the random. As can be seen in Supplemental Tables SIII, SV, SVI, and SIX, the results follow the expected statistical trends whereby higher significance corresponds to higher fold change, lower P-value, and higher Z-scores. We note that the empirical P-values are susceptible to outlier instances. For example, we observe a handful of cases among the comparisons shown in Supplemental Tables SIII, SV, SVI, and SIX in which the P-value is >1 × 10−4 for a fold change, number actual, and Z-scores that consistently yield P-values that are <1 × 10−4 for the rest of the comparisons (e.g., row 5 of Supplemental Table SVB). We also note that, while our random permutation model accounts for many local genomic biases, it does not fully account for correlated structure of annotations and epigenetic features (Bickel et al. 2010), which can lead to slightly inflated significance. As a result, we only report as significant, comparisons that have P-values < 1 × 10−4, relatively high fold change (i.e., greater than 1.5) and relatively high Z-score (i.e., greater than 10). Finally, while our one-sided upper tail P-values quantify the significance of positive enrichments (i.e., fold change > 1 and Z-score > 0), we were able to identify significant depletions of actual compared with random using fold changes that are well below unity (i.e., fold change < 0.67) and Z-scores that are negative and have a relatively large magnitude (e.g., Z-score < −10).
Data access
Bubble-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE38809.
Acknowledgments
We thank the reviewers for helpful comments and recommendations, particularly the first reviewer. This work was supported by R01-GM026108 (J.L.H. and L.D.M.) from the NIH.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.155218.113.
References
- Aladjem MI 2004. The mammalian β-globin origin of DNA replication. Front Biosci 9: 2540–2547 [DOI] [PubMed] [Google Scholar]
- Aladjem MI 2007. Replication in context: Dynamic regulation of DNA replication patterns in metazoans. Nat Rev Genet 8: 588–600 [DOI] [PubMed] [Google Scholar]
- Aladjem MI 2012. Rif1 choreographs DNA replication timing. EMBO J 31: 3650–3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aladjem MI, Fanning E 2004. The replicon revisited: An old model learns new tricks in metazoan chromosomes. EMBO Rep 5: 686–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan L, Bambara RA 2011. Eukaryotic lagging strand DNA replication employs a multi-pathway mechanism that protects genome integrity. J Biol Chem 286: 6865–6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker RA, Chambers JM, Wilks AR 1988. The New S Language. Wadsworth & Brooks/Cole, Pacific Grove, California [Google Scholar]
- Bell SP, Stillman B 1992. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 357: 128–134 [DOI] [PubMed] [Google Scholar]
- Besnard E, Babled A, Lapasset L, Milhavet O, Parrinello H, Dantec C, Marin JM, Lemaitre JM 2012. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat Struct Mol Biol 19: 837–844 [DOI] [PubMed] [Google Scholar]
- Bickel PJ, Boley N, Brown JB, Huang H, Zhang NR 2010. Subsampling methods for genomic inference. Ann Appl Stat B4: 1660–1667 [Google Scholar]
- Bielinsky AK, Gerbi SA 1998. Discrete start sites for DNA synthesis in the yeast ARS1 origin. Science 279: 95–98 [DOI] [PubMed] [Google Scholar]
- Bochman ML, Paeschke K, Zakian VA 2012. DNA secondary structures: Stability and function of G-quadruplex structures. Nat Rev Genet 13: 770–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer BJ, Fangman WL 1987. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51: 463–471 [DOI] [PubMed] [Google Scholar]
- Cadoret JC, Meisch F, Hassan-Zadeh V, Luyten I, Guillet C, Duret L, Quesneville H, Prioleau MN 2008. Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc Natl Acad Sci 105: 15837–15842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Coulombe P, Vigneron A, Stanojcic S, Ganier O, Peiffer I, Rivals E, Puy A, Laurent-Chabalier S, Desprat R, et al. 2011. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res 21: 1438–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Coulombe P, Puy A, Rialle S, Kaplan N, Segal E, Méchali M 2012a. New insights into replication origin characteristics in metazoans. Cell Cycle 1: 658–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Grégoire D, Coulombe P, Danis E, Méchali M 2012b. Genome-scale identification of active DNA replication origins. Methods 57: 158–164 [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156–159 [DOI] [PubMed] [Google Scholar]
- Czajkowsky DM, Liu J, Hamlin JL, Shao Z 2008. DNA combing reveals intrinsic temporal disorder in the replication of yeast chromosome VI. J Mol Biol 375: 12–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkwel PA, Vaughn JP, Hamlin JL 1991. Mapping of replication initiation sites in mammalian genomes by two-dimensional gel analysis: Stabilization and enrichment of replication intermediates by isolation on the nuclear matrix. Mol Cell Biol 11: 3850–3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkwel PA, Wang S, Hamlin JL 2002. Initiation sites are distributed at frequent intervals in the Chinese hamster dihydrofolate reductase origin of replication but are used with very different efficiencies. Mol Cell Biol 22: 3053–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature447: 799–816. [DOI] [PMC free article] [PubMed]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature489: 57–74. [DOI] [PMC free article] [PubMed]
- Gilbert DM 2010. Evaluating genome-scale approaches to eukaryotic DNA replication. Nat Rev Genet 11: 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlin JL, Mesner LD, Lar O, Torres R, Chodaparambil SV, Wang L 2008. A revisionist replicon model for higher eukaryotic genomes. J Cell Biochem 105: 321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA 2010. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci 107: 139–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz NH 1992. Transcription factors and the control of DNA replication. Curr Opin Cell Biol 4: 459–467 [DOI] [PubMed] [Google Scholar]
- Huberman JA, Riggs AD 1968. On the mechanism of DNA replication in mammalian chromosomes. J Mol Biol 32: 327–341 [DOI] [PubMed] [Google Scholar]
- Huppert JL, Balasubramanian S 2005. Prevalence of quadruplexes in the human genome. Nucl Acids Res 33: 2908–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon Y, Bekiranov S, Karnani N, Kapranov P, Ghosh S, MacAlpine D, Lee C, Hwang DS, Gingeras TR, Dutta A 2005. Temporal profile of replication of human chromosomes. Proc Natl Acad Sci 102: 6419–6424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH 2008. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol 26: 1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalejta RF, Hamlin JL 1996. Composite patterns in neutral/neutral two-dimensional gels demonstrate inefficient replication origin usage. Mol Cell Biol 16: 4915–4922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalejta RF, Lin HB, Dijkwel PA, Hamlin JL 1996. Characterizing replication intermediates in the amplified CHO dihydrofolate reductase domain by two novel gel electrophoretic techniques. Mol Cell Biol 16: 4923–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnani N, Taylor C, Malhotra A, Dutta A 2007. Pan-S replication patterns and chromosomal domains defined by genome-tiling arrays of ENCODE genomic areas. Genome Res 17: 865–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnani N, Taylor CM, Malhotra A, Dutta A 2010. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol Biol Cell 21: 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysan PJ, Haase SB, Calos MP 1989. Isolation of human sequences that replicate autonomously in human cells. Mol Cell Biol 9: 1026–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. 2012. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 9: 1813–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, Bickel PJ 2011. Measuring reproducibility of high-throughput experiments. Ann Appl Stat 5: 1752–1779 [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HB, Dijkwel PA, Hamlin JL 2005. Promiscuous initiation on mammalian chromosomal DNA templates and its possible suppression by transcription. Exp Cell Res 308: 53–64 [DOI] [PubMed] [Google Scholar]
- Ma C, Leu TH, Hamlin JL 1990. Multiple origins of replication in the dihydrofolate reductase amplicons of a methotrexate-resistant chinese hamster cell line. Mol Cell Biol 10: 1338–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MM, Ryan M, Kim R, Zakas AL, Fu H, Lin CM, Reinhold WC, Davis SR, Bilke S, Liu H, et al. 2011. Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Res 21: 1822–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesner LD, Hamlin JL 2009. Isolation of restriction fragments containing origins of replication from complex genomes. Methods Mol Biol 521: 315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesner LD, Crawford EL, Hamlin JL 2006. Isolating apparently pure libraries of replication origins from complex genomes. Mol Cell 21: 719–726 [DOI] [PubMed] [Google Scholar]
- Mesner LD, Dijkwel PA, Hamlin JL 2009. Purification of restriction fragments containing replication intermediates from complex genomes for 2-D gel analysis. Methods Mol Biol 521: 121–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesner LD, Valsakumar V, Karnani N, Dutta A, Hamlin JL, Bekiranov S 2011. Bubble-chip analysis of human origin distributions demonstrates on a genomic scale significant clustering into zones and significant association with transcription. Genome Res 21: 377–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norio P, Schildkraut CL 2004. Plasticity of DNA replication initiation in Epstein-Barr virus episomes. PLoS Biol 2: e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak DT, Pflumm M, Chesnokov I, Huang DW, Kelllum R, Marr J, Romanowski P, Botchan MR 1997. Association of the origin recognition complex with heterochromatin and HP1 in higher eukaryotes. Cell 91: 311–323 [DOI] [PubMed] [Google Scholar]
- Prasanth SG, Shen Z, Prasanth KV, Stillman B 2010. Human origin recognition complex is essential for HP1 binding to chromatin and heterochromatin organization. Proc Natl Acad Sci 107: 15093–15098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shareef MM, King C, Damaj M, Badagu R, Huang DW, Kellum R 2001. Drosophila heterochromatin protein 1 (HP1)/origin recognition complex (ORC) protein is associated with HP1 and ORC and functions in heterochromatin-induced silencing. Mol Biol Cell 12: 1671–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vliet PC 1996. Roles of transcription factors in DNA replication. In DNA replication in eukaryotic cells (ed. M DePamphilis), pp. 87–118. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Vassilev LT, Johnson EM 1989. Mapping initiation sites of DNA replication in vivo using polymerase chain reaction amplification of nascent strand segments. Nucleic Acids Res 17: 7693–7705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SC, Rhind N, Bechhoefer J 2010. Modeling genome-wide replication kinetics reveals a mechanism for regulation of replication timing. Mol Syst Biol 6: 404. [DOI] [PMC free article] [PubMed] [Google Scholar]