

Supplemental Digital Content is Available in the Text.
Key Words: HIV-1, CRF01AE, CRF07BC, men who have sex with men, phylogenetic tree analysis, Bayesian molecular clock analysis
Abstract
Objectives:
To investigate the epidemiological relationships between HIV-1 strains that are spread among the men who have sex with men (MSM) populations of 9 cities across China and to analyze the origins and divergence times of the major epidemic strains found in the MSM population.
Methods:
A total of 583 HIV-1-positive subjects were recruited from high-risk MSM populations in 9 cities across China between 2009 and 2011. Nucleotide sequences of 1.0-kb pro-RT regions were amplified and sequenced. Phylogenetic and Bayesian molecular clock analyses were performed.
Results:
The overall distribution of HIV-1 genotypes was as follows: CRF01_AE, 62.1%; CRF07_BC, 18.2%; subtype B (United States–European), 15.9%; subtype B', 0.7%; other recombinants, 3.1%. In addition to the 2 distinct CRF01_AE clusters [cluster 1 (n = 157, 26.9%) and cluster 2 (n = 196, 33.6%)] previously reported by our group, we identified a novel CRF07_BC cluster (cluster 3) (n = 94, 16.1%) unique to China's MSM population whose strains were homologous and could be detected in all 9 cities. These 3 lineages of HIV-1 strains (clusters 1–3) accounted for 76.7% (447 of 583) of infections among MSM in China as a whole. Clusters 1, 2, and 3 were estimated to have been introduced into the MSM population in 1999, 2001, and 2001, respectively, indicating that the newly identified CRF07_BC cluster 3 is not a young lineage. However, it spread quickly in recent years.
Conclusions:
We identified 3 distinct HIV-1 lineages (clusters 1–3) responsible for the recent upsurge of the AIDS epidemic among MSM in China. These 3 HIV-1 variants are spread widely among MSM throughout China, demonstrating remarkable founding effects.
INTRODUCTION
China has recently experienced a critical change in its HIV-1/AIDS epidemic. Historically, HIV/AIDS has predominantly affected high-risk populations, such as intravenous drug users (IDUs) and former blood and plasma donors in rural areas. However, rates of HIV-1 transmission through sexual contact have rapidly increased recently. Particularly concerning is the increasing trend of infection among men who have sex with men (MSM). According to a joint survey conducted by the Chinese Ministry of Health and UNAIDS, MSM transmission accounts for 0.3% of all HIV/AIDS cases between 1985 and 2005, and the proportion of MSM among newly identified HIV cases in China increased to 29.4% in 2011.1 In a recent large-scale national survey conducted in 2008 involving >18,000 MSM from 61 Chinese cities, HIV prevalence among MSM was 4.9% with incidence ranging between 2.6 and 5.4 per 100 person-years.2 Today, China has approximately 10–20 million MSM,3 making them one of the most important target populations for HIV prevention in China.
HIV-1 genotype distribution among MSM in China was first studied in 2005–2006 in Beijing samples (n = 45). Subtype B of US–European origin predominated among MSM (71.1%), followed by the CRF01_AE (24.4%), and CRF07_BC (4.4%) subtypes.4 Follow-up surveys of Beijing MSM revealed that subtype B percentages decreased to 41.9% in 20075 and to 20% in 2009.6 In contrast, non-B subtypes increased rapidly: CRF01_AE increased from 3.7% in 2005 to >50% in 2009; CRF07_BC increased from <5% in 2005% to 17% in 2009.6 In addition, unique recombinant forms consisting of CRF01_AE together with either subtype B or CRF07_BC were detected in 25% of samples in 2007.5 Similar trends can be observed in studies from Liaoning province7; Jiangsu province8; Shijiazhuang, capital of Hebei province9; and Zhengzhou, capital of Henan province.10 However, no nationwide molecular epidemiological survey of MSM has been conducted in China, and the relationship between regional epidemic HIV strains remains unclear.
For this study, we collected HIV pol sequences (n = 583) from newly diagnosed HIV-infected MSM between 2009 and 2011 in 9 Chinese cities. Phylogenetic and Bayesian molecular clock analyses were used to (1) clarify the epidemiological relationship between the HIV-1 strains present among MSM in different regions and (2) analyze the origins and divergence times of the major epidemic strains found in MSM populations. We discovered that 3 distinct lineages of HIV-1 strains are responsible for the recent upsurge of HIV-1 infections in these 9 cities.
MATERIALS AND METHODS
Study Subjects
In 9 Chinese cities, we studied 583 cases that involved HIV infection through homosexual contact between 2009 and 2011. Representing a variety of Chinese regions, these cities were Shenyang/Liaoning, Beijing, Jinan/Shandong, Shanghai, Nanjing/Jiangsu, Chengdu/Sichuan, Changsha/Hunan, Kunming/Yunnan, and Dongguan/Guangdong. The subjects in Shenyang, Beijing,11 and Kunming were enrolled from a prospective clinical cohort study of primary HIV-1–infected individuals (Shang, Wu, et al, unpublished data), recruitment for which was done by the categorical snowball-sampling method among high-risk MSM populations between 2009 and 2011. In addition to primary infection cases, newly diagnosed chronic infection cases in Shenyang and Beijing were also included. The Chengdu subjects were enrolled from newly diagnosed cases at the local Center for Disease Control and Prevention in 2009. All other subjects were recruited from a cross-sectional survey of high-risk MSM groups, and HIV-positive cases screened from 400 MSM in each city were recruited. All consenting individuals who met the following criteria were included in this study: male, older than 18 years of age, having at least 1 male sexual partner within 12 months before the study, and lacking other high-risk behavior. All participants completed a questionnaire administered by trained interviewers. Research personnel collected 10 mL of peripheral blood samples that were anticoagulated with EDTA-3K. Plasma was separated within 6 hours after collection, tested for antibodies and HIV-1 RNA, and frozen at −80°C for further analysis.
Ethics Statement
This study was done according to guidelines in the Helsinki II Declaration. The protocol of the study and the informed consent process were approved by the Institutional Review Board of the First Affiliated Hospital of China Medical University. All participants were volunteers and provided written informed consent for sample collection and subsequent analyses.
Viral RNA Extraction and RT-PCR Amplification and Sequencing
Viral RNA was isolated from 140 μL of plasma using a QIAamp Viral RNA kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The 1.0-kb pol region [entire protease and 256 codons of the reverse transcriptase coding gene, HXB2: 2253–3278 nucleotides (nts)] were reverse-transcribed and amplified with the SuperScript Polymerase One-Step RT-PCR System (Invitrogen, Calsbad, CA) using primers MAW-26 and RT-21, according to previously published methods.12 In the second round of polymerase chain reaction, amplification was performed with GoTaq DNA Polymerase (Promega, Madison, WI) using primers PRO-1 and RT4R. Nested polymerase chain reaction products were purified with the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) and were sequenced directly in both directions using internal walking primers made by Beijing Genomics Institute (China).
Sequence Assembly and Quality Control
The sequences were assembled with Sequencher 4.10 (Genecodes, Ann Arbor, MI) and then aligned with previously submitted sequences from our laboratory and other reference sequences from the Los Alamos database (http://hivweb.lanl.gov) using the CLUSTAL X program (available at: http://www.clustal.org/clustal2/).13 The sequences were then edited manually using Bioedit 7.09 (available at: www.mbio.ncsu.edu/bioedit/bioedit.html). All positions that contained alignment gaps were removed. To exclude experimental contamination, similarities between the pol sequences in this study and the sequence database were analyzed by applying Los Alamos HIV Database Web tools (http://www.hiv.lanl.gov).
Phylogenetic Analyses
Phylogenetic analyses were performed by applying the neighbor-joining method using the Kimura 2-parameter model with a transition-transversion ratio of 2.0. The topology of trees was tested by bootstrap analysis with 1000 replicates. Bootstrap values >70 were considered significant. Phylogenetic and molecular evolutionary analyses were conducted using MEGA 5.0 (available at: http://www.megasoftware.net/).14 Recombinant forms of virus subtypes were first analyzed with the Los Alamos Database RIP online tool (http://hivweb.lanl.gov) and then with SimPlot 3.5.1 (available at: http://sray.med.som.jhmi.edu/SCRoftware/simplot/) to define the recombination structures.
Evolutionary Analyses
Estimates of evolutionary rate and the time of the most recent common ancestor (tMRCA) for the CRF01_AE and CRF07_BC lineages were performed using the Bayesian Markov chain Monte Carlo (MCMC) inference under the relaxed log-normal molecular clock model. Relaxed-clock models have previously proved more reliable in estimating viral phylogenies and divergence dates than “strict clock” and “non-clock” methods.15 GTR nucleotide substitution models—with a gamma-distribution model of rate heterogeneity among sites (with 4 rate categories) and a constant population size model implemented in MrBayes 3.1.216—were carried out in this study. MCMC chains were run 20 million generations and sampled every 1000 steps. Bayesian MCMC output was analyzed using Tracer v1.5, and all parameters were estimated from an effective sample size >200. The trees were consolidated into a target tree using the TreeAnnotator program and scanned using the FigTree program.
Statistical Analyses
All data were analyzed using SPSS version 17.0 software packages. Differences between the genetic distances of any 2 groups were assessed using the Student t test. P values <0.05 were considered statistically significant.
RESULTS
Identification of 3 Distinct Clusters of HIV-1 Strains That Play Major Roles in the HIV-1 Epidemic Among MSM in 9 Chinese Cities
A total of 583 HIV-1 nucleotide sequences of 1.0-kb pol gene (HXB2: 2253–3278 nts) from newly diagnosed MSM between 2009 and 2011 from 9 Chinese cities were determined and genotyped by phylogenetic tree analysis. The overall distribution of Chinese MSM HIV-1 genotypes was as follows (see Table 1): CRF01_AE, 62.1% (362 of 583); CRF07_BC, 17.3% (106 of 583); subtype B (United States–European), 15.9% (93 of 583); subtype B′, 0.7% (4 of 583); other recombinant, 3.1% (18 of 583). The CRF01_AE and CRF07_BC genotypes accounted for 80.3% (468 of 583) of the HIV-1 infections among MSM in the 9 cities.
As shown in Figure 1, phylogenetic tree analyses identified a novel CRF07_BC cluster (designated cluster 3) unique to the MSM population in China, with high bootstrap confidence (86%). Two distinct CRF01_AE clusters (clusters 1 and 2) were previously reported by our group.17 CRF01_AE clusters 1 and 2 and CRF07_BC cluster 3 accounted for 26.9% (157 of 583), 33.6% (196 of 583), and 16.1% (94 of 583) of HIV-1 infections among the MSM subjects, respectively. These 3 lineages of HIV-1 strains accounted for 76.7% (447 of 583) of the MSM infections (Table 1).
FIGURE 1.

Phylogenetic tree analyses identifying 3 distinct HIV-1 phylogenetic clusters unique to MSM in China; neighbor-joining trees of 1.0-kb pol gene (HXB2: 2253–3278 nt), representing the CRF01_AE genotype (A), the CRF07_BC genotype (B), and the subtype B/B′ (C). Individual sequences are indicated by symbols corresponding to their known risk groups: MSM (circle), heterosexual (triangle), IDU (square), former plasma donor (FPD) (inverted triangle), and unknown high-risk behavior (diamond). The geographic origin of each sequence is color-coded (see inset). For panels A–C, we selected as many sequences as possible from regions with smaller sample sizes and only a few phylogenetically representative sequences from large-sample regions such as Shenyang, Beijing, and Chengdu for simplicity. These selected sequences are shown together with reference sequences previously identified in other high-risk populations in China and its neighboring countries (only IDs of reference sequences are shown in trees) and with subtype reference sequences from the Los Alamos HIV sequences database (http://hiv-web.lanl.gov/content/index). The bootstrap values of the 1000 replicates are labeled on the major branches.
TABLE 1.
The Distribution of HIV-1 Genotypes Among Men Who Have Sex With Men in 9 Cities Across China
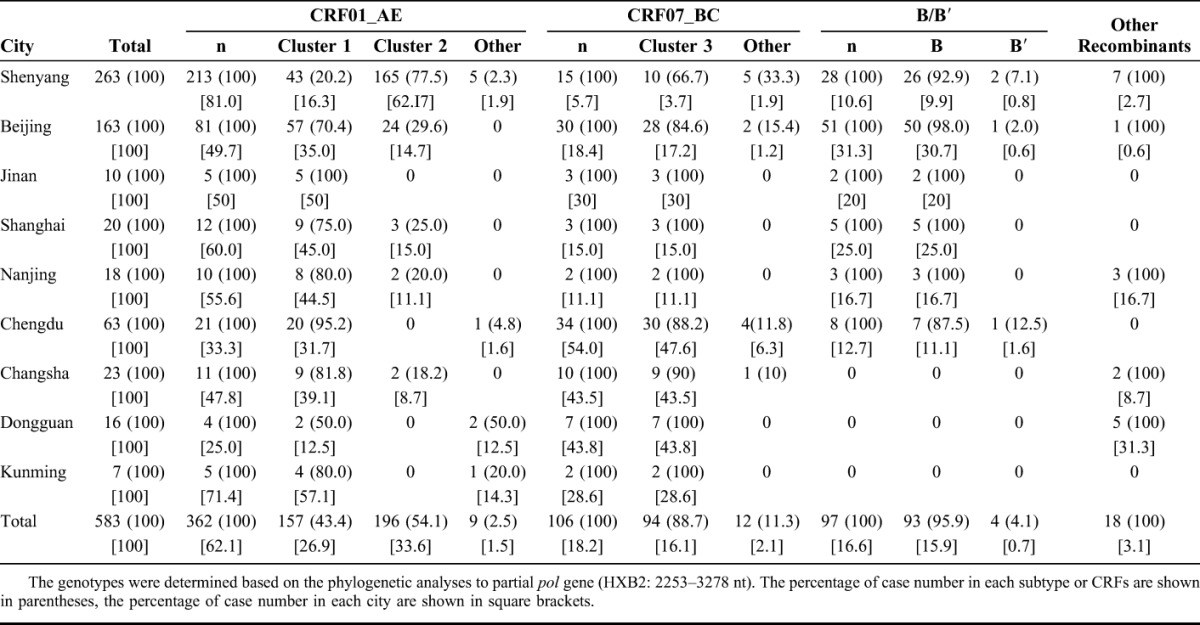
Geographical Distribution of HIV-1 Genotypes Among MSM in 9 Chinese Cities
Figure 2 illustrates the HIV-1 genotype distribution among MSM in 9 Chinese cities (Table 1). The CRF01_AE genotype was most prevalent in 7 of the 9 cities (all except Chengdu and Dongguan): prevalence of CRF01_AE ranged from 25.0% in Dongguan to 81.0% in Shenyang. As shown in Figure 1A, we detected 2 distinct phylogenetic clusters of CRF01_AE (designated China's MSM clusters 1 and 2). Among the MSM subjects, 97.5% (353 of 362) of the circulating CRF01_AE strains belonged to either cluster 1 (157 of 362, 43.4%) or cluster 2 (196 of 362, 54.1%). A small fraction of the CRF01_AE strains (9 of 362, 2.5%) share a similar phylogenetic ancestry with strains found in typical Thailand CRF01_AE groups (Table 1; Fig. 1A). The prevalence of CRF01_AE cluster 1 is higher than that of cluster 2 in most cities, except Shenyang (capital of Liaoning province, northeastern China), where cluster 2 is the predominant lineage among the CRF01_AE strains (20.2% for cluster 1 vs. 77.5% for cluster 2) and among all HIV-1 strains circulating in Shenyang (165 of 263, 62.7%). CRF01_AE cluster 2 strains were detected in Beijing (24 of 163, 14.7%), Shanghai (3 of 20, 15.0%), Nanjing (2 of 18, 11.1%), and Changsha (2 of 23, 8.7%) but not in Jinan (0 of 10), Chengdu (0 of 63), Dongguan (0 of 16), or Kunming (0 of 7) (Table 1).
FIGURE 2.
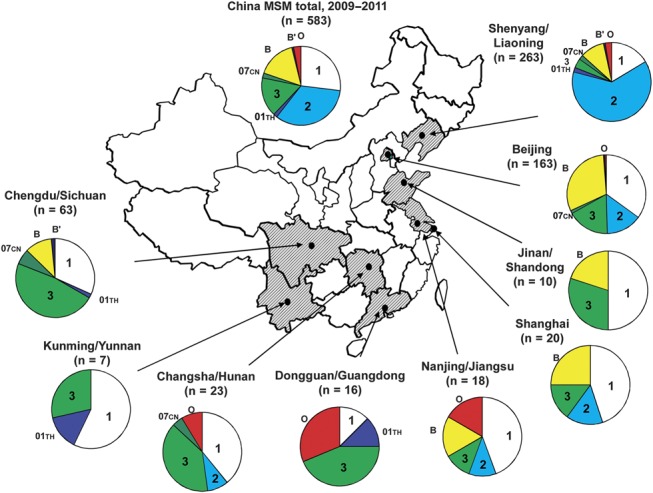
Geographic distribution of HIV-1 genotypes and 3 distinct phylogenetic clusters identified in this study. HIV-1 genotype distribution in their respective study cities are depicted on the map of China below. Symbols on the pie graphs indicate the following (also see inset): 1, CRF01_AE cluster 1; 2, CRF01_AE cluster 2; 01th, Thailand CRF01_AE; 3, CRF07_BC cluster 3; 07cn, typical CRF07_BC; B, subtype B (United States–European); B′, subtype B′ (Thailand variant of subtype B); O, other recombinant.
The HIV-1 CRF07_BC was detected among MSM in the 9 cities. The CRF07_BC cluster 3 accounted for 88.7% (94 of 106) of the CRF07_BC strains among Chinese MSM (Table 1; Fig. 1B). The prevalence of CRF07_BC cluster 3 ranged from 3.7% in Shenyang (10 of 263) to 47.6% in Chengdu (30 of 63). Chengdu and Dongguan were the only 2 cities where CRF07_BC cluster 3 prevalence (49.2% and 43.8%, respectively) was higher than CRF01_AE prevalence (31.7% and 12.5%, respectively) (Table 1).
In contrast, HIV-1 subtype B (United States–European), which is the predominant strain among MSM in Western countries,18–21 played a smaller role in China between 2009 and 2011. However, the presence of subtype B was significant in large Chinese cities: Beijing (30.7%, 50 of 163) and Shanghai (25.0%, 5 of 20) (Table 1). In contrast, subtype B was not detected in southern provincial cities: Changsha (0 of 23), Dongguan (0 of 16), and Kunming (0 of 7) (Table 1; Fig. 1C). Although sample sizes from these regions were small, the absence of subtype B may indicate that the HIV-1 epidemic among these MSM is largely influenced by locally common strains, either CRF01_AE or CRF07_BC. Different from the CRF01_AE and CRF07_BC clusters that are common across different cities, no trans-regional spread of subtype B strains was detected in this study, except for some local transmission clusters, such as those in Shenyang and Beijing (Fig. 1C). HIV-1 subtype B′, an important lineage of the viral strains responsible for the blood-borne HIV-1 epidemic in China, was rarely found (4 of 583, 0.7%) among Chinese MSM (Table 1; Fig. 1C). The geographical distribution of the HIV-1 strains and the distinct phylogenetic clusters (CRF01_AE clusters 1 and 2, and CRF07_BC cluster 3) responsible for the HIV-1 epidemic among Chinese MSM between 2009 and 2011 are schematically illustrated in Figure 2.
The genetic distances within CRF01_AE cluster 1, cluster 2, and the typical Thailand CRF01_AE groups were 0.030 ± 0.008, 0.028 ± 0.007, and 0.056 ± 0.011, respectively. This data agrees with the neighbor-joining phylogenetic tree, which shows that the sequences in CRF01_AE clusters 1 and 2 are more closely linked than those in the typical Thailand CRF01_AE groups. The genetic distance within CRF07_BC cluster 3 (0.012 ± 0.005) is the lowest among the 3 clusters and far lower than the genetic distance of the other CRF07_BC group (0.029 ± 0.006). The sequences in CRF07_BC cluster 3 were most homologous. The genetic distances within CRF01_AE clusters 1 and 2 and typical Thailand CRF01_AE groups are significantly different (P < 0.01), as are the genetic distances within CRF07_BC cluster 3 and the other CRF07_BC group (P < 0.01) (Fig. 3).
FIGURE 3.
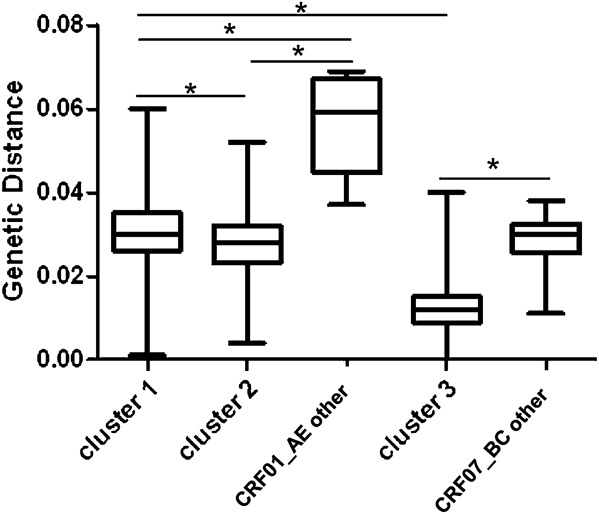
Nucleotide diversity of 3 distinct HIV-1 variants responsible for HIV-1 epidemic among MSM in China. Nucleotide diversity of CRF01_AE clusters 1 and 2 and of CRF07_BC cluster 3 are shown in comparison with other CRF01_AE and CRF07_BC strains circulating among MSM in China. Asterisks (*) indicate a statistically significant difference (P < 0.01) in nucleotide diversity between 2 indicated categories.
Evolutionary Characterization of the 3 Distinct HIV-1 Clusters Identified Among MSM in China
To determine the epidemic origins of the 3 major HIV strains among Chinese MSM, we estimated the tMRCA of the HIV-1 clusters (CRF01_AE clusters 1 and 2 and CRF07_BC cluster 3) by using the relaxed molecular clock model based on the nucleotide sequences of 1.0-kb pol gene. The analyses were carried out using nucleotide sequences of either the CRF01_AE or CRF07_BC strains determined in this study. Respective reference sequences sampled between 1990 and 2010 were downloaded from the Los Alamos HIV sequences database. The estimated evolutionary rates were 2.70 (2.05 – 3.42) ×10–3 and 1.51 (1.00 – 2.12) × 10–3 substitutions per site per year for CRF01_AE and CRF07_BC under the GTR + γ4 relaxed clock models with a constant-size model. The results were essentially similar to those previously estimated for HIV-1 CRF01_AE or CRF07_BC.21 As shown in the maximum clade credibility tree (Fig. 4), the median tMRCAs of clusters 1–3, calculated in a GTR + γ4 relaxed-clock model with a constant size model, were estimated to be 1999.1 (95% CI: 1996.2 to 2002.2), 2001.2 (95% CI: 1998.0 to 2004.3), and 2001.4 (95% CI: 1999.8 to 2004.8), respectively. Evolutionary and statistical assumptions do no significantly affect the estimated dates, and the effective sample size was >200.
FIGURE 4.
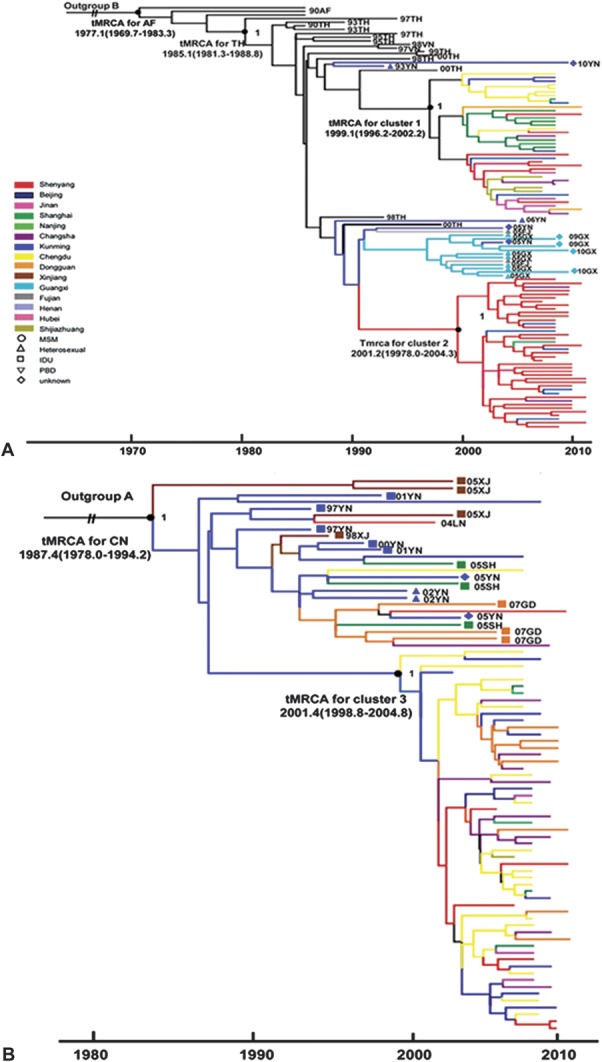
Maximum clade credibility (MCC) tree representing rooted genealogy of respective HIV-1 clusters identified among MSM in China The MCC tree was obtained by Bayesian MCMC analysis based on HIV-1 nucleotide sequences (HXB2: 2253–3278 nt) implemented in BEAST v1.6.0 (available at: http://beast.bio.ed.ac.uk/Main_Page). This particular MCC tree was constructed based on a relaxed-clock model in GTR + γ4 with a constant coalescent model. For panels A and B, we selected as many sequences as possible from regions with smaller sample sizes and only a few phylogenetically representative sequences from large-sample regions such as Shenyang, Beijing, and Chengdu for simplicity. The tree branches are colored according to their respective geographical locations and previously published sequences with known sampling times and high-risk behavior information are also included and shown with color-coded symbols as depicted in the inset. A, The medians of tMRCA with a 95% credibility interval (in parentheses) for HIV-1 CRF01_AE cluster 1 and CRF01_AE cluster 2 are 1998.0 (1995.2–2000.4) and 1999.6 (1997.6–2001.2).B, The medians of tMRCA with a 95% credibility interval (in parentheses) for HIV-1CRF07_BC cluster3 is 2000.9 (1999.0–2004.4). The distribution of the posterior probability of respective clusters is illustrated in the inset.
DISCUSSION
This study represents the first large-scale (n = 583) HIV molecular epidemiological study of MSM that included multiple regions with varying degrees of HIV prevalence: Kunming and Chengdu >10%; Shenyang, Beijing and Nanjing 5%–10%; and Jinan, Changsha, Shanghai, and Dongguan 1%–5%.22 Our study identified 3 distinct lineages of HIV-1 strains (CRF01_AE clusters 1 and 2 and CRF07_BC cluster 3) (Table 1; Fig. 2) that accounted for more than three-quarters of the MSM HIV-1 infections in 9 Chinese cities (447 of 583, 76.7%) between 2009 and 2011 (Table 1). The CRF01_AE is the most prevalent HIV-1 genotype among Chinese MSM, accounting for 62.1% (362 of 583) of HIV-1 infections, followed by CRF07_BC (106 of 583, 18.2%). Notably, the pol sequence data sets from recently published MSM studies in Beijing,5,23,24 Shijiazhuang (capital of Hebei province, near Beijing),9 and Zhengzhou (capital of Henan province, near Beijing)10 also contain these 3 variants, and the overall trend of their reported prevalence agreed with our findings (see Table S1, Supplemental Digital Content, http://links.lww.com/QAI/A417). Although the prevalence of subtype B (of United States–European origin) previously predominated among Chinese MSM,4 it accounted for only 15.9% (93 of 583) in this study. Subtype B′ (of Thai origin) played an even smaller role among Chinese MSM (4 of 583, 0.7%). The recent shift from subtype B to CRF01_AE prevalence among Chinese MSM agrees with the results from Beijing,5,23,24 Shijiazhuang,9 and Zhengzhou10 and further supports our finding that no widespread subtype B transmission cluster was detected although small local transmission clusters were found. Because of recruitment limitations, our sample sizes differed between cities and do not reflect regional HIV prevalence. Some cities yielded large samples, such as Shenyang and Beijing, whereas other heavily affected regions, such as Kunming, yielded relatively few subjects. Therefore, our estimate of HIV genotype distributions may be biased. However, we focused on the 3 distinct HIV-1 founding strains responsible for the growing HIV/AIDS epidemic among MSM in our 9 study cities, and at least 1 of the 3 clusters predominated in each of the cities.
The origins of these 3 lineages remain unknown. Database surveys found no sequences outside China that closely related to these 3. Surprisingly, we found that the vast majority of previously reported CRF01_AE sequences in heterosexual and IDU populations in Yunnan,25,26 Guangxi,27,28 and Fujian29 were on different phylogenetic branches from the 2 CRF01_AE clusters. However, many CRF01_AE sequences from the Liaoning heterosexual population7 have close phylogenetic links to MSM cluster 2. Additionally, a few CRF01_AE sequences from Yunnan30 or Guangxi28 infected cases with unknown transmission route and the heterosexual population in Liaoning7 and Beijing23 and Jiangsu IDUs31 were closely related to MSM cluster 1 (Fig. 1A). Similar to CRF01_AE cluster 1, none of the CRF07_BC sequences available in the database (n = 487) from non-MSM high-risk populations belonged to CRF07_BC cluster 3 except for a few CRF07_BC strains from Guangdong IDUs (6 of 35)32 (Fig. 1B). The close phylogenetic relationship between MSM and other high-risk population may imply the origin of 3 lineages, however, we cannot exclude the possibility that the above cases may also have homosexual contacts.
In addition, we observed indications suggesting the geographical origins of the viral strains. Although our sample sizes differed and several were insufficient for drawing definitive conclusions, we saw some patterns in the geographical distribution of these 3 lineages (Fig. 2; Table 1): (1) CRF01_AE cluster 1 was found throughout China but more frequently in the southern and eastern coastal regions [Kunming/Yunnan (4 of 7, 57.1%); Nanjing/Jiangsu (8 of 13, 61.5%); Shanghai (9 of 20, 45.0%); and Jinan/Shangdong (5 of 10, 50%)] than in other regions (χ2 = 17, P < 0.001). In contrast, (2) CRF01_AE cluster 2 was more highly concentrated in Shenyang/Liaoning in northeastern China (198 of 311, 63.7%) than in other regions (<20%) (χ2 = 198, P < 0.001). Finally, (3) CRF07_BC cluster 3 was most common in south-central provinces [Chengdu/Sichuan (30 of 63, 47.6%); Changsha/Hunan (9 of 23, 43.5%); Dongguan/Chuangdong (7 of 16, 43.8%)] (χ2 = 92, P < 0.001). These observations suggest that clusters 1, 2, and 3 may originate from southern-eastern coastal, northern, and south-central regions in China, respectively. Alternatively, the apparent distribution differences may simply reflect differences in the founding effects of the variants among the MSM populations of different regions.
In summary, the results of phylogenetic analysis based on pol sequences and the geographical distribution analysis matched the estimates in our recent MSM phylodynamic study on primary HIV infection based on viral near-full-length sequences.16 This agreement suggests that (1) CRF01_AE cluster 1 may have originated in southern/southwestern provinces and spread north along the southeastern coast, and that (2) CRF01_AE cluster 2 strains may have originated around Yunnan province, but the strains could have spread directly to the northeast. However, maximum clade credibility tree analyses (Fig. 4) revealed that the initial appearances of cluster 1 and cluster 3 strains could not be separated into a clear time gradient and that the number of some cluster 2 strains was insufficient to draw definitive time-related conclusions. These limitations do not negate the aforementioned suggestions. Rather, the lack of a clear time gradient implies that the spread of cluster 1 strains among MSM probably occurred in a short time, and that MSM from different regions across China likely had close contact. More historical samples and longer HIV sequences may be helpful to investigate the definitive origins of the 3 lineages responsible for China's MSM epidemic.
As shown in Figure 3, the nucleotide diversity of CRF07_BC cluster 3 (0.012 ± 0.005) is lower than that of CRF01_AE cluster 1 (0.030 ± 0.008) and cluster 2 (0.028 ± 0.007) (P < 0.01). The high level of genetic homogeneity of CRF07_BC cluster 3 suggests that it emerged later than clusters 1 and 2 among Chinese MSM. However, the estimated evolutionary rate was lower for CRF07_BC than CRF01_AE under the GTR + γ4 relaxed-clock models with a constant-size model. Consistent with this finding, Bayesian molecular clock analyses estimated that tMRCAs for clusters 1, 2, and 3 are 1999, 2001, and 2001, respectively (Fig. 4). To reduce the impact of uneven sample sizes in the Bayesian MCMC analysis, we selected as many sequences as possible from regions with smaller sample sizes and only a few phylogenetically representative sequences from large-sample regions, such as Shenyang, Beijing, and Chengdu. Taken together, these data indicate that the CRF07_BC cluster 3–driven epidemic among MSM occurred later than that driven by CRF01_AE clusters 1 and 2.
In this study, we detected a total of 18 other recombinant strains (18 of 583, 3.1%) that harbored recombination break points in the 1.0-kb pol gene. We noticed that B/CRF01_AE recombinants were remarkably common in Dongguan (5 of 16, 31.2%), Nanjing (3 of 18, 16.7%), and Changsha (2 of 23, 8.7%) (Table 1). The genotyping method used in this study was based on partial genome sequences, covering only 10% of the HIV-1 genome. The prevalence of recombinants may be underestimated if the break points located outside the genome region were analyzed. So the true prevalence of recombinants in Dongguan, Nanjing, and Changsha could be considerably higher than we noted. The epidemic trend of the new recombinants merits further study.
In conclusion, using a large study population with recent samples, we demonstrated that more than three-quarters of recent HIV-1 infections among Chinese MSM were caused by 3 distinct lineages of HIV-1 strains (CRF01_AE clusters 1 and 2; CRF07_BC cluster 3). These lineages are responsible for the recent upsurge in the HIV/AIDS epidemic among Chinese MSM, demonstrating a remarkable founding effect. This could, in turn, suggest that Chinese MSM engage in extremely high-risk behaviors and warrant urgent implementation of effective preventive measures to limit the epidemic.
Footnotes
Supported by the Mega Projects of the National Science Research for the 12th Five-Year Plan (2012ZX10001006) and China-Gates Foundation Cooperation Program. The funding organization had no role in the development of the study design or in the collection, analysis, and interpretation of data.
The authors have no conflicts of interest to disclose.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jaids.com).
REFERENCES
- 1.Ministry of Health of the People's Republic of China. China 2010 UNGASS Country Progress Report (2008-2009). 2010. UNGASS, http://data.unaids.org/pub/Report/2010/china_2010_country_progress_report_en.pdf [Google Scholar]
- 2.Lau JTF, Lin C, Hao C, et al. Public health challenges of the emerging HIV epidemic among men who have sex with men in China. Public Health. 2011;125:260–265 [DOI] [PubMed] [Google Scholar]
- 3.Bennett P. The theory of planned behaviour as predictor of condom use: a narrative review. Psychol Health Med. 2000;5:20 [Google Scholar]
- 4.Zhang XY, Li SW, Li XX, et al. Characterization of HIV-1 subtypes and viral antiretroviral drug resistance in men who have sex with men in Beijing, China. AIDS. 2007;21:S59–S65 [DOI] [PubMed] [Google Scholar]
- 5.Wang WH, Jiang SL, Li SW, et al. Identification of subtype B, multiple circulating recombinant forms and unique recombinants of HIV type 1 in an MSM cohort in China. AIDS Res Hum Retroviruses. 2008;24:1245–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang W, Xu J, Jiang S, et al. The dynamic face of HIV-1 subtypes among men who have sex with men in Bejiing, China. Curr HIV Res. 2011;9:136–139 [DOI] [PubMed] [Google Scholar]
- 7.Han X, Dai D, Zhao B, et al. Genetic and epidemiologic characterization of HIV-1 infection in Liaoning Province, China. J Acquir Immune Defic Syndr. 2010;53(suppl 1):S27–S33 [DOI] [PubMed] [Google Scholar]
- 8.Guo HX, Wei JF, Yang HT, et al. Rapidly increasing prevalence of HIV and Syphilis and HIV-1 subtype characterization among men who have sex with men in Jiangsu, China. Sex Transm Dis. 2009;36:120–125 [DOI] [PubMed] [Google Scholar]
- 9.Li L, Lu X, Li H, et al. High genetic diversity of HIV-1 was found in men who have sex with men in Shijiazhuang, China. Infect Genet Evol. 2011;11:1487–1492 [DOI] [PubMed] [Google Scholar]
- 10.Li L, Sun G, Li T, et al. Multiple introductions of HIV into men who have sex with men were found in Zhengzhou city, China. AIDS Res Hum Retroviruses. 2012;28:947–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang X, Chen H, Li W, et al. Precise determination of time to reach viral load set point after acute HIV-1 infection. J Acquir Immune Defic Syndr. 2012;61:448–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao B, Han XX, Dai D, et al. New trends of primary drug resistance among HIV type 1-infected men who have sex with men in Liaoning province, China. AIDS Res Hum Retroviruses. 2011;27:1047–1053 [DOI] [PubMed] [Google Scholar]
- 13.Thompson JD, Gibson TJ, Plewniak F, et al. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drummond AJ, Ho SY, Phillips MJ, et al. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574 [DOI] [PubMed] [Google Scholar]
- 17.An M, Han X, Xu J, et al. Reconstituting the epidemic history of CRF01_AE among MSM in Liaoning, northeastern China: implication in expanding MSM epidemic in China. J Virol. 2012;22:12402–12406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Babic DZ, Poljak M, Seme K, et al. Molecular epidemiology of HIV-1 subtypes based on analysis of pol sequences in Slovenia, 1996-2005. J Med Virol. 2006;78:997–1002 [DOI] [PubMed] [Google Scholar]
- 19.Eshleman SH, Husnik M, Hudelson S, et al. Antiretroviral drug resistance, HIV-1 tropism, and HIV-1 subtype among men who have sex with men with recent HIV-1 infection. AIDS. 2007;21:1165–1174 [DOI] [PubMed] [Google Scholar]
- 20.van Harmelen J, Wood R, Lambrick M, et al. An association between HIV-1 subtypes and mode of transmission in Cape Town, South Africa. AIDS. 1997;11:81–87 [DOI] [PubMed] [Google Scholar]
- 21.Lewis F, Hughes GJ, Rambaut A, et al. Episodic sexual transmission of HIV revealed by molecular phylodynamics. PloS Med. 2008;5:392–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Z. China's assessment and responses to HIV epidemic in MSM. Presented at the WHO meeting on MSM, February 8, 2009, Geneva, Switzerland
- 23.Ye JR, Lu HY, Wang WS, et al. The prevalence of drug resistance mutations among treatment-naive HIV-infected individuals in Beijing, China. AIDS Res Hum Retroviruses. 2012;28(4):418–423 [DOI] [PubMed] [Google Scholar]
- 24.Ye JR, Xin RL, Bai LS, et al. Sequence analysis of the gag-pol gene of human immunodeficiency virus type 1 of intersubtype (B′/C) recombinant strain in Beijing, China. AIDS Res Hum Retroviruses. 2011;27:331–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Lu L, Ba L, et al. Dominance of HIV-1 subtype CRF01_AE in sexually acquired cases leads to a new epidemic in Yunnan province of China. PLoS Med. 2006;3:e443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen M, Ma Y, Duan S, et al. Genetic diversity and drug resistance among newly diagnosed and antiretroviral treatment-naive HIV-infected individuals in western Yunnan: a hot area of viral recombination in China. BMC Infect Dis. 2012;12:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piyasirisilp S, McCutchan FE, Carr JK, et al. A recent outbreak of human immunodeficiency virus type 1 infection in southern China was initiated by two highly homogeneous, geographically separated strains, circulating recombinant form AE and a novel BC recombinant. J Virol. 2000;74:11286–11295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng H, Sun Z, Liang S, et al. Emergence of a new HIV-1 CRF01_AE variant in Guangxi, southern China. AIDS Res Hum Retroviruses. 2012;28(10):1352–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hai-Long H, Jian Z, Ping-Ping Y, et al. Genetic characterization of CRF01_AE full-length human immunodeficiency virus type 1 sequences from Fujian, China. AIDS Res Hum Retroviruses. 2007;23:569–574 [DOI] [PubMed] [Google Scholar]
- 30.Tu YQ, Wang MJ, Yao J, et al. Human immunodeficiency virus-1 genotypic drug resistance among volunteer blood donors in Yunnan, China. Transfusion. 2009;49:1865–1873 [DOI] [PubMed] [Google Scholar]
- 31.Guo DM, Ding N, Xu YP, et al. Near full-length genome characterization of an HIV-1 CRF01_AE strain in Jiangsu, China: evidence of two independent introductions from Fujian. AIDS Res Hum Retroviruses. 2009;25:619–623 [DOI] [PubMed] [Google Scholar]
- 32.Yang C, Liu S, Zhang T, et al. Transmitted antiretroviral drug resistance and thumb subdomain polymorphisms among newly HIV type 1 diagnosed patients infected with CRF01_AE and CRF07_BC virus in Guangdong province, China. AIDS Res Hum Retroviruses. 2012;28:1723–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]