

Abstract
Methanogens catalyze the critical methane-producing step (called methanogenesis) in the anaerobic decomposition of organic matter. Here, we present the first predictive model of global gene regulation of methanogenesis in a hydrogenotrophic methanogen, Methanococcus maripaludis. We generated a comprehensive list of genes (protein-coding and noncoding) for M. maripaludis through integrated analysis of the transcriptome structure and a newly constructed Peptide Atlas. The environment and gene-regulatory influence network (EGRIN) model of the strain was constructed from a compendium of transcriptome data that was collected over 58 different steady-state and time-course experiments that were performed in chemostats or batch cultures under a spectrum of environmental perturbations that modulated methanogenesis. Analyses of the EGRIN model have revealed novel components of methanogenesis that included at least three additional protein-coding genes of previously unknown function as well as one noncoding RNA. We discovered that at least five regulatory mechanisms act in a combinatorial scheme to intercoordinate key steps of methanogenesis with different processes such as motility, ATP biosynthesis, and carbon assimilation. Through a combination of genetic and environmental perturbation experiments we have validated the EGRIN-predicted role of two novel transcription factors in the regulation of phosphate-dependent repression of formate dehydrogenase—a key enzyme in the methanogenesis pathway. The EGRIN model demonstrates regulatory affiliations within methanogenesis as well as between methanogenesis and other cellular functions.
Methanogenic archaea (methanogens) are phylogenetically diverse, consisting of at least seven orders (Methanococcales, Methanobacteriales, Methanomicrobiales, Methanopyrales, Methanosarcinales, Methanocellales, and Methanoplasmatales) of strictly anaerobic Euryarchaeota (Liu and Whitman 2008; Sakai et al. 2008; Paul et al. 2012). Most species of methanogens are hydrogenotrophic and use hydrogen gas (H2) as the electron donor for the reduction of carbon dioxide to methane. In addition, many species can use formate in place of H2, and a few can use certain alcohols. These microorganisms contain very high levels of different types of hydrogenases and consume H2 at very high rates. Furthermore, under certain conditions the H2 uptake system can be induced to produce H2 at elevated proportions by using formate as an electron donor (Hendrickson and Leigh 2008; Lupa et al. 2008; Costa et al. 2013a). Certain species of methanogens can produce H2 by fixing nitrogen, and therefore have the potential to produce H2 using the nitrogenase system. Finally, most hydrogenotrophic methanogens are autotrophs, and assimilate CO2 by the acetyl-CoA pathway (Liu and Whitman 2008).
Methanogenesis is a form of anaerobic respiration that generates a chemiosmotic membrane potential for ATP production (Deppenmeier 2002). In hydrogenotrophic methanogenesis, H2 or formate is used to reduce CO2 to methane. Four reduction steps are interspersed with steps in which carbon units are transferred between coenzymes, and dehydration occurs as well. Electron delivery is mediated by the deazaflavin coenzyme F420 or ferredoxin (Fd). A variety of hydrogenases and/or formate dehydrogenase reduce these electron carriers. Electron flow for the final reduction step involves the cycling of two sulfhydryl-containing coenzymes between their sufhydryl and heterodisulfide forms. Recently, it has been shown that although ATP generation is chemiosmotic (driven by Na+ pumping), the phenomenon of electron bifurcation is an important aspect of energy conservation (Thauer et al. 2008; Kaster et al. 2011b). Electrons accumulate in the form of a two-electron reduced flavin in heterodisulfide reductase (Hdr). From each reduced flavin, one electron flows in the exergonic direction to reduce the heterodisulfide, and the other electron flows in the endergonic direction to reduce the Fd that carries electrons to the first reduction step in the pathway. Electron bifurcation renders the pathway cyclic, and reduced intermediates must be replenished anaplerotically by the action of the hydrogenase Eha (Lie et al. 2012; Thauer 2012).
A large body of literature exists on the biochemistry of the methanogenesis pathway (Deppenmeier 2002; Kaster et al. 2011a), yet little is known about transcriptional regulation of methanogenesis genes. Gene regulatory networks spatiotemporally regulate cellular physiology to optimize resource utilization, maintain integrity of genetic information, and contribute toward competitive fitness of the organism under changing environmental conditions. Construction of mathematical and computational global gene regulatory network models by integration of diverse high-throughput experimental data have been successful in understanding regulatory mechanisms and generating testable hypotheses (Bonneau et al. 2007; Faith et al. 2007; Kaur et al. 2010; Krouk et al. 2010). Methanococcus maripaludis S2 (M. maripaludis) is a premier model for the hydrogenotrophic methanogens with its complete genome sequence (Hendrickson et al. 2004), rapid and reliable growth in a laboratory, and existence of excellent genetic tools (Leigh et al. 2011). Extensive studies of the transcriptome (Hendrickson et al. 2007, 2008) and proteome (Xia et al. 2006, 2009) showed that genes of the methanogenic pathway are significantly regulated by H2. Our recent study on the transcriptome map of M. maripaludis generated a comprehensive list of protein-coding and noncoding RNAs for systems modeling of this organism (Yoon et al. 2011).
In this study, we have developed the first systems scale environment and gene-regulatory influence network (EGRIN) (Bonneau et al. 2007) model of a hydrogenotrophic methanogen. First, we generated a comprehensive list of coding and noncoding RNAs through comparative analysis of the complete transcriptome and a comprehensive PeptideAtlas (Deutsch 2010) for M. maripaludis—i.e., genome-wide alignments of a collection of peptides that were experimentally detected across a diverse set of tandem mass spectrometry experiments (Yoon et al. 2011; Supplemental Methods). Next, we generated a compendium of over 50 transcriptome profiles from steady-state conditions and during dynamic cellular response to a spectrum of environmental perturbations. Along with eight transcriptome data sets from our previous study (Yoon et al. 2011), a total of 58 transcriptome profiles of wild-type M. maripaludis MM901 were analyzed using the cMonkey biclustering algorithm (Reiss et al. 2006) and the Inferelator algorithm (Bonneau et al. 2006), in order to infer the set of environmental factors (EFs) and transcriptional factors (TFs, sequence-specific DNA-binding proteins that mediate regulation of mRNA transcription) that accurately modeled the dynamic transcriptional changes of genes within each bicluster. The EGRIN model was validated using 56 transcriptome data sets that were not used for the model construction: 52 new transcriptome data sets from several genetic backgrounds that modulated methanogenesis in well-understood schemes and four data sets from our previous work (Costa et al. 2013b). We demonstrated that this EGRIN model accurately predicted transcriptional responses of all genes in the network and can be used as a framework to formulate novel hypotheses regarding gene functions and regulation. Specifically, we used the modular organization of genes in EGRIN biclusters to delineate how the methanogenesis genes are coregulated with other genes from different biological processes. Further, we analyzed regulatory influences on these modules to uncover novel regulators of methanogenesis, two (MMP1100 and MMP0719) of which we subsequently validated through new environmental and genetic perturbation experiments that confirmed the predicted direct and indirect regulatory influences of MMP1100 and MMP0719 on formate dehydrogenase.
Results and Discussion
Experimental design
We used a systems approach to investigate the dynamic regulation of all genes in M. maripaludis under differing environmental conditions that modulate H2 utilization/production. Our basic premise here is to iteratively perturb, observe, and model cellular responses to characterize the gene regulatory circuits that coordinate genes involved in methanogenesis and H2 utilization/production with other aspects of physiology. We selected culture conditions that separate the varying effects of different environmental factors on H2 utilization/production and methanogenesis (Fig. 1) and collected RNA samples to capture global transcriptional responses. We performed these experiments in two different modes:
Cellular responses to single environmental perturbations. In order to test the effects of H2 availability, the proportion of H2 in the input gas was rapidly increased to 110 mL/min (H2-excess) or decreased to 21 mL/min (H2-limiting). In this experiment, growth rate was proportional to H2 availability (Fig. 1A). Cell density increased linearly and gradually after the transition from H2-limiting to H2-excess conditions (P-value < 0.0001) and decreased similarly when H2 limiting conditions were restored (P-value < 0.0001). In contrast, CH4 production increased and decreased much more rapidly. This response reflects a marked difference in growth yield on a per-CH4 basis under the two H2 conditions (Costa et al. 2013b).
Cellular responses to combinatorial environmental perturbations. In the above experiments, the changes in H2 were accompanied by changes in growth rate and cell density, which can have confounding consequences for global transcriptional responses. Therefore, it was important to perform additional experiments in which growth rate and cell density were held constant in order to isolate the effects of environmental variables such as H2 (Supplemental Table S3). In these experiments, we operated the bioreactor in chemostat mode by simultaneously changing two essential nutrients in the culture media—e.g., H2 and phosphate (P) or H2 and nitrogen (N) (Fig. 1B,C). The initial culture condition of H2-excess/P-limitation was switched to H2-limiting/P-excess as the experiment progressed. In this way, the cell growth was limited by P and H2 before and after perturbation, respectively. Growth rates were held constant across all experiments by keeping the dilution rates constant in the chemostat. All cultures had cell densities (OD660) between 0.6 and 0.8.
Figure 1.

Chemostats of environmental perturbations for time-series arrays. Four separate growth experiments for time-series array data in chemostats were performed for M. maripaludis MM901 (A,B), and five chemostat runs were performed for gene deletion strains MM901Δmtd, MM901ΔfruAΔfrcA, MM901ΔMMP1447, MM901ΔMMP0719, and MM901ΔMMP1100 (C). Before perturbation, cell cultures were allowed to reach steady state (constant growth rate and cell density). The perturbation for each experiment was as follows: (A) The proportion of H2 in the gas entering the chemostat was rapidly increased from limiting to excess (left) or decreased from excess to limiting (right). (B,C) The proportion of H2 entering the chemostat was rapidly decreased from excess to limiting and, simultaneously, the concentration of ammonium or phosphate was rapidly increased from limiting to excess. Samples were taken 30 min before perturbation, immediately after the perturbation, and after 5, 10, 20, 30, 45, 60, 90, 120, 180, and 300 min. In each plot, culture time is shown on the x-axis, cell density (red line) in OD660 on the left y-axis, and CH4 (black line) in the output gas on the right y-axis. Above each plot, culture conditions before and after perturbation are shown for excess (red triangle) and limiting (inverted green triangle) of a nutrient.
The EGRIN model was constructed based on a compendium of 58 transcriptome profiles of wild-type M. maripaludis MM901; these included eight samples from a growth curve experiment (previously published) (Yoon et al. 2011), 48 from time-series sampling of the H2 perturbation experiments described above (Fig. 1A,B), and two from steady-state cultures using formate as an electron donor in the presence of H2. EGRIN predictions of global transcriptional changes in response to new environmental perturbations were tested by using data from 56 transcriptome measurements that were not included in building the original EGRIN model. These experiments included 52 transcriptome profiles from gene deletion strains (Fig. 1C) and four from steady-state cultures grown with formate as an electron donor “without” H2 (Costa et al. 2013b). Culture conditions and samples for transcriptome analysis are summarized in Supplemental Table S4.
Biclustering analysis reveals complex transcriptional regulatory patterns of genes involved in the methanogenesis pathway
We performed the network inference in two steps to first uncover the modular organization of genes based on their conditional coregulation and, second, to infer the regulatory influences responsible for regulation and intercoordination of gene expression within each of these modules. First, using the cMonkey algorithm we discovered biclusters, i.e., conditionally coregulated sets of genes that share conserved cis-regulatory motifs in their promoters (Reiss et al. 2006). The cMonkey algorithm uses functional associations such as operon information, shared phylogenetic histories, as well as protein–protein and protein–DNA interactions to discover modules of conditionally coregulated genes that are not only coexpressed across some environments but also share conserved cis-regulatory motifs in their promoters. In order to discover regulatory programs associated with methanogenesis, the experiments were designed to probe responses to changes in factors such as H2, P, and N that are known to affect this process. Notably, by allowing genes to be grouped into multiple biclusters, the algorithm is able to discover different regulatory programs for the same gene. Before model building, 203 genes (∼11%) that did not show significant expression changes across most of the experimental conditions were filtered out as described previously (Reiss et al. 2006). The remaining 1661 genes, including unannotated transcripts from the transcriptome architecture analysis (Yoon et al. 2011), that changed significantly in at least a subset of experiments, were grouped into 166 biclusters. (Supplemental Fig. S1; Supplemental Table S5). Altogether, 149 of 166 biclusters were of high quality, as their mean residual values were less than 0.45; residual of a bicluster is a metric for significant gene coexpression among member genes (Reiss et al. 2006).
We identified six biclusters that were enriched for known genes of the methanogenesis pathway (Fig. 2; Table 1) (hypergeometric P-value < 0.05, Methods). Among these six biclusters (Supplemental Fig. S2), four were clearly dominated by genes that play an integral role in methanogenesis (bc_0114, bc_0009, bc_0019, and bc_0035). The fifth bicluster (bc_0133) is composed mainly of genes for the energy-conserving hydrogenase Eha. Our recent results (Lie et al. 2012) show that Eha does not function in methanogenesis at a level stoichiometrically equal to the core steps, but anaplerotically supplements electron bifurcation as a means of supplying electrons to formylmethanofuran dehydrogenase. The sixth bicluster (bc_0010) consists of genes for early biosynthetic steps that are linked to methanogenesis; these steps comprise a biosynthetic pathway of CO2 fixation starting with a methyl group of methyltetrahydromethanopterin (CH3-H4MPT) (Fig. 2). Interestingly, the results of biclustering analysis revealed that different steps of methanogenesis were separated into several biclusters. The first four biclusters were distinguished by the presence of genes that responded positively to H2 limitation. mRNA levels of these genes were generally down-regulated as the cultures transitioned from H2-limiting to H2-excess conditions, up-regulated in a transition from H2 excess to H2 limitation, and up-regulated in both transitions to H2 limitation with cell density being kept constant. More specifically, biclusters bc_0114 and bc_0009 both contained genes that were the most noticeably increased by H2 limitation, such as F420-dependent methylene-H4-methanopterin dehydrogenase (mtd), methylene-H4-methanopterin reductase (mer), subunits of F420-reducing hydrogenase (fruAG and frcB), and formate dehydrogenase (fdhC and two operons for fdhAB). These enzymes represent the four steps of methanogenesis that use F420 as an electron carrier, and all were suggested previously to be regulated by H2 limitation, based on transcriptome analysis of steady-state samples (Hendrickson et al. 2007). The first three sets of genes were included in bc_0114 while formate dehydrogenase was in bc_0009. These biclusters also revealed previously unknown affiliations. bc_0114 and bc_0009 shared genes encoding subunits of F420-nonreducing hydrogenase (cysteine-containing, vhcGB) and a hypothetical protein (MMP1378). The bc_0114 contained genes encoding subunits of F420-nonreducing hydrogenase (selenocysteine-containing, vhuDGAUB), a subunit of formylmethanofuran dehydrogenase (selenocysteine-containing, fwdB), and a subunit of heterodisulfide reductase (hdrA). bc_0009 contained subunits of Hdr (hdrC2B2) and a gene involved in the biosynthesis of coenzyme F420 (cofH). These results show that our network model was able to recapitulate known regulation of key methanogenesis genes under limiting H2 conditions and identify novel associations with other genes, including hypothetical proteins. While many genes of methanogenesis are regulated by hydrogen, these newly discovered associations could be key to revealing additional factors that can alter methanogenesis.
Figure 2.

Differential regulation of methanogenesis genes. Methanogenesis pathway of M. maripaludis together with corresponding bicluster membership information is shown. Methanogenesis from CO2 occurs in four reduction steps, two C-1 transfer steps, and one dehydration step. Note that the Fd that donates electrons to Fmd/Fwd can be reduced in two ways—by electron bifurcation from Hdr or by the Eha hydrogenase. Dotted arrows indicate coupling of metabolic steps with membrane ion gradients. Biosynthetic CO2 fixation to acetylCoA and pyruyvate is also shown. Colors in enzyme designations denote bicluster membership as indicated in the color key in the lower left corner (see Fig. 4 for their member genes). The main methanogenesis pathway genes are included in four biclusters, bc_0114 (red), bc_0009 (pink), bc_0019 (green), and bc_0035 (purple). The other two biclusters, bc_0133 (brown) and bc_0010 (blue), include genes involved in reactions that are related to methanogenesis such as electron bifurcation and carbon fixation (see text for details). Transcriptional changes of member genes of each of the biclusters are shown in Supplemental Figure S2. Genes encoding enzymes: (Cdh) carbon monoxide dehydrogenase/acetylCoA synthase; (Eha) energy-conserving hydrogenase A; (Ehb) energy-conserving hydrogenase B; (Fdh) formate dehydrogenase; (Fmd/Fwd) formyl-methanofuran dehydrogenase; (Fru/c) F420-reducing hydrogenase; (Ftr) formyl-methanofuran-H4-methanopterin formyltransferase; (Hdr) heterodisulfide reductase; (Hmd) H2-dependent methylene-H4-methanopterin dehydrogenase; (Mch) methenyl-H4-methanopterin cyclohydrolase; (Mcr) methyl-coenzyme M reductase; (Mer) methylene-H4-methanopterin reductase; (Mtd) F420-dependent methylene-H4-methanopterin dehydrogenase; (Mtr) methyl-H4-methanopterin-coenzyme M methyltransferase; (Por) pyruvate oxidoreductase; (Vhu/c) F420-nonreducing (Hdr-associated) hydrogenase. Metabolites: (Fd) ferredoxin; (MFR) methanofuran; (H4MPT) tetrahydromethanopterin; (CoM) coenzyme M.
Table 1.
Methanogenesis biclusters
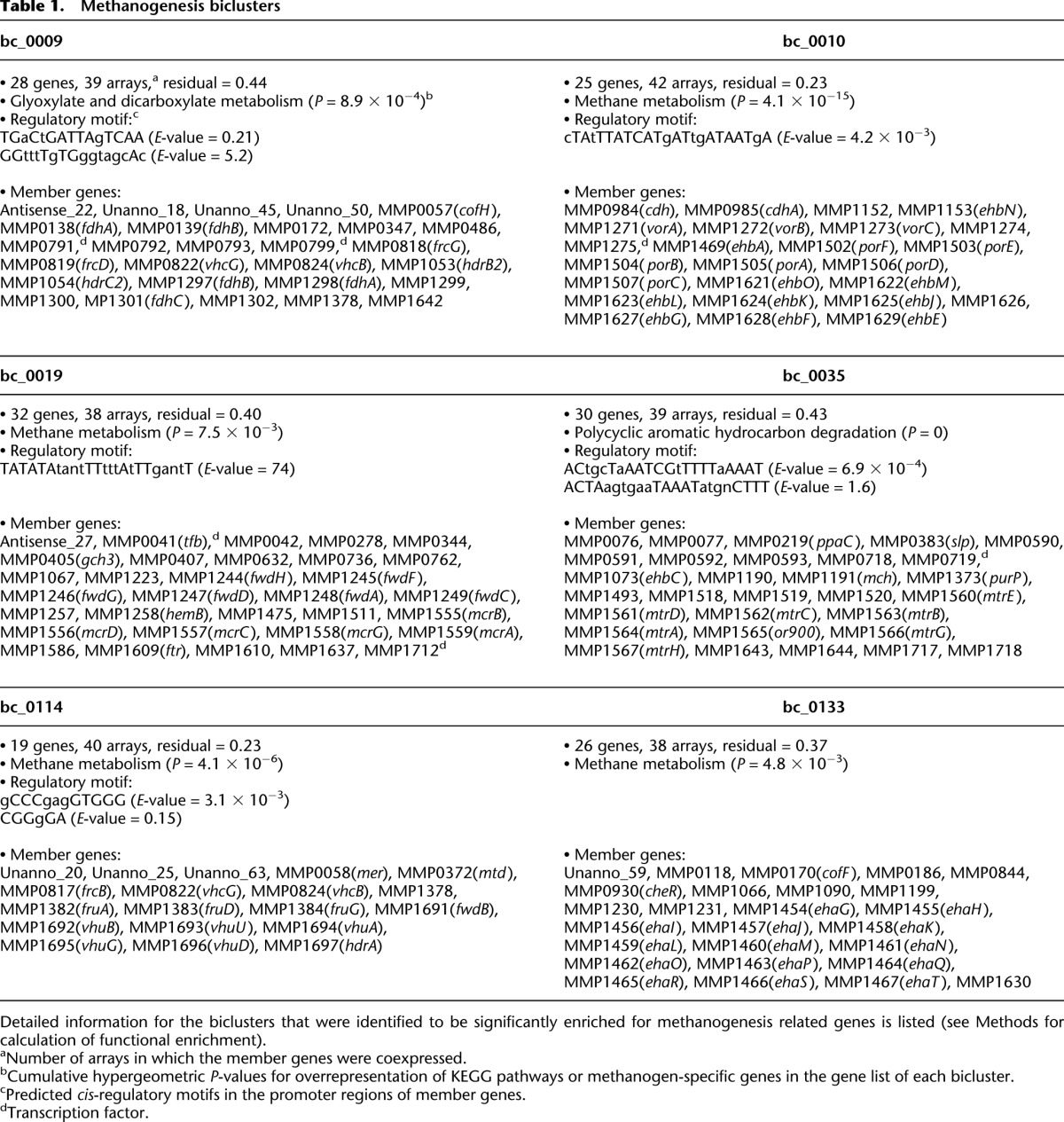
The remaining steps in methanogenesis are affiliated with the other two methanogenic biclusters. The bc_0019 contains genes for methyl-coenzyme M reductase (mcrBDCGA), formyltransferase (ftr), and subunits of formylmethanofuran dehydrogenase (fwdHFGDAC) other than fwdB, which is in bc_0114 (Table 1). The bc_0035 contains the sodium-translocating methyltransferase (mtrEDCBA-GH) and the methenyl-H4-methanopterin cyclohydrolase mch. The separation of methyl-coenzyme M reductase (mcrBDCGA) from the sodium-translocating methyltransferase (mtrEDCBA-GH) is particularly striking since they are present in two neighboring operons and catalyze the final two steps in methanogenesis. This partitioning might indicate functional nuances in associations with other bicluster members such as electron carriers. It is possible that this regulatory scheme may be necessary to maintain redox homeostasis during methanogenesis by separating regulation of the nonredox pathway (Mtr) from the redox pathway (Mcr). In addition, the differential regulation of these adjacent steps might be necessary to accommodate distinct environment-dependent roles for Mcr, which is cytoplasmic, and Mtr, which is membrane bound and capable of directly generating a chemiosmotic membrane gradient. The correlation of the expression patterns of the first operon genes (mcrB and mtrE) was 0.76 throughout the samples, compared to 0.99 between genes within an operon. Their expressions were highly correlated in the four time-series cultures of the wild-type strain (r > 0.93) except for a transition from an H2-excess to an H2-limiting condition (r = 0.77). However, their patterns varied with chemostats of gene deletion mutants: Δmtd (r = 0.94), ΔMMP0719 (r = 0.88), ΔMMP1100 (r = 0.74), ΔMMP1447 (r = 0.65), and ΔfruΔfrc (r = 0.65). Interestingly, a putative transcript (Antisense_27) was also present in bc_0019. Antisense_27 is cotranscribed with its flanking genes MMP1635 encoding a redox-active disulfide protein and MMP1637 encoding a hypothetical protein (r > 0.96) and is antisense to MMP1636 encoding a major facilitator transporter (Yoon et al. 2011). These observations suggest that conditions that favor expression of genes encoding formylmethanofuran dehydrogenase, methyl-coenzyme M reductase, and tetrahydromethanopterin formyltransferase also favor expression of the redox-active disulfide protein and the hypothetical protein, while inhibiting, via antisense transcription, expression of the major facilitator transporter.
In two cases, subunits of the same enzyme were in separate biclusters. hdrA was in a separate bicluster from hdrC2B2, and fwdB was in a separate bicluster from fwdHFGDAC. In the latter case, the separation is consistent with different gene expression patterns, especially in the growth-curve experiment (r = −0.85) (Supplemental Fig. S2). In agreement with the biclustering results, in M. maripaludis, hdrA and fwdB are adjacent to the genes for the Vhu hydrogenase, while the remaining subunits of Hdr and Fwd are encoded elsewhere. Furthermore, the transcriptome architecture analysis had previously indicated that hdrA and fwdB are organized as a single operon with the vhu genes (MMP1691 to 1697) (Yoon et al. 2011). This pattern of coexpression and gene organization may have to do with the organization of the respective enzymes in a functional complex. We showed previously that Hdr, Fwd, and Vhu, which function in the bifurcated flow of electrons from hydrogen to heterodisulfide and to CO2 for reduction to formylmethanofuran, are physically associated (Costa et al. 2010). Of the three subunits of Hdr, HdrA, which contains the electron bifurcating center, likely forms the immediate contact with Vhu. Similarly, FwdB, which harbors the redox-active site of Fwd, may contact HdrA. Hence, the bicluster organization may reflect the enzyme subunits that are most closely associated, physically and functionally, with electron bifurcation from hydrogen.
Strikingly, three putative novel transcripts (potentially protein-encoding; Supplemental Table S2) were present in bc_0114 (Table 1). BLASTN and BLASTX searches of these transcripts against the NCBI nonredundant (nr) database did not produce any significant alignments, and no hits were found for protein-sequence similarity searches (Finn et al. 2011) against protein family databases such as Pfam, TIGRFAM, Gene3D, and Superfamily; yet biclustering analysis suggested that they played a role in methanogenesis. Furthermore, the three novel transcripts have motifs resembling a consensus ribosome binding site (gCCCgagGTGGG), which verifies that those putative novel transcripts are bona fide genes (Supplemental Fig. S2).
M. maripaludis contains two membrane-bound energy-conserving hydrogenases, Eha and Ehb. The genes encoding these two enzymes showed markedly different expression patterns (eha was in bc_0133 and ehb was in bc_0010), indicating that they are differentially regulated (Supplemental Fig. S2). The ehb genes were highly up-regulated after H2 perturbations, and they were clustered with genes encoding subunits of acetyl-CoA synthase/carbon monoxide dehydrogenase (Cdh), pyruvate oxidoreductase (Por), and 2-oxoisovalerate oxidoreductase (Vor), all anabolic oxidoreductases that catalyze early biosynthetic steps using CO2 and reduced Fd. It has been suggested (Porat et al. 2006; Xia et al. 2006; Major et al. 2010) that Ehb is the primary source of low-potential electrons for carbon assimilation, and our results strongly support this hypothesis. Additional members of bc_0010 included a gene encoding a relative of AMP-dependent acetyl-CoA synthetase gene (MMP1274) and a hypothetical protein (MMP1275) (Table 1). These genes were adjacent to the vor operon, and might have a role in this process.
We recently showed that Eha plays an anaplerotic role that both sustains the methanogenic pathway and replaces intermediates used for biosynthesis (Lie et al. 2012; Thauer 2012). In addition, Eha is thought to serve as an auxiliary source of anabolic electrons in the absence of Ehb (Porat et al. 2006; Xia et al. 2006; Major et al. 2010). Nevertheless, the regulation of Eha was markedly different from that of Ehb. No significant changes in gene expressions were observed for the eha operon when H2 was perturbed. Furthermore, in a transcriptomic and proteomic analysis, the expression of eha genes was not affected by the deletion of ehb genes (Xia et al. 2006). These results are consistent with the anaplerotic replenishment of methanogenic intermediates as the primary function of Eha. Thus, exploration of EGRIN recapitulated known biological phenomena and patterns associated with methanogenesis, and more importantly, it uncovered novel genes and regulatory associations that will drive hypothesis-driven investigations into novel aspects of this important process. The interactive exploration of all biclusters to enable similar discoveries is possible online at the following links:
Regulation of methanogenesis is distributed across five subnetworks each linked to a different set of biological processes
To find out which biological processes were associated with methanogenesis, we identified biclusters that share three or more genes with the methanogenesis-associated biclusters (Fig. 3). The network of biclusters distinctively separated into five subnetworks: among six biclusters enriched with methanogenesis genes, two of them (bc_0009 and bc_0114) were related to each other. Most of the genes for FwdB, Mtd, Mer, Fru, Frc, Vhu, Vhc, and Hdr formed a single subnetwork, indicating their shared regulatory pattern (bc_0009 and bc_0114). Genes encoding FwdHFGDAC, Ftr, and Mcr were present in bc_0089 as well as bc_0019. The latter bicluster also contains genes for ABC transporters, including a molybdenum/tungsten transporter that may provide the metal for the cofactor of Fmd or Fwd, a phosphate transporter, and a variety of hypothetical genes. MMP1243, which neighbors the fwd operon in the opposite strand and encodes a UBA/THIF-type NAD/FAD-binding protein, was also a shared gene. Genes encoding Mch and Mtr (bc_0035) were connected to bc_0126, which contained a two-component response regulator (MMP1304). Genes encoding membrane-bound hydrogenase Ehb (bc_0010) were linked to CO2 fixation via an acetyl-CoA pathway (bc_0017), further supporting the role of Ehb in providing anabolic electrons. The other membrane-bound hydrogenase, Eha (bc_0133) was linked to chemiosmotic ATP production (bc_0107) and to chemotaxis and flagellar biosynthesis (bc_0098), suggesting that motility and chemotaxis are coordinated with hydrogen utilization and energy flux. In the related species Methanocaldococcus jannaschii, flagellum biosynthesis was found to respond to hydrogen conditions (Mukhopadhyay et al. 2000). Similarly, M. maripaludis may have a motile and chemotactic response, perhaps toward hydrogen, which responds to energy conditions. Thus, the distribution of genes across biclusters revealed a systems level perspective on the regulatory, and perhaps metabolic, dependencies of methanogenesis with other aspects of physiology such as motility and transport. In the subsequent section, we provide orthogonal evidence for the intercoordination of these interlinked pathways via regulation by a shared set of TFs and EFs.
Figure 3.

Biclusters related to the methanogenesis. Each panel shows methanogenesis-related bicluster(s) and associated biclusters. (A) bc_0009 and bc_0114, (B) bc_0133, (C) bc_0019, (D) bc_0035, and (E) bc_0010. Colored boxes denote biclusters that are related to the methanogenesis as shown in Figure 2 and Supplemental Figure S2 (same color scheme). Biclusters that share three or more genes with methanogenesis-related biclusters are linked to these biclusters for identifying functional associations and are shown as gray-colored boxes. Each rectangle inside the bicluster boxes indicates member genes. Blue rounded squares of “A  B” show member genes common in the biclusters of A and B. Genes are colored according to their category from KEGG pathways—methane and hydrogen metabolism (red), motility (amber), oxidative phosphorylation (baby blue), ABC transporters (yellow), CO2 fixation (green), other functions (gray), and hypothetical genes (dark gray). Genes that are adjacent to member genes of a bicluster, but belong to the other connected bicluster, are in white. Transcription factor (TF) genes including putative ones are shown in red lettering. Arrows above gene(s) indicate direction and span of transcription units determined in the previous study (Yoon et al. 2011). Un_XX means a transcript that did not match any protein sequence in the nr data set, and As_XX denotes a transcript antisense to annotated genes. Detailed information of the biclusters can be found in Supplemental Figure S1 and Supplemental Table S5.
B” show member genes common in the biclusters of A and B. Genes are colored according to their category from KEGG pathways—methane and hydrogen metabolism (red), motility (amber), oxidative phosphorylation (baby blue), ABC transporters (yellow), CO2 fixation (green), other functions (gray), and hypothetical genes (dark gray). Genes that are adjacent to member genes of a bicluster, but belong to the other connected bicluster, are in white. Transcription factor (TF) genes including putative ones are shown in red lettering. Arrows above gene(s) indicate direction and span of transcription units determined in the previous study (Yoon et al. 2011). Un_XX means a transcript that did not match any protein sequence in the nr data set, and As_XX denotes a transcript antisense to annotated genes. Detailed information of the biclusters can be found in Supplemental Figure S1 and Supplemental Table S5.
Construction of a predictive model of global gene regulation of methanogenesis
In order to infer regulatory mechanisms responsible for coregulation of methanogenesis with disparate aspects of physiology, we used the Inferelator algorithm (Bonneau et al. 2006). Inferelator uses a regression-based approach to model the expression changes of genes in each bicluster as a function of corresponding (steady-state data) or preceding (time-course data) changes in one or more TFs and/or EFs. In other words, the causal regulatory influences in the EGRIN model are inferred through regression analysis of expression-level changes in bicluster genes against corresponding (or time-lagged) concentration changes across EFs and mRNA levels of TFs. While this strategy does not fully capture mechanistic detail of transcriptional regulation, it does generate meaningful associations that can drive further targeted analyses in the context of gene functions, genomic locations, physical interactions, and evolutionary associations among a specific set of interconnected genes in the EGRIN model to generate experimentally testable hypotheses. The Inferelator discovered the most probable set of regulatory influences from 57 TFs and two EFs that sufficiently explained expression changes of genes within each of the cMonkey-identified biclusters. The collection of the complete set of regulatory influences interconnected regulation of genes across all biclusters of M. maripaludis into a unified EGRIN model. Using this approach, we discovered that individual and combinatorial regulatory influences of 46 (out of 57 predicted) TFs (Supplemental Table S6) and two EFs (H2 and formate) accurately recapitulated experimentally observed transcriptional changes in 90% of the genes in the entire genome. In toto, the EGRIN model includes a set of 1227 EF and TF regulatory influences that interlink the regulation of 1661 genes (90% of all genes in the genome) that are organized into 166 biclusters.
Consistent with the biclustering results, genes involved in the methanogenesis pathway were predicted to be under the regulatory influences of multiple TFs and EFs (Fig. 4A). As expected, H2 was predicted to negatively regulate biclusters (bc_0114 and bc_0009), which showed the most marked response to H2 conditions. Interestingly, biclusters bc_0019 and bc_0035 were predicted to be under different regulatory influences, although their member genes involved in consecutive metabolic steps—the mcr operon (member of bc_0019) and the mtr operon (bc_0035)—are adjacent in the genome. As explained before, the differential regulation of these adjacent steps might reflect redox homeostasis and/or distinct environment-dependent functional differences between these two operons.
Figure 4.

Environment and gene regulatory influence network for methanogenesis. (A) Environment and gene regulatory influence of methanogenesis biclusters is shown as a network diagram. One of the striking influences is the activation of three of the six methanogenesis related biclusters (bc_0009, bc_0019, and bc_0114) by MMP1100. H2 on the other hand, repressed bc_0009 and bc_0114. Regulatory influences of TFs and EFs on biclusters are associated with influence weight calculated by Inferelator. Influence weights can be used to filter most probable regulatory influences. For methanogenesis, network influences with weight less than 0.1 were not included in the analysis. Rectangles designate biclusters (colored the same as in Figs. 2, 3); circles denote transcription factors (red) and environmental factors (pale green); and triangles indicate AND logic gates of regulatory influences. Red arrows and green blunt-headed lines represent positive and negative influences, respectively, with their width being proportional to the influence weight. (B) The EGRIN model accurately recapitulates transcriptional changes within each bicluster, just from measured values of TFs and EFs. For selected biclusters, comparison of predicted mean expression values with the corresponding measured state is shown as line plots in the top panels. Prediction of the bicluster mean expression level in the time course conditions and steady-state conditions are shown as solid black and blue dashed lines, respectively. Corresponding measured expression is shown as dashed red lines. Correlation between predicted and measured relative transcript level changes for each bicluster is shown as a scatterplot in the bottom panels. Corresponding correlation coefficient is given at top, left and denoted by R.
Intercoordination of different steps is accomplished by shared regulators in a combinatorial scheme
We found that the intercoordinated regulation of genes across multiple biclusters was accomplished through combinatorial regulatory schemes by several TFs regulating each other. Interestingly, MMP1100, a TrmB family TF, was predicted to coregulate genes in biclusters 9, 19, and 114, which are involved in the majority of the steps in methanogenesis, except for those catalyzed by Mch and Mtr (Fig. 4A). This is evident in how the expression of MMP1100 perfectly tracks with the expression of the putative regulatory targets in the methanogenesis-related biclusters (Supplemental Fig. S3). Additional regulators are predicted to act in a combinatorial scheme to uniquely alter regulation of genes within each of these biclusters; e.g., regulation of bc_0114 by the TF MMP1023 and regulation of bc_0009 by the TF MMP0629. Furthermore, TFs that are coregulated with genes in a bicluster interlink the network of regulation across biclusters; e.g., the TF MMP1275 is a member of bc_0010 and a regulator of bc_0035. Likewise, MMP0719 is a member of bc_0035 and a regulator of bc_0114. MMP1100, a regulator of biclusters 9, 19, and 114, is itself in bicluster 124 (Supplemental Table S5) that also contains the anabolic genes including cdh, cdhD, cdhB, acsA, and acd. This may indicate that MMP1100 is coregulated with carbon fixation genes and regulates methanogenesis. Based on the measured values of TFs and EFs, we used regulatory influences in the EGRIN model to predict average transcriptional changes of genes within each bicluster. This analysis showed that the model accurately recapitulated the global transcriptional changes in biclusters across the entire EGRIN model. The predicted and observed global transcriptional changes for the methanogenesis biclusters is illustrated in Figure 4B.
Experimental verification of EGRIN predictions uncover phosphate-dependent regulation of formate dehydrogenase
Since MMP1100 and MMP0719 were predicted to play key regulatory roles in methanogenesis, we tested the effect of knocking out these TFs. We focused on genes encoding formate dehydrogenase active-site subunits (fdhB, MMP0139 and MMP1297), members of bc_0009, as target genes. Although MMP1100 and MMP0719 could interact with H2 as an environmental signal, other possibilities include P and N, which were inversely varied with H2 in time-course transition experiments. As an indication that P is involved, we found that for ΔMMP1100, the level of P in the medium supplied to the chemostat in order to achieve P-limited conditions (0.18 mM PO42− to maintain a steady-state OD660 of 0.76) was different from that required for the wild-type strain (0.12 mM). We therefore plotted the levels of MMP0139 and MMP1297 mRNAs over time-course transitions from PO42--limited conditions to H2-limited conditions (Fig. 5A,B). Compared with the wild type, both MMP0139 and MMP1297 were expressed at elevated levels in ΔMMP0719 (paired t-test P = 0.043 and P = 0.0003, respectively) and ΔMMP1100 (P = 0.016 and P = 0.00003, respectively), especially after transition to H2 limitation and PO42− excess conditions. Interestingly, regulation of MMP1100 itself was also affected in the ΔMMP0719 genetic background, being expressed at lower levels, particularly after the transition. In addition to providing experimental validation for EGRIN-predicted regulation of formate dehydrogenase, these results suggest a model in which MMP1100 is a transcriptional repressor that responds to excess PO42− to modulate formate dehydrogenase levels. Additionally, MMP0719 is a transcriptional activator that coregulates MMP1100 and the fdh genes (Fig. 5C).
Figure 5.

Roles of the transcriptional regulators MMP0719 and MMP1100. Transcriptional changes of formate dehydrogenase subunits in TF knockouts MMP1100 and MMP0719 during transition from P-limited to H2-limited conditions confirm EGRIN predictions. (A) barplot of fdhB2 (MMP0139) mRNA levels versus time in the wild-type (WT) (brown), MMP0719 knockout (dark green), and MMP1100 knockout (dark yellow) during transition from P-limiting to H2-limiting conditions. Expression of MMP1100 is also shown as blue dotted line in both genetic backgrounds. (B) Barplot of fdhB1 (MMP1297) mRNA changes, similar to A. (C) Based on the experimental confirmation of EGRIN predictions, we developed a model for the regulation of formate dehydrogenase. According to this model, MMP1100 modulates the activity of MMP1297 in a PO4-dependent manner, while MMP0719 activates both MMP1100 and MMP1297. (D) Predictive power of the EGRIN network model evaluated over the training (left) and new transcriptome data (right). Histogram of RMSD error between the predicted and measured response for 166 biclusters evaluated over 58 conditions in the training (left) and 56 conditions in the new data sets that were not used for the model construction (right). Similar median values (0.42 for training and 0.44 for new data set) indicate that our model performed equally well on both data sets.
We evaluated the power of the EGRIN model to predict global transcriptional responses of M. maripaludis over 56 new transcriptome data sets that were not used for the model construction. These new data sets included transcriptional responses of Δmtd, ΔfruAΔfrcA, ΔMMP1447, ΔMMP0719, and ΔMMP1100 to changes in H2 and PO4 (Fig. 1C; Supplemental Table S4). The predicted expression state of each bicluster based on the levels of TFs and EFs were compared with the corresponding measured states both in the training data set and in the new data by using root mean square deviation (RMSD) to estimate the predictive power. The predictive powers of the M. maripaludis EGRIN were quite similar over training data (RMSD = 0.42) and new data (RMSD = 0.44) (Fig. 5D). Thus, the small and similar RMSD values in both training and new data sets highlighted that the overall expression state is well-predicted and that the M. maripaludis EGRIN network model accurately captured causal regulatory relationships across genes and biclusters to reveal a regulatory landscape of methanogenesis.
Conclusion
In this study, we built the first predictive model of global H2 regulation of methanogenesis in a hydrogenotrophic methanogen. The model revealed important aspects of methanogenesis regulation and related metabolic processes. Methanogenesis was generally up-regulated with H2 limitation, which clearly has a broad impact on transcriptional regulation. H2 had an especially marked effect on genes contained in biclusters 0114 and 0009, and genes that were most markedly affected were those that are associated with F420, consistent with previous observations (Hendrickson et al. 2007) and with the central role of this coenzyme. In addition, three steps associated with coenzyme F420 were found to be transcriptionally coregulated with other, non-F420-associated steps of methanogenesis. Also interesting was the observation that in two cases (Hdr and Fwd), different subunits of a single enzyme were under separate regulatory influences. This appears to reflect the association of these enzymes into multi-enzyme complexes that are important for electron flow and bifurcation centering on the heterodisulfide reductase. Reflecting this separation of methanogenesis into multiple biclusters, at least five regulatory mechanisms are predicted to control methanogenesis (Figs. 2,3). Each mechanism links a key step of methanogenesis with a different aspect of metabolism. Similarly, regulation of Ehb membrane-bound hydrogenase is uniquely coupled with enzymes associated with carbon assimilation, supporting its role in using H2 to supply electrons for biosynthesis. Many cellular processes were transcriptionally coordinated with methanogenesis (Fig. 3). Chemotaxis, flagellar synthesis, and ATP generation were affiliated with Eha. As expected, the synthesis of certain ABC transporters was associated with several steps, including the metal-containing formymethanofuran dehydrogenases and carbon fixation. Strikingly, additional genes of previously unknown functions including three novel coding RNAs and one antisense transcript may also be involved in methanogenesis (Supplemental Table S2).
We have previously demonstrated the power of the EGRIN approach to characterize the biology of an organism by providing insights into environmental context-dependent dynamic interactions (functional and regulatory) among nearly all genes in the genome (including noncoding RNAs and genes of unknown function). In this study, we extended this model to methanogenic archaea to gain insights into the regulation of methanogenesis. It should be noted that in its current instantiation this model does not account for all types of regulatory mechanisms, including signal transduction and allosteric control. However, this is not entirely a limitation of the modeling algorithms, but more a function of our limited understanding of many pre- and post-transcriptional regulatory mechanisms. For instance, incorporation of regulation by RNases and ncRNAs can be partly attributed to our limited understanding of their mechanism of action. Once characterized, we can develop appropriate strategies to incorporate novel regulatory mechanisms, as was done with miRNA regulation (Plaisier et al. 2011, 2012). Ultimately, the EGRIN model is just a powerful instrument for generating hypotheses that should be further characterized through iterative experimentation and modeling (Pang et al. 2013).
Methanogenesis, like all aspects of metabolism, is not an insulated process. It has complex interdependencies with many other cellular processes, partly due to shared and limited resources including metabolic intermediates or the general energy demand. By observing the dynamic molecular changes when M. maripaludis transitions across varied environment-dependent states of methanogenesis, we have captured the complex interrelationships among known genes of methanogenesis and other genes of known and unknown function. The comparative analyses of these dynamic changes have revealed the multifaceted modular organization even within the known components of methanogenesis, and shed insight into causal influences of EFs and TFs on this process. These systems level interactions within the EGRIN model could also be a reflection of dynamic, complex, and coupled changes in the natural environment of this organism. Conversely, the model could also help to characterize the microbial ecology of methanogens. From the perspective of metabolic engineering for industrial applications, the first systems scale predictive model for regulation in a hydrogenotrophic methanogen provides a map for potential systems level consequences resulting from targeted manipulation of specific steps in methanogenesis.
Methods
Bacterial strains and growth conditions
M. maripaludis MM901, wild-type strain S2 with an in-frame deletion of the uracil phosphoribosyltransferase gene (Costa et al. 2010), and all gene deletion strains were grown by continuous culture in a 1-L fermenter (New Brunswick Scientific) at 37°C (Haydock et al. 2004). Medium and gas compositions were modified from those for excess conditions (Xia et al. 2009; Supplemental Table S3).
The standard gassing regime was a 110-mL/min H2, 40-mL/min CO2, 35-mL/min Ar, and 15-mL/min H2S/Ar mixture (1:99). For shifts from a H2-excess to a H2-limited condition, H2 was lowered from standard 110 mL/min to 21 mL/min and Ar was raised from standard 35 mL/min to 125 mL/min. For shifts from P-limited to P-excess conditions, phosphate was raised from 0.12 mM to the standard 0.8 mM. Shifts from N-limited to N-excess conditions were achieved by raising NH4+ from 2.8 mM to the standard 10 mM (Fig. 1). The dilution rate was held constant at 0.083 h−1. For time-series array data, cultures before perturbation were allowed to reach steady state. We rapidly changed concentration(s) of H2 and/or a nutrient, and sampled at 30 min before perturbation, immediately after the perturbation, and after 5, 10, 20, 30, 45, 60, 90, 120, 180, and 300 min. Culture samples (1.5 mL) were rapidly removed from the chemostat vessels by syringe and cell pellets collected by microcentrifugation, immediately frozen in an ethanol-dry ice bath, and stored at −80°C. Rates of CH4 production were measured for all cultures as described before (Lie et al. 2012).
For single environmental perturbations, we used a constant dilution rate in a chemostat (Haydock et al. 2004) until the culture reached steady state (defined as constant cell density). Subsequently, we rapidly increased or decreased the proportion of H2 in the input gas, 110 mL/min or 21 mL/min, respectively (Fig. 1A). A sample was collected prior to the perturbation and subsequently, in a nonlinear time course, multiple samples were collected to capture the early, middle, and late stages of the cellular response. For combinatorial environmental perturbations, we first equilibrated the cells to a H2-excess and P-limited (or N-limited) condition and sampled as the culture transitioned to a H2-limited and P-excess (or N-excess) condition (Fig. 1B,C). Total RNA was prepared from each of these samples, labeled, hybridized to a high-density tiling microarray, scanned, and analyzed for global transcriptional changes.
Construction of gene deletion mutants
In-frame deletions of the genes mtd (MMP0372), frcA (MMP0820), and fruA (MMP1382) were previously constructed in the vector pCRprtneo (Hendrickson and Leigh 2008). The inserts, which contained ∼500 bp of each upstream and downstream flanking DNA, were digested out and recloned into pCRuptneo (Costa et al. 2010). Plasmid DNA was transformed into the strain MM901, and merodiploids were resolved to make the mutants containing the deletions Δmtd (strain MM1283) and ΔfruAΔfrcA (strain MM1280) (Lie et al. 2012). Mutants containing the deletions ΔMMP1100 (strain MM1323) and ΔMMP0719 (strain MM1321) were made by PCR amplifying 500 bp of the flanking regions of each target gene. The 3′ end of each upstream product contained the start codon followed by the restriction site AscI. The 5′ end of each downstream product contained AscI, additional base(s), and the stop codon. The fragments were digested with AscI, ligated, PCR amplified, cloned into pCRuptneo, and introduced into MM901 as above. To make MM901ΔMMP1447 Pnif∷eha (strain MM1322) (Costa et al. 2013b), regions flanking MMP1447 were PCR amplified and cloned into the plasmid pHW40 containing Pnif (Chaban et al. 2007). The product was PCR amplified, ligated into pCRuptneo, and introduced into MM901 as above. The complete replacement of all wild-type alleles with the introduced mutant constructs was confirmed by Southern blot or PCR.
Construction of high-resolution tiling microarray, RNA hybridization, and image analysis
A whole-genome high-resolution tiling array was designed to contain 60K 60-mer probes with strand-specific sequences and manufacturer's controls (SurePrint G3 8 × 60K, Agilent Technologies). Probes were tiled every 60 nt for M. maripaludis (GenBank genome accession: NC_005791) (Hendrickson et al. 2004). Most of the ORFs (96.9%) had over five probes (mean: 15.8 ea, std: 8.9) to provide statistically significant coverage over the entire region of the ORF.
Total RNA was prepared by using the mirVana miRNA isolation kit (Applied Biosystems) according to the manufacturer's instructions. Total RNA from each sample was compared against a reference RNA pool that was generated in bulk from a mid-log phase culture of MM901 (Yoon et al. 2011). Total RNA from samples and reference were directly labeled with Cyanine 3 (Cy3) or Cyanine 5 (Cy5) dyes (Molecular Probes and Kreatech BV), and were hybridized to the tiling array and washed according to the array manufacturer's instructions. The arrays were scanned by a Microarray Scanner (Agilent Technologies) and spot finding was done using Feature Extraction software (Agilent Technologies). Labeling dye for sample versus reference was flipped to have two differentially labeled replicates per sample.
Signal intensities and local backgrounds were determined by using Feature Extraction software (Agilent Technologies). Raw intensity signals from each slide were processed by the SBEAMS-microarray pipeline (Marzolf et al. 2006) (http://www.SBEAMS.org/microarray), where resultant data was median normalized and subjected to significance of microarray (SAM) and variability and error estimates (VERA) analysis. Each data point was assigned a significance statistic, λ, using maximum likelihood (Ideker et al. 2000).
Construction of environmental and gene regulatory influence network
In our previous study of the transcriptome architecture of M. maripaludis, we identified 62 transcripts that did not overlap any previously annotated coding sequences and 29 transcripts that were antisense to annotated genes (Yoon et al. 2011). To develop a mechanistically accurate EGRIN model, those transcripts were appended to the list of annotated genes. We identified 57 known and putative transcription factors (TFs), 32 from the initial genome annotation, one from a literature (Kaster et al. 2011a), and an additional 24 from databases of DBD (Wilson et al. 2008) and ArchaeaTF (Wu et al. 2008; Supplemental Table S6).
The cMonkey integrated biclustering algorithm was applied to identify subsets of genes that were coregulated under certain culture conditions (Reiss et al. 2006). The inputs to cMonkey were 58 transcriptome profiles of wild-type M. maripaludis MM901 (Supplemental Table S4), upstream regions of all genes, and functional association networks, including operon predictions from MicrobesOnline and functional protein interactions from EMBL String databases (Szklarczyk et al. 2011). Briefly, cMonkey iteratively prioritizes the grouping of genes with similar expression profiles, supported by additional evidence of coregulation such as the existence of similar cis-regulatory motifs in their promoter regions (detected de novo using the MEME algorithm) (Bailey and Elkan 1994) and functional associations between genes (here we used only the integrated functional association network provided by STRING database) (Szklarczyk et al. 2011). cMonkey first creates seed clusters and then optimizes them to create biclusters by adding or removing genes and conditions after calculating coexpression measures, searching for motifs and additional evidences of coregulation. At each stage it computes the probability of being a member of the bicluster for each gene or condition sampled from the conditional probability distribution. cMonkey biclusters are sets of genes that are putatively coregulated in subsets of the experimental conditions. The algorithm allows genes to be members of multiple coregulated gene groups—a property that is consistent with how biology operates—thereby allowing the discovery of combinatorial regulation of the same genes by multiple EFs and/or TFs.
Following the protocol of Bonneau et al. (2007), transcriptional influences of each bicluster were inferred using the Inferelator (Bonneau et al. 2006). The algorithm attempts to predict the mean expression levels of the genes in each bicluster via regularized linear regression and variable selection with 10-fold cross-validation. It does so by using a sparse subset of linear combinations of measured expression level changes in TFs and the experimental record of EF changes as linear predictors. Whereas the original Inferelator utilized L1-constrained regression (Tibshirani 1996), which required preclustering of highly correlated predictors into “TF groups” (see Bonneau et al. 2006 for details) prior to the inference, we modified the procedure slightly to utilize the “softer” “elastic net” linear constraint (Zou and Hastie 2005). This procedure effectively groups correlated predictors “during” the variable selection process, thereby eliminating the predictor preclustering step and enabling the variable selection for each bicluster independently from the list of highly correlated predictors. We verified that this modified version of the algorithm generated nearly identical predictions to those reported previously (Bonneau et al. 2006, 2007), including all of the experimentally verified predictions in Bonneau et al. (2007) (data not shown). The resulting EGRIN model was visualized as a network in Cytoscape (Cline et al. 2007) and was explored using the Gaggle framework (Shannon et al. 2006).
Enrichment analysis of KEGG pathways
For functional enrichment analysis, a list of available KEGG annotated functions for Methanococcus maripaludis S2 genes were collected from the KEGG database. Genes represented in each of the specific KEGG pathways were compared with the members of each bicluster to find statistically significant enrichment of pathways. P-values for overrepresentation of KEGG pathways or methanogen-specific genes in the gene list of each bicluster were calculated using the cumulative hypergeometric distribution and were corrected for multiple hypothesis testing by the Bonferroni method (Sheskin 2007).
Data access
The microarray data have been deposited in the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE42115, GSE42126, GSE42130, GSE42143, GSE42159, GSE42162, GSE42164–GSE42167. All of the data from this study are available at http://baliga.systemsbiology.net/enigma/. In addition, a network model is available at the following URL: http://networks.systemsbiology.net/mmp.
Acknowledgments
This work was supported by the U.S. Department of Energy, Award Nos. DE-FG02-07ER64327 and DG-FG02-08ER64685 (to N.S.B.); the Office of Science (BER), U.S. Department of Energy, Award No. DE-FG02-08ER64685 (to J.A.L.); and the National Science Foundation MRI, Grant No. 0923536 (to R.L.M.). The work conducted by ENIGMA-Ecosystems and Networks Integrated with Genes and Molecular Assemblies (http://enigma.lbl.gov), a Scientific Focus Area Program at Lawrence Berkeley National Laboratory, was supported by the Office of Science, Office of Biological and Environmental Research, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The work of S.H.Y. was supported in part by KRIBB and the Korean Ministry of Science, ICT & Future Planning (NRF-2012-C1AAA001-2012M1A2A2026559).
Author contributions: N.S.B. and J.A.L. conceived and organized the project. S.H.Y., N.S.B., and J.A.L. designed the experiments, analyzed the data, and wrote the manuscript. D.J.R. contributed to construction of biclusters and the EGRIN model, and assessed the significance of the EGRIN model. M.P. did microarray experiments. J.B., K.C.C., and T.J.L. performed fermentations and constructed gene deletion mutants. J.S. constructed the PeptideAtlas. M.H. and R.L.M. helped with construction of the PeptideAtlas. S.T. contributed to bicluster analysis, EGRIN model validation, and manuscript preparation.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.153916.112.
Freely available online through the Genome Research Open Access option.
References
- Bailey TL, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 [PubMed] [Google Scholar]
- Bonneau R, Reiss DJ, Shannon P, Facciotti M, Hood L, Baliga NS, Thorsson V 2006. The Inferelator: An algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol 7: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau R, Facciotti MT, Reiss DJ, Schmid AK, Pan M, Kaur A, Thorsson V, Shannon P, Johnson MH, Bare JC, et al. 2007. A predictive model for transcriptional control of physiology in a free living cell. Cell 131: 1354–1365 [DOI] [PubMed] [Google Scholar]
- Chaban B, Ng SY, Kanbe M, Saltzman I, Nimmo G, Aizawa S, Jarrell KF 2007. Systematic deletion analyses of the fla genes in the flagella operon identify several genes essential for proper assembly and function of flagella in the archaeon, Methanococcus maripaludis. Mol Microbiol 66: 596–609 [DOI] [PubMed] [Google Scholar]
- Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. 2007. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2: 2366–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa KC, Wong PM, Wang T, Lie TJ, Dodsworth JA, Swanson I, Burn JA, Hackett M, Leigh JA 2010. Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proc Natl Acad Sci 107: 11050–11055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa KC, Lie TJ, Jacobs MA, Leigh JA 2013a. H2-independent growth of the hydrogenotrophic methanogen Methanococcus maripaludis. MBio 4: e00062-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa KC, Yoon SH, Pan M, Burn JA, Baliga NS, Leigh JA 2013b. Effects of H2 and formate on growth yield and regulation of methanogenesis in Methanococcus maripaludis. J Bacteriol 195: 1456–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deppenmeier U 2002. The unique biochemistry of methanogenesis. Prog Nucleic Acid Res Mol Biol 71: 223–283 [DOI] [PubMed] [Google Scholar]
- Deutsch EW 2010. The PeptideAtlas Project. Methods Mol Biol 604: 285–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS 2007. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol 5: e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR 2011. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res 39: W29–W37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydock AK, Porat I, Whitman WB, Leigh JA 2004. Continuous culture of Methanococcus maripaludis under defined nutrient conditions. FEMS Microbiol Lett 238: 85–91 [DOI] [PubMed] [Google Scholar]
- Hendrickson EL, Leigh JA 2008. Roles of coenzyme F420-reducing hydrogenases and hydrogen- and F420-dependent methylenetetrahydromethanopterin dehydrogenases in reduction of F420 and production of hydrogen during methanogenesis. J Bacteriol 190: 4818–4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson EL, Kaul R, Zhou Y, Bovee D, Chapman P, Chung J, Conway de Macario E, Dodsworth JA, Gillett W, Graham DE, et al. 2004. Complete genome sequence of the genetically tractable hydrogenotrophic methanogen Methanococcus maripaludis. J Bacteriol 186: 6956–6969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson EL, Haydock AK, Moore BC, Whitman WB, Leigh JA 2007. Functionally distinct genes regulated by hydrogen limitation and growth rate in methanogenic Archaea. Proc Natl Acad Sci 104: 8930–8934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson EL, Liu Y, Rosas-Sandoval G, Porat I, Soll D, Whitman WB, Leigh JA 2008. Global responses of Methanococcus maripaludis to specific nutrient limitations and growth rate. J Bacteriol 190: 2198–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Thorsson V, Siegel AF, Hood LE 2000. Testing for differentially-expressed genes by maximum-likelihood analysis of microarray data. J Comput Biol 7: 805–817 [DOI] [PubMed] [Google Scholar]
- Kaster AK, Goenrich M, Seedorf H, Liesegang H, Wollherr A, Gottschalk G, Thauer RK 2011a. More than 200 genes required for methane formation from H2 and CO2 and energy conservation are present in Methanothermobacter marburgensis and Methanothermobacter thermautotrophicus. Archaea 2011: 973848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaster AK, Moll J, Parey K, Thauer RK 2011b. Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc Natl Acad Sci 108: 2981–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur A, Van PT, Busch CR, Robinson CK, Pan M, Pang WL, Reiss DJ, DiRuggiero J, Baliga NS 2010. Coordination of frontline defense mechanisms under severe oxidative stress. Mol Syst Biol 6: 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krouk G, Mirowski P, LeCun Y, Shasha DE, Coruzzi GM 2010. Predictive network modeling of the high-resolution dynamic plant transcriptome in response to nitrate. Genome Biol 11: R123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh JA, Albers SV, Atomi H, Allers T 2011. Model organisms for genetics in the domain Archaea: Methanogens, halophiles, Thermococcales and Sulfolobales. FEMS Microbiol Rev 35: 577–608 [DOI] [PubMed] [Google Scholar]
- Lie TJ, Costa KC, Lupa B, Korpole S, Whitman WB, Leigh JA 2012. Essential anaplerotic role for the energy-converting hydrogenase Eha in hydrogenotrophic methanogenesis. Proc Natl Acad Sci 109: 15473–15478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Whitman WB 2008. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann NY Acad Sci 1125: 171–189 [DOI] [PubMed] [Google Scholar]
- Lupa B, Hendrickson EL, Leigh JA, Whitman WB 2008. Formate-dependent H2 production by the mesophilic methanogen Methanococcus maripaludis. Appl Environ Microbiol 74: 6584–6590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major TA, Liu Y, Whitman WB 2010. Characterization of energy-conserving hydrogenase B in Methanococcus maripaludis. J Bacteriol 192: 4022–4030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzolf B, Deutsch EW, Moss P, Campbell D, Johnson MH, Galitski T 2006. SBEAMS-Microarray: Database software supporting genomic expression analyses for systems biology. BMC Bioinformatics 7: 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay B, Johnson EF, Wolfe RS 2000. A novel pH2 control on the expression of flagella in the hyperthermophilic strictly hydrogenotrophic methanarchaeaon Methanococcus jannaschii. Proc Natl Acad Sci 97: 11522–11527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang WL, Kaur A, Ratushny AV, Cvetkovic A, Kumar S, Pan M, Arkin AP, Aitchison JD, Adams MW, Baliga NS 2013. Metallochaperones regulate intracellular copper levels. PLoS Comput Biol 9: e1002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul K, Nonoh JO, Mikulski L, Brune A 2012. “Methanoplasmatales,” Thermoplasmatales-related archaea in termite guts and other environments, are the seventh order of methanogens. Appl Environ Microbiol 78: 8245–8253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisier CL, Bare JC, Baliga NS 2011. miRvestigator: Web application to identify miRNAs responsible for co-regulated gene expression patterns discovered through transcriptome profiling. Nucleic Acids Res 39: W125–W131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisier CL, Pan M, Baliga NS 2012. A miRNA-regulatory network explains how dysregulated miRNAs perturb oncogenic processes across diverse cancers. Genome Res 22: 2302–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat I, Kim W, Hendrickson EL, Xia Q, Zhang Y, Wang T, Taub F, Moore BC, Anderson IJ, Hackett M, et al. 2006. Disruption of the operon encoding Ehb hydrogenase limits anabolic CO2 assimilation in the archaeon Methanococcus maripaludis. J Bacteriol 188: 1373–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss DJ, Baliga NS, Bonneau R 2006. Integrated biclustering of heterogeneous genome-wide datasets for the inference of global regulatory networks. BMC Bioinformatics 7: 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai S, Imachi H, Hanada S, Ohashi A, Harada H, Kamagata Y 2008. Methanocella paludicola gen. nov., sp. nov., a methane-producing archaeon, the first isolate of the lineage ‘Rice Cluster I', and proposal of the new archaeal order Methanocellales ord. nov. Int J Syst Evol Microbiol 58: 929–936 [DOI] [PubMed] [Google Scholar]
- Shannon PT, Reiss DJ, Bonneau R, Baliga NS 2006. The Gaggle: An open-source software system for integrating bioinformatics software and data sources. BMC Bioinformatics 7: 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheskin D. 2007. Handbook of parametric and nonparametric statistical procedures. Chapman & Hall/CRC, Boca Raton, FL. [Google Scholar]
- Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, et al. 2011. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39: D561–D568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauer RK 2012. The Wolfe cycle comes full circle. Proc Natl Acad Sci 109: 15084–15085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R 2008. Methanogenic archaea: Ecologically relevant differences in energy conservation. Nat Rev Microbiol 6: 579–591 [DOI] [PubMed] [Google Scholar]
- Tibshirani R 1996. Regression shrinkage and selection via the lasso. J R Stat Soc Ser B Methodol 58: 267–288 [Google Scholar]
- Wilson D, Charoensawan V, Kummerfeld SK, Teichmann SA 2008. DBD-taxonomically broad transcription factor predictions: New content and functionality. Nucleic Acids Res 36: D88–D92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Wang S, Bai J, Shi L, Li D, Xu Z, Niu Y, Lu J, Bao Q 2008. ArchaeaTF: An integrated database of putative transcription factors in Archaea. Genomics 91: 102–107 [DOI] [PubMed] [Google Scholar]
- Xia Q, Hendrickson EL, Zhang Y, Wang T, Taub F, Moore BC, Porat I, Whitman WB, Hackett M, Leigh JA 2006. Quantitative proteomics of the archaeon Methanococcus maripaludis validated by microarray analysis and real time PCR. Mol Cell Proteomics 5: 868–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, Wang T, Hendrickson EL, Lie TJ, Hackett M, Leigh JA 2009. Quantitative proteomics of nutrient limitation in the hydrogenotrophic methanogen Methanococcus maripaludis. BMC Microbiol 9: 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SH, Reiss DJ, Bare JC, Tenenbaum D, Pan M, Slagel J, Moritz RL, Lim S, Hackett M, Menon AL, et al. 2011. Parallel evolution of transcriptome architecture during genome reorganization. Genome Res 21: 1892–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Hastie T 2005. Regularization and variable selection via the elastic net. J R Stat Soc Ser B Methodol 67: 301–320 [Google Scholar]