

Supplemental Digital Content is available in the text.
Key Words: Hereditary spastic paraplegia, Length-dependent axonal degeneration, Transgenic rat, Tubulovesicular structures
Abstract
Hereditary spastic paraplegias (HSPs) are characterized by progressive spasticity and weakness in the lower extremities that result from length-dependent central to peripheral axonal degeneration. Mutations in the non-imprinted Prader-Willi/Angelman syndrome locus 1 (NIPA1) transmembrane protein cause an autosomal dominant form of HSP (SPG6). Here, we report that transgenic (Tg) rats expressing a human NIPA1/SPG6 mutation in neurons (Thy1.2-hNIPA1G106R) show marked early onset behavioral and electrophysiologic abnormalities. Detailed morphologic analyses reveal unique histopathologic findings, including the accumulation of tubulovesicular organelles with endosomal features that start at axonal and dendritic terminals, followed by multifocal vacuolar degeneration in both the CNS and peripheral nerves. In addition, the NIPA1G106R mutation in the spinal cord from older Tg rats results in an increase in bone morphogenetic protein type II receptor expression, suggesting that its degradation is impaired. This Thy1.2-hNIPA1G106R Tg rat model may serve as a valuable tool for understanding endosomal trafficking in the pathogenesis of a subgroup of HSP with an abnormal interaction with bone morphogenetic protein type II receptor, as well as for developing potential therapeutic strategies for diseases with axonal degeneration and similar pathogenetic mechanisms.
INTRODUCTION
Hereditary spastic paraplegias (HSPs) are a group of diverse genetic disorders that are clinically characterized by insidiously progressive spasticity and weakness affecting the lower extremities (1). Although the number of HSP loci exceeds 40, neurologic examination of HSP patients typically shows common deficits referable to a length-dependent central-peripheral distal axonopathy involving the longest axons of the corticospinal tract and dorsal column and to a lesser degree of the peripheral nerves (2–4). Therefore, animal models of HSP are potentially important tools for expanding our understanding of molecular and cellular mechanisms involved in distal axonopathy, so-called dying-back neuropathy (5, 6).
The functions of several HSP-causing genes have been extensively studied; kinesin and spastin are implicated in microtubule function or transport, whereas others such as paraplegin and HSP60 are involved in mitochondria function that is vital for energy-dependent maintenance of axonal transport (7–12). Collectively, these and other observations put membrane trafficking (i.e. fast axonal transport) to the forefront of the central dogma of length-dependent axonal degeneration.
An important group of HSP-associated mutations are found in proteins localized to the endosomal protein traffic compartment, implicating involvement of membrane shaping/remodeling in the pathophysiology of the disease (13). This endosomal group includes atlastin-1, non-imprinted PraderWilli/Angelman syndrome 1 (NIPA1), spastin, and spartin, which are inhibitors of bone morphogenetic protein type II receptor (BMPRII) signaling (14–16). Non-imprinted PraderWilli/Angelman syndrome 1 encodes an integral membrane protein, and the mutations are responsible for one of the autosomal dominant forms of HSP (i.e. SPG6). It has been shown that fly larvae lacking the Drosophila melanogaster homologue of NIPA1, spichthyin, show significantly increased numbers of synaptic boutons at neuromuscular junctions (17), suggesting that NIPA1 normally acts to inhibit synaptic overgrowth at the neuromuscular junction. Non-imprinted PraderWilli/Angelman syndrome 1 and spichthyin are predominantly endosomal proteins that are also found in the Golgi complex and at the plasma membrane, where they are subject to clathrin-mediated endocytosis (16–19). Overexpression of NIPA1 facilitates internalization of BMPRII. Thus, NIPA1 is considered an inhibitor of BMP signaling by binding to the type II BMP receptor and promoting its endocytic internalization and degradation in lysosomes (16, 17). Although these studies have provided important clues to understanding the molecular interactions between NIPA1 and BMPRII, a mammalian animal model is needed to study the evolution of the disease process and the significance of impaired endosomal trafficking/membrane remodeling to the length-dependent distal axonopathy. Moreover, such a model will be essential for the development of clinically relevant therapeutic interventions.
Here, we report a transgenic (Tg) rat model (Thy1.2-hNIPA1G106R), in which a missense human NIPA1 mutant cDNA (hNIPA1G106R) is expressed specifically in neural cells. These Tg rats show a significantly earlier onset (4–6 weeks) and more robust behavioral phenotype than previously reported autosomal dominant HSP rodent models. Histopathologic analyses revealed progressive length-dependent and multifocal vacuolar degeneration in both the CNS and peripheral nerves. The hallmark pathologic change is the abnormal accumulation of tubulovesicular (TV) membranous organelles at the axonal and dendritic nerve endings as the earliest abnormality. Ultrastructural and immunohistochemical characteristics of TV membranous organelles reveal their endosomal identity (i.e. they are referred to interchangeably as endosomes or TV membranous organelles). In addition, accumulation of these structures into large aggregates is seen within distal axons, dendrites, and cell bodies. These large aggregates of densely packed endosomes are surrounded by double delimiting membranes and, subsequently, they turn into large vacuoles presumably via fusions with autophagosomes. Furthermore, we show that, in the spinal cord, NIPA1G106R missense mutation resulted in an almost 2-fold increase in BMPRII expression, in agreement with the previous in vitro studies. Our results provide further support for the proposed model of NIPA1 action on BMPRII trafficking.
MATERIALS AND METHODS
Animals
All experiments using Tg rats are reviewed and approved by the Institutional Animal Care and Use Committees at The Ohio State University and The University of Texas Southwestern Medical Center at Dallas. The G106R mutation was introduced to human NIPA1 cDNA by site-directed mutagenesis (Gene Editor kit, Promega, Madison, WI), and then a transgene construct was created using the murine Thy1.2 promoter (gift from Dr. Pico Caroni [20]). Founder rats were generated by pronuclear injection of transgene into the fertilized eggs of Sprague-Dawley rats.
Antibodies
A polyclonal rabbit NIPA1-specific antibody was raised by polypeptides CFKEFNFNLGEMNKSNMKTD-COOH (hNIPA1 311–329 residues). Peptide synthesis and antibody production were carried by 21st Century Biochemicals, Inc (Marlboro, MA). All commercial antibodies used are included in Supplemental Digital Content 1, http://links.lww.com/NEN/A510.
Expression Study
Poly(A)-RNA was harvested from organs of 3-week-old Tg and wild-type rats using STAT-60 (Tel-Test, Friendswood, TX) and Qiagen RNA extraction kit (Qiagen, Valencia, CA). Polymerase chain reaction (PCR) primer sequences and the detailed protocol for RT-PCR are described in Supplemental Digital Content 1, http://links.lww.com/NEN/A510. ABI-prism 7700 HT (Applied Biosystems, Foster City, CA) was used for quantitative PCR analysis. For protein detection, protein extracts were applied to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot analysis using anti-NIPA1, anti-BMPRII, anti–phosphorylated Smad 1/5/8, and anti–β-actin antibodies. ImageJ Software was used for quantification of proteins.
Primary Cortical Neuron Culture and Transfection
Cortical neuronal cultures from newborn (postnatal day 0 [P0]) and P2 Sprague-Dawley wild-type rats were prepared with modifications, as previously described (21). Detailed culture conditions and RNA oligonucleotide sequencesused for targeting rat NIPA1 are described in Supplemental Digital Content 1, http://links.lww.com/NEN/A510. The stimulation of primary cortical neurons by BMP9 and measurement of phosphorylated Smad were performed with modifications, as previously described (22) (Supplemental Digital Content 1, http://links.lww.com/NEN/A510).
Behavioral Studies
The horizontal ladder gait (23) and inclined grid climbing (24) analyses were applied. Detailed descriptions for each study are in Supplemental Digital Content 1, http://links.lww.com/NEN/A510.
Neuropathology
Histopathologic studies were carried out in 10 Thy1.2-hNIPA1G106R Tg rats at the age of 4 weeks (n = 3), 6 weeks (n = 2), 12 weeks (n = 2), and 30 to 40 weeks (n = 3) and in wild-type age-matched control rats. Lumbar, sacral, and cervical segments of spinal cord, dorsal root ganglia from L4-S1 levels, midsciatic and distal tibial nerves, and soleus, lumbrical, and intrinsic foot muscles were studied. Additional spinal cord segments and brain were fixed in 10% formalin and processed for paraffin embedding. Immunohistochemistry was done on paraformaldehyde-fixed and cryoprotected spinal cord and brain tissue from Tg and age-matched control rats. Detailed protocols for semithin and ultrastructural analyses and immunohistochemistry are described in Supplemental Digital Content 1, http://links.lww.com/NEN/A510.
Morphometry
Three Tg and 3 age-matched wild-type rats, at 38 weeks of age, were used for quantitative analysis. Five randomly selected areas of 1-μm-thick toluidine blue–stained cross sections from midsciatic and distal tibial nerves were photographed using a 60× objective. Axon diameters and numbers of myelinated nerves were determined using Adobe Illustrator CS4 software (Adobe Systems Inc., San Jose, CA). The myelinated fiber densities (mean ± SD per square millimeter of endoneurial area) in each group were calculated, and axon diameter histograms were generated. A total of 2,080 and 6,180 fibers were counted for each genotype (wild-type and Tg, respectively).
Electrophysiology
The amplitudes and conduction velocities from tail motor, sciatic, and mixed tail sensory nerves were recorded. Detailed methods of nerve conduction studies are described in Supplemental Digital Content 1, http://links.lww.com/NEN/A510.
In Situ Hybridization
Using human NIPA1 cDNA, a riboprobe template vector was cloned and sense and antisense digoxigenin-UTP–labeled riboprobes were made (DIG RNA labeling kit; Roche Applied Science, Indianapolis, IN). Hybridized riboprobe is visualized with alkaline phosphatase–conjugated anti-digoxigenin antibody and NBT/BCIP (nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl-phosphate; Roche Applied Science). Protocols are described in Supplemental Digital Content 1, http://links.lww.com/NEN/A510.
Statistical Analysis
Student t test (immunoblot), 1-way analysis of variance with Tukey post hoc analysis (histologic analysis), and repeated-measures 2-way analysis of variance with Bonferroni post hoc analysis (for weight, behavioral, and electrophysiologic analyses) were performed using JMP9 Software (SAS Institute, Inc., Cary, NC). A value of p < 0.05 was considered significant.
RESULTS
Generation of Thy1.2-hNIPA1G106R Tg Rats
We used the neuron-specific mouse Thy1.2 promoter (20) to express human NIPA1G106R mutant cDNA in rats (Fig. 1A). From the initial 13 founders, 2 representative lines (A and B) were selected based on their phenotype and expression of the transgene for further characterization. Line A presented with an earlier-onset more robust phenotype than line B and was used throughout the studies. Figure 1B shows that the transgene is specifically expressed within neural tissues as compared with ubiquitous expression of endogenous rat NIPA1 mRNA. This expression pattern is consistent with previous reports in mice (18) and humans (25). Figure 1C shows that human transgene expression does not affect the expression of endogenous rat NIPA1 mRNA. Figure 1D shows that the estimated amount of NIPA1 protein (consisting of a ∼34-kDa and a ∼40-kDa band [as shown in Fig. 7A]) is not different between the brains of wild-type and Thy1.2-hNIPA1G106R Tg rats at different ages.
FIGURE 1.
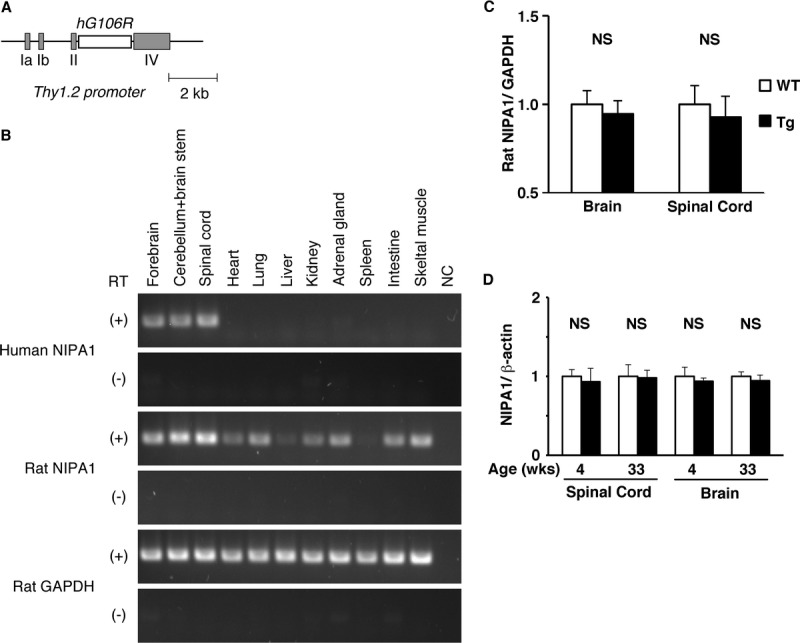
Thy1.2-hNIPA1G106R transgenic (Tg) rat. (A) The transgene construct was created using the mouse Thy1.2 promoter and human NIPA1G106R mutant cDNA. (B) RT-PCR of the transgene, human non-imprinted Prader-Willi/Angelman syndrome locus 1 (NIPA1) (first row), endogenous rat NIPA1 (third row), and glyceraldehyde-3-phosphate dehydrogenase ([GAPDH] fifth row) in major organs from Thy1.2-hNIPAG106R rat at 4 weeks of age. Transgene expression was detected specifically in the brain and spinal cord compared with ubiquitous expression of endogenous NIPA1. NC, negative control (absence of RNA template); RT, reverse transcriptase. (C) Quantitative PCR of endogenous NIPA1 mRNA expression in the brain and spinal cord between age-matched (4 weeks) wild-type (WT) and Thy1.2-hNIPAG106R rats (n = 3 in each group). WT NIPA1 expression standardized by GAPDH was set as 1. There was no apparent difference in endogenous NIPA1 expression between WT and Thy1.2-hNIPAG106R Tg rats. NS, not significant. (D) Age-matched young (4 weeks) and old (33 weeks) WT and Thy1.2-hNIPA1G106R Tg rats (n = 3 in each group) were compared for NIPA1 protein expression (standardized by β-actin is set as 1). There was no apparent difference detected in the amount of NIPA1 between WT and Thy1.2-hNIPA1G106R Tg rats at different ages.
FIGURE 7.
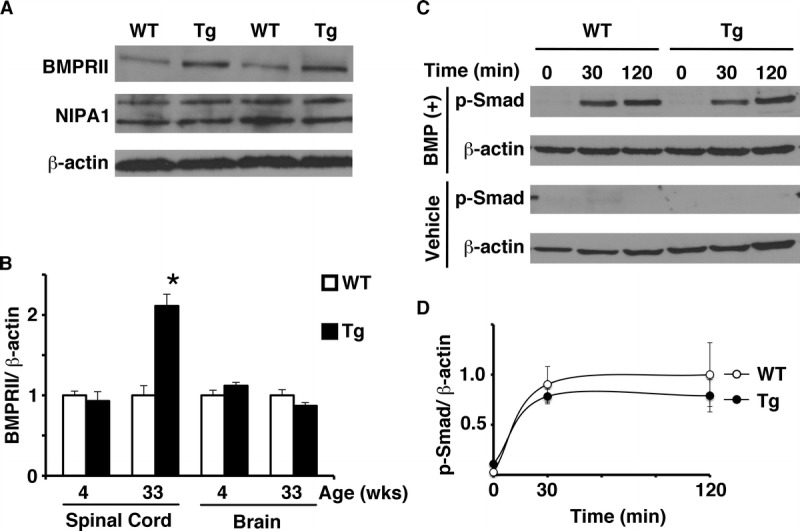
Thy1.2-hNIPA1G106R transgenic (Tg) rat spinal cord protein. (A) Immunoblot of spinal cord protein extracts from age-matched (33 weeks) wild-type (WT) and Tg rats with anti–non-imprinted Prader-Willi/Angelman syndrome locus 1 (NIPA1), bone morphogenetic protein type II receptor (BMPRII), and β-actin (loading control) antibodies. NIPA1 was detected as an approximately 34-kDa and an approximately 40-kDa band. (B) Age-matched young (4 weeks) and old (33 weeks) WT and Tg rats (n = 3 in each group) were compared for BMPRII expression. WT BMPRII expression standardized by β-actin was set as 1. BMPRII was significantly increased (∼2-fold) in the spinal cords only from the older Tg rats versus age-matched WT rats (* p < 0.01). No apparent difference in the amount of BMPRII was detected in brains from either young or old Tg rats. (C) Immunoblot of nuclear fraction protein extracts of primary cortical nerve culture derived from WT and Tg rats with anti–phosphorylated Smad (p-Smad) and –β-actin antibodies, in response to either bone morphogenetic protein 9 (BMP9) (BMP[+]) or vehicle. Time from the application of either BMP9 or vehicle is shown (in minutes). Increased p-Smad was seen only in the extracts of nerve cells exposed to BMP9, whereas no detectable response was seen in vehicle controls. (D) Time course of p-Smad activation in response to BMP9 onto primary rat cortical neuron culture. Response of p-Smad at 120-minute exposure to BMP9 ligand in WT nerve culture, standardized by β-actin was set as 1. No apparent difference in the response of p-Smad to BMP ligand was seen between WT and Tg rat primary cortical nerve culture (data set was triplicated; p = 0.42).
In the next set of experiments, we demonstrated that the NIPA1 transgene is specifically expressed in brain neurons via in situ mRNA hybridization analysis (Supplemental Digital Content 2, http://links.lww.com/NEN/A511) and immunohistochemistry (Supplemental Digital Content 3, http://links.lww.com/NEN/A512). In wild-type neurons, a uniformly low level of NIPA1 signal was observed (Supplemental Digital Content 3A-C, http://links.lww.com/NEN/A512). In contrast, there was intense immunoreactivity of NIPA1 in numerous neurons in Thy1.2-hNIPA1G106R Tg rat brains (Supplemental Digital Content 3D-F, http://links.lww.com/NEN/A512).
Behavioral Characterization of Thy1.2-hNIPA1G106R Tg Rats
Thy1.2-hNIPA1G106R Tg rats show age-dependent progressive gait deficits characterized by poorly coordinated hindlimb movements, trunk instability, and difficulty in initiating locomotion (Supplemental Digital Content 4, http://links.lww.com/NEN/A513). The earliest abnormality is detected by the air-lift reflex in hindlimbs, characterized by the pronounced adduction, often causing the hindlimbs to cross (26), whereas wild-type rats held their hindlimbs apart with paw external rotation (Supplemental Digital Content 5A-C, http://links.lww.com/NEN/A514). Typically, the paws rotated internally, inverted, and clasped each other; these abnormal limb postures are similar to those described by Lalonde and Strazielle (27). The onset of this abnormal hindlimb posturing was detected as early as 4 weeks of age, with an average onset of 7 weeks, and regular stepping was lost at approximately 6 to 8 months of age. Severe muscle atrophy was evident in hindlimb muscles at that age (data not shown).
The progressive gait difficulty and hindlimb weakness were quantified using horizontal ladder walking (23) and inclined grid climbing (24). Thy1.2-hNIPA1G106R Tg rats showed an age-dependent decline in locomotion and progressive hindlimb weakness compared with age-matched wild-type littermates (Supplemental Digital Content 5D-E, http://links.lww.com/NEN/A514). Transgenic rats showed significantly slower weight gain (Supplemental Digital Content 5F, http://links.lww.com/NEN/A514), although they did not develop weight loss until regular stepping was lost. Supplemental Digital Content 6, http://links.lww.com/NEN/A515, shows horizontal ladder walking; Supplemental Digital Content 7, http://links.lww.com/NEN/A516, shows inclined grid climbing.
Central Neurodegeneration in Thy1.2-hNIPA1G106R Tg Rats
By 6 weeks of age, there was axonal and dendritic vacuolar degeneration within the gray matter of anterior horns from sacral and lumbar spinal cord segments; neuronal cell body involvement in the spinal cord and dorsal root ganglia became evident at that time point (Supplemental Digital Content 8, http://links.lww.com/NEN/A517). Some neurons showed peripherally localized multiple homogeneous areas that subsequently appeared to evolve into multiple vacuoles. At the ultrastructural level, branched-tubular profiles appeared dispersed within dendritic or axonal extensions, suggesting fragmentation of smooth endoplasmic reticulum (Supplemental Digital Content 9, http://links.lww.com/NEN/A518).
At 12 weeks of age, many neurites contained large aggregates of osmiophilic material, some of which appeared partially digested within large autophagic vacuoles (Fig. 2A). Higher magnification revealed densely packed TV membranous organelles ranging from 40 to 110 nm in diameter (short tubular axis), with most falling into the 40- to 80-nm range. These aggregates were surrounded by double delimiting membranes (Fig. 2B); neuronal shrinkage and cell body death with vacuolar degeneration were common (Supplemental Digital Content 10, http://links.lww.com/NEN/A519). Based on the ultrastructural features of TV membranous structures, we hypothesized that they represent early endosomes. Figure 3 demonstrates that early endosome marker EEA1 and the endosomal membrane protein NIPA1 are colocalized with these aggregates of TV organelles within the neurites and neuronal cell bodies, strongly suggestive of their endosomal identity.
FIGURE 2.
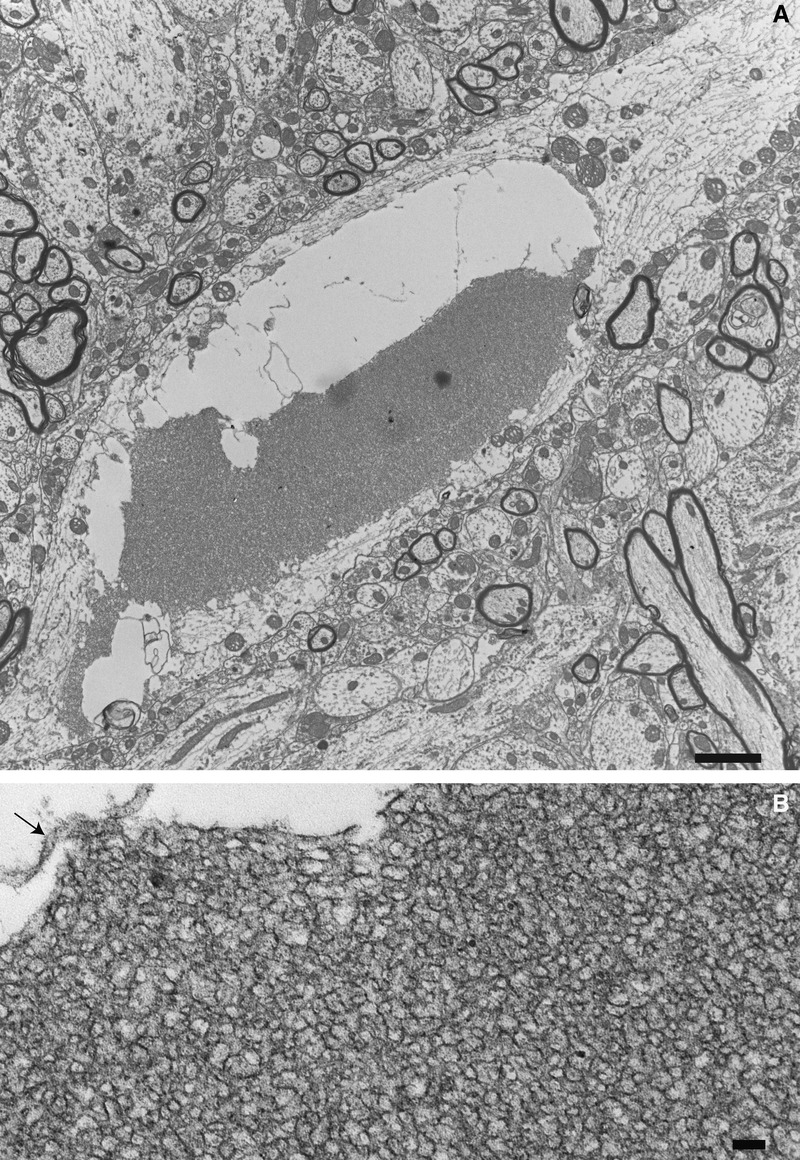
Thy1.2-hNIPA1G106R rat morphology. (A) A neurite within the anterior horn of the lumbar cord of a 12-week-old Thy1.2-hNIPA1G106R rat showing a partially digested large aggregate of a tubulovesicular structure within an autophagic vacuole. Scale bar = 2 μm. (B) Higher magnification shows the elongated tubular morphology of these aggregates. Arrow indicates the remnants of the surrounding double membrane. Scale bar = 100 nm.
FIGURE 3.
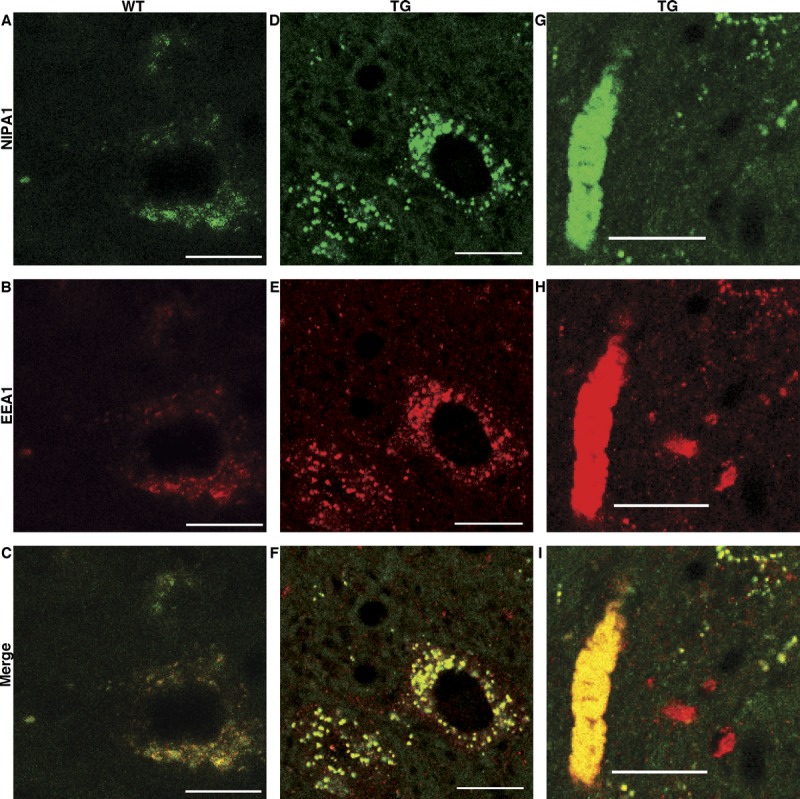
(A–I) Panel of confocal images showing colocalization of non-imprinted Prader-Willi/Angelman syndrome locus 1 (NIPA1) (green; A, D, G) with an endosomal marker EEA1 (red; B, E, H) signals in the spinal cord from wild-type (WT) and Thy1.2-hNIPAG106R transgenic (Tg) rats at 31 weeks of age. Compared with a WT motor neuron (C), NIPA1-EEA1 colocalization shows a coarse granular pattern around the nucleus of a motor neuron (F) and in a large axonal aggregate of a tubulovesicular structure (I) in the Tg rat. Scale bar = 20 μm.
Vacuolar degeneration in occasional axons of descending corticospinal tracts at the sacral and lumbar levels and the ascending dorsal column and spinocerebellar tracts at the cervical levels were detected with increasing age; they became more marked at 40 weeks of age (not shown). The most prominent axonal degeneration was in the external regions of the anterior and lateral funiculus, and there was also axonal degeneration in the medial aspects of the posterior column. In paraformaldehyde- or formalin-fixed, paraffin-embedded sections from 12- to 30-week-old animals, neuronal dropout in the ventral horns was evident compared with those from age-matched controls (Fig. 4A, B). The brainstem showed the most prominent changes with focal areas of neuropil loosening and vacuolization and homogeneous-appearing eosinophilic spheroid bodies (Fig. 4C, D), which likely correspond to large aggregates of TV organelles.
FIGURE 4.
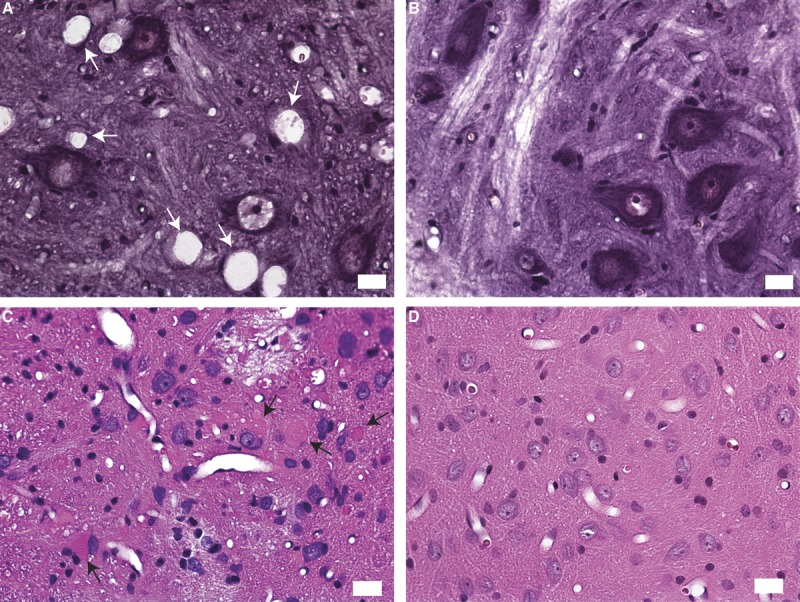
Thy1.2-hNIPA1G106R transgenic (Tg) histopathology. (A, B) Paraformaldehyde-fixed, hematoxylin and eosin–stained frozen sections from the lumbar cord showing vacuolization and neuronal dropout at 12 weeks of age from a Tg rat (A) compared with those from an age-matched control (B). (C) A paraffin-embedded, hematoxylin and eosin–stained section of the nucleus gracilis region of the brainstem from a 30-week-old Tg rat showing homogeneous-appearing eosinophilic spheroid bodies (arrows) and vacuolar changes. (D) An age-matched control. Scale bar = 20 μm.
Peripheral Neurodegeneration in Thy1.2-hNIPA1G106R Tg Rats
At 4 weeks of age, before a detectable clinical phenotype, light microscopic examination of the intrinsic foot muscles revealed a few fibers undergoing Wallerian-like degeneration within the intramuscular nerve bundles. At the ultrastructural level, the hallmark pathologic finding, accumulations of TV organelles was present at the presynaptic terminals of some neuromuscular junctions. Dense accumulations of TV organelles were more common in older animals (Supplemental Digital Content 11A, http://links.lww.com/NEN/A520). Partially denervated neuromuscular junctions were encountered at 4 weeks, and the denervation became more prominent with age (Supplemental Digital Content 11B, http://links.lww.com/NEN/A520). In addition, rare axons of intramuscular nerves had clusters of TV structures sequestered by double membranes that presumably were formed by invaginating Schwann cell–axon membranous networks (Supplemental Digital Content 11C, http://links.lww.com/NEN/A520), as previously described (28).
At the light microscopic level, sciatic nerves showed no abnormalities until 6 weeks of age when large intra-axonal osmiophilic spheroid bodies were recognized in cross and longitudinal sections. These predegenerative changes were more common at 12 weeks of age in the sciatic and tibial nerves and were less frequent in the proximal ventral and dorsal roots (Supplemental Digital Content 12, http://links.lww.com/NEN/A520). At the ultrastructural level, densely packed TV structures encircled by double membranes appeared compressed, and the vast majority showed a uniform diameter of approximately 30 to 40 nm (Fig. 5). These aggregates varied in size and, in some axons, were multiple (Fig. 5A); whereas in others, they occupied almost the entire cross-sectional area as a single aggregate (Fig. 5B).
FIGURE 5.
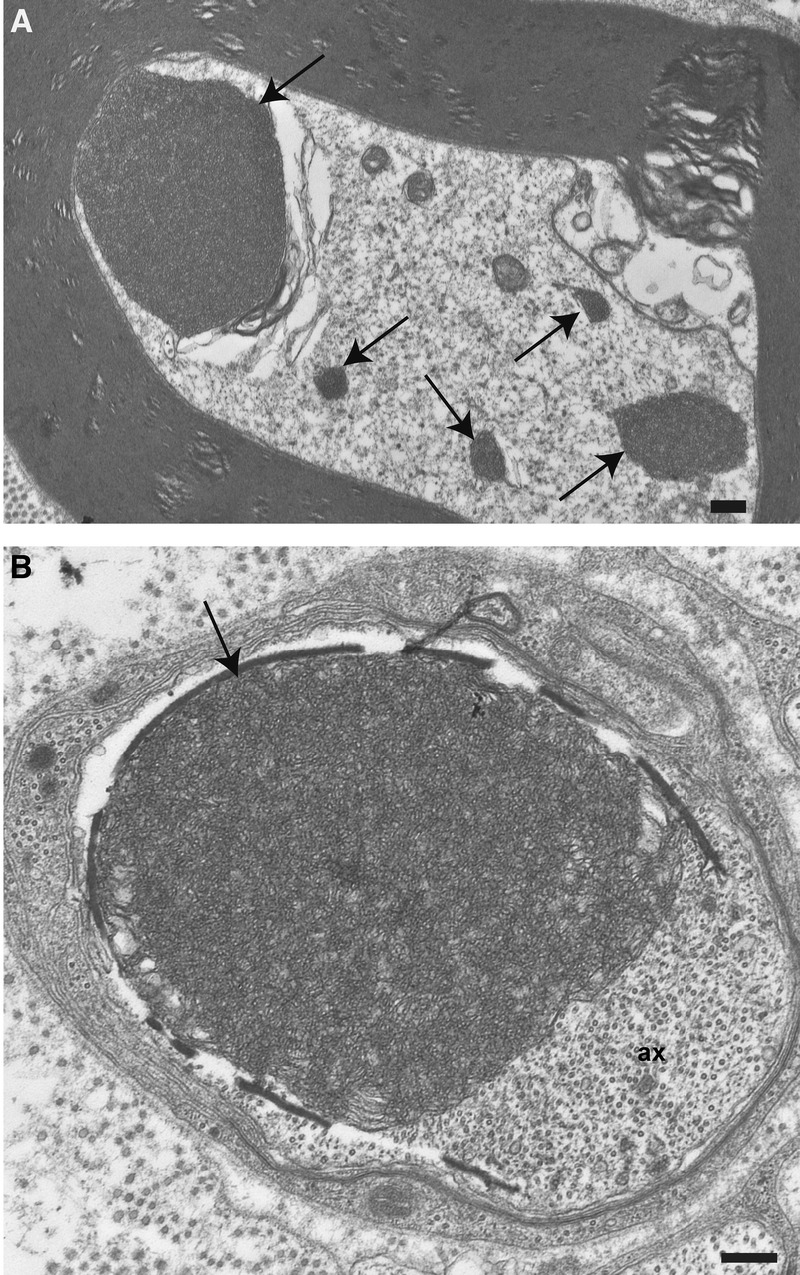
Electron microscopy of Thy1.2-hNIPA1G106R rat peripheral nerve. (A) Cross sections of axons from sciatic nerve at 12 weeks of age showing multiple aggregates of tubulovesicular profiles (arrows). (B) A single large aggregate (arrow) displaying axonal contents (ax). Scale bar = 500 nm.
Multifocal vacuolar degeneration along the axons occurred at subsequent ages. By light microscopy, scattered axons from the sciatic and tibial nerves showed large vacuoles, displacing the axonal contents to the periphery before myelin collapse. Granulovacuolar disintegration of the axonal contents was present at the ultrastructural level (not shown). Acute Wallerian degenerative changes and myelin ovoids as the final stage were also noted in numerous fibers. Myelinated and unmyelinated fiber loss with a nonrandom preservation of some fibers increased with age in both proximal and distal sciatic nerves (Fig. 6A–D). Thinly myelinated small regenerating fibers in clusters were also common.
FIGURE 6.
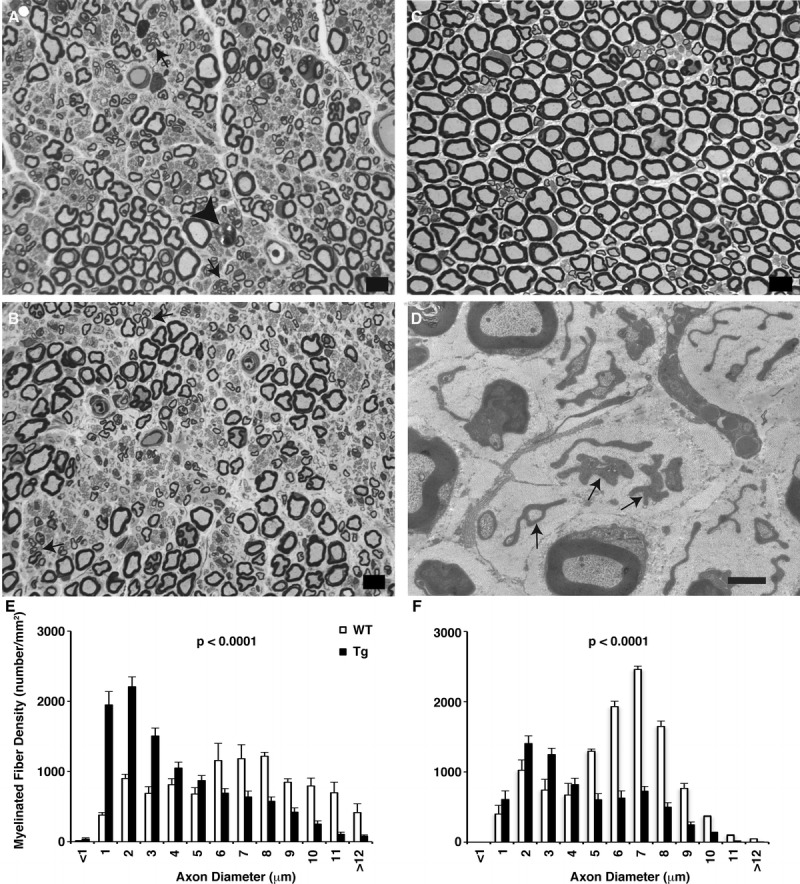
Thy1.2-hNIPA1G106R transgenic (Tg) rat peripheral nerve. (A) One-micrometer-thick toluidine blue–stained plastic sections from the proximal sciatic nerve of a Tg rat showing large myelinated fiber loss. Arrows point to clusters of thinly myelinated regenerating axonal sprouts; a fiber undergoing Wallerian degeneration is marked with an arrowhead. (B) Fiber loss was more prominent distally in the tibial nerve from a Tg rat. (C) Wild-type (WT) proximal sciatic nerve. Scale bar = 10 μm. (D) At the ultrastructural level, there were empty stacks of Schwann cell processes (arrows), some engulfing collagen, because of unmyelinated axon loss in the Tg nerve. Scale bar = 2 μm. (E, F) Myelinated fiber size distribution histograms of sciatic (E) and tibial (F) nerves from WT and Tg rats at 40 weeks of age. The composite histograms were derived from 3 rats in each group. Loss of large myelinated fibers (i.e. myelinated fibers with axon diameter >5 μm) in the Tg was more prominent distally in the tibial branch (F) compared with the proximal sciatic nerve (E). The prominent increase in the small myelinated fiber population seen at both levels reflects ongoing regeneration.
Comparisons of the midsciatic and distal tibial nerves showed distal accumulation of multifocal vacuolar degeneration along the axons, giving rise to a length-dependent distal nonterminal axonopathy. Composite histograms of myelinated axon size distribution in the sciatic and tibial nerves from Thy1.2-hNIPA1G106R Tg rats and age-matched controls at 38 weeks of age demonstrate that, in Tg rats, there is a significant loss in the large myelinated fiber population (axon diameter, >5 μm) whereas the small-diameter fibers are increased resulting from ongoing axonal regeneration compared with those in controls (Fig. 6E, F). The reduction in large-diameter myelinated fiber densities, both in the sciatic (3,636.49 ± 275.01/mm2 vs 6,990.54 ± 333.39/mm2 in control; p < 0.0001) and the tibial nerves (2,859.77 ± 331.93/mm2 vs 8,602.83 ± 243.19/mm2 in control; p < 0.0001), was highly significant. Moreover, the extent of large myelinated fiber loss was more prominent in the (distal) tibial (67%) versus (proximal) sciatic (48%) nerves, thereby providing quantitative evidence for the length-dependent feature of the disease process.
Electrophysiologic Studies
Electrophysiologic studies were carried out at monthly intervals between 8 and 24 weeks of age. Thy1.2-hNIPA1G106R Tg rats showed reduced sensory and motor amplitudes of sciatic motor and tail sensory nerves and slowing of conduction velocities of sciatic, tail sensory, and tail motor nerves (Supplemental Digital Content 13, http://links.lww.com/NEN/A522), confirming peripheral sensory and motor neuropathy.
BMPRII Expression
The proposed mechanism by which NIPA1 inhibits BMP signaling involves downregulation of BMP receptors by promoting endocytosis and lysosomal degradation. Disease-associated mutant versions of NIPA1 alter the trafficking of BMPRII and are less efficient at promoting BMPRII degradation than wild-type NIPA1. To assess whether NIPA1G106R mutant protein has an effect on BMPRII, we carried out Western blot analysis, comparing spinal cord and brains from mutant and wild-type rats at different ages. There was an almost 2-fold increase (p < 0.01) in the amount of BMPRII protein in the spinal cords from older Tg rats; there was, however, no apparent difference in the brains compared with those from wild-type rats (Fig. 7A, B). Neither the spinal cord nor the brain from 4-week-old young Tg rats showed increased BMPRII compared with those from wild-type rats.
We were unable to detect the baseline phosphorylated Smad (p-Smad) from tissue extracts (brain and spinal cord) by Western blot analysis (data not shown). Therefore, we analyzed p-Smad in response to BMP stimulation using wild-type and Tg-derived primary cortical neuronal cultures. In response to BMP application, a BMP-specific p-Smad response was present in both wild-type and Tg preparations, with no apparent difference in the quantity of p-Smad (p = 0.42) (Fig. 7C, D).
DISCUSSION
We report a novel rodent model for SPG6, Thy1.2-hNIPA1G106R Tg rat, which has a distinct histopathologic phenotype leading to a length-dependent central-peripheral distal axonopathy. The morphologic hallmark of the model is the accumulation of endosomal/tubulovesicular organelles at axonal and dendritic terminals followed by vacuolar degeneration. Subsequently, there is a rapidly evolving process of multifocal intraneuronal accumulation of these structures within peripheral and CNS axons and dendrites. The aggregates are sequestered by a double limiting membrane, as seen in autophagy. As the disease progresses, multifocal vacuolar degeneration starts in distal axons and dendritic extensions and extends pari passu in a centripetal direction with concomitant cell body involvement.
The neuropathologic characteristics of Thy1.2-hNIPA1G106R Tg rats have general implications for other diseases and models characterized by endosomal changes, including neuroaxonal or neuritic dystrophies. Among these are the infantile neuroaxonal dystrophy mouse models, cytosolic Ca2+-independent phospholipase A2–deficient (iPLA2β-/-) mice (29–31) and iPLA2β–mutant mice (32). Similar to iPLA2β-/- mice, Thy1.2-hNIPA1G106R rats develop spheroids (seen as osmiophilic large bodies in 1-μm plastic sections), which appear as large accumulations of densely packed TV membranous structures. However, the types of organelle accumulation in iPLA2β-/- mice are heterogeneous, composed of TV structures, similar to what we observed in our model, as well as branched tubular structures having a smaller diameter than TV structures, degenerating abnormal mitochondria, amorphous matrix, and sheets of membranes (30–32). The branched configuration of the structures seen in iPLA2β-/- mice is suggestive of smooth endoplasmic reticulum origin and was previously observed in several experimental neuropathologic processes, for example, vitamin E deficiency (33), central-peripheral distal terminal axonopathies induced by zinc pyridinethione (34) and 2,4-dithiobiuret (35), and in patients with infantile neuroaxonal degeneration (36). The TV organelles and autophagic changes in our model are also reminiscent of changes described in axonal and dendritic nerve terminal swellings in aging and diabetic mouse sympathetic ganglia (37). A distinct feature of our model is that the organelle accumulation/spheroids uniformly appear to be composed of TV structures with elongated tubular shape morphology and they are positive for the early endosomal marker EEA1 and NIPA1. Moreover, in our model, once the TV organelles accumulate, they are rapidly degraded, leaving behind large empty vacuoles; such a feature is not pronounced in either the Tg or the toxic neuropathy models of infantile neuroaxonal degeneration.
We do not know the exact mechanisms of how the deficiency of iPLA2β or the presence of mutant NIPA1 protein leads to the accumulation of membranous structures in neurons. Thus far, no known functional relationship between NIPA1 and iPLA2β has been reported. The iPLAβ is localized to mitochondria, neurites, and axon terminals (38, 39), and it has been proposed that deficiency of iPLA2β can lead to insufficient remodeling and degeneration of mitochondrial inner membranes and presynaptic membranes (31). Non-imprinted Prader-Willi/Angelman syndrome locus 1 protein is reported to be localized to early endosomes, cell surface, and Golgi apparatus but not mitochondria (18, 19). The iPLA2β is known to hydrolyze the sn-2 ester bound in phospholipids, including glycerophospholipids such as phosphatidylcholine and phosphatidylethanolamine (40), and to have a wide range of biologic functions including remodeling of membrane phospholipids (41). It is interesting to note that zinc pyridinethione–induced peripheral neuropathy, previously reported to be a model for infantile neuroaxonal dystrophy (34), has the ability to form stable interactions with membrane phospholipid phosphatidylethanolamine (42), therefore interfering with the normal membrane turnover/turnaround at the nerve terminals (43). The uniform feature of the organelle accumulation in our model suggests a more restricted cellular function for NIPA1, specifically downregulating BMPRII compared with iPLA2β.
The relationship between dysregulated BMP signaling and other HSP molecules was confirmed previously in in vitro (15) and in vivo studies using a zebrafish atlastin-1 knockdown model (14). A similar interaction was shown with NIPA1 protein interacting with BMPRII to facilitate receptor-mediated endocytosis and lysosomal degradation of BMPRII (i.e. downregulation of BMP signaling) (16). Based on studies using Drosophila, it was proposed that downregulation of BMPRII is an important function of NIPA1 that prevents the synaptic overgrowth at the neuromuscular junction (17). As we predicted in our Tg model, NIPA1G106R mutant protein has an effect on BMPRII expression levels, resulting in an almost 2-fold increase in the amount of BMPRII in the spinal cord in older Tg rats, consistent with previous in vitro studies (16). However, we did not find a difference in the amount of BMPRII from either brain or spinal cord from young Tg rats. This may be related to the tissue burden of the pathologic changes, which is higher in the older than younger rats and more pronounced in the spinal cord than in the brain tissue. In our studies, the acute response to BMP9, measured by p-Smad, was not different between wild-type and Thy1.2-hNIPA1G106R Tg primary nerve cultures, as previously reported (16). The age- and tissue-specific dysregulation of BMPRII in Thy1.2-hNIPA1G106RTg rats may reflect the cumulative pathologic process; in addition, the analysis of downstream effects might be limited for detection using our assay, which relies on acute response to BMP. Further studies are needed to clarify the underlying molecular pathways involving BMPRII and NIPA1.
We hypothesize that accumulation of TV structures in Thy1.2-hNIPA1G106R Tg rats is caused by impaired trafficking of endosomes. Based on this hypothesis, the evolution of the neurodegeneration in Thy1.2-hNIPA1G106R rats is illustrated in Figure 8. The NIPA1G106R mutation leads to the accumulation of TV structures at the axonal and dendritic nerve endings, as well as in the neuronal cell bodies. This accumulation is likely caused by impaired transport and/or impaired recycling of endosomes. The adaxonal membranes originating from oligodendrocytes and Schwann cells will invaginate into axons, encircling and then sequestering these TV structures. Glial cells are known to actively participate in the removal of unwanted/accumulated or damaged organelles from axons for selective phagocytosis, a process suggestive of an early axonal disease, as previously proposed (28). However, this compensatory mechanism eventually fails to remove the burden of accumulated TV structures. Tubulovesicular structures that failed to be removed (and are unable to be transported) likely undergo an autodigestive process that results in the formation of vacuoles. Axons and dendrites distal to the location of vacuoles are degenerated, giving way to the formation of nascent axon/neurite tips, and the progression of pathology extends pari passu in a centripetal direction with concomitant cell body involvement. Even though we see neuronal cell body involvement in the spinal cord, the disease process clearly has the predominant feature of a length-dependent distal axonal disease. We also note that, in our model, axonal and dendritic sprouting/regeneration remains active, based on the presence of a prominent increase in the small myelinated fiber population in the peripheral nerves and marked neuritic proliferation in the spinal cord gray matter.
FIGURE 8.
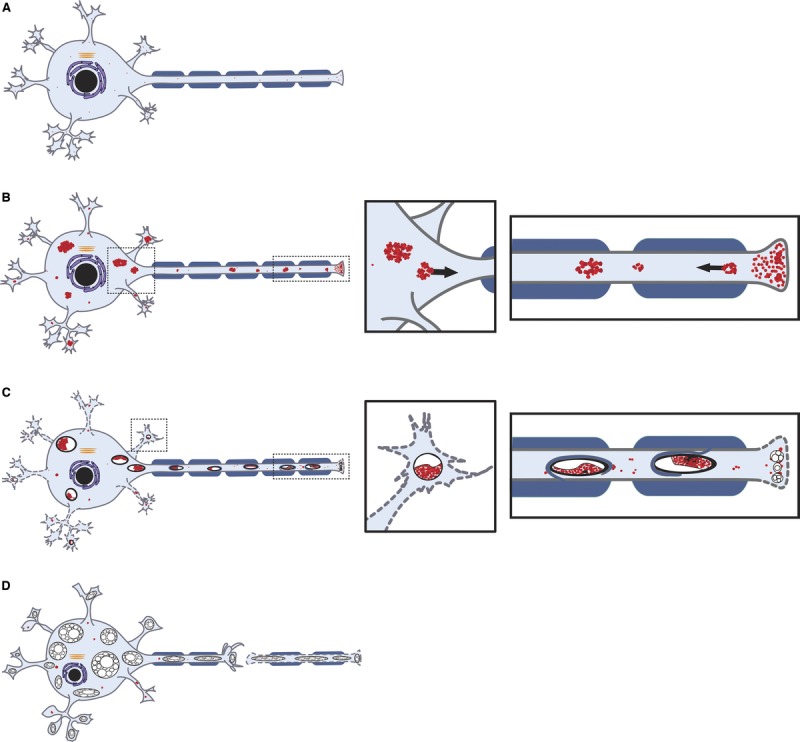
Schematic illustration depicts the evolution of neurodegeneration in autosomal dominant hereditary spastic paraplegia (SPG6-HSP). (A) Normal spinal motor neuron. At the early stages of the disease process, the tubulovesicular membranous structures with endosomal identity accumulated at the axonal and dendritic nerve endings, along the distal axons and in the neuronal cell bodies. This accumulation was likely caused by an impaired transport and/or impaired recycling of endosomes. (B–D) A rapidly evolving multifocal intraneuronal accumulation of these aggregates sequestered by a double limiting membrane for autophagy was observed within axons (B) and dendrites (C). Multifocal vacuolar degeneration then occurred in distal axons and dendritic extensions extending pari passu in a centripetal direction with concomitant cell body involvement (C, D).
Prader-Willi/Angelman syndrome patients with a NIPA1 deletion (i.e. lacking a copy of the NIPA1 gene) seldom develop clinical HSP, and all the reported SPG6 mutations are so far single amino acid substitutions, suggesting that SPG6-HSP is less likely to be caused by haploinsufficiency but more likely to be a “toxic gain of function.” In agreement, in vitro NIPA1 protein studies cause retention of wild-type NIPA1 in the endoplasmic reticulum, resulting in less efficient degradation of BMPRII (16). Our results are also consistent with the assumption that NIPA1G106R mutation serves as a “toxic gain of function,” instead of haploinsufficiency. Although we detected abundant NIPA1 immunoreactivity in neurons and in aggregates of TV structures colocalizing with EEA1, we did not detect differences in the expression of endogenous rat NIPA1 mRNA or total amount of NIPA1 protein between wild-type and Tg rats. This might be related to the disease process leading to multifocal vacuolar degeneration, suggesting that the aggregates of TV structures/endosomes are rapidly degraded, leaving large vacuoles behind. This unique histopathologic feature might explain why we could not see increased mutant NIPA1 protein levels at the detection level of Western blot analysis.
We believe that our Thy1.2-hNIPA1G106R Tg rats can serve as a valuable tool for developing therapeutic trials for HSPs because of their early, simultaneous-onset, and robust behavioral and histologic phenotype. Further research is needed to reveal how mutant NIPA1 protein triggers impaired endosomal trafficking, axonal degeneration, and its interaction with other HSP-related molecules. Furthermore, understanding of the molecular cascade disturbed in HSP gene mutations will provide further insight into the mechanisms involving endosomal trafficking and axonal degeneration that are also seen in motor neuron disease and in some acquired toxic/metabolic and hereditary peripheral neuropathies.
ACKNOWLEDGMENTS
We thank the Spastic Paraplegia Foundation for financial support (to Odelia Ghodsizadeh and Yaz Kisanuki). We thank Dr. Pico Caroni for Thy1.2-promoter vector and Ms. Anna Hughes and Mr. Minhaj Uddin from Nationwide Children’s Hospital, Pathology, for their expert technical assistance in ultrastructural studies.
Footnotes
Send correspondence and reprint requests to: Yaz Y. Kisanuki, MD, Department of Neurology, Division of Neurogenetics, The Ohio State University, 682 Biomedical Research Tower, 460 W. 12th Ave, Columbus, OH 43210; E-mail: yasushi.kisanuki@osumc.edu and Zarife Sahenk, MD, PhD, Department of Neurology and Pediatrics, The Ohio State University, The Research Institute at Nationwide Children’s Hospital, 700 Children’s Drive, Columbus, Ohio 43205; E-mail: zarife.sahenk@nationwidechildrens.org
This work was supported by the Spastic Paraplegia Foundation (Odelia Ghodsizadeh and Yaz Kisanuki).
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.jneuropath.com).
REFERENCES
- 1. Harding AE. Hereditary spastic paraplegias. Semin Neurol 1993; 13: 333– 36 [DOI] [PubMed] [Google Scholar]
- 2. Salinas S, Proukakis C, Crosby A, et al. Hereditary spastic paraplegia: Clinical features and pathogenetic mechanisms. Lancet Neurol 2008; 7: 1127– 38 [DOI] [PubMed] [Google Scholar]
- 3. Deluca GC, Ebers GC, Esiri MM. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol 2004; 30: 576– 84 [DOI] [PubMed] [Google Scholar]
- 4. Kumar KR, Sue CM, Burke D, et al. Peripheral neuropathy in hereditary spastic paraplegia due to spastin (SPG4) mutation—A neurophysiological study using excitability techniques. Clin Neurophysiol 2012; 123: 1454– 59 [DOI] [PubMed] [Google Scholar]
- 5. Schaumburg HH, Wisniewski HM, Spencer PS. Ultrastructural studies of the dying-back process: 1. Peripheral nerve terminal and axon degeneration is systemic acrylamide intoxication. J Neuropathol Exp Neurol 1974; 33: 260. [DOI] [PubMed] [Google Scholar]
- 6. Spencer PS, Schaumburg HH. Ultrastructural studies of the dying-back process. J Neuropathol Exp Neurol 1977; 36: 300. [DOI] [PubMed] [Google Scholar]
- 7. Reid E, Kloos M, Ashley-Koch A, et al. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am J Hum Genet. 2002; 71: 1189– 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hazan J, Fonknechten N, Mavel D, et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet 1999; 23: 296– 303 [DOI] [PubMed] [Google Scholar]
- 9. Casari G, De Fusco M, Ciarmatori S, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 1998; 93: 973– 83 [DOI] [PubMed] [Google Scholar]
- 10. Hansen JJ, Durr A, Cournu-Rebeix I, et al. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am J Hum Genet 2002; 70: 1328– 32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ochs S. Local supply of energy to the fast axoplasmic transport mechanism. Proc Natl Acad Sci USA 1971; 68: 1279– 82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cai Q, Davis ML, Sheng Z-H. Regulation of axonal mitochondrial transport and its impact on synaptic transmission. Neurosci Res 2011; 70: 9– 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blackstone C, O’kane CJ, Reid E. Hereditary spastic paraplegias: Membrane traffic and the motor pathway. Nat Rev Neurosci 2011; 12: 31– 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fassier C, Hutt JA, Scholpp S, et al. Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat Neurosci 2010; 13: 1380– 87 [DOI] [PubMed] [Google Scholar]
- 15. Zhao J, Hedera P. Hereditary spastic paraplegia-causing mutations in atlastin-1 interfere with BMPRII trafficking. Mol Cell Neurosci 2013; 52: 87– 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsang HTH, Edwards TL, Wang X, et al. The hereditary spastic paraplegia proteins NIPA1, spastin and spartin are inhibitors of mammalian BMP signaling. Hum Mol Genet 2009; 18: 3805– 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X, Shaw WR, Tsang HTH, et al. Drosophila spichthyin inhibits BMP signaling and regulates synaptic growth and axonal microtubules. Nat Neurosci 2007; 10: 177– 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goytain A, Hines RM, El-Husseini A, et al. NIPA1(SPG6), the basis for autosomal dominant form of hereditary spastic paraplegia, encodes a functional Mg2+ transporter. J Biol Chem 2007; 282: 8060– 68 [DOI] [PubMed] [Google Scholar]
- 19. Zhao J, Matthies DS, Botzolakis EJ, et al. Hereditary spastic paraplegia-associated mutations in the NIPA1 gene and its Caenorhabditis elegans homolog trigger neural degeneration in vitro and in vivo through a gain-of-function mechanism. J Neurosci 2008; 28: 13938– 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Caroni P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods 1997; 71: 3– 9 [DOI] [PubMed] [Google Scholar]
- 21. Caughlan A, Newhouse K, Namgung U, et al. Chlorpyrifos induces apoptosis in rat cortical neurons that is regulated by a balance between p38 and ERK/JNK MAP kinases. Toxicol Sci 2004; 78: 125– 34 [DOI] [PubMed] [Google Scholar]
- 22. Lopez-Coviella I, Mellott TM, Kovacheva VP, et al. Developmental pattern of expression of BMP receptors and Smads and activation of Smad1 and Smad5 by BMP9 in mouse basal forebrain. Brain Res 2006; 1088: 49– 56 [DOI] [PubMed] [Google Scholar]
- 23. Soblosky JS, Colgin LL, Chorney-Lane D, et al. Ladder beam and camera video recording system for evaluating forelimb and hindlimb deficits after sensorimotor cortex injury in rats. J Neurosci Methods 1997; 78: 75– 83 [DOI] [PubMed] [Google Scholar]
- 24. Ramón-Cueto A, Cordero MI, Santos-Benito FF, et al. Functional recovery of paraplegic rats and motor axon regeneration in their spinal cords by olfactory ensheathing glia. Neuron 2000; 25: 425– 35 [DOI] [PubMed] [Google Scholar]
- 25. Rainier S, Chai J-H, Tokarz D, et al. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6). Am J Hum Genet 2003; 73: 967– 71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Whishaw IQ, Schallert T, Kolb B. An analysis of feeding and sensorimotor abilities of rats after decortication. J Comp Physiol Psychol 1981; 95: 85– 103 [DOI] [PubMed] [Google Scholar]
- 27. Lalonde R, Strazielle C. Brain regions and genes affecting limb-clasping responses. Brain Res Rev 2011; 67: 252– 59 [DOI] [PubMed] [Google Scholar]
- 28. Spencer PS, Thomas PK. Ultrastructural studies of the dying-back process. II. The sequestration and removal by Schwann cells and oligodendrocytes of organelles from normal and diseases axons. J Neurocytol 1974; 3: 763– 83 [DOI] [PubMed] [Google Scholar]
- 29. Shinzawa K, Sumi H, Ikawa M, et al. Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: A model of human neurodegenerative disease. J Neurosci 2008; 28: 2212– 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Malik I, Turk J, Mancuso DJ, et al. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am J Pathol 2008; 172: 406– 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beck G, Sugiura Y, Shinzawa K, et al. Neuroaxonal dystrophy in calcium-independent phospholipase A2β deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J Neurosci 2011; 31: 11411– 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wada H, Yasuda T, Miura I, et al. Establishment of an improved mouse model for infantile neuroaxonal dystrophy that shows early disease onset and bears a point mutation in Pla2g6. Am J Pathol 2009; 175: 2257– 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Southam E, Thomas PK, King RH, et al. Experimental vitamin E deficiency in rats. Morphological and functional evidence of abnormal axonal transport secondary to free radical damage. Brain 1991; 114: 915– 36 [DOI] [PubMed] [Google Scholar]
- 34. Sahenk Z, Mendell JR. Ultrastructural study of zinc pyridinethione–induced peripheral neuropathy. J Neuropathol Exp Neurol 1979; 38: 532– 50 [DOI] [PubMed] [Google Scholar]
- 35. Sahenk Z. Distal terminal axonopathy produced by 2,4-dithiobiuret: Effects of long-term intoxication in rats. Acta Neuropathol 1990; 81: 141–47. [DOI] [PubMed] [Google Scholar]
- 36. Cowen D, Olmstead EV. Infantile neuroaxonal dystrophy. J Neuropathol Exp Neurol 1963; 22: 175– 236 [DOI] [PubMed] [Google Scholar]
- 37. Schmidt RE, Parvin CA, Green KG. Synaptic ultrastructural alterations anticipate the development of neuroaxonal dystrophy in sympathetic ganglia of aged and diabetic mice. J Neuropathol Exp Neurol 2008; 67: 1166– 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ong W-Y, Yeo J-F, Ling S-F, et al. Distribution of calcium-independent phospholipase A2 (iPLA 2) in monkey brain. J Neurocytol 2005; 34: 447– 58 [DOI] [PubMed] [Google Scholar]
- 39. Seleznev K, Zhao C, Zhang XH, et al. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J Biol Chem 2006; 281: 22275– 88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res 2009; 50: S237– 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Strokin M, Sergeeva M, Reiser G. Docosahexaenoic acid and arachidonic acid release in rat brain astrocytes is mediated by two separate isoforms of phospholipase A2 and is differently regulated by cyclic AMP and Ca2+. Br J Pharmacol 2003; 139: 1014– 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guthery E, Seal LA, Anderson EL. Zinc pyrithione in alcohol-based products for skin antisepsis: Persistence of antimicrobial effects. Am J Infect Control 2005; 33: 15– 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sahenk Z, Mendell JR. Axoplasmic transport in zinc pyridinethione neuropathy: Evidence for an abnormality in distal turn-around. Brain Res 1980; 186: 343– 53 [DOI] [PubMed] [Google Scholar]