

Abstract
The adaptive immune response selectively expands B- and T-cell clones following antigen recognition by B- and T-cell receptors (BCR and TCR), respectively. Next-generation sequencing is a powerful tool for dissecting the BCR and TCR populations at high resolution, but robust computational analyses are required to interpret such sequencing. Here, we develop a novel computational approach for BCR repertoire analysis using established next-generation sequencing methods coupled with network construction and population analysis. BCR sequences organize into networks based on sequence diversity, with differences in network connectivity clearly distinguishing between diverse repertoires of healthy individuals and clonally expanded repertoires from individuals with chronic lymphocytic leukemia (CLL) and other clonal blood disorders. Network population measures defined by the Gini Index and cluster sizes quantify the BCR clonality status and are robust to sampling and sequencing depths. BCR network analysis therefore allows the direct and quantifiable comparison of BCR repertoires between samples and intra-individual population changes between temporal or spatially separated samples and over the course of therapy.
Healthy humans have ∼3 × 109 B-cells in the peripheral blood, and this population consists of a repertoire of distinct B-cells expressing different B-cell receptors (BCRs) necessary to bind diverse antigens and produce an effective humoral immune response. BCRs consist of two identical heavy-chain (IGH) and two identical light-chain proteins, where the antigen-binding regions are highly diversified (Tonegawa 1983; Woof and Burton 2004). BCR diversity is generated in a number of ways. The IGH gene locus encodes for multiple distinct copies of the variable (V), diversity (D), and joining (J) gene segments (Jung et al. 2006), with functional IGH BCR genes generated by site-specific V-D-J recombination (Latchman 2005; Schatz and Swanson 2010). The imprecise joining of the V-D-J gene segments leads to random deletion and insertion of nucleotides during recombination events, resulting in sequence diversification at the junctional regions (Fig. 1A). Rearranged BCR genes are further diversified by helper T-cell-mediated somatic hypermutation (SHM) through the action of activation-induced cytosine deaminase. Through clonal affinity selection for enhanced antigen binding, non-germ-line SHM-mediated variation contributes significantly to the diversification of the mature B-cell repertoire (Brezinschek et al. 1995; Dorner et al. 1998; Weinstein et al. 2009; Batrak et al. 2011).
Figure 1.

Sequencing of B-cell receptor repertoires. (A) Representation of the genomic rearrangement process during V-D-J recombination to generate the heavy-chain B-cell receptor. B-cell receptor amplification was performed by reverse transcription on total RNA by single J region primer, and subsequent multiplex PCR amplification. (B) The percentage of reads corresponding to the highest expressed B-cell receptor sequence for each sample, separated into sample type: healthy individuals, chronic lymphocytic leukemia patients (CLL), and human lymphoblastoid cell lines (LCL). Two-sided t-tests were performed between the sample subsets, with the P-values indicated above. (C) Percentage of sequences shared between runs for technical repeats for (1) the RT-PCR and resequencing (RT-PCR repeats, green bars), and (2) the 454 sequencing from the same RT-PCR product (sequencing repeats, purple bars). For each sample, two repeats were performed and the percentage of reads shared between the repeats is shown (each repeat is compared with the other, so two bars are shown per sample).
The diversification and selection dynamics of BCR repertoires in healthy individuals and those with infection, autoimmunity, immunodeficiency, or B-cell malignancies remain poorly understood but can have important clinical implications. For example, the majority of B-cells in individuals with B-cell malignancies typically express a single dominant clonal BCR sequence (Arber 2000; Campbell et al. 2008), and continued intraclonal tumor evolution by SHM in patients with B-cell lymphomas has been observed (Stamatopoulos et al. 1996; Bagnara et al. 2006; Volkheimer et al. 2007). Importantly, patients with chronic lymphocytic leukemia (CLL) with mutated BCR sequences in the tumor clone compared with the germ line have a prognostically inferior survival rate and requirement of early treatment compared with those with unmutated malignant clones (Caligaris-Cappio and Ghia 2008). The BCR sequence repertoire of an individual therefore represents a surrogate of their B-cell clonality status in health and disease, with the potential to give new insights into the adaptive immune response as well as providing diagnostic and prognostic power when used clinically.
Previous studies have mainly produced descriptive analyses of the BCR populations. Isoelectric focusing (IEF) spectrotype methods (Williamson et al. 1973; Rieben et al. 1996; Satoh et al. 1996) preceded the advent of sequencing technologies (Arnaout et al. 2011) and are not quantitative. The potential size of the human repertoire is estimated to be 1011 unique BCR sequences; therefore, deep, high-throughput sequencing is necessary for sampling this repertoire robustly and to identify different subsets of BCRs (Dimitrov 2010; Benichou et al. 2012). There are several methods for isolation, amplification, and sequencing of B-cell repertoires. Multiplex PCR amplification, using degenerate PCR primers complementary to germ-line V and J segments have been designed and validated previously (van Dongen et al. 2003; Lukowsky et al. 2006; Bruggemann et al. 2007; Evans et al. 2007; van Krieken et al. 2007; Vargas et al. 2008), used in numerous biological studies (Sanchez et al. 2003; Campbell et al. 2008; Boyd et al. 2009, 2010; Krause et al. 2011; Jager et al. 2012; Lev et al. 2012; Maletzki et al. 2012), and optimized for clinical use (McClure et al. 2006; Harris et al. 2012; Sproul and Goodlad 2012), although the potential for biased PCR amplification remains. The 5′ rapid amplification of cDNA ends (5′ RACE) has also been used (Bertioli 1997; Freeman et al. 2009; Varadarajan et al. 2011; Warren et al. 2011), but can suffer from low efficiency and high levels of nonspecific amplification, contamination by short fragments from RNA degradation, or incomplete cDNA synthesis. Both methods utilize PCR and therefore have a risk of systematic over/under-representation of immunoglobulin sequences either through different primer annealing or different amplification efficiencies of the distinct V families (Sandberg et al. 2005).
Previous studies have qualitatively shown diverse IGH repertoires in healthy patients contrasting with clonal populations in malignancies (Sanchez et al. 2003; Campbell et al. 2008; Boyd et al. 2009; Carulli et al. 2011; Logan et al. 2011; Maletzki et al. 2012) and have also shown that distinct subsets of B-cells within the same individual have distinct repertoires (Wu et al. 2010). To date, next-generation sequencing (NGS) of BCRs have primarily focused on classifying the IGHV, D, and J recombination frequencies to understand the diversity of the BCR repertoire (Stewart et al. 1997; Sanchez et al. 2003; Campbell et al. 2008; Boyd et al. 2009, 2010; Weinstein et al. 2009; Wu et al. 2010; Jager et al. 2012; Lev et al. 2012; Maletzki et al. 2012). However, computational assignment of V-D-J sequences to reference databases results in many incompletely assigned IGHV, D, and J genes, even when the germ-line alleles are known (Weinstein et al. 2009). This is most likely due to SHM masking the identity of the germ-line genes present in the NGS, or the existence of allelic variation relative to the reference IGH genes. Further, investigation of V-D-J gene usage frequencies utilizes only part of the BCR sequence diversity, with important information about the V-D-J joining regions and mutational relationships not considered.
Here, we propose that analysis of the BCR sequence relationships using the full BCR V-D-J sequence is more informative for human BCR repertoire analysis than V-D-J gene classification. We show that human BCR repertoire diversity can be interpreted through full V-D-J genotype diversity using BCR networks, previously shown to be an intuitive way for understanding B-cell repertoires in zebrafish (Ben-Hamo and Efroni 2011). In such networks, the lowest level of organization in a population of B-cells, namely, independent B-cells, is represented by sparse networks, whereas highly developed (connected) networks most likely result from clonal expansions of B-cells arising through antigenic exposure or B-cell malignancies (Ben-Hamo and Efroni 2011). Using degenerate PCR-based methods we focus on sequencing RNA populations to maximize analysis of functionally rearranged BCRs rather than any nonfunctional first BCR allele defective rearrangements present in the genomic DNA from B-cell populations, but with the disadvantage that unequal numbers of RNA molecules per cell have the potential to inflate or deflate detected B-cell populations in the repertoire. Through sequencing the BCRs from samples with clonally expanded B-cell populations (peripheral blood from patients with CLL and human lymphoblastoid cell lines [LCLs]) as well as diverse BCR populations from peripheral blood from healthy individuals, we show that network analysis provides a robust framework to understand vast sequencing repertoires by sequence relationships that clearly distinguish between B-cells quantitatively using network measures. This framework is complimentary to existing phylogenetic methods, and we show, for the first time, B-cell tumor clone evolution over the course of therapy. These methods are robust to sampling and sequencing depths as well as different sequencing technologies, thereby allowing the direct comparison of multiple tumor samples from the same and different patients.
Results
Next-generation sequencing of IgH variable genes
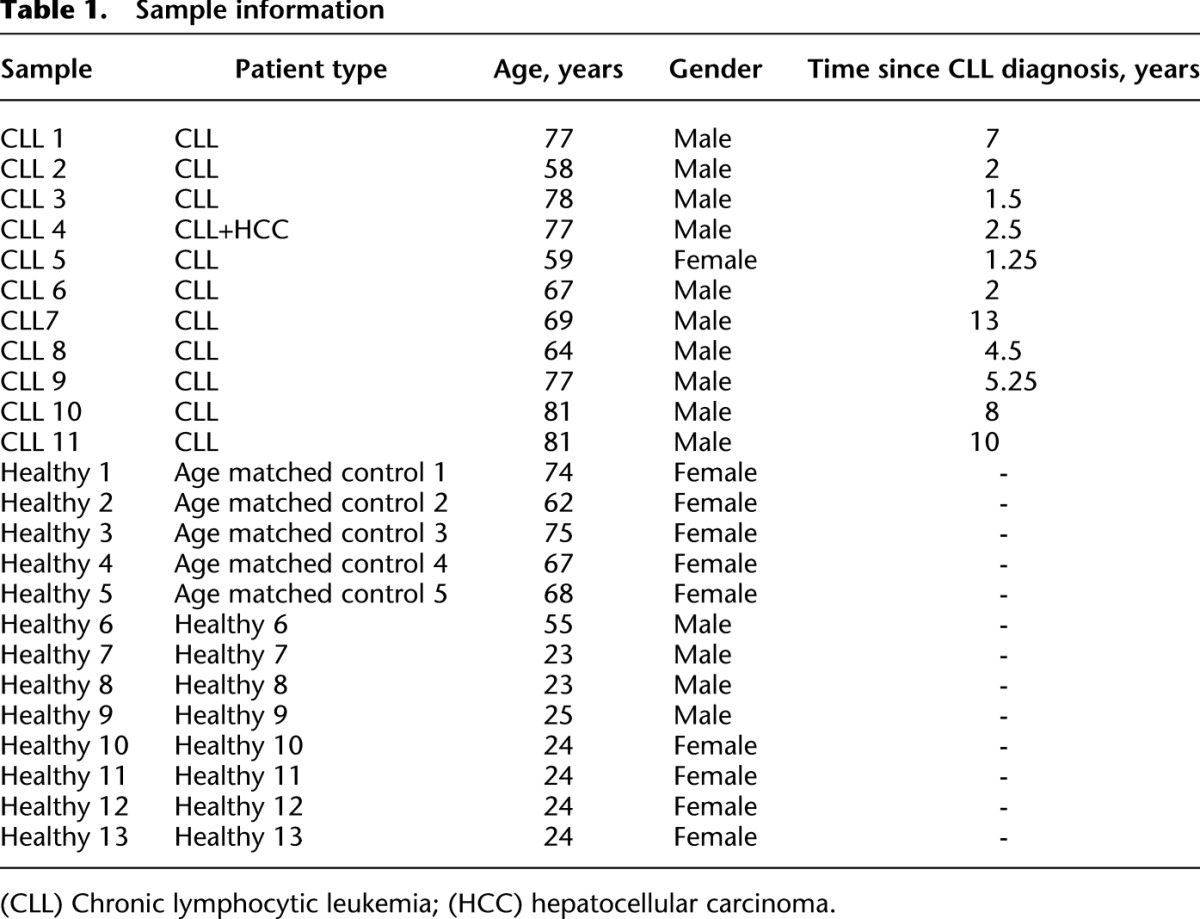
We amplified by RT-PCR the expressed rearranged IGHV-D-J loci from mRNA from human B-cell populations using the consensus IGHJ primer and FR1 or FR2 IGHV family primers (Fig. 1A; Supplemental Table S1; van Dongen et al. 2003). Peripheral blood (PB) samples from 13 healthy individuals, 11 CLL patients, and eight LCLs yielded PCR products of expected sizes (310–360 bp for FR1 and 250–295 bp for FR2 primed samples) and were 454 sequenced (Table 1). Samples yielded an average of 42,324 sequencing reads after filtering for quality and presence of IGH sequence (Supplemental Table S2). Two additional samples from CLL patient A (pre- and post-treatment) were sequenced on the MiSeq platform. We also analyzed the BCR 454 sequence data sets from Boyd et al. (2009), which includes three healthy individuals and five patients with clonal blood disorders (Supplemental Table S6).
Table 1.
Sample information
The combined per-base error-rate for the RT-PCR and sequencing process for the 454 platform was similar to other studies (Wang et al. 2007; Boyd et al. 2009) (1.74 × 10−4, of which homopolymeric indels and nonhomopolymeric errors accounted for 59.7% [1.04 × 10−4] and 40.3% [7.04 × 10−5] of the total error-rate, respectively). Similarly, the combined per-base error-rate for MiSeq was 2.06 × 10−4.
To initially assess the clonality of our samples, we determined the percentage of reads identical to the most abundant BCR sequence in each sample. The percentage of reads corresponding to the highest expressed BCR sequence in each of the CLL and LCL samples (range 39.3%–87.8% and 35.2%–78.7%, respectively) were significantly higher than that of PB from healthy individuals (range 0.10%–14.0%) with a P-value of <0.001 (Fig. 1B). There was no significant difference in the percentage of identical reads between the LCL and CLL patient samples (P-value = 0.0594). Therefore, we confirm that the healthy individuals represent diverse BCR populations, whereas the LCL and CLL samples represent more restricted or clonal BCR populations. Sanger and MiSeq sequencing confirmed that the dominant clonal sequences from the CLL samples were identical to that from 454 sequencing (excluding homopolymeric indels) (Supplemental Fig. S2).
Validation of sequencing to represent the B-cell populations
To assess the reproducibility of the RT-PCR sequencing method to sample the BCR repertoire, we compared the number of overlapping sequences from two types of technical repeats on a range of samples: (1) repeating the RT-PCR and resequencing (RT-PCR repeats) and (2) repeating the 454 sequencing from the same RT-PCR product (sequencing repeats). The percentage of the sequences shared (no more than 1-bp difference) between sequencing runs was calculated using all-against-all alignments. This showed over 98% and 30% reproducibility for LCL and healthy PB samples, respectively (Fig. 1C), probably due to the increased probability of resampling more abundant BCR types in LCLs. It is clear that the sequencing overlaps between RT-PCR repeats (Fig. 1C, green bars) are not significantly different from those between sequencing repeats (Fig. 1C, purple bars) (P-value = 0.738 by paired t-test), suggesting that our RT-PCR amplification and sequencing depth is sufficient to be representative of the major clonal BCR population in the sample.
Comparison between independent primer sets suggests limited primer bias
To assess whether multiplex PCR methods cause significant PCR amplification bias, we determined the correlation between IGHV gene usages for samples independently amplified by the FR1 and/or FR2 primer sets. The IGHV gene usage frequency distributions for both healthy individuals and LCLs resemble those seen by Arnaout et al. (2011) (Supplemental Fig. S3A,B). IGHV gene usage frequencies between FR1 and FR2 amplified samples from eight LCL samples and four healthy individual samples are highly correlated (R-value = 0.984, Supplemental Fig. S3C) suggesting that there is minimal primer amplification bias. Performing RT-PCR repeats using the FR1 primer set measures the stochastic effect of resampling the RNA populations, and again we found a strong correlation between IGHV gene usage frequencies between replicates (R-value = 0.972, Supplemental Fig. S3D). Importantly, for both of these comparisons, we see a strong linear correlation between IGHV gene usage frequencies when percentages are >5% of the overall population, but BCR sequences at a frequency of 0%–5% show some effect of stochastic sampling, irrespective of primer set usage. Therefore, overall, we do not see significant primer amplification bias under the conditions used here.
Limitations of V-D-J classification
We classified the V-D-J genes for each read by sequence similarity to germ-line sequences from the ImMunoGeneTics database (IMGT) (Lefranc et al. 2009; Supplemental Fig. S4). The majority of sequences could be classified to their most closely related reference sequences for IGHV and IGHJ genes (average of 99.8% and 96.1%, respectively). Substantially fewer IGHD were identifiable (average of 40.5%) due to the shorter sequence length and potential insertions and deletions within the joining regions between the V-D-J boundaries, which has been noted in previous studies (Weinstein et al. 2009). Incomplete V-D-J gene classification may be due to SHM masking the identity of the germ-line genes present in individuals and/or the existence of allelic variants of reference IGH (Boyd et al. 2010). We find no significant difference between the percentage of classified V, D, and J genes of our data set compared with that of Boyd et al. (2009) (Supplemental Fig. S4). To overcome limitations of IGHV-D-J gene classification, we propose that the use of complete V-D-J sequence information and mutational relationships would be a more informative and robust framework for BCR repertoire analysis and B-cell population structure than simple V-D-J gene classification.
BCR sequences naturally organize into networks based on sequence diversity
For each sample, filtered and trimmed 454 or MiSeq sequences were used directly to generate a sequence network (Fig. 2A). Each vertex represents a different sequence, and the number of identical BCR sequences defines the vertex size. Edges are created between vertices that differ by one nucleotide. Clusters are groups of interconnected vertices. Differences in network architectures are clearly seen by comparing B-cell populations from healthy individuals and LCLs. In LCLs, the majority of 454 sequences fall within a small number of clusters as these samples are predominantly comprised of a small number of large B-cell clone types (Fig. 2B,i). In contrast, healthy individuals have sparsely connected networks where most sequences are unique, thus yielding small vertices indicative of high BCR sequence diversity in the sampled repertoire (Fig. 2B,ii). From healthy individuals, 4.8%–32.2% of BCR sequences fall within clusters of three or more reads, with the largest cluster representing 16.7% (4023 reads) of the total population in healthy individual 10. Sequences within a cluster are most likely related to a single V-D-J BCR progenitor. Alignment of sequences within the clusters shows that the nucleotide differences are distributed along the length of the 454 sequences (Supplemental Fig. S5A–C). In all of the healthy individual samples, mutations significantly occur within the complementary determining regions (CDRs), known to be hotspots for somatic hypermutation (Lin et al. 1997) compared with the framework regions (FWRs) (P-value = 0.000338, Supplemental Fig. S5D). As expected, the LCLs showed no significant difference between the mutational proportions of the CDR and FWR regions. Mismatches are not found primarily at the V-D or D-J boundary, suggesting that cluster sequences are derived from the same B-cell precursor.
Figure 2.

B-cell receptor repertoires from different samples. (A) Schematic diagram showing the method by which the sequencing networks are generated: Each vertex represents a unique sequence, where the relative size of the vertex is proportional to the number of 454 sequencing reads that were identical to the vertex sequence. Edges are created between vertices that differ by one base (indel or substitution). The vertex colors correspond to the relative abundance of the corresponding sequences, where red, orange, and yellow indicates observation of a sequence in >90%, between 40%–90%, and <40% of the reads in the sample, respectively. (B) Comparison of BCR sequence networks between (i) a typical LCL sample and (ii) a typical healthy individual. (C) BCR sequence networks of CLL patients with (i) extensive clonal enlargement and (ii) limited clonal expansion. (D) BCR sequence networks of CLL patient 5 showing expansion of two dominant clusters. (E) Networks generated from sequencing data set from Boyd et al. (2009) of (i) healthy donor 1, (ii) patient 2 with follicular lymphoma (FL), and (iii) patient 3 with FL and small lymphocytic lymphoma (SLL).
The maximum CLL vertex sizes differ between samples (39.2%–99.5% of total sequences), suggesting that large but variable-sized subsets of B-cells express the predominant BCR sequence(s), surrounded by BCR variants (including total process errors) of the dominant sequence. Of note, the extent of cluster-size diversity is different between CLL samples, with some displaying extensive clonal enlargement (Fig. 2C,i), whereas others have more limited clonal expansion (Fig. 2C,ii) and expansion of two dominant clusters (Fig. 2D). Similar networks were generated from Boyd et al.'s (2009) data set (Fig. 2E). Therefore, the magnitude of connectivity of different samples varies between individual patients with CLL. In all cases, however, the CLL sequence networks are clearly distinct from the largely sparsely connected age-matched healthy individuals.
Population measures capture network and sample diversity
We next aimed to quantify BCR population measures to allow comparison and interpretation of B-cell repertoire dynamics and biology. We investigated several parameters to describe different aspects of the B-cell populations. The Gini Index is an unevenness measure. When applied to the vertex size distribution for a given sample, these measures indicate the overall clonal nature of a sample, and when applied to the cluster-size distributions, these measures indicate the overall SHM of a sample. The maximum cluster size is the percentage of reads corresponding to the largest cluster and indicates the degree of clonal expansion of a sample. To assess the possibility of dual clonal expansions, we include a measure of the second maximum cluster size as a percentage of reads in a sample.
The LCL samples, due to the more restricted repertoires and highly connected clusters, yield high cluster and vertex Gini Indices (averages of 0.94 and 0.80, range 0.91–0.97 and 0.62–0.91, respectively) (Fig. 3A), suggesting a high unevenness of the size distributions. In contrast, B-cell networks of healthy individuals occupy a distinct region of the Gini Index vertex and cluster space (averages of 0.21 and 0.05, range 0.10–0.39 and 0.03–0.11, respectively). The CLL samples occupy a spatial range between healthy individuals and LCL B-cell population extremes with a low vertex (between 0.62 and 0.97), and cluster Gini Indices (between 0.15 and 0.83), indicating B-cell clonal expansions. There is, however, considerable variation between the cluster Gini Indices, with CLL patients 1, 10, and 11 having low-cluster Gini Indices, indicative of a highly expanded dominant cluster or dominant clones. Of note, one healthy individual (healthy individual 10) has a more developed network as defined by an increase in connectivity and vertex sizes, resulting in higher vertex and cluster Gini Indices (Fig. 3A, point a). This was also verified by independent sequencing using the FR2 primer set (Supplemental Fig. S6). Further, the highest expressed BCR sequence for healthy individual 10 has 90.6% sequence identity with the closest germ-line IGHV gene (16 mismatches in 243 bp of alignment) suggesting that this B-cell clone has undergone SHM.
Figure 3.

Measures differentiating between B-cell receptor populations. (A) Cluster Gini Index plotted against vertex Gini Index for 13 healthy individual samples, 11 chronic lymphocytic leukemia (CLL), and eight human lymphoblastoid cell line (LCL) samples. Point (a) corresponds with healthy individual 10. The red box and gray dashed box distinguish between the regions occupied between diverse and clonal populations, respectively. (B) The second maximum cluster sizes plotted against the maximum cluster sizes. The red, gray-dashed, and black solid boxes distinguish between the regions occupied between unexpanded populations, monoclonal expanded populations, and biclonally expanded populations, respectively. (C) B-cell receptor networks for the titration of a chronic lymphocytic leukemia clonal sample into healthy peripheral blood from the data set from Boyd et al. (2009), and (D) the corresponding number of reads corresponding to the leukemic clone (green) and the maximum cluster size of each dilution (gray). The solid horizontal line shows the mean maximum cluster size for healthy individuals from this data set (0.52% of total reads), and the dashed horizontal lines show the mean ±SD of maximum cluster size for healthy individuals for this data set. (E) Correlation between the Gini Index and the length of time since chronic lymphocytic leukemia (CLL) diagnosis for each patient in our data set, with corresponding R2-value.
We generated networks from the sequences derived from Boyd et al. (2009) to validate these population measures on independent BCR sequence data. We show that the clonal populations of the patients with CLL, small lymphocytic lymphoma (SLL), and/or follicular lymphoma (FL) are distinct from the diverse populations of healthy individuals (Supplemental Fig. S7A), occupying equivalent regions of the cluster and vertex Gini Index graphs to samples within this study. Therefore, we conclude that the Gini Index population measure robustly separates distinct B-cell populations into different regions based on the clonal nature of the sample.
We then determined whether we could separate monoclonal expansions, biclonal expansions, and diverse B-cell populations using the maximum cluster sizes and second maximum cluster sizes (Fig. 3B). We show that the CLL and LCL samples have maximum cluster sizes >30% of the total reads compared with maximum cluster sizes of healthy individual samples of <20%. However, the LCLs and CLLs collectively occupy two distinct regions in this space. One group exhibits a single dominant clonal sequence, where the remainder of the clusters are <5% of the total reads (Fig. 3B, surrounded by the dashed line). The second group of samples has two dominant clusters above 40% and 20% of the total reads, respectively. The CLL patient 5 repertoire comprises two dominant clonal groups, each utilizing different V-D-J genes ([IGHV3-66*03/IGHD6-19*01/IGHJ3*02] and [IGHV6-1*01/IGHD3-3*01/IGHJ4*02], respectively), where the two clones are unlinked and represented by completely different BCR sequences (Fig. 2D; Supplemental Fig. S8). Limited polyclonal expansions were also observed in 5/8 of the LCL samples, reflecting that EBV transformation of peripheral B-cells frequently results in polyclonal LCLs. Using the data set from Boyd et al. (2009), we show the same phenomenon of polyclonal expansions in a subset of samples (patients with CLL/SLL and FL/SLL, Fig. 2E,iii) where the maximum cluster sizes are >35% and second maximum cluster sizes are >19% of the total reads (Supplemental Fig. S7B). Therefore, the polyclonal status of the tumor samples can be determined using B-cell network reconstruction and analysis.
An important requirement of this approach is that the network diversity measures must not be highly dependent on the depth of sequencing (scale invariant) and volume of PB sample. If a given diversity measure is scale invariant for B-cell networks, then the network diversity measure should be the same regardless of the depth of sampled sequences, i.e., a subset of 454 sequences should yield the same network diversity measure as the full set of sequences. We tested all of the proposed population measures as a function of sequencing depth by randomly sampling different proportions of the sequence data for each sample, followed by calculation of the corresponding network parameters for both the vertex and cluster-size distributions for the LCL, CLL, and healthy samples. All of the proposed measures showed little variation at different sample sizes, even when subsampling was as low as 20% of the original data size (Supplemental Fig. S9). Below 20%, small deviations in the Gini Index measures are seen. As these network measures had minimal standard deviation over all sub-sampling ranges, they are therefore robust parameters for intersample comparison.
Minimal effect of sequencing errors on network properties
We determined whether clusters were likely to be due to the process of somatic hypermutation or sensitive to or generated through sequencing error of unique amplified BCR sequences. For a given BCR sequenced multiple times, such as when multiple B-cells express identical BCRs, we estimated the expected number of vertices comprising a cluster that could be due to sequencing error given our experimentally derived PCR and sequencing error-rates. We find that all of the samples have cluster sizes greater than that expected due to error alone, even at twice the measured error-rate (Supplemental Fig. S10). Therefore, the connectivity patterns of networks predominantly reveal differences in clonal expansions of B-cell populations rather than total sequencing errors. We propose that clusters identified in BCR networks are therefore derived from B-cells that share a common pro-B-cell progenitor with rearranged V-D-J that have subsequently expanded and diversified.
Directly comparing the Gini Index measures of V-D-J sequences from samples amplified independently by distinct primer sets (FR1 or FR2 primer sets) showed a strong positive linear correlation between the two primer sets, with R-values of 0.999 and 0.996, respectively, for the vertex and cluster size diversities (Supplemental Fig. S11A). This supports a lack of PCR or sampling bias or an effect of sequencing errors for independent RT-PCRs with FR1 compared with FR2 primer sets. Further, we find that networks generated to include edge lengths of up to five base changes faithfully retain the network architecture for both the LCL and healthy individual samples (Supplemental Fig. S11B).
BCR repertoire network properties relate to CLL development
To assess the sensitivity of this analysis method, we use the titration experiment from Boyd et al. (2009), in which serial 10-fold dilutions of a known clonal CLL PB sample into normal peripheral blood was performed. We find 90.9% of all reads in the undiluted sample fall within the leukemic cluster (Fig. 3C,D). Using our methods, we can detect the leukemic clonal sequences at dilutions as low as 1:100,000. When the leukemic cluster sequences are unknown, detection of expanded clones relies on detecting the maximum cluster size that is significantly different from that of healthy individuals. We see significant increases in maximum cluster size above that of the healthy individual in CLL dilutions of 1:100 or less. Therefore, deep sequencing of BCR repertoires potentially allows the detection of a clonal lymphoid population in a background of polyclonal cells without prior knowledge of the leukemic sequence types by comparing to healthy control BCR clonality (Sayala et al. 2007).
We therefore sought to understand the relationship between the BCR population measures and the CLL clinical information for each patient. Interestingly, there was a strong correlation between the length of time since CLL diagnosis and the vertex Gini Index (Fig. 3E). This suggests that longer disease times lead to larger vertices representing larger tumor clonal populations, in agreement with previous studies (Kelly et al. 2002; Hayes et al. 2010).
By BCR deep-sequencing and network analysis, we hypothesize that we can follow the dynamics of dominant clonal populations at multiple time points. To test this, we sampled a stage B CLL patient (patient A) during the course of therapy. A pretreatment sample was taken immediately before entering the second cycle of Chlorambucil treatment, and the second sample was taken 28 d later. The BCRs were sequenced on the MiSeq platform, yielding 40,414 and 36,197 high-quality reads, respectively. The most abundant BCR sequences in the two samples were identical. Network construction shows a single dominant cluster at both time points, which decreases from 86% to 53% of total reads, reflecting the reduction in WBC (Fig. 4A). This corresponds to the vertex Gini Index reducing from 0.892 to 0.713 and the cluster Gini Index reducing from 0.244 to 0.138. A composite network was generated of all of the sequences from the dominant clusters in both time-points, where the vertex sizes correspond to frequencies of each BCR at either the pre- or post-treatment samples (Fig. 4B). The proportions of each unique BCR sequence between the pre- and post-treatment samples give a strong linear relationship (R-value = 0.999995) (Fig. 4C), where the post-treatment proportional frequencies are 62% of those in the pretreatment sample, indicating that all BCR clones within the leukemic cluster are equally affected by this treatment.
Figure 4.

B-cell leukemic clonal evolution. (A) The B-cell sequence networks for patient A with chronic lymphocytic leukemia for samples (i) prior to and (ii) after second cycle of Chlorambucil treatment, separated by 1 mo with corresponding white blood cell counts. (B) All sequences from the dominant clusters from both temporal samples were used to generate a composite network, and the differential frequencies at each time point are indicated by the relative vertex sizes. (C) Correlation between the proportional frequencies of each unique BCR within the dominant clones of patient A with corresponding R-value and linear regression equation. (D) An unrooted maximum parsimony tree was generated showing the relationships between sequences that were observed at least six times between the pre- and post-treatment samples, where the branch lengths are proportional to the number of varying bases (evolutionary distance). The tip colors show the relative difference in sequence abundance between the different time points, where green indicates observation of sequence primarily in the pretreatment sample, blue indicates predominant observations in the post-treatment sample, and white indicates no change in frequency. Clones 1 and 2 refer to examples of BCRs observed only in the pre- or post-treatment samples, respectively.
To understand the evolution of the leukemic cluster over time, a maximum parsimony tree was fitted (Schliep 2011) encompassing sequences that were observed at least six times for the two samples (Fig. 4D). Bootstrapping was performed to evaluate the reproducibility of the trees, showing strong tree support (>95% certainty for all branches). A total of 122/231 of the BCR sequences are unique to the pretreatment sample (e.g., Fig. 4, Clone 1) compared with 13/231 unique to the post-treatment sample (e.g., Fig. 4, Clone 2). However, the sequences that are primarily observed in the post-treatment sample show divergence from the dominant leukemic clone, suggesting leukemic cluster BCR evolution during treatment (e.g., Clone 2). A similar analysis was performed on an independent data set from Boyd et al. (2009) for a patient with chronic lymphocytic leukemia and small lymphocytic lymphoma samples separated by 3 mo (Supplemental Fig. S12) showing distinct clonal diversification patterns.
Discussion
Deep sequencing of B-cell and T-cell repertoires offers the potential for quantitative understanding of the adaptive immune system in health and disease. Here, we use deep sequencing of B-cell receptor V-D-J population frequencies and novel analyses of BCR repertoires at the level of clonal populations. The observation of frequent multiple identical BCR sequences in tumors and much lower frequency in identical BCR sequences in PB from healthy individuals suggests that we rarely sequence multiple identical RNA molecules from a single B-cell. Therefore, clusters of related sequences are likely to represent BCRs from clonal expansions of evolutionarily related B-cells, whereas naïve B-cell populations form singletons in sparsely connected networks. The effects of RT-PCR or sequencing error and amplification bias on our analysis, often of concern for deep sequencing, are minimal. We show a strong linear correlation between the network parameters of samples that have been RT-PCR amplified using different primer sets to distinct regions of the IGH variable RNA transcript, suggesting that the PCR methods here have limited primer or amplification bias. We confirm the dominant clonal sequences for the CLL patients by Sanger sequencing, and show that in all cases the samples have cluster sizes notably greater than that expected due to the measured total process error-rate. Therefore, these observed V-D-J clusters are likely to have undergone mutational processes greater than process errors.
We define for the first time B-cell V-D-J sequence population measures that describe the clonality of the sequences and quantify both the effect of B-cell sequence diversification (cluster size) and clonal proliferation (vertex size) using the Gini Index as an unevenness measure. The maximum and second maximum cluster size is used to assess dual clonal expansions. If the B-cell network from limited sequencing is a random sample of the entire circulating peripheral blood BCR repertoire, then a scale invariant diversity measure should also capture the predominant structure of the unsampled network. We show that network structures combined with these population measures discriminate between B-cell repertoires of different clonalities in health and disease. These measures are robust to variations in sequencing and sampling depth and different filtering strategies, and are applicable to independently produced data sets (Boyd et al. 2009). Using different primer sets, sequencing depths, and sequencing technologies, the samples still cluster according to the clonal nature of the samples, occupying the equivalent distinct regions of Gini Index and maximum/second maximum graphs. Therefore, this analytical strategy is applicable to any BCR deep-sequencing technology.
We observed variation between the BCR repertoires in healthy individuals and in CLL. One healthy individual showed a more developed network, defined by an increase in connectivity, with corresponding higher Gini Index values and larger maximum cluster sizes compared with the other healthy individuals. This clone was not germ line in sequence and could be a result of antigen-specific memory B-cell expansion or an undiagnosed malignant transformation. Variation in network structures between individual healthy zebrafish BCR repertoires was also observed in the study by Ben-Hamo and Efroni (2011), where higher connectivity suggested an immune response within the individual. Similarly in CLL, assessing the maximum and second maximum cluster sizes, we identify patients with more than one BCR clonal expansion, where the two dominant clones have different V-D-J gene usages. This may be due to either the expansion of two distinct malignant B-cell transformations or separate antigen-stimulated B-cell clonal expansion unrelated to CLL. These methods used in time-series may allow the distinction between antigen-driven positive selection in CDRs compared with malignant-driven expansion (Supplemental Fig. S5). Multiple separate clonal B-cell populations have been observed in previously published data in a subset of patients identified by different V and J chain usages (Hsi et al. 2000; Boyd et al. 2009), but the clinical significance of these findings are not known.
Time-dependent evolution of BCR networks may, however, provide a powerful means of assessing B-cell tumor evolution and response to therapy as well as the dynamics of a healthy B-cell repertoire. The CLL vertex Gini Index is correlated with the time an individual has been living with CLL (Fig. 3E). This coupled with the observation of in vivo evolution of BCR clones in CLL (Fig. 4) suggests that BCR sequencing in CLL may provide an additional prognostic value for the disease. Divergent evolution from a common leukemic ancestor has previously been observed in CLL, possibly through the accumulation of driver mutations with selective advantages in growth over other subclones (Campbell et al. 2008). Hypermutations within the IGH locus may also play a driver role in clonal expansions (Ghia and Caligaris-Cappio 2006). Therefore, BCR sequencing and subsequent network and evolutionary analysis may play an important role in identifying population changes. However, an evolutionary model for BCR diversity in health and disease, similar to the models used in infectious disease phylogenetics is needed to fully explore these possibilities. Nevertheless, for the first time we show the short-term effect of therapy on the B-cell repertoire in CLL and demonstrate how networks lend themselves to phylogenetic approaches. These methods are sensitive and informative for characterizing of B-cell populations in health and B-cell malignancies.
Methods
Samples
Peripheral blood mononuclear cells (PBMCs) were isolated from 10 mL of whole blood from healthy volunteers and CLL patients using Ficoll gradients (GE Healthcare). Total RNA was isolated using TRIzol and purified using RNeasy Mini Kit (Qiagen), including on-column DNase digestion according to the manufacturer's instructions. Total RNA was also isolated from 1 × 106 cells from human lymphoblastoid cell lines (LCLs) from the HapMap project (The International HapMap Consortium 2007). Research was approved by relevant institutional review boards and ethics committees (07/MRE05/44).
RT-PCR
RT-PCR reagents were purchased from Invitrogen. The FR1 and FR2 primer sets used (supplied by Sigma Aldrich) are described by van Dongen et al. (2003) and in Supplemental Table S1. Reverse transcription was performed using 500 ng of total RNA mixed with 1 μL of JH reverse primer (1 μM), 1 μL of dNTPs (0.25 mM), and RNase free water added to make a total volume of 11 μL. This was incubated for 5 min at 65°C and 4 μL of First strand buffer, 1 μL of DTT (0.1 M), 1 μL of RNaseOUT Recombinant Ribonuclease Inhibitor, and 1 μL of SuperScript III reverse transcriptase (200 units/μL) were added. RT was performed at 50°C for 60 min before heat-inactivation at 70°C for 15 min. PCR amplification of cDNA (5 μL of the RT product) was performed with the JH reverse primer and the FR1 or FR2 forward primer set pools (0.25 μM each) using 0.5 μL of Phusion High-Fidelity DNA Polymerase (Finnzymes), 1 μL of dNTPs (0.25 mM), 1 μL of DTT (0.25 mM) per 50 μL of reaction. The following PCR program was used: 3 min at 94°C, 35 cycles of 30 sec at 94°C, 30 sec at 60°C, and 1 min at 72°C, with a final extension cycle of 7 min at 72°C on an MJ Thermocycler.
Sequencing methods
454 libraries were prepared using standard Roche 454 Rapid Prep protocols incorporating 10-base multiplex identifier (MID) tags and sequenced using a 454 GS FLX Titanium (Roche) or by 250-bp paired-ended MiSeq (Illumina). Raw 454 or MiSeq reads were filtered for base quality (median >32) using the QUASR program (http://sourceforge.net/projects/quasr/) (Watson et al. 2013). MiSeq forward and reverse reads were merged together if they contained an identical overlapping region of >65 bp, or otherwise discarded. Non-immunoglobulin sequences were removed and only reads with significant similarity to reference IGHV genes from the IMGT database (Lefranc et al. 2009) using BLAST (Altschul et al. 1990) were retained (1 × 10−10 E-value threshold). Primer sequences were trimmed from the reads, and sequences retained for analysis only if both primer sequences were identified and if sequence lengths were >255 bp or 195 bp for FR1 and FR2 primed samples, respectively, for 454, or both forward and reverse reads >110 bp for MiSeq. FR1-primed PCR samples from CLL patients were also Sanger sequenced.
Per-base error quantification
The same PCR protocol and read quality filtering was used to amplify beta actin, beta hemoglobin, and GAPDH genes from two healthy individuals (amplicon sizes of 150 bp and 340 bp, respectively). The sequence representing the majority of the reads for each sample was classified as the “true” gene sequence for that individual to account for individual allelic variation. Any differences between this sequence and the reads were considered to be a PCR and/or sequencing error and classified as homopolymeric indels (occurring in a region of two or more consecutive identical bases), nonhomopolymeric indels, or mismatches.
Reference-based V-D-J assignment
BLAST (Altschul et al. 1990) was used to align the 454 sequences against known BCR sequences from the ImMunoGeneTics (IMGT) database (Lefranc et al. 2009). Due to the difference in length of the different gene families, different BLAST E-value thresholds were used for the IGHV, IGHD, and IGHJ genes (10−70, 10−3, and 10−20, respectively).
Network assembly and analysis
The network generation algorithm is summarized in Figure 2A and Supplemental Figure S1. Briefly, each vertex represents a unique sequence, where the relative size of the vertex is proportional to the number of sequence reads identical to the vertex sequence. Edges were calculated between vertices that differed by single nucleotide non-indel differences. The network analyses were performed using igraph implemented in R (http://igraph.sourceforge.net/index.html). The distribution of mismatches within a single network cluster were determined by aligning the sequence representing the largest vertex with the sequences to which it is connected, and the positions of mismatches were determined along the sequences. Two-sided t-tests were performed in R.
Diversity measure calculations
The Gini index was calculated by ordering the cluster sizes from the largest to smallest and creating a cumulative frequency distribution, where 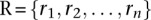
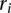 is the cumulative size of all of the largest clusters until the ith largest cluster and normalized such that
is the cumulative size of all of the largest clusters until the ith largest cluster and normalized such that 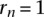 . The Gini index is
. The Gini index is
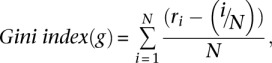 |
where 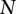 is the number of clusters (Morrow 1977).
is the number of clusters (Morrow 1977).
Estimation of cluster sizes due to sequencing error
The Poisson distribution can estimate the expected number of reads containing i errors from the (central) vertex of size n reads, given an estimated error rate. The expected number of sequences with i errors is n.pi, where
and 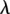 is the expected number of mutations per read. A cluster is defined as a set of interconnected vertices in which edges are generated between vertices that differ by a single base. A vertex v is only included in a cluster when the minimum distance from v to any of the sequences in the cluster containing the central vertex is one. Thus, all of the sequencing errors at i=1 generate vertices that have edges connecting to the central vertex. At i >1, a vertex with a set of mutations
is the expected number of mutations per read. A cluster is defined as a set of interconnected vertices in which edges are generated between vertices that differ by a single base. A vertex v is only included in a cluster when the minimum distance from v to any of the sequences in the cluster containing the central vertex is one. Thus, all of the sequencing errors at i=1 generate vertices that have edges connecting to the central vertex. At i >1, a vertex with a set of mutations 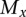 will be connected to the cluster only if there exists a vertex in the cluster with a set of mutations
will be connected to the cluster only if there exists a vertex in the cluster with a set of mutations 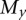 such that
such that
(i.e., there is only one mutation in 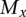 that is not in
that is not in 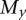 ). Therefore, the probability of vertices due to i sequencing errors is estimated by drawing
). Therefore, the probability of vertices due to i sequencing errors is estimated by drawing 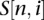 samples from a multinomial distribution, for which the probability of the possible vertices that could connect to the cluster is given by
samples from a multinomial distribution, for which the probability of the possible vertices that could connect to the cluster is given by
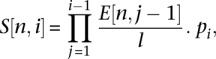 |
where l is the length of the sequence and 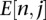 is the estimated number of vertices that are in the cluster that are at a distance of j from the central node. Here, we draw 1000 independent samples from the multinomial distribution to estimate the average number of vertices at distances i from the central vertex and, therefore, the cluster size due to sequencing error can be estimated by summing over the expected number of vertices at all i,
is the estimated number of vertices that are in the cluster that are at a distance of j from the central node. Here, we draw 1000 independent samples from the multinomial distribution to estimate the average number of vertices at distances i from the central vertex and, therefore, the cluster size due to sequencing error can be estimated by summing over the expected number of vertices at all i, 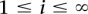 .
.
Temporal evolution of dominant clones
Sequences from the dominant clusters that were observed at least six times for the two samples underwent a multiple alignment using the ClustalW2 algorithm (www.ebi.ac.uk/Tools/clustalw2/index.html) with default parameters. A phylogenetic tree was fitted using the unrooted parsimony methods implemented in R (http://cran.r-project.org/web/packages/phangorn/). Model tests were performed on different substitution models, for which the JC+G+I substitution model was found to be optimum and thus used here. A total of 1000 bootstrap samples of individual nucleotides in the multiple alignment was used to assess the reproducibility of the phylogenetic trees. The proportional difference in expression, diff(i), of a given sequence i between month 0 and month 3 was calculated by the difference in expression between the two time points and divided by the sum of the expression of the sequence over both times:
where 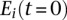 and
and 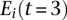 is the expression of sequence i at 0 mo and 3 mo, respectively.
is the expression of sequence i at 0 mo and 3 mo, respectively.
Data access
The IGH sequences discussed can be found under accession number ERP002120 in the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena/).
Acknowledgments
This work was supported by the Wellcome Trust. We thank the Cambridge Cancer Trials Centre and nurse specialists Gwyn Stafford, Rosie Tween, Lisa Walbridge, and Joanna Baxter, and the patients and staff of Addenbrooke's Haematology Translational Research Laboratory and Cambridge Blood and Stem Cell Biobank which is supported by the BRC.
Author contributions: R.J.M.B-R., A.L.P., and P.K. designed the study; R.J.M.B-R. performed experiments and analyzed the data; G.A.F. and G.S.V. provided patient samples; R.R. performed the 454 sequencing; B.J.H, G.A.F., and G.S.V. provided advice for the project; R.J.M.B-R., A.L.P., and P.K. wrote the paper, and all authors reviewed and approved the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.154815.113.
Freely available online through the Genome Research Open Access option.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ 1990. Basic local alignment search tool. J Mol Biol 215: 403–410 [DOI] [PubMed] [Google Scholar]
- Arber DA 2000. Molecular diagnostic approach to non-Hodgkin's lymphoma. J Mol Diagn 2: 178–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaout R, Lee W, Cahill P, Honan T, Sparrow T, Weiand M, Nusbaum C, Rajewsky K, Koralov SB 2011. High-resolution description of antibody heavy-chain repertoires in humans. PLoS ONE 6: e22365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnara D, Callea V, Stelitano C, Morabito F, Fabris S, Neri A, Zanardi S, Ghiotto F, Ciccone E, Grossi CE, et al. 2006. IgV gene intraclonal diversification and clonal evolution in B-cell chronic lymphocytic leukaemia. Br J Haematol 133: 50–58 [DOI] [PubMed] [Google Scholar]
- Batrak V, Blagodatski A, Buerstedde JM 2011. Understanding the immunoglobulin locus specificity of hypermutation. Methods Mol Biol 745: 311–326 [DOI] [PubMed] [Google Scholar]
- Ben-Hamo R, Efroni S 2011. The whole-organism heavy chain B cell repertoire from Zebrafish self-organizes into distinct network features. BMC Syst Biol 5: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benichou J, Ben-Hamo R, Louzoun Y, Efroni S 2012. Rep-Seq: Uncovering the immunological repertoire through next-generation sequencing. Immunology 135: 183–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertioli D 1997. Rapid amplification of cDNA ends. Methods Mol Biol 67: 233–238 [DOI] [PubMed] [Google Scholar]
- Boyd SD, Marshall EL, Merker JD, Maniar JM, Zhang LN, Sahaf B, Jones CD, Simen BB, Hanczaruk B, Nguyen KD, et al. 2009. Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing. Sci Transl Med 1: 12ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd SD, Gaeta BA, Jackson KJ, Fire AZ, Marshall EL, Merker JD, Maniar JM, Zhang LN, Sahaf B, Jones CD, et al. 2010. Individual variation in the germline Ig gene repertoire inferred from variable region gene rearrangements. J Immunol 184: 6986–6992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brezinschek HP, Brezinschek RI, Lipsky PE 1995. Analysis of the heavy chain repertoire of human peripheral B cells using single-cell polymerase chain reaction. J Immunol 155: 190–202 [PubMed] [Google Scholar]
- Bruggemann M, White H, Gaulard P, Garcia-Sanz R, Gameiro P, Oeschger S, Jasani B, Ott M, Delsol G, Orfao A, et al. 2007. Powerful strategy for polymerase chain reaction-based clonality assessment in T-cell malignancies. Report of the BIOMED-2 Concerted Action BHM4 CT98-3936. Leukemia 21: 215–221 [DOI] [PubMed] [Google Scholar]
- Caligaris-Cappio F, Ghia P 2008. Novel insights in chronic lymphocytic leukemia: Are we getting closer to understanding the pathogenesis of the disease? J Clin Oncol 26: 4497–4503 [DOI] [PubMed] [Google Scholar]
- Campbell PJ, Pleasance ED, Stephens PJ, Dicks E, Rance R, Goodhead I, Follows GA, Green AR, Futreal PA, Stratton MR 2008. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci 105: 13081–13086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carulli G, Marini A, Ciancia EM, Bruno J, Vignati S, Lambelet P, Cannizzo E, Ottaviano V, Galimberti S, Caracciolo F, et al. 2011. Discordant lymphoma consisting of splenic mantle cell lymphoma and marginal zone lymphoma involving the bone marrow and peripheral blood: A case report. J Med Case Reports 5: 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov DS 2010. Therapeutic antibodies, vaccines and antibodyomes. MAbs 2: 347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner T, Brezinschek HP, Foster SJ, Brezinschek RI, Farner NL, Lipsky PE 1998. Delineation of selective influences shaping the mutated expressed human Ig heavy chain repertoire. J Immunol 160: 2831–2841 [PubMed] [Google Scholar]
- Evans PAS, Pott C, Groenen PJTA, Salles G, Davi F, Berger F, Garcia JF, van Krieken JHJM, Pals S, Kluin P, et al. 2007. Significantly improved PCR-based clonality testing in B-cell malignancies by use of multiple immunoglobulin gene targets. Report of the BIOMED-2 Concerted Action BHM4-CT98-3936. Leukemia 21: 207–214 [DOI] [PubMed] [Google Scholar]
- Freeman JD, Warren RL, Webb JR, Nelson BH, Holt RA 2009. Profiling the T-cell receptor β-chain repertoire by massively parallel sequencing. Genome Res 19: 1817–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghia P, Caligaris-Cappio F 2006. The origin of B-cell chronic lymphocytic leukemia. Semin Oncol 33: 150–156 [DOI] [PubMed] [Google Scholar]
- Harris S, Bruggemann M, Groenen PJTA, Schuuring A, Langerak AW, Hodges E 2012. Clonality analysis in lymphoproliferative disease using the BIOMED-2 multiplex PCR protocols: Experience from the EuroClonality group EQA scheme. J Hematopathol 5: 91–98 [Google Scholar]
- Hayes GM, Busch R, Voogt J, Siah IM, Gee TA, Hellerstein MK, Chiorazzi N, Rai KR, Murphy EJ 2010. Isolation of malignant B cells from patients with chronic lymphocytic leukemia (CLL) for analysis of cell proliferation: Validation of a simplified method suitable for multi-center clinical studies. Leuk Res 34: 809–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsi ED, Hoeltge G, Tubbs RR 2000. Biclonal chronic lymphocytic leukemia. Am J Clin Pathol 113: 798–804 [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449: 851–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager U, Fridrik M, Zeitlinger M, Heintel D, Hopfinger G, Burgstaller S, Mannhalter C, Oberaigner W, Porpaczy E, Skrabs C, et al. 2012. Rituximab serum concentrations during immuno-chemotherapy of follicular lymphoma correlate with patient gender, bone marrow infiltration and clinical response. Haematologica 97: 1431–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung D, Giallourakis C, Mostoslavsky R, Alt FW 2006. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol 24: 541–570 [DOI] [PubMed] [Google Scholar]
- Kelly LM, Yu JC, Boulton CL, Apatira M, Li J, Sullivan CM, Williams I, Amaral SM, Curley DP, Duclos N, et al. 2002. CT53518, a novel selective FLT3 antagonist for the treatment of acute myelogenous leukemia (AML). Cancer Cell 1: 421–432 [DOI] [PubMed] [Google Scholar]
- Krause JC, Tsibane T, Tumpey TM, Huffman CJ, Briney BS, Smith SA, Basler CF, Crowe JE Jr 2011. Epitope-specific human influenza antibody repertoires diversify by B cell intraclonal sequence divergence and interclonal convergence. J Immunol 187: 3704–3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman D. 2005. Gene regulation: A eukaryotic perspective, 5th ed. Advanced Text Series. Taylor & Francis Group, NY. [Google Scholar]
- Lefranc MP, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, Wu Y, Gemrot E, Brochet X, Lane J, et al. 2009. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res 37: D1006–D1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev A, Simon AJ, Bareket M, Bielorai B, Hutt D, Amariglio N, Rechavi G, Somech R 2012. The kinetics of early T and B cell immune recovery after bone marrow transplantation in RAG-2-deficient SCID patients. PLoS ONE 7: e30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MM, Zhu M, Scharff MD 1997. Sequence dependent hypermutation of the immunoglobulin heavy chain in cultured B cells. Proc Natl Acad Sci 94: 5284–5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan AC, Gao H, Wang C, Sahaf B, Jones CD, Marshall EL, Buno I, Armstrong R, Fire AZ, Weinberg KI, et al. 2011. High-throughput VDJ sequencing for quantification of minimal residual disease in chronic lymphocytic leukemia and immune reconstitution assessment. Proc Natl Acad Sci 108: 21194–21199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukowsky A, Marchwat M, Sterry W, Gellrich S 2006. Evaluation of B-cell clonality in archival skin biopsy samples of cutaneous B-cell lymphoma by immunoglobulin heavy chain gene polymerase chain reaction. Leuk Lymphoma 47: 487–493 [DOI] [PubMed] [Google Scholar]
- Maletzki C, Jahnke A, Ostwald C, Klar E, Prall F, Linnebacher M 2012. Ex-vivo clonally expanded B lymphocytes infiltrating colorectal carcinoma are of mature immunophenotype and produce functional IgG. PLoS ONE 7: e32639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure RF, Kaur P, Pagel E, Ouillette PD, Holtegaard CE, Treptow CL, Kurtin PJ 2006. Validation of immunoglobulin gene rearrangement detection by PCR using commercially available BIOMED-2 primers. Leukemia 20: 176–179 [DOI] [PubMed] [Google Scholar]
- Morrow JS 1977. Toward a more normative assessment of maldistribution: The Gini Index. Inquiry 14: 278–292 [PubMed] [Google Scholar]
- Rieben R, Frauenfelder A, Nydegger UE 1996. Spectrotype analysis of human ABO antibodies: Evidence for different clonal heterogeneity of IgM, IgG, and IgA antibody populations. Vox Sang 70: 104–111 [DOI] [PubMed] [Google Scholar]
- Sanchez ML, Almeida J, Gonzalez D, Gonzalez M, Garcia-Marcos MA, Balanzategui A, Lopez-Berges MC, Nomdedeu J, Vallespi T, Barbon M, et al. 2003. Incidence and clinicobiologic characteristics of leukemic B-cell chronic lymphoproliferative disorders with more than one B-cell clone. Blood 102: 2994–3002 [DOI] [PubMed] [Google Scholar]
- Sandberg Y, van Gastel-Mol EJ, Verhaaf B, Lam KH, van Dongen JJ, Langerak AW 2005. BIOMED-2 multiplex immunoglobulin/T-cell receptor polymerase chain reaction protocols can reliably replace Southern blot analysis in routine clonality diagnostics. J Mol Diagn 7: 495–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Akizuki M, Yamagata H, Nakayama S, Homma M 1996. Restricted heterogeneity and changing spectrotypes in autoantibodies to La/SS-B. Autoimmunity 24: 229–236 [DOI] [PubMed] [Google Scholar]
- Sayala HA, Rawstron AC, Hillmen P 2007. Minimal residual disease assessment in chronic lymphocytic leukaemia. Best Pract Res Clin Haematol 20: 499–512 [DOI] [PubMed] [Google Scholar]
- Schatz DG, Swanson PC 2010. V(D)J recombination: Mechanisms of initiation. Annu Rev Genet 45: 167–202 [DOI] [PubMed] [Google Scholar]
- Schliep KP 2011. phangorn: Phylogenetic analysis in R. Bioinformatics 27: 592–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sproul AM, Goodlad JR 2012. Clonality testing of cutaneous lymphoid infiltrates: Practicalities, pitfalls and potential uses. J Hematopathol 5: 69–82 [Google Scholar]
- Stamatopoulos K, Kosmas C, Stavroyianni N, Loukopoulos D 1996. Evidence for immunoglobulin heavy chain variable region gene replacement in a patient with B cell chronic lymphocytic leukemia. Leukemia 10: 1551–1556 [PubMed] [Google Scholar]
- Stewart JJ, Lee CY, Ibrahim S, Watts P, Shlomchik M, Weigert M, Litwin S 1997. A Shannon entropy analysis of immunoglobulin and T cell receptor. Mol Immunol 34: 1067–1082 [DOI] [PubMed] [Google Scholar]
- Tonegawa S 1983. Somatic generation of antibody diversity. Nature 302: 575–581 [DOI] [PubMed] [Google Scholar]
- van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, Delabesse E, Davi F, Schuuring E, Garcia-Sanz R, et al. 2003. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 17: 2257–2317 [DOI] [PubMed] [Google Scholar]
- van Krieken JH, Langerak AW, Macintyre EA, Kneba M, Hodges E, Sanz RG, Morgan GJ, Parreira A, Molina TJ, Cabecadas J, et al. 2007. Improved reliability of lymphoma diagnostics via PCR-based clonality testing: Report of the BIOMED-2 Concerted Action BHM4-CT98-3936. Leukemia 21: 201–206 [DOI] [PubMed] [Google Scholar]
- Varadarajan N, Julg B, Yamanaka YJ, Chen H, Ogunniyi AO, McAndrew E, Porter LC, Piechocka-Trocha A, Hill BJ, Douek DC, et al. 2011. A high-throughput single-cell analysis of human CD8+ T cell functions reveals discordance for cytokine secretion and cytolysis. J Clin Invest 121: 4322–4331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas RL, Felgar RE, Rothberg PG 2008. Detection of clonality in lymphoproliferations using PCR of the antigen receptor genes: Does size matter? Leuk Res 32: 335–338 [DOI] [PubMed] [Google Scholar]
- Volkheimer AD, Weinberg JB, Beasley BE, Whitesides JF, Gockerman JP, Moore JO, Kelsoe G, Goodman BK, Levesque MC 2007. Progressive immunoglobulin gene mutations in chronic lymphocytic leukemia: Evidence for antigen-driven intraclonal diversification. Blood 109: 1559–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW 2007. Characterization of mutation spectra with ultra-deep pyrosequencing: Application to HIV-1 drug resistance. Genome Res 17: 1195–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren RL, Freeman JD, Zeng T, Choe G, Munro S, Moore R, Webb JR, Holt RA 2011. Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res 21: 790–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson SJ, Welkers MRA, Depledge DP, Coulter E, Breuer JM, de Jong MD, Kellam P 2013. Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos T R Soc B 368: 1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JA, Jiang N, White RA 3rd, Fisher DS, Quake SR 2009. High-throughput sequencing of the zebrafish antibody repertoire. Science 324: 807–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson AR, Salaman MR, Kreth HW 1973. Microheterogeneity and allomorphism of proteins. Ann N Y Acad Sci 209: 210–224 [DOI] [PubMed] [Google Scholar]
- Woof JM, Burton DR 2004. Human antibody-Fc receptor interactions illuminated by crystal structures. Nat Rev Immunol 4: 89–99 [DOI] [PubMed] [Google Scholar]
- Wu YC, Kipling D, Leong HS, Martin V, Ademokun AA, Dunn-Walters DK 2010. High-throughput immunoglobulin repertoire analysis distinguishes between human IgM memory and switched memory B-cell populations. Blood 116: 1070–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]