

Abstract
Identifying the pathways that are significantly impacted in a given condition is a crucial step in understanding the underlying biological phenomena. All approaches currently available for this purpose calculate a P-value that aims to quantify the significance of the involvement of each pathway in the given phenotype. These P-values were previously thought to be independent. Here we show that this is not the case, and that many pathways can considerably affect each other's P-values through a “crosstalk” phenomenon. Although it is intuitive that various pathways could influence each other, the presence and extent of this phenomenon have not been rigorously studied and, most importantly, there is no currently available technique able to quantify the amount of such crosstalk. Here, we show that all three major categories of pathway analysis methods (enrichment analysis, functional class scoring, and topology-based methods) are severely influenced by crosstalk phenomena. Using real pathways and data, we show that in some cases pathways with significant P-values are not biologically meaningful, and that some biologically meaningful pathways with nonsignificant P-values become statistically significant when the crosstalk effects of other pathways are removed. We describe a technique able to detect, quantify, and correct crosstalk effects, as well as identify independent functional modules. We assessed this novel approach on data from four experiments involving three phenotypes and two species. This method is expected to allow a better understanding of individual experiment results, as well as a more refined definition of the existing signaling pathways for specific phenotypes.
The correct identification of the signaling and metabolic pathways involved in a given phenotype is a crucial step in the interpretation of high-throughput genomic experiments. Most approaches currently available for this purpose treat the pathways as independent. In fact, pathways can affect each other's P-values through a phenomenon we refer to as crosstalk. This crosstalk may be due to the regulatory interactions among different pathways or to the gene overlap among pathways. In this work, we will use the term crosstalk to refer to the effect that pathways exercise on each other due to the presence of overlapping genes. Although it is intuitive that various pathways could influence each other, especially when they share genes, the presence and extent of this phenomenon have not been rigorously studied and, most importantly, there is no currently available technique able to quantify the amount of such crosstalk. There are three major categories of methods that aim to identify significant pathways: enrichment analysis (e.g., Fisher's exact test–hypergeometric) (Tavazoie et al. 1999; Draghici et al. 2003); functional scoring (e.g., GSEA) (Mootha et al. 2003; Subramanian et al. 2005); and topology-based methods (e.g., impact analysis) (Draghici et al. 2007; Tarca et al. 2009). Another classification of gene set analysis methods is based on the definition of the null hypothesis and divides the methods into competitive and self-contained (Goeman and Bühlmann 2007; Nam and Kim 2008). In this work, we focus on competitive methods, and in particular on the Fisher's exact test, although the problems identified likely apply also for self-contained methods.
Here we show that the results of all these methods are affected by crosstalk effects and that this phenomenon is related to the structure of the pathways. We propose the first approach that can (1) detect crosstalk when it exists, (2) quantify its magnitude, (3) correct for it, resulting in a more meaningful ranking among pathways in a specific biological condition, and (4) identify novel functional modules that can play an independent role and have different functions than the pathway they are currently located on. This method is expected to allow a better understanding of individual experiment results, as well as a more refined definition of the existing signaling pathways for specific phenotypes.
Crosstalk effects
In order to demonstrate the existence and assess the extent of crosstalk effects, we conducted a systematic exploration of this phenomenon. We constructed a reference set of genes from all genes present in at least one KEGG signaling pathway (2963 genes at the time). Then, each pathway was used as a “bait,” choosing from it a number of genes that would make it significant at a chosen significance level (α = 0.01 after Bonferroni correction). Other random genes were selected up to a constant number (n = 100) of “differentially expressed” (DE) genes (see Supplemental Material for details). Under these circumstances, the research hypothesis is true for the bait, while the null hypothesis is true for all other pathways. We repeated this selection 1000 times for each pathway Pi, and each time, we computed the Fisher's exact test P-value (Tavazoie et al. 1999), SPIA (impact analysis) (Tarca et al. 2009), and GSEA (Subramanian et al. 2005) P-values for all pathways from the KEGG database (Kanehisa et al. 2004). With these results, we constructed the empirical distributions of the false discovery rate (FDR)-corrected P-values corresponding to each prey Pj. The distributions of the P-values for all three methods are severely skewed toward zero, showing that all methods produce a significant number of false positives due to crosstalk effects (Supplemental Fig. S1). We hypothesized that crosstalk is mostly due to the common genes between pathways. If this were true, we would expect to see a strong coupling between pairs of pathways with many genes in common and a weak coupling between pathways that do not share any genes. In order to test this hypothesis, we calculated the Jaccard similarity index (Jaccard 1910) for each pair of pathways, as well as the Pearson correlation between the Jaccard index and the P-values of the preys (Figs. 1, 2). The data shows a very strong correlation (Pearson correlation index of 0.87 for Fisher's exact test, 0.62 for GSEA, and 0.83 for SPIA), which confirms our hypothesis that the crosstalk can be explained by the presence of genes that are involved in more than one pathway.
Figure 1.

Pathway coupling in the Fisher's exact test P-values. (Left panel) A number of random genes were chosen from a “bait” pathway i such that its Fisher's exact test P-value is 0.01. Other genes were chosen randomly from all other pathways (acting as preys), up to a constant number (n = 100). The elements [i, j], where i ≠ j, represent the mean of the distribution of P-values for 1000 random trials using pathway i as bait and pathway j as prey. The data show that a considerable number of pathways influence each other through a “coupling” of the P-values. For instance, row 3 of the matrix shows that when pathway 3 is chosen to be significant, several other pathways (e.g., columns 57 to 70) also tend to be significant (dark shades of blue represent significant P-values). (Right panel) Each point represents the average of the P-values of all the random trials for pairs with the same Jaccard index. The lines represent the fitting of linear and quadratic models. Both models show a strong dependence between the P-value coupling and the Jaccard index. Similar results were obtained for GSEA and impact analysis (see Fig. 2).
Figure 2.

Pathway coupling in the impact analysis (left panel) and GSEA (right panel). Each point represents the mean of all P-values of all random trials for pairs with the same Jaccard index. The lines represent the fitting of linear and quadratic models. Both models show a strong dependence between the P-value coupling and the Jaccard index.
Results
Crosstalk analysis and correction
The method we propose for correcting for crosstalk effects takes as input a set of reference pathways and a list of genes that are DE in the given condition. The crosstalk analysis is composed of three steps. The first step is the computation of a crosstalk matrix, i.e., the analysis of pairwise crosstalk interactions between pathways in the given condition, resulting in a heat map showing the P-value of each pathway when the genes from each other pathway are removed. This matrix allows the visualization of various cases of crosstalk effects and provides information on the extent of crosstalk effects in the condition analyzed. The second step is the module detection that allows the identification of subpathways that appear to be involved in the specific condition, studied independently of the pathway they belong to. The final step is the maximum impact estimation, where the biological impact of each gene is assigned to only one of the pathways to which the gene belongs, for those genes that belong to more than one pathway. The result of the last two steps is essentially a new set of pathways that can include (1) original pathways as found in the literature, (2) novel functional modules that might be relevant to the given condition, and (3) pathways modified by the removal of a such a functional module. The new set of pathways is not affected by crosstalk effects, and new P-values are computed for each pathway, building a ranked list from which the false positives due to crosstalk have been removed (see Methods).
Fat remodeling in mice
We include here the results of the crosstalk analysis in an experiment investigating cellular and metabolic plasticity of white fat tissue (WAT), where the classical overrepresentation analysis (ORA) produced a number of false positives, and failed to rank highly the pathways that were known to be involved in the given condition. In this experiment, the chronic activation of WAT beta-adrenergic receptors by certain physiological and pharmacological conditions transforms the tissue into one resembling brown fat, a thermogenic organ (Granneman et al. 2005; Li et al. 2005; Mottillo et al. 2007). The data set was obtained from a microarray analysis of white fat from mice treated with low dose (0.75 nmol/h) CL 316,243 (CL) for 0, 3, and 7 d. Here we discuss only the list of DE genes coming from the comparison between expression levels of genes at days 3 and 0 (for the comparison between days 7 and 0, see Supplemental Material).
The top 20 pathways ranked by ORA and their associated FDR-corrected P-values are shown in Figure 3A. In this figure, pathways highlighted in red represent pathways not related to the phenomenon in analysis, while pathways highlighted in green are those for which we know, with reasonable confidence, that they are involved in the given phenomenon. The white background indicates pathways for which we do not have conclusive information on their involvement (or lack of) with the phenomenon in analysis. The three most significant pathways in the comparison between days 3 and 0 were Parkinson's, Alzheimer's, and Huntington's diseases. The fourth pathway in the ranked list is Leishmaniasis. The first three pathways describe degenerative diseases of the central nervous system that have no connection to fat remodeling. Leishmaniasis describes the signaling involved in a disease spread by the bite of certain species of sand flies. Clearly, this pathway is also unlikely to give insights about the fat remodeling phenomenon. While other pathways, such as Phagosome (Newman et al. 1982), PPAR Signaling (Granneman et al. 2005), and Cell cycle (Lee et al. 2012), are definitely more related to the phenomenon of fat remodeling, their presence in the middle of a ranked list dominated by false positives (six false positives in the 10 pathways significant at 1%) illustrates how the crosstalk effects make the classical ORA unable to find the truly relevant pathways.
Figure 3.

The results of the ORA analysis in the fat remodeling experiment for the comparison between days 3 and 0, before (A) and after (B) correction for crosstalk effects. All P-values are FDR corrected. The lines show the significance thresholds: (blue) 0.01, (yellow) 0.05. Pathways highlighted in red represent pathways not related to the phenomenon in analysis, while pathways highlighted in green are those for which we know, with reasonable confidence, are involved in the given phenomenon. The white background indicates pathways for which we do not have conclusive information on their involvement (or lack of ) with the phenomenon in analysis. (A) The top 20 pathways resulting from classical ORA before correction for crosstalk. The top four pathways are not related to fat remodeling. (B) The top 20 pathways after correction for crosstalk. Pathways ranked 1, 3, and 5 are modules that are functioning independently of the rest of their pathways in this particular condition. Starred pathways are pathways edited by removing such modules. Note the lack of any obvious false positive above the significance threshold(s).
In order to analyze and eliminate the crosstalk effects, we computed the crosstalk matrix as described in the Methods section. The analysis of the matrix illustrates some interesting examples of crosstalk effects. Figure 4 represents a detail of the entire matrix. In this figure, the high significance of Parkinson's (bright red in row 1, column 1) disappears when the crosstalk due to Alzheimer's is eliminated (green in row 1, column 2). This indicates that Parkinson's is a false positive, since its significance is due exclusively to genes from Alzheimer's. Furthermore, the high significance of Alzheimer's (bright red in row 2, column 2) also disappears when the crosstalk effect of Parkinson's is eliminated (green in row 2, column 1). This means that Alzheimer's significance is also due only to the genes in common with Parkinson's. Essentially, the analysis tells us that the genes in common between the two pathways are activated independently of either pathway, which suggests that these genes constitute an independent functional module. The same phenomenon involves the Cardiac Muscle Contraction and Huntington's disease pathways. The same independent functional module is responsible for the changes shown in areas marked with a in Figure 4.
Figure 4.

Detail of the crosstalk matrix: comparison between days 3 and 0 in the CL treatment. Areas marked with a correspond to functional modules that are activated independently from the pathways they belong to. The cell marked with b corresponds to a specific part of the TLR pathway that is responsible for the immune response to host genetic material. Cells on the diagonal contain the P-values of the classical ORA, ordered from the most significant one to the least significant one. The cell Pi,j contains the P-value of pathway Pi after the effect of Pj is removed. The color of each cell represents the P-value: bright red for P-values close to zero, bright green for P-values close to 1.
An inspection of these genes and their signaling mechanisms reveals that this module is composed by genes present in mitochondria, organelles involved in all pathways above. The fact that this module is strongly activated in this fat remodeling experiment that is not related to any of the above conditions (Alzheimer's, Parkinson's, Huntington's), suggests that this should be considered as an independent pathway, dedicated to mitochondrial activity. Figure 5 shows a representation of this new pathway. In order to investigate the involvement of mitochondria in this condition, epididymal white fat of control and CL-treated (CL-7d) mice were stained with fluorescent Alexa-555 conjugated to streptavidin and imaged by spinning disc confocal microscopy. Figure 6 shows a comparison between the control (left) and CL-treated mice (right). The right panel of this figure shows a massive generation of new mitochondria after 7 days of treatment, demonstrating in vivo that, indeed, the mitochondrial pathway is central in this experiment.
Figure 5.

Mitochondrial activity pathway. This independent functional module is responsible for the incorrect identification of the pathways Parkinson's disease, Alzheimer's disease, Huntington's disease, and Cardiac Muscle Contraction by the classical ORA.
Figure 6.

Epididymal white adipose tissue of a control mouse (left) and a mouse treated with CL for 7 d (right). Treatment with CL for 7 d triggered massive mitochondrial biogenesis, demonstrating in vivo that indeed, the mitochondrial pathway is central in this experiment. White bar, 20 μm.
Another very interesting phenomenon can be observed in Figure 4 (circle b). Here, the Toll-like Receptor Signaling (TLR) pathway becomes more significant when the Rig-I Like Receptor Signaling (RLR) pathway (not significant on its own) is removed. The TLR pathway is the generic pathway involved in the immune response. The RLR pathway is the antiviral innate immunity pathway, which includes the mechanisms specifically aimed at the detection of exogenous DNA or RNA. In essence, the crosstalk analysis tells us that, in the fat remodeling experiment, the immune system has been activated, but this immune response is not due to the presence of foreign genetic material. This is exactly what happens here. The CL treatment causes the death of some white fat cells (Granneman et al. 2005). In turn, this causes an immune response in which macrophages are required to dispose of the dead cells (Li et al. 2001). Such subtle distinctions between various triggers that activated the immune response are not possible with any classical analysis methods, and it is remarkable that a data analysis method was able to provide this type of insight.
We then applied the proposed maximum impact estimation described in the Methods section to the data. The corrected P-values are shown in Figure 3B. The ranking based on these crosstalk corrected P-values is greatly improved. The most significant pathway is now the newly discovered mitochondrial pathway shown in Figure 5 and validated by the in situ hybridization shown in Figure 6. The new P-values also indicate the Phagosome pathway as one of the pathways related to this phenomenon (Newman et al. 1982). Third in the list is an independent module shared by Cell Cycle and Oocyte Meiosis. This can be thought of as a pathway related to the creation of new cells. Finally, the true involvement of the PPAR signaling pathway in the phenomenon of fat remodeling has been previously demonstrated (Granneman et al. 2005). After removing the influence of the mitochondrial crosstalk, the Parkinson's, Alzheimer's, and Huntington's pathways are not significant anymore (now ranked 60th, 61st, and 54th, respectively) (data not shown). Also, after removing the crosstalk from Phagosome, Leishmaniasis is not significant anymore (now ranked 62nd) (data not shown).
Cervical ripening
The second data set analyzed was obtained from a recent study that investigated the transcriptome of uterine cervical ripening in human pregnancy before the onset of labor at term (Hassan et al. 2009). The tissue analyzed is the human uterine cervix, the lower part of the uterus extending from the isthmus of the uterus into the vagina. It is mainly composed of smooth muscle and extracellular matrix, which consists of collagen, elastin, proteoglycans, and glycoproteins (Uldbjerg et al. 1983b; Leppert 1995). The uterine cervix has an essential function in the maintenance of pregnancy and also in parturition (Hassan et al. 2006, 2009, 2010). Cervical ripening is a critical component of the common terminal pathway of parturition, which includes the extensive remodeling of the cervix (Hassan et al. 2009). Disorders of cervical ripening can lead to premature or protracted cervical change, complicating term (e.g., protracted dilatation or arrest of dilatation) or preterm gestations (e.g., premature cervical dilation in the second trimester) (Hassan et al. 2009). The state of cervical ripening has traditionally been assessed by clinical examination (Bishop score or its modifications) (Bishop 1964), which includes the digital examination of the cervix for its consistency, dilatation, effacement, and position. This method has also been used to predict the likelihood that a patient would go into spontaneous labor. The goal of this experiment was to examine the relationship between human cervical ripening and the cervical transcriptome, aiming to improve our understanding of the biology of cervical ripening at term. This study included pregnant women who underwent elective C-section at term with an unripe (n = 11) or ripe cervix (n = 11). Cervical biopsies were obtained from these women transvaginally, from the anterior lip of the uterine cervix following C-section. Microarray analysis was performed on RNA isolated from these cervical tissue specimens using Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays (Hassan et al. 2009).
On this data set, we performed the comparison between gene expression levels from cervical tissues obtained from women with an unripe or ripe cervix using the classical ORA. The results are shown in Figure 7A. Pathways with a P-value smaller than 0.05 after FDR correction were Focal adhesion, ECM-receptor interaction, Amoebiasis, Cell adhesion molecules (CAMs), Small cell lung cancer, and Dilated cardiomyopathy.
Figure 7.

The results of the ORA for the cervical ripening experiment, before (A) and after (B) the correction for crosstalk effects. All P-values are FDR corrected. The lines show the significance thresholds: (blue) 0.01, (yellow) 0.05. (A) The top 20 pathways reported by ORA before correction for crosstalk. Pathways such as Amoebiasis and Small Cell Lung Cancer are not related to this phenotype. (B) The top 20 pathways reported by ORA after the crosstalk analysis. After the correction, neither Amoebiasis nor Small Cell Lung Cancer are significant anymore. At the same time, Cell Adhesion Molecules and the Integrin-mediated ECM Signaling have an increased significance. Starred pathways are pathways edited by removing such a module.
There is plenty of experimental evidence that biological processes described by the pathways Focal Adhesion, ECM-Receptor Interaction, and Cell Adhesion Molecules are related to cervical ripening. The relation between these pathways and the phenomenon in analysis was revealed by studies on humans and animals showing the involvement of extracellular matrix metabolism and cell adhesion molecules in cervical ripening (Uldbjerg et al. 1983a,b; Leppert et al. 1986; Leppert 1995; Mahendroo et al. 1999). However, the pathway Amoebiasis describes the biological process of infection from a parasite that invades the intestinal epithelium. Amoeba infection involves the parasite attachment to the intestinal mucus layer, followed by disruption and death of host epithelial cells. This process is completely unrelated to the physiological condition of cervical ripening in term pregnancy. The same is true for the Small Cell Lung Cancer pathway. Clearly, the top ranked pathways include some describing complex phenomena that are unrelated to the studied condition. Also, the significant pathways known to be involved in the process of cervical ripening are somewhat general pathways describing cellular interactions.
The analysis of the crosstalk matrix (see Supplemental Fig. S6) shows that there is an independent functional module among the top three pathways in the ranking. This novel module includes the genes present in the interaction between the cellular transmembrane protein integrin and three important ECM components—collagen, laminin, and fibronectin. The KEGG pathways involved in the identification of this pathway are Focal adhesion, ECM-receptor interaction, and Amoebiasis. Henceforth, we will refer to this pathway as the Integrin-Mediated ECM Signaling (Fig. 8).
Figure 8.

The novel Integrin-Mediated ECM Signaling. This new module was found to be independently activated and statistically significant in two different conditions: hormone treatment of post-menopausal women and cervical ripening in normal pregnancies. Genes shown in red were found to be differentially expressed in the hormone treatment experiment.
Very interestingly, the independent functional module found in this condition is, in fact, the exact same module found in the hormone treatment experiment described in the Supplemental Material. Interestingly, the KEGG pathways involved in the identification of this functional module are slightly different between the two phenotypes. While in this phenotype this module was found from the interaction of Focal adhesion, ECM-receptor interaction, and Amoebiasis, in the hormone treatment the last pathway is replaced by Pathways in Cancer. The fact that the same module was found to be activated and statistically significant in two different phenotypes, from the interaction of different sets of canonical pathways, further supports the idea that this module describes an independent mechanism and should therefore be considered as an independent pathway.
Further analysis of the crosstalk matrix shows that the Small Cell Lung Cancer loses significance when the crosstalk effects of the first three pathways are removed (bright green loss of significance in first three columns of row 5 in Supplemental Fig. S6). This allows us to conclude that it is a false positive in the classical ORA, with its ORA significance due exclusively to crosstalk effects.
The ranking of pathways with the P-values corrected for crosstalk by our analysis is shown in Figure 7B. The first pathway is Integrin-mediated ECM Signaling with an FDR-corrected P-value of 2.9 × 10−13. Cell Adhesion Molecules is now the second in ranking, with an FDR-corrected P-value of 0.004. The false positives in the classical ORA results, Amoebiasis and Small Cell Lung Cancer, are not significant anymore. The biological significance of the pathway Dilated Cardiomiopathy may be linked to the fact that 10%–15% of the uterine cervix is constituted of smooth muscle, and cervical ripening involves alterations of this component. The last significant pathway at the 5% significance threshold is Leukocyte Transendothelial Migration. Although human and animal studies (Hassan et al. 2009) have shown that cervical ripening does not require activation of a typical inflammatory response and influx of inflammatory cells into the cervix, the significance of this pathway may reflect the beginning of later inflammatory events typical of parturition (Word et al. 2007; Timmons et al. 2009).
In addition to the data sets described above, we analyzed a data set coming from the comparison of expression levels at days 7 and 0 in the fat remodeling experiment, a data set investigating the effect of various types of hormones on the endometrium of healthy post-menopausal women who underwent hysterectomy (Hanifi-Moghaddam et al. 2007), as well as a data set requested by one of the reviewers, investigating the correlation between gene expression values “with MiniMental Status Examination (MMSE) and neurofibrillary tangle (NFT)” in subjects with Alzheimer's disease (Blalock et al. 2004). The latter data set was analyzed to identify significant pathways from both KEGG and Reactome databases (see the Supplemental Results section in the Supplemental Material).
Conclusions
These results show that the novel approach proposed for the detection and correction for crosstalk effects allows not only the elimination of most of the false positives present in the results of the classical ORA but also the identification of novel functional subpathways that are specifically involved in the condition studied, giving useful insights on the phenomenon in analysis that are not captured by existing techniques.
We assessed this novel approach on data from four experiments involving three phenotypes and two species. In all cases, this approach was able to eliminate most false positives, as well as correctly identify as significant pathways that had been biologically proven to be involved in the given condition, yet not found to be significant by the classical analysis. We also found several independent functional modules, including a mitochondrial activity module active in different stages of fat remodeling in mice, and an Integrin-mediated ECM signaling found to be involved in hormone treatment in post-menopausal women and cervical ripening in pregnant women. Interestingly, the later module was extracted independently from the crosstalk interactions of two different groups of pathways, in the two conditions analyzed.
This approach is a departure from the current paradigm that considers the pathways as static models, independent of the phenotype. In the view proposed here, various specific modules, or subpathways, can be dynamically linked to specific conditions. When such independent functional modules are identified in independent conditions, such as the integrin-mediated ECM signaling above, these modules could be considered as candidate new pathways.
Methods
Pathway analysis in the presence of overlapping pathways: The crosstalk matrix
Our goal is to develop an approach that can detect and quantify the crosstalk between pathways. The main issue we are trying to address here is the fact that, in the presence of overlapping pathways (i.e., for all pathways databases available today), crosstalk phenomena increase the probability of false positives, i.e., increase the number of pathways reported as significant but that in reality are not interesting (borrowing terminology from Brad Efron, we call pathways that have lesser biological significance “not interesting” even though they might be statistically significant with a large enough sample size). To better understand the approach we are going to present, let us briefly review the classical Fisher's exact test approach described above. Figure 9A represents the contingency table used for assessing the significance of a pathway Pi by the classical overrepresentation approach. The table divides genes as either being in the pathway or not, versus being considered DE or not DE (NDE); ni represents the number of DE genes on Pi, while n represents the total number of DE genes; and mi represents the number of NDE genes on Pi, while m represents the total number of NDE genes. It follows that ni + mi = ⎪Pi⎪ represents the number of genes on Pi, while with n + m we represent the total number of genes.
Figure 9.
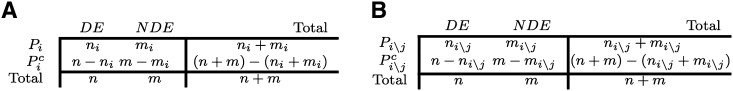
A comparison of the classical overrepresentation analysis (A) with the crosstalk matrix analysis proposed here (B). (A) The standard overrepresentation approach contingency table: ni + mi and n + m represent, respectively, the number of genes belonging to pathway Pi and the total number of genes. ni and n represent, respectively, the number of differentially expressed genes belonging to pathway Pi and the total number of DE genes. (B) Contingency table for the overrepresentation approach, taking into account the overlap between pairs of pathways; Pi\j represents the set of elements in Pi excluding the intersection with Pj; with the notations ni\j + mi\j we represent the total number of genes that are in pathway Pi but not in pathway Pj, and with ni\j the number of DE genes that are in pathway Pi but not in pathway Pj.
The reasoning behind the ORA is that if the number of DE genes on a pathway is much higher than expected by chance, then the pathway is likely to be biologically interesting. In order to take into account the effect of the overlap on the significance of the two pathways, we consider the effect of the removal of the overlapping part on the significance of the pathways. This is achieved as follows: Let us consider two overlapping pathways Pi and Pj. With the notation Pi\j we define the set of elements in Pi excluding the intersection with Pj; in the same way, with the notations ni\j + mi\j we represent the number of genes that are in pathway Pi but not in pathway Pj, and with ni\j the number of DE genes that are on pathway Pi but not in pathway Pj. We then consider the contingency table shown in Figure 9B, whose bottom margin is identical to that of Figure 9A.
With this contingency table, we compute for every pair of pathways [i, j] the P-value of Pi\j. Since this computation yields a k × k matrix, where k is the number of pathways, the results are most conveniently represented using a heat map of the negative log P-values. Each cell (i, j) of this matrix characterizes the significance of pathway Pi when we remove the effect of pathway Pj. The rows and the columns are ordered by the original P-values of the pathways, which are placed on the diagonal. We will refer to this matrix as the crosstalk matrix. This matrix is useful for identifying the effects of crosstalk among pathways.
An example of the crosstalk matrix can be found in Figure 10. We will refer to the part of the matrix above the horizontal significance threshold as the significance strip. The nonsignificance strip will be the part below the horizontal significance threshold. The significance quadrant will be the part of the significance strip to the left of the significance threshold. Using these terms, we can identify and discuss several interesting phenomena that are not captured by any of the existing pathway analysis methods.
Figure 10.

Example of a crosstalk matrix. On the diagonal, we find the classical overrepresentation analysis, ordered by P-value. The blue line represents the 0.01 significance level, while the black line represents the 0.05 significance level. The P-values in the matrix have been log-transformed (base 10 log), and the sign of the result has been inverted. The color of the cell represents the P-value: bright red for P-values close to zero, bright green for P-values close to 1.
A first interesting case is when a pathway Pi is reported as significant by the classical analysis, but it loses its significance when the effect of another pathway Pj is removed. This is represented, in the crosstalk matrix, by a nonsignificant P-value (green square) in the significance strip. In this case, Pi is unlikely to be biologically meaningful, since its significance is most likely due to a crosstalk from Pj.
A second interesting case is when a pathway Pi that is not significant for the classical analysis becomes significant when the crosstalk effect of another pathway Pj is removed. This is represented in the crosstalk matrix by a significant P-value (red square) in the nonsignificant strip. The meaning of this is that pathway Pj was masking the significance of Pi, indicating that a phenomenon likely to be biologically meaningful is happening in the part of Pi which is not in common with Pj.
A third and last interesting case is a symmetric (with respect to the diagonal) decrease in significance of pathways in the significance quadrant. This indicates the presence of an independent functional submodule, common to both Pi and Pj, that is responsible for their significance. Note that the activity of this module is tightly related to the condition studied.
The maximum impact estimation: An expectation maximization technique for the assessment of the significance of signaling pathways in the presence of crosstalk
The crosstalk matrix is a useful tool for the interpretation of the effect of crosstalk between pathways. However, the ultimate goal of the analysis of signaling pathways is to provide a meaningful ranking among pathways, as well as a P-value quantifying the likelihood that a certain pathway is involved in the phenomenon in analysis. Here, we developed a correction method for the ranking of pathways that takes into account the overlaps between pathways.
The main idea is that if there is no crosstalk, then there is no ambiguity in the ORA significance calculations. In such a case, if genes in a pathway are overrepresented, it cannot be a false positive caused by crosstalk. Our approach is therefore to infer an underlying pathway impact matrix where each gene contributes to one and only one pathway and hence is devoid of crosstalk, and then to perform the ORA analysis using that impact matrix. Since this underlying pathway impact matrix is not observed directly, it is inferred through likelihood-based methods, and estimated using the EM algorithm. The corrected ranking is computed using ORA analysis with the underlying pathway impact matrix, shown as follows.
Let us consider the DE indicator vector Y, representing the differential expression of genes, and the membership matrix X describing the membership of each gene in each one of k pathways P1…Pk. The vector Y is defined as follows:
and each cell Xi,j of the matrix X is defined as follows:
The matrix Y⎪X obtained by combining the vector Y with the X matrix is shown in the example in Figure 11.
Figure 11.

Example of a DE/membership matrix. Column Y represents the indicator of differential expression of the various genes (1 for the n DE genes and 0 for the m NDE). Column Pj represents the membership indicator for pathway j. Row gi describes gene i in terms of its differential expression and its membership in the various pathways.
In many analysis methods, the membership matrix X is also interpreted as the impact matrix: If Xij = 1, then gene gi impacts pathway Pj. In ORA, for example, each gene is considered to have the same full impact on all pathways the gene belongs to. Crosstalk effects result from the fact that a gene can belong to more than one pathway, but in principle, it can potentially have a different biological impact on each such pathway. Our aim is to identify the pathway where the biological impact of such a shared gene is maximum. We do so by estimating the maximum impact pathway using an expectation maximization approach (see Supplemental Material).
Identification of independent functional modules
The maximum impact estimation procedure alone is not able to identify overlapping modules responsible for the entire significance of other pathways, as in the situations represented by case 3 in the section describing the crosstalk matrix. In such cases, the overlap should be considered as a separate pathway that is more likely to be biologically meaningful in the condition under analysis. An additional step is needed in order to correctly deal with this situation. In this additional step, we extract certain significant overlaps from the list of pathways and include them in the list as independent functional modules. An independent functional module is a module for which there is evidence of an activity independent of the pathways it resides in, for the given condition. If an independent module is found in more than one, possibly unrelated, condition, this module is considered as a candidate novel pathway.
A module must satisfy certain conditions in order to be treated as an independent functional module. Let us assume that we are analyzing the overlap between the pathways Pi and Pj; the first condition is that both pathways are significant (after FDR correction for multiple comparisons) at a certain threshold α. The threshold α is the significance threshold chosen by the user. Typical values for this threshold are 0.01 and 0.05. This condition limits the search to the significance quadrant of the crosstalk matrix. The second condition is that the overlap Pi∩j itself must be significant at α (after FDR correction). The third condition is that the subpathways obtained by removing the overlap from both original pathways, indicated by Pi\j and Pj\i, must not be significant at α (after FDR correction). If we denote with p(P) the P-value of a generic pathway P, then the conditions can be summarized as follows:
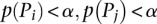
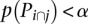
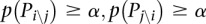
This pairwise procedure might yield modules that are similar one to each other, for example, in cases where a module is contained in three or more pathways. That could be solved with a three-way or n-way search, but we opted for another approach for limiting the number of new modules. Once all interesting pairwise modules are created, we test for similarity among modules. The index used for similarity is a modified Jaccard similarity index mJS defined as follows:
where M1 and M2 are two modules obtained with the search criteria explained above. We merge any two modules whose similarity is greater than a certain threshold st. Once the modules are merged, the similarity among all the modules (including the newly created one) is computed again, and the merging procedure is applied again until there are no more modules that can be merged.
These newly created modules are removed from all pathways with which they overlap, and this list of modified pathways is used in the EM procedure. For the data sets analyzed in this work, we used an st threshold of 0.25. The procedure used to select this threshold can be found in the Supplemental Material.
After applying the module discovery and the EM approach, the result is a modified membership matrix that can be used to perform the desired type of analysis. This matrix now includes three types of pathways: (1) original pathways as found in the literature; (2) novel functional modules that are impacted in the given condition independently from the pathways they belong to; and (3) the pathways from which such independent modules have been removed. If the same independent module is found in several conditions—in other words, if this module is active independently from its parent pathways in several different phenotypes—such a module should be considered a good candidate for a novel pathway.
Additional aspects of the crosstalk correction, such as a detailed description of the module detection step, false negative rate, and pathway size bias, can be found in the Methods section of the Supplemental Material (module detection step and false negative rate), and in Supplemental Figures S11 and S12 (pathway size bias).
Acknowledgments
This work has been partially supported by the following grants: NIH RO1 RDK089167, R42 GM087013, and NSF DBI-0965741 (to S.D.), by the Robert J. Sokol Endowment in Systems Biology, and by the Perinatology Research Branch, Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH, DHHS. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NSF, NIH, or any other of the funding agencies.
Author contributions: M.D. and S.D. identified the problem, conceived the study, and wrote the manuscript. M.D. and Z.X. developed the crosstalk correction based on the EM. S.D., M.D., P.H.W., and A.T. developed the crosstalk matrix analysis. J.G.G. and R.G.M. performed the fat remodeling experiment, helped interpret the results, and edited the fat remodeling experiment section in the manuscript. R.B. helped interpret the results and edited the hormone treatment experiment section in the manuscript. N.G.T. helped interpret the results and edited the cervical ripening section in the manuscript. P.H.W. edited the crosstalk matrix analysis and the crosstalk correction section in the manuscript. S.D., P.H.W., and R.R. supervised the study.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.153551.112.
References
- Bishop EH 1964. Pelvic scoring for elective induction. Obstet Gynecol 24: 266–268 [PubMed] [Google Scholar]
- Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW 2004. Incipient Alzheimer's disease: Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci 101: 2173–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghici S, Khatri P, Martins RP, Ostermeier GC, Krawetz SA 2003. Global functional profiling of gene expression. Genomics 81: 98–104 [DOI] [PubMed] [Google Scholar]
- Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C, Georgescu C, Romero R 2007. A systems biology approach for pathway level analysis. Genome Res 17: 1537–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeman JJ, Bühlmann P 2007. Analyzing gene expression data in terms of gene sets: Methodological issues. Bioinformatics 23: 980–987 [DOI] [PubMed] [Google Scholar]
- Granneman JG, Li P, Zhu Z, Lu Y 2005. Metabolic and cellular plasticity in white adipose tissue I: Effects of β3-adrenergic receptor activation. Am J Physiol Endocrinol Metab 289: E608–E616 [DOI] [PubMed] [Google Scholar]
- Hanifi-Moghaddam P, Boers-Sijmons B, Klaassens AHA, van Wijk FH, den Bakker MA, Ott MC, Shipley GL, Verheul HAM, Kloosterboer HJ, Burger CW, et al. 2007. Molecular analysis of human endometrium: Short-term tibolone signaling differs significantly from estrogen and estrogen plus progestagen signaling. J Mol Med (Berl) 85: 471–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan SS, Romero R, Haddad R, Hendler I, Khalek N, Tromp G, Diamond MP, Sorokin Y, Malone J 2006. The transcriptome of the uterine cervix before and after spontaneous term parturition. Am J Obstet Gynecol 195: 778–786 [DOI] [PubMed] [Google Scholar]
- Hassan SS, Romero R, Tarca AL, Nhan-Chang C-L, Vaisbuch E, Erez O, Mittal P, Kusanovic JP, Mazaki-Tovi S, Yeo L, et al. 2009. The transcriptome of cervical ripening in human pregnancy before the onset of labor at term: Identification of novel molecular functions involved in this process. J Matern Fetal Neonatal Med 22: 1183–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan SS, Romero R, Tarca AL, Nhan-Chang C-L, Mittal P, Vaisbuch E, Gonzalez JM, Chaiworapongsa T, Ali-Fehmi R, Dong Z, et al. 2010. The molecular basis for sonographic cervical shortening at term: Identification of differentially expressed genes and the epithelial-mesenchymal transition as a function of cervical length. Am J Obstet Gynecol 203: 472.e1–472.e14 [DOI] [PubMed] [Google Scholar]
- Jaccard P 1910. Étude anatomique de bois comprimés. Bull Soc Vaud Sci Nat 37: 547–579 [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okunom Y, Hattori M 2004. The KEGG resource for deciphering the genome. Nucleic Acids Res 32: 277–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y-H, Petkova AP, Mottillo EP, Granneman JG 2012. In vivo identification of bipotential adipocyte progenitors recruited by β3-adrenoceptor activation and high-fat feeding. Cell Metab 15: 480–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppert PC 1995. Anatomy and physiology of cervical ripening. Clin Obstet Gynecol 38: 267–279 [DOI] [PubMed] [Google Scholar]
- Leppert PC, Cerreta JM, Mandl I 1986. Orientation of elastic fibers in the human cervix. Am J Obstet Gynecol 155: 219–224 [DOI] [PubMed] [Google Scholar]
- Li M, Carpio DF, Zheng Y, Bruzzo P, Singh V, Ouaaz F, Medzhitov RM, Beg AA 2001. An essential role of the NF-κ B/Toll-Like Receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. J Immunol 166: 7128–7135 [DOI] [PubMed] [Google Scholar]
- Li P, Zhu Z, Lu Y, Granneman JG 2005. Metabolic and cellular plasticity in white adipose tissue II: Role of peroxisome proliferator-activated receptor-α. Am J Physiol Endocrinol Metab 289: E617–E626 [DOI] [PubMed] [Google Scholar]
- Mahendroo MS, Porter A, Russell DW, Word RA 1999. The parturition defect in steroid 5α-reductase type 1 knockout mice is due to impaired cervical ripening. Mol Endocrinol 13: 981–992 [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. 2003. PGC-1 α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273 [DOI] [PubMed] [Google Scholar]
- Mottillo EP, Shen XJ, Granneman JG 2007. Role of hormone-sensitive lipase in β-adrenergic remodeling of white adipose tissue. Am J Physiol Endocrinol Metab 293: E1188–E1197 [DOI] [PubMed] [Google Scholar]
- Nam D, Kim S-Y 2008. Gene-set approach for expression pattern analysis. Brief Bioinform 9: 189–197 [DOI] [PubMed] [Google Scholar]
- Newman SL, Henson JE, Henson PM 1982. Phagocytosis of senescent neutrophils by human monocyte-derived macrophages and rabbit inflammatory macrophages. J Exp Med 156: 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim JS, Kim CJ, Kusanovic J P, Romero R 2009. A novel signaling pathway impact analysis (SPIA). Bioinformatics 25: 75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM 1999. Systematic determination of genetic network architecture. Nat Genet 22: 281–285 [DOI] [PubMed] [Google Scholar]
- Timmons BC, Fairhurst A-M, Mahendroo MS 2009. Temporal changes in myeloid cells in the cervix during pregnancy and parturition. J Immunol 182: 2700–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldbjerg N, Ekman G, Malmström A, Olsson K, Ulmsten U 1983a. Ripening of the human uterine cervix related to changes in collagen, glycosaminoglycans, and collagenolytic activity. Am J Obstet Gynecol 147: 662–666 [DOI] [PubMed] [Google Scholar]
- Uldbjerg N, Ulmsten U, Ekman G 1983b. The ripening of the human uterine cervix in terms of connective tissue biochemistry. Clin Obstet Gynecol 26: 14–26 [DOI] [PubMed] [Google Scholar]
- Word RA, Li X-H, Hnat M, Carrick K 2007. Dynamics of cervical remodeling during pregnancy and parturition: Mechanisms and current concepts. Semin Reprod Med 25: 69–79 [DOI] [PubMed] [Google Scholar]