

Abstract
Notwithstanding their biological importance, Y chromosomes remain poorly known in most species. A major obstacle to their study is the identification of Y chromosome sequences; due to its high content of repetitive DNA, in most genome projects, the Y chromosome sequence is fragmented into a large number of small, unmapped scaffolds. Identification of Y-linked genes among these fragments has yielded important insights about the origin and evolution of Y chromosomes, but the process is labor intensive, restricting studies to a small number of species. Apart from these fragmentary assemblies, in a few mammalian species, the euchromatic sequence of the Y is essentially complete, owing to painstaking BAC mapping and sequencing. Here we use female short-read sequencing and k-mer comparison to identify Y-linked sequences in two very different genomes, Drosophila virilis and human. Using this method, essentially all D. virilis scaffolds were unambiguously classified as Y-linked or not Y-linked. We found 800 new scaffolds (totaling 8.5 Mbp), and four new genes in the Y chromosome of D. virilis, including JYalpha, a gene involved in hybrid male sterility. Our results also strongly support the preponderance of gene gains over gene losses in the evolution of the Drosophila Y. In the intensively studied human genome, used here as a positive control, we recovered all previously known genes or gene families, plus a small amount (283 kb) of new, unfinished sequence. Hence, this method works in large and complex genomes and can be applied to any species with sex chromosomes.
Y chromosomes play a major role in sexual reproduction by harboring master sex-determination genes in many species and male fertility factors in most of them (Bull 1983; Carvalho et al. 2009; Kaiser and Bachtrog 2010; Ezaz and Graves 2012; Hughes and Rozen 2012). Analysis of their origin and evolution has revealed unexpected biological phenomena (Rozen et al. 2003; Carvalho and Clark 2005; Koerich et al. 2008; Lemos et al. 2008; Murtagh et al. 2012), as well as general principles of evolutionary genetics, including the role of recombination and sex-antagonistic genes (Rice 1996; Charlesworth and Charlesworth 2000; Zhou and Bachtrog 2012). However, despite their importance, little is known about Y chromosomes because in many species they are heterochromatic, being composed of highly repetitive DNA that cannot be fully assembled with current technologies (Carvalho et al. 2003; Hoskins et al. 2007). The same issues apply to W chromosomes in ZZ/ZW sex-determination systems (Bull 1983; International Chicken Genome Sequencing Consortium 2004). Mammalian Y chromosomes contain a large euchromatic portion that nonetheless is also very repetitive; in a few species (human, chimp, and macaque), its sequence is nearly complete, owing to painstaking BAC mapping and sequencing (Skaletsky et al. 2003; Hughes and Rozen 2012). These formidable achievements demanded a huge investment of time and resources and placed these Y chromosomes apart (in all other species, only fragmentary assemblies are available, at best). A similar effort successfully assembled the less repetitive portion of the D. melanogaster heterochromatin (Hoskins et al. 2007). It is telling that even in the finished human genome most heterochromatic regions remain unassembled (International Human Genome Sequencing Consortium 2004).
Although it is not possible to fully assemble heterochromatic Y chromosomes, Y-linked genes can nonetheless be assembled even if they are deeply buried within repetitive DNA, and this partial genomic data is very informative (Carvalho et al. 2000; Carvalho and Clark 2005; Koerich et al. 2008; Murtagh et al. 2012). In “whole genome shotgun” projects (WGS), which comprise the majority of recent genome projects, the euchromatic portion of chromosomes assemble into large and easily studied scaffolds, whereas heterochromatic regions are represented by thousands of small unmapped scaffolds (International Chicken Genome Sequencing Consortium 2004; Hoskins et al. 2007; Levy et al. 2007). Exons of heterochromatic genes and other islands of unique sequence are faithfully assembled but appear as isolated scaffolds because the repeat-laden introns and intergenic regions cannot be assembled. Further assembly fragmentation in the Y-chromosome is caused by its low coverage (compared to the autosomes) (Carvalho et al. 2003), a consequence of its hemizygosity. A major obstacle to the study of the Y chromosome is to identify among the many unmapped scaffolds those that are Y-linked. This has been done by a combination of computational methods that suggest candidates and a PCR test to confirm Y-linkage (Carvalho et al. 2000; Carvalho and Clark 2005; Koerich et al. 2008; see Chen et al. 2012 for W-linkage). The experimental verification is labor intensive when applied to hundreds of scaffolds but is necessary owing to the high rate of false positives of current computational methods. Nearly all known Drosophila Y-linked genes were identified using this approach (Carvalho et al. 2000; Carvalho and Clark 2005; Carvalho et al. 2009; Krsticevic et al. 2010). When technically feasible, Y-linked scaffolds can be identified by the preparation of separate male and female DNA libraries before WGS sequencing, as these scaffolds would contain only male reads (Krzywinski et al. 2004). This approach is not possible for the majority of the available genome sequences because they employed mixed-sex libraries (also, in mammals, sequencing of a single homogametic female is common practice).
Here we show that Y chromosome sequences can be identified with a simple, efficient, and inexpensive method (Y chromosome Genome Scan or YGS) (Fig. 1) suitable for all genome projects that include the heterogametic sex, and apply it to Drosophila and humans.
Figure 1.

Outline of the YGS (Y chromosome Genome Scan) method. Y-linked sequences can be efficiently identified by a comparison of the assembled genome with inexpensive short-reads obtained from female DNA: The Y-linked sequences should get no match, whereas autosomal and X-linked sequences should be nearly completely matched. Efficient removal of all types of repetitive sequences is critical because they are shared between the Y chromosome and the female DNA, and was accomplished by a straight comparison of the short DNA words (k-mers) present in the assembled genome and female short-reads. We successfully applied the YGS method to two very different genomes, D. virilis and human.
Results
Identification of Y-linked scaffolds in Drosophila virilis
The Drosophila virilis genome was sequenced in 2007 using unsexed embryos (Drosophila 12 Genomes Consortium 2007). A previous search of the orthologs of the D. melanogaster Y-linked genes identified six genes on the D. virilis Y chromosome (Koerich et al. 2008). Here, we apply a single lane of Illumina short-read sequencing to adult female DNA (∼22 Gbp; ∼85-fold coverage of the genome; current cost US ∼$2000). When comparing these female reads with the available genome, the Y-linked scaffolds should get no match if all types of repetitive sequences are masked. This comparison can in principle be implemented using standard programs such as RepeatMasker and BLAST, but they do not allow the fine-tuning needed for this particular purpose. Using a computer program tailored for this application (“YGS.pl”) (Carvalho and Clark 2008; Methods), we first built a list (implemented as a bit-array) of all overlapping 15-bp sequences (“15-mers”) present in the female reads. Then we built a list of all 15-mers present in only one copy in the assembled D. virilis genome. This list of single-copy 15-mers efficiently removes identical repeats that would escape usual repeat masking programs (e.g., from gene duplications), while preserving the useful information provided by single-copy variants of transposable elements, segmental duplications, and other repeats (Krsticevic et al. 2010; Dennis et al. 2012; Hughes and Rozen 2012). Finally, we compared the two lists and obtained for each scaffold the proportion of unmatched single-copy k-mers; to increase resolution we optionally removed k-mers that are likely to be sequencing errors (Methods). As shown in Figures 2A and 3A, the result is clear-cut: The distribution is sharply bimodal; the right peak (centered at 95%–100% unmatched k-mers) corresponds to the Y chromosome, whereas the left peak (centered at 0%–5% unmatched k-mers) corresponds to the X and autosomes. The few previously known Y-linked scaffolds of D. virilis, and ∼800 new ones (totaling 8.5 Mbp), were cleanly identified (Figs. 2A, 3A). We tested by PCR a sample of 15 candidate scaffolds from the right peak, and all were Y-linked (Supplemental Table S1). Additional validation of the method is provided by the known D. virilis Y-linked genes: All 52 scaffolds that encode them are located in the right peak. Fifteen scaffolds (out of 1186) produced ambiguous results, containing above ∼25% and below ∼80% unmatched k-mers. These intermediate scaffolds are small (average size of 2.7 kb and range of 1.1–5.8 kb), very repetitive (with many BLASTN hits in the D. virilis genome), and amount to 40 kb (0.02% of the genome size). They probably originate from misassembled repeats. At any rate, essentially all D. virilis genome sequences can be safely classified as Y-linked or not Y-linked without experimental verification.
Figure 2.
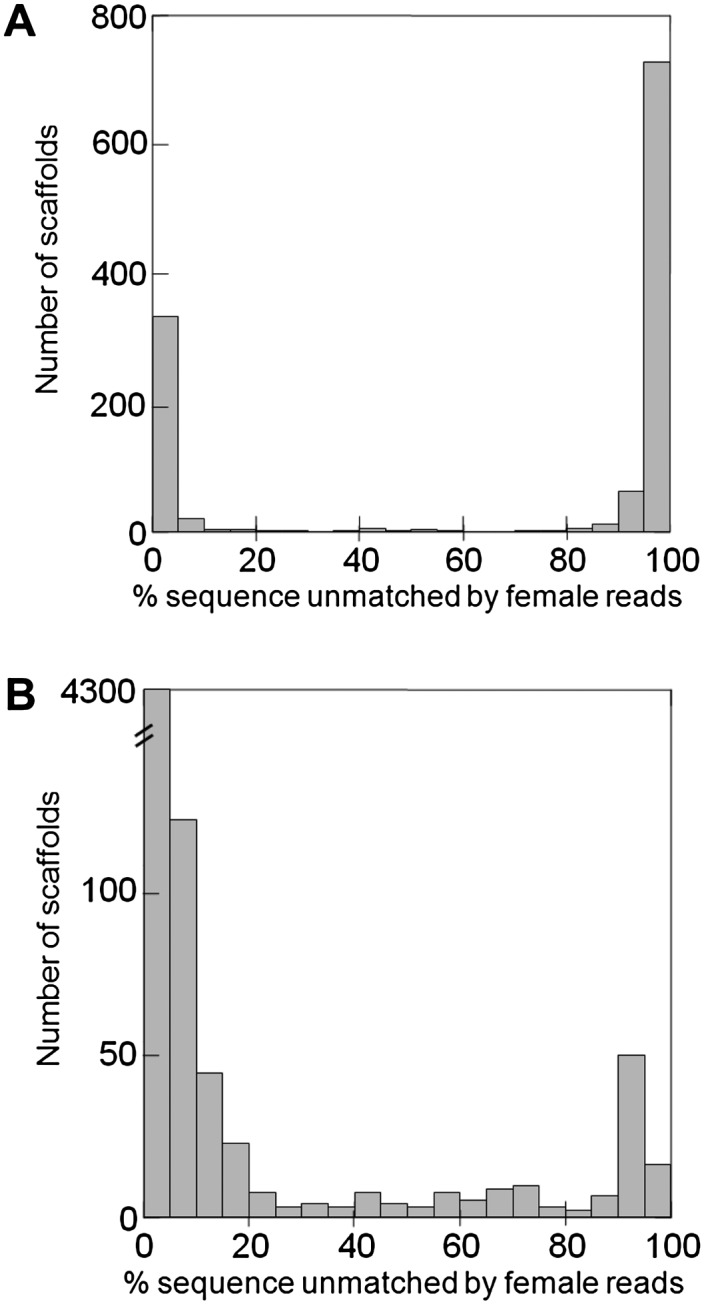
Identification of Y-linked scaffolds in the D. virilis (A) and human (B) genomes. The peak on the right corresponds to Y-linked scaffolds, and the peak on the left corresponds to autosomal and X-linked scaffolds. (Abscissa) Proportion of scaffold sequence not matched by female short reads (in percentage unmatched single-copy k-mers); (ordinate) number of scaffolds. (A) D. virilis genome, CA assembly (1186 scaffolds, 165 Mbp of sequence excluding gaps; see Methods). There are 807 Y-linked scaffolds (total sequence: 8.5 Mbp); only a few were known before. (B) Human genome, HuRef assembly (Levy et al. 2007; 4606 scaffolds, 2796 Mbp of sequence excluding gaps). There are 119 Y-linked scaffolds (total sequence: 18.4 Mbp); as expected, most of their sequence was already known to belong to the Y chromosome (Skaletsky et al. 2003).
Figure 3.

Identification of Y-linked scaffolds in the D. virilis (A) and human (B) genomes (scatter plot). The figure complements Figure 2A,B, adding the scaffold size and mapping. Each dot represents one scaffold in the assembled genomes; red dots are scaffolds previously known to be Y-linked, and blue dots are unmapped or not Y-linked scaffolds. (Abscissa) proportion of scaffold sequence not matched by female short reads (in percentage unmatched single-copy k-mers); (ordinate) scaffold size. (A) D. virilis genome (CA assembly).There are 15 intermediate scaffolds (defined as those having between 25% and 80% unmatched single-copy k-mers); they are all small (total size: 40.4 kb ; 0.02% of the genome), and they seem to originate from misassembled repeats (see Results). (B) Human genome (HuRef assembly). There are 51 intermediate scaffolds; all are Y-linked and many of them could be traced to misassembled segmental duplications involving the Y chromosome, such as the two marked with arrows (accessions DS486171 and DS486351; see Results).
We searched for protein coding genes among the newly identified Y-scaffolds, and we found four new functional genes, 11 pseudogenes, and ∼10 scrambled copies of the mitochondrial genome. These mitochondrial copies are very similar and show signs of misassembly, so further studies (e.g., Krsticevic et al. 2010) are needed to clarify whether or not any mitochondria-derived gene is functional. We focus here on the four functional genes. GJ19835 belongs to the M20 dipeptidase gene family; phylogenetic and synteny analyses strongly suggest that its ortholog was lost in the melanogaster group of species, being present in all other sequenced Drosophila; its D. pseudoobscura ortholog GA25180 (which is autosomal) is expressed only in males (Supplemental Fig. S1). The three other genes, GJ19633, GJ11126, and GJ18574, have autosomal orthologs in D. melanogaster (CG11719, CG2964, and JYalpha), which are expressed only in males and encode, respectively, a conserved sperm tail structural protein, a male-specific pyruvate kinase present in the sperm proteome, and a sperm-specific subunit of the Na,K-ATPase (Supplemental Figs. S2–S4). Hence, these four genes fit the general pattern of Drosophila Y-linked genes, formerly autosomal male-specific genes transposed to the Y-chromosome (Carvalho et al. 2009). In order to determine when they were acquired by the Y-chromosome, we investigated other Drosophila species using PCR and synteny analysis as described in Koerich et al. (2008). Three genes (GJ19835, GJ19633, and GJ11126) moved to the Y-chromosome after the split between D. melanogaster and D. virilis (Fig. 4; Supplemental Figs. S1–S4; Supplemental Table S2). In contrast, we found that GJ18574/JYalpha belong to the ancestral Drosophila Y-chromosome; its autosomal location in D. melanogaster resulted from a Y-to-autosome movement in the ancestor of the melanogaster subgroup of species.
Figure 4.

Gene gains and losses in the Drosophila Y. Gene location (Y-linkage vs. autosomal/X-linkage) was determined by PCR. Direction of the movements (gene gains, red arrows; gene losses, blue arrows) was inferred by synteny and parsimony (Supplemental Figs. S1–S4). For the six ancestral genes (dashed arrows), there is no close outgroup for inferring the direction (gain versus loss) (Koerich et al. 2008). Genes were labeled with the names of the D. melanogaster (JYalpha, CG11719, CG2964) or D. virilis (GJ19835) orthologs. Data for genes JYalpha (abridged as “JYa”), GJ19835, CG11719, and CG2964 came from the present study, and the remaining genes from Koerich et al. (2008).
Identification of Y-linked scaffolds in the human genome
The YGS method worked very well in D. virilis and other Drosophila species (not shown), but these genomes are fairly small and not very rich in repetitive DNA. Furthermore, they were sequenced using inbred strains, whereas in outbred genome sequences (e.g., humans and many other organisms) males and females will differ not only in the sex chromosomes but also at polymorphic sites. We found that the YGS method cleanly identify the Y chromosome in the reference human genome (Fig. 5). As a more stringent and realistic test we applied it to a WGS assembly. Since the human Y has been intensively studied (Skaletsky et al. 2003; Hughes and Rozen 2012), the main point here is to test whether previously known Y sequences could be identified. We downloaded the WGS assembly “HuRef,” which came from a male with Great Britain ancestry (Levy et al. 2007), and the Illumina short reads from 36 Great Britain women, produced by the 1000 Genomes Project (The 1000 Genomes Project Consortium 2010). We then applied the same general procedures described above for D. virilis, except that we increased k-mer size from 15 to 18 to allow for the larger genome size (Methods). Again the distribution of matching is bimodal, with Y-linked scaffolds centered around 95% unmatched single-copy k-mers, and non-Y centered below 5% (Figs. 2B; 3B). The Y chromosome amounts to ∼2% of the total genome sequence in humans (International Human Genome Sequencing Consortium 2004) and ∼19% in Drosophila (Carvalho et al. 2009), but its impressive appearance in Figure 2A when compared to Figure 2B basically reflects a more fragmented assembly in Drosophila. More relevant differences are the prominence and origin of the “intermediate scaffolds” in the human genome. We classified 51 scaffolds as intermediate (i.e., with ambiguous position in Fig. 3B), and three of them are quite large (DS486460, 269 kb; DS486351, 885 kb; DS486171, 3.9 Mbp). Also differently from Drosophila, many of these intermediate scaffolds do not have tens or hundreds of BLASTN hits in the genome but instead have one or a few hits. As detailed in the next section, we found that all intermediate scaffolds are Y-linked, and that many of them were caused by misassembled segmental duplications involving the Y chromosome. Note, however, that segmental duplications are perfectly classified (as Y- or not Y-linked) by the YGS method if they were correctly assembled (Fig. 5).
Figure 5.

Test of the YGS method in the reference human genome. Each dot represents a human chromosome in the reference assembly (International Human Genome Sequencing Consortium 2004). (Blue dots) Autosomes and the X; (red dot) Y chromosome. The stars are the X-linked (blue star) and Y-linked (red star) copies of a 3.9-Mbp segmental duplication (“XTR”); the two copies are 98.8% identical (Ross et al. 2005). The region is continuous in the X chromosome (accession NT_011651.17; coordinates 11754754–15671817) and interrupted by a large insertion in the Y (Hughes and Rozen 2012); we used only the homologous region of the Y-linked copy (accession NT_011896.9; coordinates 268439–3453096 and 3751260–3967080). Note that the two copies of this segmental duplication are correctly identified as Y and not Y-linked, despite their very high sequence identity. (Abscissa) Proportion of scaffold sequence not matched by female short reads (in percentage unmatched single-copy k-mers); (ordinate) chromosome size.
On the whole, we found 119 Y-linked HuRef scaffolds (18.4 Mbp), which contain all known single-copy human Y-linked genes and at least one representative of the multicopy ones (below). A sequence comparison is needed to distinguish among the 119 scaffolds those scaffolds that are covered by the reference Y from those containing new sequences. We searched potentially new Y-linked sequences using two conservative and complementary approaches (Methods). We found that most of the scaffolds (85 of 119) and the vast majority of the sequence (18.165 Mbp of 18.371 Mbp) are entirely contained (or nearly so) within the reference Y sequence, whereas 34 small scaffolds (totaling 206 kb) are primarily composed of new sequence (Supplemental Fig. S5; Supplemental Table S4, column 5). These 34 scaffolds contain mostly repeats (76% of the sequence; RepeatMasker search), including known types of Y satellite sequences (Supplemental Table S4); six of them (DS487348, DS487367, DS487401, DS488597, DS489331, and DS490176) match the few known sequences (accessions AY598345–AY598347, AC068123) of the large heterochromatic Yq12 band, which comprises 40 Mbp and has not been sequenced (Skaletsky et al. 2003; Jehan et al. 2007; Hughes and Rozen 2012). The Yq12 band (and the Y centromere) are the likely source of many of these 34 scaffolds; and hence, they may provide entry points for the heterochromatic regions of the human Y. Within the 85 scaffolds that closely match the reference Y, we found five small regions of new sequence (in scaffolds DS486512, DS486519, DS486628, and DS486277; total size: ∼76 kb) (Supplemental Table S3). Four regions are located in or very near the end of both the HuRef and reference Y scaffolds; dot plots show a good alignment of both assemblies until the end of the reference scaffold, followed by the new sequence present only in the HuRef scaffold (Supplemental Fig. S6). Hence, these new sequences likely partially fill gaps in the reference Y sequence. The fifth new region (scaffold DS486227, approximate position 1628481–1637920) is an 11-kb insertion in the middle of the reference scaffold NT_011875 (Supplemental Table S3). We later found that this insertion is a polymorphism covered by high-quality sequence (fosmid AC236424, obtained as part of the Human Genome Structural Variation Project [Kidd et al. 2008] and BAC clone AC245170). The rediscovery of this region is another proof of the method's power. As commonly seen in human Y chromosome euchromatic sequences (Skaletsky et al. 2003), these five regions contain repeats and duplications from other chromosomes (Supplemental Table S3). All BLASTX hits came from incomplete copies of genes from other chromosomes, frequently with in-frame stop codons, so they are clearly pseudogenes. A BLASTN search against the NCBI human EST database does not show any transcript that came from these regions (not shown). In short, there is no sign of functional genes. These newly found euchromatic sequences may be useful targets for finishing. On the whole, we found 283 kb of new sequence (∼1% of the reference Y). Using the 1000 Genomes Project reads, we confirmed that they are present in other males and absent in females; hence, they are newly found pieces of the human Y instead of artifacts or rare polymorphisms in the HuRef assembly (Fig. 6; Methods).
Figure 6.

New human Y chromosome sequences. Snapshot of the IGV browser (Thorvaldsdóttir et al. 2012) showing the end of scaffold DS486227, which contains 32 kb of sequence not included in the reference Y. The three tracks show the coverage (linear scale, range 0–100) of the alignment between DS486227 and male short reads (upper), male reads that align to the scaffold but not to the reference Y (middle; “differential male reads”), and female reads. Note that the full region aligns with male reads (upper track), which confirms its existence in males other than the HuRef donor; it does not align to female reads (bottom track), which confirms Y-linkage. From position 2315 kb until the end of the scaffold (2350 kb), DS486227 aligns to the “differential male reads” (middle track); this shows that it is new Y sequence. All 119 putative Y-linked scaffolds were similarly inspected and displayed a pattern of Y-linkage. A few of them (e.g., DS486227) reveal new sequences. The zero coverage regions in the upper tracks correspond to the sequence gaps in the DS486227 scaffold.
Intermediate scaffolds in the human data and segmental duplications
In contrast with Drosophila, in the human genome some scaffolds that produce ambiguous results are quite large and do not seem to result from trivial causes such as misassembled highly repeated sequences. While investigating their origin, we considered four hypotheses: (1) Binomial sampling error; (2) autosomal or X-linked scaffolds displaced toward the Y-peak (i.e., the low-match region in the right side of Fig. 3B) due to sequencing errors or rare polymorphisms in the HuRef assembly; (3) chimeric scaffolds involving Y-linked and non-Y-linked sequences; and (4) Y-linked scaffolds displaced toward the autosomal/X-peak (i.e., the high-match region in the left side of Fig. 3B) due to incomplete detection of repetitive k-mers. Binomial sampling error is quantitatively irrelevant: Due to the high depth of female reads (38×), the proportion of unmatched k-mers expected by chance in an autosomal scaffold is very small (approximately 10−38, assuming a Poisson distribution, but this ignores polymorphism and sequencing bias). Regarding sequencing errors/rare polymorphisms, their effect was virtually eliminated by the use of male short reads to validate k-mers (see Methods, Removal of Sequencing Errors in the Assembled Genomes section). Regarding chimeric scaffolds, we have not found any example in Sanger-based genome projects (such as HuRef), but it remains a possibility (see below, Application of YGS to Genomes Assembled from Short Reads section). The last possible cause of intermediate scaffolds, Y-linked scaffolds displaced toward the autosomal/X-peak due to incomplete detection of repetitive k-mers, proved to be relevant for the human genome. Specifically, we could trace a number of intermediate scaffolds to misassembled (collapsed) segmental duplications in the HuRef assembly.
Segmental duplications (SD) are regions larger than 1 kb that are not transposable element copies and have sequence identity >90% (International Human Genome Sequencing Consortium 2004). They are common in the human genome and create major problems because they frequently are collapsed, particularly in unfinished WGS assemblies (Bailey et al. 2001; Alkan et al. 2011), but also in the finished human genome (Dennis et al. 2012). SD would be perfectly classified by our procedures if they were correctly assembled (Fig. 5). On the other hand, collapsed segmental duplications will cause repetitive k-mers to be classified as single-copy; if one SD copy is located in the Y chromosome and the other is X-linked or autosomal, the corresponding scaffold will get many matches from female reads in “single-copy” k-mers and will be displaced toward the autosomal/X peak, the exact amount of displacement depending on how much non-SD sequence it contains. Note also that the assembled sequence probably will contain patches of the different copies of the SD.
If the above hypothesis for the origin of intermediate scaffolds is correct, then they should be Y-linked. Indeed, as shown in the previous section, alignment with male and female short reads showed that all intermediate scaffolds are Y-linked. Furthermore, when we examined a sample of intermediate scaffolds, we found several cases of collapsed segmental duplications involving the Y chromosome. For example, a BLASTN search of scaffold DS487199 (53% unmatched) under high stringency (word size of 128) detects only one full copy in the HuRef assembly (i.e., itself), but four full copies in the reference human assembly: the Y-linked NT_113819 (with 100% identity), and three autosomal (NT_113888, NT_113889, and NT_113958; all above 97% identity). Analogous results were obtained for DS488767 (59% unmatched) and other scaffolds. By far the most serious case of misassembled SD we found in the HuRef assembly was the X-transposed region (“XTR”), a 3.9-Mbp block that transposed from the X to the Y ∼5 million years (Myr) ago; the X and Y copies have 98.8% identity (Page et al. 1984; Ross et al. 2005). As acknowledged by Levy et al. (2007), the X and Y copies of this region are collapsed in the HuRef assembly; we found that most of the XTR sequence ended up in two large Y-linked scaffolds plus 26 small scaffolds (BLASTN search; not shown). Seven of the 51 intermediate scaffolds, including the two largest (DS486171, 3.9 Mbp, 14% unmatched k-mers; DS486351, 957 kb 75%) trace to this misassembled segmental duplication. When we added the missing region of the X chromosome to the HuRef assembly (e.g., accession NT_011651.17, between positions 11946708–1554301) and repeat the whole analysis with the YGS method, the DS486171 and DS486351 scaffolds got the typical low matching of Y-chromosome sequences (85% and 93% unmatched; as expected, the number of single-copy k-mers decreased, e.g., from ∼1,750,000 to 295,000 in DS486171). This clearly illustrates the effect of collapsed segmental duplications on the detection of Y-linked scaffolds. It is worth noting that misassembled SD are not the sole source of intermediate scaffolds; all six scaffolds that came from the heterochromatic band Yq12 are intermediate (Supplemental Table S4). These six scaffolds came from highly repetitive regions (Jehan et al. 2007; see also Supplemental Table S4, column 6), so as in Drosophila, these highly repetitive regions can also give ambiguous results, possibly because they have unassembled copies in other chromosomes.
Coverage of the reference Y chromosome sequence by the 119 HuRef Y-linked scaffolds
It is important to know what proportion of the Y-linked genes (and other important sequences) we can expect to detect by the application of the YGS method to a draft assembly. Note that this proportion reflects both the quality of the WGS assembly (Alkan et al. 2011) and the efficiency of the YGS method. In order to answer this question for the HuRef assembly, we did a BLASTN search of the coding sequences of all known human Y-linked genes (of the male-specific region), against the 119 Y-linked HuRef scaffolds detected by the YGS method (we took one representative in the case of multicopy genes). All 28 genes (Skaletsky et al. 2003; Hughes and Rozen 2012) are present among the 119 scaffolds, 20 of them with an essentially complete coding sequence. The ZFY gene, the worst case, was missing 20% of its sequence.
Application of YGS to genomes assembled from short reads
Nearly all recent genome projects employed short reads (Illumina, 454, Solid) instead of the older Sanger reads, which were used in the genome assemblies analyzed above. This makes it important to know how the YGS method would perform on short-read genome assemblies. The higher coverage allowed by the short reads can produce an assembly with fewer gaps in single-copy regions, which is particularly useful for assembling Y-linked scaffolds (due to their inherently lower coverage when compared to the autosomes), but the shorter reads increase fragmentation and possibly the chance of chimerism and other misassemblies. For example, short-read assemblies are more prone to misassemble segmental duplications (Alkan et al. 2011). We did a preliminary analysis of the D. kikkawai genome, which was assembled using only short reads (S Richards, J Qu, H Jiang, M Batterton, K Blankenburg, S Gubbala, Y Han, J Jayaseelan, D Kalra, C Kovar, et al., unpubl.; accession number AFFH00000000.2), and we found the typical bimodal distribution of matches (Supplemental Fig. S7). The four previously known Y-linked genes of D. kikkawai are contained in scaffolds with a very high proportion of unmatched k-mers, except for a piece of the kl-2 gene (Supplemental Table S5). This gene was split into three scaffolds, two with the typical match rate of the Y-chromosome, and one intermediate (KB459848; 15.7% unmatched k-mers). The latter seems to contain a misassembly, being composed by one contig that contains the kl-2 gene and has the typical signature of Y-linkage (AFFH02007563; 99.8% unmatched k-mers); a second Y-linked contig (AFFH02007564; 83% unmatched); and 19 other contigs that behave as autosomal or X-linked (AFFH02007565–AFFH02007583; average: 0.6% unmatched). This pattern suggests that KB459848 is chimeric, or that it is part of a misassembled segmental duplication (such as the DS486171 scaffold of the human assembly) (Supplemental Fig. S5); settling this point would require a finished genome sequence. Given the increased risk of chimerism and misassembly of segmental duplications, it may be safer to run YGS with both scaffolds and contigs when dealing with short-read assemblies. Despite these limitations, YGS will be a useful tool for these assemblies as well, since all known Y-linked genes of D. kikkawai would have been detected.
Discussion
The study of Y chromosomes has been hampered by their fragmentation after genome sequencing and by the difficulty of identifying their pieces among the many unmapped scaffolds. Previous studies used methods such as “staggered TBLASTN hits” (Carvalho et al. 2000; 2001), “low-shotgun depth” (Carvalho et al. 2003; Carvalho and Clark 2005), and “testis ESTs × armU” (Vibranovski et al. 2008), which had a false positive rate of 40%–65% and an unknown rate of false negatives (none of them was carried exhaustively). This high false-positive rate made experimental confirmation of Y-linkage obligatory and time consuming. In comparison, the YGS method classified nearly all scaffolds of the D. virilis genome assembly as Y-linked or not Y-linked, without false positives and false negatives, making the laborious and time-consuming experimental confirmation of Y-linkage much simpler (we did it only with the few scaffolds that contain functional genes, and all proved to be Y-linked). As a cautionary note, we should keep in mind that the Y chromosome differs greatly among species, so more experimental validation may be necessary in some genomes or assemblies. In the human genome, some ambiguity was introduced by misassembled segmental duplication, which caused some Y-linked scaffolds to be located between the Y and autosomal/X peaks of the distribution (Figs. 2B, 3B). Hence, particularly when dealing with these complex genomes, scaffolds that are suspiciously distant from the “autosomal/X peak” of the distribution should be considered as probably Y-linked, even if they are distant from the “Y peak” (e.g., scaffold DS486171) (Fig. 3B). Another assembly-related problem is that draft genomes, where the YGS method will be most useful, will have sequence gaps, which obviously will limit the identification of Y-linked sequences. Despite these problems, if the assembly we examined (HuRef) belonged to some poorly known mammalian genome, all its single-copy Y-linked genes and at least one representative of each multicopy gene would have been discovered. In sum, the YGS method worked nearly perfectly with a Drosophila genome, and worked well in a complex genome. It also worked well in a preliminary experiment with the D. kikkawai genome, which was assembled from short reads. Hence, it seems that the problem of identification of Y-linked scaffolds is solved. As discussed below, application of this method has yielded interesting biological insights for both the D. virilis and the human genome. Since it is efficient, simple, and inexpensive, it seems likely that more will come in the near future.
JYalpha, a gene involved with reproductive isolation, is part of the ancestral Drosophila Y chromosome
GJ18574, the ortholog of JYalpha, is one of the four new genes we found in the D. virilis Y chromosome. We found that it belonged to the ancestral Drosophila Y chromosome, and it moved to an autosome (the chromosome 4 of D. melanogaster) in the ancestor of the melanogaster subgroup (Fig. 4). Masly et al. (2006) found a second, more recent JYalpha movement: It transposed from chromosome 4 to 3R in the ancestor of D. simulans (Supplemental Fig. S8). As a result, certain D. melanogaster/D. simulans hybrids lack the gene and are sterile. The finding of a gene involved in reproductive isolation in the ancestral Drosophila Y chromosome suggests a simple explanation for the key role of this chromosome in several cases of reproductive isolation between Drosophila species (e.g., Pantazidis et al. 1993; Sweigart 2010), especially because sex-chromosomes are much more prone than autosomes to cause imbalances in hybrids (Long et al. 2012). It will be interesting to verify whether gene movements to/from the Y chromosome occurred in these species.
Gene gains outnumber gene losses by 11-fold in Drosophila
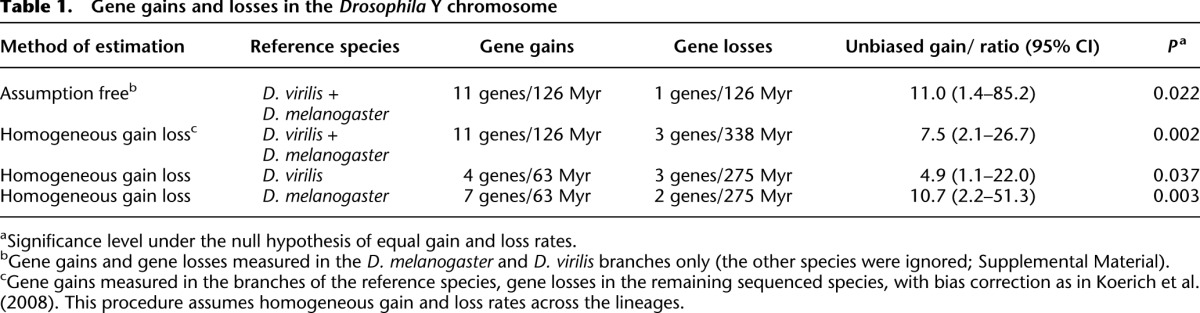
Current theory on Y chromosome evolution states that X and Y chromosomes evolved from an ordinary pair of autosomes after one of them acquired a male-determining function and became a proto-Y. Massive gene loss on the proto-Y would follow, generating a typical Y chromosome in which the few remaining genes are mostly shared with the X (Bull 1983; Rice 1996; Charlesworth and Charlesworth 2000). Although this theory is very well-supported in mammals and other groups (e.g., Skaletsky et al. 2003), the evidence in Drosophila does not support it (Carvalho et al. 2009). For example, a previous study with ten Drosophila species (Koerich et al. 2008) estimated that the rate of gene gain is 10.7-fold higher than the rate of gene loss (95% confidence interval: 2.2–51.3). At that time, the Y-linked gene content was well-known only in D. melanogaster; therefore, gene gains could only be measured in this lineage and gene losses only in the other species. Hence, the 10.7 estimate requires the assumption that the rates of gene gain and gene loss are homogeneous across lineages (Koerich et al. 2008) and may be incorrect if this assumption is wrong. Given the D. virilis data reported here, we can now directly measure gene gain and gene loss rates in the same lineages, without the above assumption. Namely, there were four gene gains and zero gene losses across 63 Myr in the D. virilis lineage, and the corresponding values in the D. melanogaster lineage are seven gene gains and one gene loss across 63 Myr (Fig. 4). These values imply a gene gain/gene loss ratio of 11.0 (P = 0.022, Poisson regression) (Table 1; Supplemental Methods). Similar results were obtained by applying the method described in Koerich et al. (2008) to the combined D. virilis and D. melanogaster data or to each species separately (Table 1) and by approximate Bayesian computation (Supplemental Fig. S9). Hence, independent data from two species, analyzed by different methods, support the conclusion that the gene content of the Drosophila Y indeed increased, at least in the last ∼63 Myr. This suggests that the canonical model of Y chromosome evolution, based on gene degeneration and loss, is not valid for all organisms (Carvalho et al. 2009).
Table 1.
Gene gains and losses in the Drosophila Y chromosome
Completeness of the euchromatic sequence of the human Y
The total amount of new sequence we found in the human Y chromosome is small (283 kb), and no new large scaffold was found, in accordance with the conclusion that the reference Y chromosome sequence is essentially complete. However, it may be argued that the methods we used are overly conservative or that the HuRef assembly is too incomplete; and if this is true, then our data would say little about the completeness of the reference Y. In order to evaluate this possibility, we repeated the k-mer comparison procedure (see Methods, Detection and Validation of New Human Y-linked Sequences section), removing from the reference Y one of its most elusive regions, a 450-kb euchromatic island buried within the pericentromeric repeats and found only in 2005 (coordinates 13193955–13748578 of NC_000024.9) (Kirsch et al. 2005). This mimics a missing sequence in the reference Y and was indeed its actual situation before 2005. We found that the missing sequence would have been immediately detected; a large portion of the 450 kb is covered by four HuRef scaffolds, all with a moderate or high proportion of unmatched k-mers (DS488544, 4.7 kb, 77% unmatched; DS487120, 10.4 kb, 66%; DS486616, 40.3 kb, 82%; DS486460, 269.5 kb, 74.2%). Thus, it is unlikely that another region similar to this 450-kb euchromatic island is missing in the reference human Y sequence. This is relevant since only a small portion the 40-Mbp Yq12 band has been sequenced (Skaletsky et al. 2003; Jehan et al. 2007; Hughes and Rozen 2012). Also remarkable is the small total size of the scaffolds likely derived from these heterochromatic regions (34 scaffolds; 206 kb), confirming its extremely low sequence complexity. However, we cannot exclude the possible existence of segmental duplications with extreme sequence identity; such regions can be completely missed in the HuRef and other WGS assemblies and even in the finished human genome. For example, a segmental duplication originated four copies of the SRGAP2 genes (Dennis et al. 2012), which sequence divergences range from 0.014% to 0.58%; a BLASTN search of the SRGAP2 mRNA (NM_015326) detected only one full copy and very small additional pieces in the HuRef assembly (not shown). Until recently, the whole region was also collapsed in a single copy in the reference human genome sequence.
Identification of Y-linked sequences in complex genomes and segmental duplications
The YGS method worked nearly perfectly in D. virilis and probably will do the same in other Drosophila-like genomes. In mammals (and possibly other complex genomes) there will be the issue of SD misassembly, a problem that affects many genomic analyses such as gene mapping, and polymorphism patterns (Bailey et al. 2001; Dennis et al. 2012). Detection of Y-linked scaffolds through female short-read sequencing is one more on this list. SD are much more common and longer in complex genomes (e.g., primates) than in Drosophila or in Caenorhabditis (Samonte and Eichler 2002; Fiston-Lavier et al. 2007) and are more frequently misassembled in WGS projects that use short reads, when compared to Sanger-based projects (Alkan et al. 2011) such as HuRef. These factors should be taken into account while interpreting the distribution of matching (e.g., Fig. 3); and in most cases, one may want to consider as candidate to Y-linkage those scaffolds having a relevant proportion of unmatched k-mers instead of only those close to 100%. The cutoff we chose for this “relevant proportion of unmatched k-mers” (25% for small scaffolds) is conservative, so we certainly missed some Y-linked sequence. For the HuRef assembly, the missing amount is small: The really doubtful scaffolds are the small ones that are below the 25% cutoff but not too close to the autosomal/X peak (e.g., <10 kb and with the proportion of unmatched k-mers between 5% and 25%); there are 173 such scaffolds, amounting to 740 kb (0.03% of the human genome). In other words, the vast majority of the sequence could be reliably identified as Y- or not Y-linked, even in an unfinished assembly of a complex genome.
Other limitations of the YGS method and alternative approaches
In the previous section we addressed the issue of segmental duplications, which probably is the main limitation of the YGS method. Here we discuss other limitations as well as alternative approaches.
Pseudoautosomal regions (PAR), when present, cannot be detected by the YGS method, because they are fully shared between the X and Y chromosomes.
Several genome projects, including virtually all mammalian genome projects in recent years, sequenced only the homogametic sex to increase the coverage of the X (or Z) chromosome (e.g., silkworm and rat) (Rat Genome Sequencing Project Consortium 2004; Xia et al. 2004). In these cases, the study of the Y (or W) chromosomes would require a separate sequencing effort targeting the heterogametic sex.
Assembly quality is expected to affect the detection of Y-linked scaffolds. Unassembled sequences obviously cannot be mapped, and chimeric scaffolds may produce ambiguous results if they misjoin Y-linked and autosomal (or X-linked) sequences, as possibly exemplified by the KB459848 scaffold of D. kikkawai (see Results). Sequencing errors create noise if they were not efficiently removed during the assembly, which may partially obscure the difference between Y-linked and not Y-linked small scaffolds; this problem is much ameliorated by k-mer validation (Supplemental Fig. S16). Contaminant scaffolds are spuriously detected as Y-linked if control male reads are not used (Supplemental Fig. S9). Assemblers differ in their performance, sometimes dramatically (e.g., Supplemental Fig. S14), but unfortunately it is difficult to obtain a reliable measure of assembly quality without a reference genome (Salzberg et al. 2012). Thus, as in all analyses of unfinished genomes, the YGS performance may be affected by assembly errors that are not easy to predict in advance.
Finally, it is worth considering alternative methods for the detection of Y-linked scaffolds. We already mentioned that the best strategy is to generate and sequence separate male and female libraries (Krzywinski et al. 2004; Carvalho et al. 2009). Two recent studies showed that comparative genome hybridization can identify X- and Y-linked scaffolds. Baker and Wilkinson (2010) labeled male and female DNA of stalk-eyed flies with different dyes and hybridized them to arrays of probes representing EST sequences. This method allowed identification of both X- and Y-linked ESTs, due to the copy number differences between males and females. He et al. (2012) used chromosomal deletions and a conceptually similar design to place the unmapped scaffolds of D. melanogaster into the heterochromatic regions of all chromosomes, including the Y. The identification of both X- and Y-linked scaffolds is an attractive feature of this approach. On the other hand, the requirements of sequence uniqueness are much higher for DNA hybridization than for the direct sequence comparison employed by YGS: He et al. (2012) could design probes for < 20% of the unmapped scaffolds, whereas ∼99% of the D. virilis scaffolds could be reliably classified as Y-linked or not Y-linked (Fig. 3A). It will be very interesting to apply YGS to D. melanogaster and to directly compare the two mapping approaches.
Methods
Genomic sequences
We used the CAF1.2 (Drosophila 12 Genomes Consortium 2007) and the CA assemblies of Drosophila virilis (see Comparison of the D. virilis Assemblies, below) and two versions of the human genome, the finished version Build 37 (GRCh37.p9; International Human Genome Sequencing Consortium 2004) and the unfinished WGS assembly HuRef (Levy et al. 2007). We also used the D. kikkawai short-read assembly (S Richards, J Qu, H Jiang, M Batterton, K Blankenburg, S Gubbala, Y Han, J Jayaseelan, D Kalra, C Kovar, et al., unpubl.; accession number AFFH00000000.2).
Short reads
D. virilis Illumina female reads were produced at Macrogen in an Illumina HiSeq 2000 sequencer (one lane, 100-bp single-end; 22 Gbp of raw sequence or ∼85-fold coverage of the genome after filtering) from the same inbred strain used in the genome project (Drosophila 12 Genomes Consortium 2007). D. kikkawai females were similarly sequenced, except for the lower coverage (20-fold).
To reduce the interference of polymorphism in the human data, we used the Illumina short reads from the available 36 females of Great Britain ancestry (The 1000 Genomes Project Consortium 2010), two FASTQ files from each (listed in Supplemental Data File S1, Illumina_reads.txt), which yield ∼38-fold coverage after filtering. Male reads were used to detect contaminant scaffolds (Supplemental Fig. S9) and to remove sequencing errors in the HuRef assembly (below; Supplemental Fig. S11), they came from 40 males of Great Britain ancestry (44-fold coverage after filtering) (The 1000 Genomes Project Consortium 2010).
We used a high coverage of female reads to test the limits of the method, but pilot experiments with Drosophila (0.6-fold coverage) (Carvalho and Clark 2008) and humans (fivefold coverage from a single female) achieved good results (Supplemental Fig. S12). Low coverage introduces significant binomial sampling error and hence reduces resolution for small scaffolds. It is difficult to provide a more precise guidance on the minimum female sequencing coverage required to accurately detect Y-linked scaffolds because it depends on many factors such as genome properties (e.g., amount of segmental duplications), assembly quality (e.g., fragmentation), and the error rates in the genome and female short reads (e.g., Supplemental Fig. S14).
We found that removal of sequencing errors in the female short reads is important in order to achieve a good resolution. This was done using two criteria: (1) masking low quality bases; and (2) removing k-mers that are too rare in comparison to the genomic coverage, as detailed in Kelley et al. (2010). Specifically, for the D. virilis short reads, the sequencing errors were filtered by masking bases with phred score <20 and removing 15-mers that are present less than 5 times. For the human short reads, we used a less stringent filtering (phred score <10; frequency cut-off of 2), attempting to preserve rare variants. Although we initially did the quality filtering and frequency cutoff with our YGS.pl program, we found that the program Jellyfish (Marçais and Kingsford 2011) can do both operations much faster, which is essential in the case of large data sets (e.g., human genome). Jellyfish can output in FASTA format all k-mers found after filtering; this FASTA file then is read by our YGS.pl program to build the bit-array.
Description of the YGS method and computer implementation
The genome sequence of males and females differ only by the Y chromosome. This, and the availability of inexpensive sequencing suggested that the pieces of the Y chromosome in assembled genomes can be identified by the lack of matches to female short reads (Carvalho and Clark 2008). While doing this genome vs. female comparison, identical repeats of all types (e.g., transposable elements, segmental duplications) should be removed from the genome sequence because they will cause spurious matches to female reads, but variants in these repeats (called “sequence family variants” or “singly unique nucleotide”) contain valuable information (Krsticevic et al. 2010; Dennis et al. 2012; Hughes and Rozen 2012) and should be preserved along with all single-copy sequences. Preserving repeat variants is particularly important because the Y chromosome is repeat-rich; many of its “single-copy” regions actually are rare variants of some repeat (e.g., decayed transposable elements). Despite much effort, we could not achieve this selective masking using BLAST (due to its built-in heuristics), and tools such as RepeatMasker are designed to broadly mask known repeats. A straight comparison based on short DNA words (k-mers) achieved the goal of a genome vs. female comparison that uses repeat variants and removes all types of identical repeats. We implemented it using bit-arrays, which can compactly store the presence (coded as “1”) or absence (“0”) of each k-mer. For our purposes, each k-mer and its reverse complement are equivalent; we stored whichever comes first lexicographically. Two bit-arrays were initially built, one for the k-mers present in the female short reads (“F”) and one for k-mers that occurred more than once in the assembled genome (repetitive k-mers, stored in bit-array “R”). In the final run, a third bit-array was obtained for each scaffold of the assembled genome (“G”) and compared to the two previous ones using logical operations: G NOT R yields a bit-array with single-copy k-mers of the scaffold (“GSC”); GSC NOT F yields the desired comparison between the assembled genome (one scaffold a time) and the female short reads. We then obtained the proportion of unmatched single-copy k-mers for each scaffold. Optionally, as described in a section below, a fourth bit-array was built for removal of sequencing errors in the assembled genomes. The k-mer size should be large enough so that identical k-mers seldom occur by chance in the assembled genome or are generated by sequencing errors in the female reads (Kelley et al. 2010; Li et al. 2010). On the other hand, run time and memory usage scale by a factor of 4k, which is the number of elements of the bit-array. We used k = 15 for Drosophila, and k = 18 for humans, which are typical values used for filtering errors in short reads from these genomes (Kelley et al. 2010; Li et al. 2010). The main sign of insufficient k-mer size is the displacement of the peak containing the Y-linked scaffolds toward the autosomal/X-linked peak, which reduces the resolution of the method. We observed this effect when we analyzed the human genome using k = 16 instead of k = 18 (Supplemental Fig. S13, panels A,B). On the other hand, little additional resolution was gained by increasing k from 15 to 17 with the Drosophila data (Supplemental Fig. S13, panels C,D). So we suggest as initial values k = 15 for insect-like genomes (although a higher value would not harm) and k = 18 for vertebrate-like genomes. Going beyond k = 18 seems unnecessary and would require ∼160 Gb of RAM memory. The final run of the Drosophila data (using k = 15) required ∼9 h and 4 Gb of memory in a 2.33 GHz Linux machine; the human data (using k = 18) required 15 d and 40 Gb. Since each scaffold is processed separately in the final run, faster results are easily obtained by using multiple machines. Computer code (Supplemental Data File S2 YGS.pl) was implemented in Perl making extensive use of the Bit∷Vector module (S Beyer, unpubl.; http://search.cpan.org/dist/Bit-Vector/) and runs in a single processor.
As commented previously, the program Jellyfish (Marçais and Kingsford 2011) was used to filter the short reads. The program YGS.pl (Supplemental Data File S2) then does the main steps of the YGS method: extraction of k-mers from the filtered short reads, extraction of repetitive k-mers from the genomic scaffolds, and the comparison between the genomic and short-read k-mers. Each step requires a separate run. A sample script with the commands used in all steps of the YGS method is included in the Supplemental Material.
Although primarily designed to detect Y-linked (or W-linked) scaffolds, YGS seems to be a useful tool for detection of contaminant (Supplemental Fig. S10A) and low-quality scaffolds (Supplemental Fig. S14), and for assembly comparisons/detection of new sequences in the presence of large amounts of repetitive DNA (Supplemental Fig. S5).
Detection of contaminant scaffolds
Most assembled genomes contain, at least in the initial releases, a small amount of contaminant sequences such as bacteria and yeast (e.g., Alkan et al. 2011). The YGS method will pinpoint any sequence of the assembled genome not present in the female reads, and hence some of the Y-linked scaffolds may actually be contaminants. We investigated this possibility in the HuRef assembly by a variant of the YGS method: By using male (instead of female) short reads, all legitimate sequences (and only them) will be matched, whereas contaminant sequences will remain largely unmatched. This procedure assumes that the contaminant is absent from the male reads, which is reasonable (except in cases such as a widespread virus, but in this case, being present in females, it will not be detected as Y-linked).
As shown in Supplemental Figure S10A, this procedure worked very well: Five HuRef scaffolds stood out, and BLAST analysis showed that all are bacterial sequences (99% identity to a plasmid of Streptomyces clavuligerus). They were removed from all subsequent analyses (the accession numbers are: DS487629, DS489113, DS490066, DS490212, and DS490583).
The above analysis was done including all k-mers (repetitive and single-copy). When we repeated it using only the single-copy k-mers, we observed an increased proportion of unmatched k-mers in legitimate scaffolds, particularly on the small ones (Supplemental Fig. S10B). This increase is expected because both sequencing errors and rare polymorphisms usually generate single-copy k-mers. Irrespective of their origin, these unmatched k-mers most likely will not match the female short reads, and if present in an autosomal (or X-linked) scaffold, will displace it toward the region of Y-linked scaffolds. This suggested that male short reads could be used not only to detect contaminant scaffolds but also to remove sequencing errors (and rare polymorphisms) from the legitimate scaffolds in order to improve the separation between Y- and not Y-linked scaffolds (see next section).
Because we did not have male short reads from D. virilis, we searched for contaminant scaffolds in both D. virilis assemblies by a BLASTN search against a large database of bacterial genomes plus Saccharomyces cerevisae (the latter is used to feed Drosophila and is a common contaminant). Contamination was irrelevant in the CA assembly (we found one 200-bp piece of a vector inside a 1.3-Mbp scaffold), which was used for most analyses reported in this paper. The CAF1.2 assembly contains 39 small contaminant scaffolds, and all have a relevant amount (13%–100%) of bacterial sequences (vector sequences in many cases). They are identified in the Supplemental Data File S10 virCAF12_scafs_classification.pdf and were excluded from all subsequent analyses. As expected, they behaved as Y-linked sequences when compared to female short reads (not shown).
Removal of sequencing errors in the assembled genomes
Sequencing errors in the assembled genomes (and rare polymorphisms in the HuRef donor) are expected to increase the proportion of unmatched k-mers, and hence displace autosomal/X-linked scaffolds toward the Y peak. We initially did not expect it to be a problem because sequencing errors should be rare, even in unfinished genomes. Indeed, in both the D. virilis and HuRef assemblies, when we exclude the Y-linked scaffolds, the genome-wide proportion of single-copy k-mers unmatched by female reads is low (D. virilis CA assembly: 0.15%; HuRef: 0.26%). However, we also found that in small scaffolds this proportion is substantially higher: For the non-Y-linked scaffolds smaller than 5 kb, it is 6.5% and 2.2% (D. virilis CA and human HuRef assemblies, respectively) (see Supplemental Fig. S10B). This inverse association with size probably has two causes: (1) In small scaffolds a larger share of the sequence came from the edges, which tend to have lower coverage (and hence higher error rate); and (2) traces containing errors will cause assembly breaks and result in smaller scaffolds. Whatever the cause, we found that removal of these sequencing errors yield a cleaner result for small scaffolds. Given the data that are available, we did the sequence error removal differently in Drosophila and human genomes, but the principle is the same.
In the human data, we had access to Illumina short reads of 40 Great Britain males (The 1000 Genomes Project Consortium 2010); k-mers not represented there were deemed as sequencing errors in the assembled genome (or rare polymorphisms in the HuRef donor) and removed from the analysis. Namely, we built an additional bit-array storing the k-mers from male short reads (“M”); the bit-array containing the validated single-copy (VSC) k-mers of each scaffold is obtained by GSC AND M; then VSC NOT F yields the desired assembled genome vs. female short reads comparison. This procedure indeed yields a clearer separation between Y- and non-Y-linked scaffolds (Supplemental Fig. S11). It removed 0.2% of the single-copy k-mers across the genome and 4.5% in the case of small scaffolds.
When male reads are not available (as in D. virilis), sequencing errors in the assembled genome can be partially removed by identifying regions of low quality, e.g., by using the quality values from the consensus sequences or from the Sanger traces used to build them. We chose the last option because the consensus quality values of the D. virilis assemblies were not available. We first downloaded from the NCBI trace archive the same Sanger traces used to assemble the D. virilis sequence (Drosophila 12 Genomes Consortium 2007) and filtered them at a phred score of 20 (i.e., all bases with >1% error rate were masked). Then we used the filtered Sanger traces to validate the genomic k-mers, analogously to the use of male reads in the human data (see above). In the D. virilis CA assembly, this validation procedure removed 0.2% of the single-copy k-mers across the genome and 4.6% in the case of small scaffolds (<5 kb). As a control, we repeated the whole procedure using unfiltered Sanger traces; as expected, these values drop to nearly zero (genome-wide: 0.03% ; small scaffolds: 0.2%). The effect of the validation in the D. virilis CA assembly was clear (Supplemental Fig. S15) but perhaps less dramatic than in the human genome (Supplemental Fig. S11); a major benefit of this analysis is that it allowed us to detect the problems of the CAF1.2 assembly (next section).
Comparison of the D. virilis assemblies
We used two assemblies, CAF1.2 (available at ftp://ftp.flybase.net/genomes/Drosophila_virilis/dvir_r1.2_FB2012_01/fasta/dvir-all-chromosome-r1.2.fasta.gz) and CA (http://goo.gl/qLrG0). The D. virilis CAF1.2 is the assembly used in the Drosophila 12 Genomes Consortium (2007) and in FlyBase (McQuilton et al. 2012). It is derived from two primary assemblies done with the Arachne and the Celera assemblers (Drosophila 12 Genomes Consortium 2007; Zimin et al. 2008). While doing the removal of sequencing errors (see previous section), we noticed that the CAF1.2 assembly has a very large number of low quality, small scaffolds. Namely, the CAF1.2 assembly has 13,530 scaffolds, and only 2261 have >80% valid single-copy k-mers (Supplemental Fig. S14A); among these 2261, only 1056 have >50 valid single-copy k-mers. As detailed in the previous section, we did this comparison using Sanger traces filtered at a phred score of 20 (i.e., 1% error), so ∼11,300 scaffolds (i.e., 13,530 minus 2261) contain a substantial amount of low quality sequence. Nearly all low-quality scaffolds are small (average size 1527 bp; two have ∼100 kb); they are very rich in simple repeats: 46% of their sequence is masked by DUST (Morgulis et al. 2006), perhaps reflecting the huge amount of satellite DNA in the D. virilis genome (Bosco et al. 2007). Removal of low quality scaffolds and those containing less than 50 single-copy k-mers results in an assembly with 1056 scaffolds and 170 Mbp (90% of the total sequence, 189 Mbp) and yields a clean separation between Y and not Y-linked scaffolds (Supplemental Fig. S16B). Supplemental Data File S10 virCAF12_scafs_classification.pdf lists all 13,530 CAF1.2 scaffolds along with their classification (Y-linked, not Y-linked, intermediate, contaminant, low-quality, and small).
Although the quality filtering solved at least part of the CAF1.2 problems, it is undesirable to rely only on such data, so we searched for alternative assemblies. Unfortunately, the original Arachne and Celera assemblies (Drosophila 12 Genomes Consortium 2007; Zimin et al. 2008) are no longer available (G Sutton, pers. comm.), but the assembly evaluation team of the Drosophila 12 Genomes Consortium produced in 2005 different assemblies of D. virilis (using different assemblers) based on exactly the same Sanger traces. We examined two of them, “CA” (Celera Assembler) and “AR” (Arachne), using the same procedures we did with the CAF1.2 assembly. We found that the AR assembly was similar to CAF1.2, containing a large number of small, low quality scaffolds (not shown), which are absent from the CA assembly (Supplemental Fig. S14, panels C,D). Unless otherwise noted, we used the CA assembly in our analyses to avoid the use of filtered data. It is similar to the filtered CAF1.2 assembly both in number of scaffolds (1186) and in size (165 Mbp; all reported assembly sizes exclude gaps). The main results (namely, the identification of four new Y-linked genes and 11 pseudogenes) were the same with both assemblies, and all tested candidates are represented in the CAF1.2 assembly (Supplemental Table S1).
Sequence finishing and nomenclature of the new D. virilis Y-linked genes
Due to the low coverage of the Y chromosome (Carvalho et al. 2003) and its abundance of repetitive sequences, the sequences of almost all Y-linked genes have gaps and sequencing errors, and different exons of the same gene are scattered in several scaffolds (Carvalho et al. 2000; Koerich et al. 2008). We corrected these problems by directly sequencing the products from polymerase chain reactions with reverse transcription (RT–PCR) in the four new D. virilis Y-linked genes and in the majority of their Y-linked orthologs in other species. Their finished sequences were submitted to GenBank (see Data Access).
Given their fragmentary assembly, the Y-linked genes usually are incompletely annotated (or not annotated at all) during the automatic annotation step of genome projects (Carvalho et al. 2000; Koerich et al. 2008), and this also happened in D. virilis. When available, we maintained the original names even when the original annotation covers a small portion of the gene. Namely, for the four D. virilis genes, the original annotations were as follows: GJ19835 was missing >50% of the gene (at the N terminus); GJ19633 was complete; GJ11126 was missing one-third of the sequence (at the N terminus); and the D. virilis ortholog of the JYalpha was missing one-third of the sequence and split in two genes (GJ18574 and GJ18410); we used the name GJ18574 because it covers a larger region.
Detection and validation of new human Y-linked sequences
119 HuRef scaffolds were identified as Y-linked due to a low match to the female short reads (Supplemental Table S4). A sequence comparison is needed to distinguish among the 119 scaffolds those scaffolds covered by the reference Y from those containing new sequences. This comparison should take into account that as an unfinished sequence, HuRef scaffolds contain gaps, collapsed repeats, and possibly large-scale misassemblies, such as inversion of contigs within scaffolds, so we do not expect perfect matching with the reference Y; these discrepancies should not be confounded to “new sequence.” We initially tried Nucmer (Delcher et al. 2002) to compare the 119 scaffolds to the reference Y, but the very high repeat content of the Y-linked sequences created problems. We also considered using RepeatMasker, but as we suspected and later confirmed, it would broadly mask most new Y sequence (70% of the 283 kb we found). We searched potentially new Y-linked sequences using two complementary approaches described below. As described in Results, these approaches were sensitive enough to detect even an 11-kb piece of new sequence in the middle of the 2.3-Mbp scaffold DS486277.
First, we ran the YGS.pl program on the 119 Y-linked scaffolds, using the k-mers extracted from the reference Y sequence (accession NC_000024.9) instead of k-mers from female short reads. This procedure should distinguish Y-linked HuRef scaffolds covered by the reference Y from those containing new sequences (the latter having a high proportion of unmatched k-mers) and is insensitive to the more trivial discrepancies mentioned above. As detailed in Results (see also Supplemental Fig. S5; Supplemental Table S4, column 5), the majority of the scaffolds (85 of 119) and of the sequence (18.165 Mbp of 18.371 Mbp) are entirely contained (or nearly so) within the reference Y sequence. Thirty-four small scaffolds are mostly composed of new sequence (>50% unmatched single-copy k-mers) and likely came from the Yq12 and other heterochromatic regions of the Y chromosome, which are not included into the reference Y sequence (Results). A few of the 85 Y-linked scaffolds that closely match the reference Y have a moderate amount of unmatched k-mers (∼10%) (Supplemental Fig. S5), which suggests that they too may contain a small amount of new sequence. The four largest are DS486171, DS486512, DS486519, and DS486628. The largest (DS486171) contains the misassembled XTR region; further analysis (below) shows that all its unmatched regions correspond to patches of the misassembled X-chromosome sequence, so it does not contain new sequence. The three next in size (DS486512, DS486519, DS486628) were shown to contain new sequence (below).
The preceding approach may not detect a rather large amount of new sequence if it is inside a large scaffold, nor does it show where the new sequence is located within the scaffolds. The second approach addressed these issues. We used the BWA program (Li and Durbin 2009) to align the short reads from 40 Great Britain males (44-fold coverage of the genome) (The 1000 Genomes Project Consortium 2010) to the HuRef assembly under very high stringency (allowing at most one mismatch or one indel), and using BamTools (Barnett et al. 2011), extracted from the BAM file the reads that aligned to the 119 Y-linked scaffolds. We then aligned these reads to the reference Y sequence (using the same BWA parameters), and now extracted the reads that did not align. These “differential male reads” represent the regions of the 119 Y-linked scaffolds not covered by the reference Y and are present in other males (i.e., potentially new regions of the Y chromosome). In order to locate them, we aligned the differential male reads to the 119 scaffolds and used SAMtools (Li et al. 2009) to count the number of reads per scaffold (Supplemental Table S4, column 6). Finally, the HuRef assembly was aligned to female reads to confirm the Y-linkage of the 119 scaffolds. Scaffolds with a high number of hits of differential male reads possibly contain new sequence; we confirmed this using the Integrative Genomics Viewer (IGV) browser (Thorvaldsdóttir et al. 2012) to visualize the location and coverage of the three types of reads (male, differential male, female) within each of the 119 scaffolds (e.g., Fig. 6).
We found that all 34 HuRef scaffolds that were deemed before as “new sequence” (Supplemental Fig. S5) also have a large number of hits from differential male reads (Supplemental Table S4, column 6), which confirms that they contain new Y-linked sequence. Among the 85 scaffolds that closely match the reference Y, we found three patterns of hits of differential male reads: (1) scaffolds that have very few or zero hits from the differential male reads; these scaffolds are completely subsumed into the reference Y; (2) scaffolds that contain the misassembled XTR region, characterized by a large number of hits from the differential male reads, unevenly distributed (DS486171, DS486351, etc.) (Supplemental Table S4); they also have many hits from female reads in the same region of differential male reads, so we dismissed them; and (3) scaffolds that contain a discrete region with a large number of hits from the differential male reads, evenly distributed and devoid of significant coverage by female reads (e.g., Fig. 6). The latter pattern provides unequivocal evidence of new Y-linked sequences; we found it in five regions, belonging to four scaffolds. Three of them were already detected with the previous approach (DS486628, DS486519, and DS486512); the remaining two regions are part of a large scaffold (DS486277), so their presence was inconspicuous in Supplemental Figure S5. Four additional scaffolds contain small patches (< 2 kbp) of new sequence (DS486286, DS486428, DS486288, and DS487109) and were not further studied.
The above procedures control for all reasonable sources of artifacts. Two different approaches (Supplemental Table S4, columns 5,6) yield similar results (namely, both identified the 34 small scaffolds, plus scaffolds DS486512, DS486519, and DS486628 as containing new Y sequences). The k-mer comparison (Supplemental Table S4, column 5) is insensitive to large scale misassemblies (contig inversions, etc.), which are common in repetitive regions. The alignment of male differential reads (Supplemental Table S4, column 6) is insensitive to contaminant DNA, plain sequence errors in HuRef (which might mimic new sequence), and also to rare alleles/new mutations present in the HuRef donor because the male reads would not match them. The visualization of the alignment of the three types of short reads (male, differential male, and female) (Fig. 6) allowed the separation of differences between HuRef and the reference Y due to new sequence from artifacts caused by misassemblies (the later have hits against female reads in the same region of the differential male reads).The five newly found regions that extend the reference Y scaffolds are summarized in Supplemental Table S3.
Data access
The new nucleotide sequences reported in this manuscript have been submitted to GenBank (http://www.ncbi.nlm.nih.gov/genbank/) as Third Party Annotation sequences under the accession numbers TPA: BK008736–BK008744.
Acknowledgments
We thank L. Farinelli, G. Marçais, S. Beyer, P. Green, E. Eichler, G. Sutton, J. Miller, A. Zimin, J. Felsenstein, M. Khuner, E. Walkup, J. McGill, R. Giansiracusa, C. Berg, M. Bahia-Schlee, B. Lemos, A. Peixoto, M. Luz, P. Paiva, G. Nassar, E. Dupim, and L. Koerich for valuable suggestions and help. We thank NACAD/UFRJ and GS/UW for computational facilities, and the Drosophila Species Stock Center (U.C. San Diego) for the Drosophila strains. This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPq, Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro-FAPERJ, Guggenheim Foundation, FIC-NIH grant TW007604 (A.B.C.) and NIH grant GM64590 (A.G.C. and A.B.C.). A.B.C. conceived the work, performed the research, and analyzed the data; A.G.C. suggested the use of human data; A.B.C. and A.G.C. wrote the paper. A.B.C. dedicates this paper to the memory of Alexandre Peixoto, outstanding scientist and human being.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.156034.113.
References
- The 1000 Genomes Project Consortium 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Sajjadian S, Eichler EE 2011. Limitations of next-generation genome sequence assembly. Nat Methods 8: 61–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE 2001. Segmental duplications: Organization and impact within the current human genome project assembly. Genome Res 11: 1005–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RH, Wilkinson GS 2010. Comparative Genomic Hybridization (CGH) reveals a neo-X chromosome and biased gene movement in stalk-eyed flies (genus Teleopsis). PLoS Genet 6: e1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, Marth GT 2011. BamTools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27: 1691–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco G, Campbell P, Leiva-Neto JT, Markow TA 2007. Analysis of Drosophila species genome size and satellite DNA content reveals significant differences among strains as well as between species. Genetics 177: 1277–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ. 1983. Evolution of sex determining mechanisms. Benjamin/Cummings, Advanced Book Program, Menlo Park, CA. [Google Scholar]
- Carvalho AB, Clark AG 2005. Y chromosome of D. pseudoobscura is not homologous to the ancestral Drosophila Y. Science 307: 108–110 [DOI] [PubMed] [Google Scholar]
- Carvalho AB, Clark AG 2008. Efficient identification of Drosophila Y-chromosome sequences by short-read sequencing. In The 49th Annual Drosophila Research Conference, Abstract 112, p. 124. The Genetics Society of America, San Diego, CA [Google Scholar]
- Carvalho AB, Lazzaro BP, Clark AG 2000. Y chromosomal fertility factors kl-2 and kl-3 of Drosophila melanogaster encode dynein heavy chain polypeptides. Proc Natl Acad Sci 97: 13239–13244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AB, Dobo BA, Vibranovski MD, Clark AG 2001. Identification of five new genes on the Y chromosome of Drosophila melanogaster. Proc Natl Acad Sci 98: 13225–13230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AB, Vibranovski MD, Carlson JW, Celniker SE, Hoskins RA, Rubin GM, Sutton G, Adams M, Myers EW, Clark AG 2003. Y chromosome and other heterochromatic sequences of the Drosophila melanogaster genome: How far can we go? Genetica 117: 227–237 [DOI] [PubMed] [Google Scholar]
- Carvalho AB, Koerich LB, Clark AG 2009. Origin and evolution of Y chromosomes: Drosophila tales. Trends Genet 25: 270–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D 2000. The degeneration of Y chromosomes. Philos Trans R Soc London Ser B 355: 1563–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Bellott DW, Page DC, Clark AG 2012. Identification of avian W-linked contigs by short-read sequencing. BMC Genomics 13: 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher A, Phillippy A, Carlton J, Salzberg S 2002. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res 30: 2478–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MY, Nuttle X, Sudmant PH, Antonacci F, Graves TA, Nefedov M, Rosenfeld JA, Sajjadian S, Malig M, Kotkiewicz H, et al. 2012. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell 149: 912–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218 [DOI] [PubMed] [Google Scholar]
- Ezaz T, Graves JA 2012. Foreword: Sex and sex chromosomes—New clues from nonmodel species. Chromosome Res 20: 1–5 [DOI] [PubMed] [Google Scholar]
- Fiston-Lavier AS, Anxolabehere D, Quesneville H 2007. A model of segmental duplication formation in Drosophila melanogaster. Genome Res 17: 1458–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Caudy A, Parsons L, Rosebrock A, Pane A, Raj S, Wieschaus E 2012. Mapping the pericentric heterochromatin by comparative genomic hybridization analysis and chromosome deletions in Drosophila melanogaster. Genome Res 22: 2507–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins RA, Carlson JW, Kennedy C, Acevedo D, Evans-Holm M, Frise E, Wan KH, Park S, Mendez-Lago M, Rossi F, et al. 2007. Sequence finishing and mapping of Drosophila melanogaster heterochromatin. Science 316: 1625–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Rozen S 2012. Genomics and genetics of human and primate Y chromosomes. Annu Rev Genomics Hum Genet 13: 83–108 [DOI] [PubMed] [Google Scholar]
- International Chicken Genome Sequencing Consortium 2004. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432: 695–716 [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium 2004. Finishing the euchromatic sequence of the human genome. Nature 431: 931–945 [DOI] [PubMed] [Google Scholar]
- Jehan Z, Vallinayagam S, Tiwari S, Pradhan S, Singh L, Suresh A, Reddy HM, Ahuja YR, Jesudasan RA 2007. Novel noncoding RNA from human Y distal heterochromatic block (Yq12) generates testis-specific chimeric CDC2L2. Genome Res 17: 433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser VB, Bachtrog D 2010. Evolution of sex chromosomes in insects. Annu Rev Genet 44: 91–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Schatz MC, Salzberg SL 2010. Quake: Quality-aware detection and correction of sequencing errors. Genome Biol 11: R116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. 2008. Mapping and sequencing of structural variation from eight human genomes. Nature 453: 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch S, Weiss B, Miner TL, Waterston RH, Clark RA, Eichler EE, Munch C, Schempp W, Rappold G 2005. Interchromosomal segmental duplications of the pericentromeric region on the human Y chromosome. Genome Res 15: 195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerich LB, Wang X, Clark AG, Carvalho AB 2008. Low conservation of gene content in the Drosophila Y chromosome. Nature 456: 949–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krsticevic FJ, Santos HL, Januario S, Schrago CG, Carvalho AB 2010. Functional copies of the Mst77F gene on the Y chromosome of Drosophila melanogaster. Genetics 184: 295–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski J, Nusskern DR, Kern MK, Besansky NJ 2004. Isolation and characterization of Y chromosome sequences from the African malaria mosquito Anopheles gambiae. Genetics 166: 1291–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos B, Araripe LO, Hartl DL 2008. Polymorphic Y chromosomes harbor cryptic variation with manifold functional consequences. Science 319: 91–93 [DOI] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, et al. 2010. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20: 265–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M, Vibranovski MD, Zhang YE 2012. Evolutionary interactions between sex chromosomes and autosomes. In Rapidly evolving genes and genetic systems (ed. Singh RS, et al.), pp. 101–114. Oxford University Press, Oxford, UK [Google Scholar]
- Marçais G, Kingsford C 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27: 764–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masly JP, Jones D, Noor MA, Locke J, Orr HA 2006. Gene transposition as a cause of hybrid sterility in Drosophila. Science 313: 1448–1450 [DOI] [PubMed] [Google Scholar]
- McQuilton P, St Pierre SE, Thurmond J 2012. FlyBase 101–the basics of navigating FlyBase. Nucleic Acids Res 40: D706–D714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgulis A, Gertz EM, Schaffer AA, Agarwala R 2006. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J Comput Biol 13: 1028–1040 [DOI] [PubMed] [Google Scholar]
- Murtagh VJ, O'Meally D, Sankovic N, Delbridge ML, Kuroki Y, Boore JL, Toyoda A, Jordan KS, Pask AJ, Renfree MB, et al. 2012. Evolutionary history of novel genes on the tammar wallaby Y chromosome: Implications for sex chromosome evolution. Genome Res 22: 498–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page DC, Harper ME, Love J, Botstein D 1984. Occurrence of a transposition from the X-chromosome long arm to the Y-chromosome short arm during human evolution. Nature 311: 119–123 [DOI] [PubMed] [Google Scholar]
- Pantazidis AC, Galanopoulos VK, Zouros E 1993. An autosomal factor from Drosophila arizonae restores normal spermatogenesis in Drosophila mojavensis males carrying the D. arizonae Y chromosome. Genetics 134: 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rat Genome Sequencing Project Consortium 2004. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493–521 [DOI] [PubMed] [Google Scholar]
- Rice WR 1996. Evolution of the Y sex chromosome in animals. Bioscience 46: 331–343 [Google Scholar]
- Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, Platzer M, Howell GR, Burrows C, Bird CP, et al. 2005. The DNA sequence of the human X chromosome. Nature 434: 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H, Marszalek JD, Minx PJ, Cordum HS, Waterston RH, Wilson RK, Page DC 2003. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature 423: 873–876 [DOI] [PubMed] [Google Scholar]
- Salzberg SL, Phillippy AM, Zimin A, Puiu D, Magoc T, Koren S, Treangen TJ, Schatz MC, Delcher AL, Roberts M, et al. 2012. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res 22: 557–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samonte RV, Eichler EE 2002. Segmental duplications and the evolution of the primate genome. Nat Rev Genet 3: 65–72 [DOI] [PubMed] [Google Scholar]
- Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, Pyntikova T, Ali J, Bieri T, et al. 2003. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423: 825–837 [DOI] [PubMed] [Google Scholar]
- Sweigart AL 2010. Simple Y-autosomal incompatibilities cause hybrid male sterility in reciprocal crosses between Drosophila virilis and D. americana. Genetics 184: 779–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP 2012. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibranovski MD, Koerich LB, Carvalho AB 2008. Two new Y-linked genes in Drosophila melanogaster. Genetics 179: 2325–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, Zhou Z, Lu C, Cheng D, Dai F, Li B, Zhao P, Zha X, Cheng T, Chai C, et al. 2004. A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science 306: 1937–1940 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Bachtrog D 2012. Sex-specific adaptation drives early sex chromosome evolution in Drosophila. Science 337: 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin AV, Smith DR, Sutton G, Yorke JA 2008. Assembly reconciliation. Bioinformatics 24: 42–45 [DOI] [PubMed] [Google Scholar]