

Abstract
Acetaminophen (APAP) overdose is the most frequent cause of acute liver failure in the US and many western countries. It is well known that APAP induces mitochondrial damage to trigger centrilobular necrosis. Emerging evidence suggests that autophagic removal of damaged mitochondria may protect against APAP-induced liver injury. Electron and confocal microscopy analysis of liver tissues revealed that APAP overdose triggers unique biochemical and pathological zonated changes in the mouse liver, which includes necrosis (zone 1), mitochondrial spheroid formation (zone 2), autophagy (zone 3) and mitochondrial biogenesis (zone 4). In this graphic review, we discuss the role of autophagy/mitophagy in limiting the expansion of necrosis and promoting mitochondrial biogenesis and liver regeneration for the recovery of APAP-induced liver injury. We also discuss possible mechanisms that could be involved in regulating APAP-induced autophagy/mitophagy and the formation of mitochondrial spheroids.
Keywords: Autophagy, Mitophagy, Mitochondrial spheroid, Acetaminophen, Liver injury
Highlights
-
•
Acetaminophen induces zonated biochemical and pathophysiological changes in mouse liver.
-
•
Damaged mitochondria can be removed via selective autophagy/mitophagy or formation of mitochondrial spheroids to attenuate acetaminophen-induced liver injury.
-
•
Pharmacological modulating autophagy maybe a novel potential therapeutic approach for treating acetaminophen-induced liver injury.
Introduction
Mechanisms of acetaminophen hepatotoxicity: role of mitochondrial damage
Acetaminophen (APAP) is a widely used antipyretic and analgesic drug in the US. At therapeutic doses APAP is a safe drug, but an overdose can cause severe liver injury and even acute liver failure in animals and man. APAP overdose is the most frequent cause of acute liver failure of any etiology in the US and many western countries. APAP is also one of the most studied hepatotoxic drugs worldwide [1], and thus considerable progress has been made in understanding the mechanisms of APAP-induced liver injury. Today, it is well known that N-acetyl-p-benzoquinone imine (NAPQI), a highly reactive metabolite that is generated from the metabolism of APAP by the cytochrome P450 system (such as CYP2E1), plays a key role in APAP-induced hepatotoxicity. NAPQI depletes cellular glutathione (GSH) and reacts with many cellular proteins, including mitochondrial proteins, to form protein adducts (AD), which are critical to trigger mitochondrial damage and subsequent necrosis. It has been recognized that APAP overdose causes mitochondrial dysfunction such as inhibition of mitochondrial respiration, mitochondrial oxidant stress and peroxynitrite formation, mitochondrial DNA damage, release of mitochondrial intermembrane space proteins such as apoptosis inducing factor (AIF) and endonuclease G (Endo G), which translocate to the nucleus and cause nuclear DNA fragmentation and eventual opening of the membrane permeability transition pore (MPT). While it remains to be tested for Endo G knockout (KO) mice, we have previously shown that AIF KO mice are more resistant to APAP-induced liver injury [2]. In addition to the protein adduct-initiated mitochondrial damage, early oxidant stress also promotes c-jun-N-terminal kinase (JNK) activation. Activated phosphorylated-JNK translocates to mitochondria and amplifies the mitochondrial oxidant stress, which eventually leads to the MPT pore opening, membrane potential collapse, ATP depletion and necrotic cell death [1,3–5]. Pharmacological inhibition of JNK using SP600125, a specific JNK inhibitor, significantly attenuates APAP-induced liver injury. Interestingly, while knockdown of either JNK1 or JNK2 using antisense oligonucleotides showed protection against APAP-induced liver injury, it was later shown that JNK2 KO mice had higher mortality rates compared to the wild type mice up to 48 h after APAP treatment [6–8]. This was due to the impaired hepatocyte proliferation and repair in JNK2 KO mice after APAP treatment [8]. Therefore, the effects of JNK inhibitors need to be carefully evaluated on both injury mechanisms and regeneration and potentially selective JNK1 inhibitors may be considered for therapeutic interventions in the future.
More recently, we showed that mitochondrial dynamics could also be involved in APAP-induced necrosis because APAP induced translocation of Drp1, a mitochondrial fission molecule, to mitochondria [9]. More importantly pharmacological inhibition of Drp1 attenuated APAP-induced necrosis. The translocation of Drp1 to mitochondria induced by APAP seemed to be mediated by the receptor interacting protein kinase 3 (Rip3), and deletion of Rip3 in the mouse liver resulted in reduced early phase APAP-induced liver injury [9].
Adaptive response to cellular stress: autophagy/mitophagy
While the detrimental mechanisms induced by APAP have been well studied, little is known about the cellular adaptive mechanisms that may attenuate APAP-induced liver injury. Cells may protect themselves by removing damaged mitochondria using a mechanism called autophagy. Autophagy is an evolutionary conserved catabolic process that degrades cellular proteins and organelles, a process involved in the formation of double-membrane autophagosomes that deliver cargos into lysosomes [10]. Enclosed cargos are then degraded inside the lysosomes and can be recycled as sources for the synthesis of new macromolecules or to generate ATP for cell survival. In addition to non-selective bulk autophagy, emerging evidence now support that autophagy can also be selective. Selective autophagy for peroxisomes (pexophagy), endoplasmic reticulum (erphagy), ribosomes (ribophagy), lipid droplets (lipophagy), invading microbes (xenophagy) and protein aggregates have been reported. Selective autophagy is mediated by a series of autophagy receptor complexes that can bridge cargo to the autophagy machinery. In mammalian cells, one common tag for autophagic recognition is ubiquitin, and autophagy receptor proteins such as p62/SQSTM1, NBR1, NDP52 or optineurin bind to ubiquitin and LC3 protein through their LIR (LC3 interacting region) [11–13]. Among the types of selective autophagy, autophagic removal of damaged/excess mitochondria (termed mitophagy) has been well studied. In yeast, the mitochondrial outer membrane protein Atg32 serves as a mitochondrial receptor for selective mitophagy, in which Atg32 binds with Atg11 and then later further recruits Atg8-PE to the mitochondria [14,15]. While no Atg32 homologs have been identified in mammalian cells, several mitochondrial outer membrane proteins such as NIX and FUNDC1 that have a LIR motif and directly interact with LC3 have been implicated in selective mitophagy [16–19]. In addition to NIX and FUNDC1, perhaps the best studied signaling pathway for selective mitophagy in mammalian cells is the PINK1 (PTEN-induced putative protein kinase 1)-Parkin mediated mitophagy. In this model, PINK1 is stabilized on depolarized mitochondria, which further recruits the ubiquitin E3 ligase Parkin to the mitochondria from the cytosol [19,20]. Parkin then ubiquitinates a subset of mitochondrial proteins, including Miro, Mitofusin1/2, hFis1, VDAC1 and Tom20 as well as others to promote mitophagy [21,22]. It has been suggested that Parkin-mediated mitochondrial protein ubiquitination can subsequently recruit an autophagy receptor such as p62 to mitochondria. Once on the mitochondria, p62 further recruits LC3-positive autophagosomes to p62–ubiquitin decorated mitochondria for their degradation, but the exact role of p62 in mitophagy is still controversial [19,23,24]. Mitochondria are extended reticular-shaped organelles; thus, it is a great challenge for autophagic removal of such elongated structures. It is thus not surprising that work from both mammalian cells and yeast suggest that mitochondrial fission can facilitate mitophagy [25,26]. Moreover, Parkin can also promote mitophagy by inducing mitochondrial fragmentation through proteasome-mediated Mfn1/2 degradation [27,28]. It should be noted that most of the molecules regulating mitophagy and mitochondrial dynamics have not been studied in the liver, and their roles in liver cell necrosis remain to be investigated. The possible scheme for APAP-induced mitophagy is illustrated in Fig. 1.
Fig. 1.
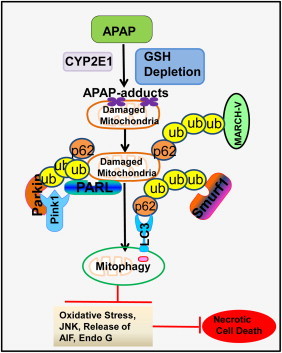
Possible mechanisms and functions of APAP-induced mitophagy. APAP is metabolized by CYP2E1 to generate NAPQI, which depletes cellular GSH to induce oxidative stress. Following GSH depletion, NAPQI adducts cellular proteins by targeting their cysteine residues, and some of them are mitochondrial proteins. Increased oxidative stress and mitochondrial protein adducts trigger mitochondrial damage. Damaged mitochondria then could be removed through selective mitophagy involving the autophagy receptor complex: mitochondrial ubiquitination–p62–LC3. Mechanistically, damaged depolarized mitochondria inactivate the mitochondrial protease PARL to prevent PARL-mediated degradation of PINK1. Increased PINK1 then recruits Parkin to mitochondria to trigger the ubiquitination of mitochondrial proteins. In addition to Parkin, several other ubiquitin E3 ligases such as smurf1 and MARCH-V may also promote mitochondrial ubiquitination. The removal of damaged mitochondria through mitophagy may attenuate mitochondria-mediated oxidative stress, JNK activation and the release of mitochondrial cell death factors such as apoptosis inducing factor.
As a cellular catabolic process, autophagy is activated when cells lack nutrients and energy. Two key sensors in response to the changes of cellular nutrients and energy are the mammalian target of rapamycin complex 1 (mTORC1) and the AMP-activated protein kinase (AMPK). Both mTORC1 and AMPK regulate the activity of ULK1 (yeast Atg1) complex, the most upstream component of the core autophagy machinery that is composed of ULK1, Atg13, FIP200 and Atg101 [29]. mTORC1 negatively regulates autophagy by directly phosphorylating ULK1 (S757) and inhibiting ULK1 activity [30,31]. Conversely, AMPK phosphorylates ULK1 at different sites (S317, S467, S555, T574, S637 and S777) and activates ULK1 to promote autophagy [31,32]. Interestingly, phosphorylation of ULK1 on S757 by mTORC1 disrupts the interaction between AMPK and ULK1. Since mTORC1 is activated when anabolic inputs are presented such as amino acids, glucose and growth factors, the phosphorylation of ULK1 by mTORC1 to prevent AMPK-mediated ULK1 activation will ensure that autophagy is blocked to allow the synthesis of proteins and lipids for cell proliferation. Intriguingly, when cellular energy is depleted, AMPK can also suppress mTORC1 activity by phosphorylation of TSC2 and raptor, two essential regulators of mTORC1 [33,34]. Once activated, ULK1 further phosphorylates Beclin1 on S14 to activate the Beclin1 complex which includes Atg14L, p150 and the class-III PI3 kinase VPS34 [35]. This ULK1-mediated phosphorylation of Beclin 1 enhances the autophagy specific VPS34 kinase activity containing Atg14L and is required for autophagy activation [35]. To add more layers of complexity to this regulation, AMPK also regulates the function of Beclin 1 complex by directly phosphorylating VPS34 and Beclin 1. It is known that Beclin1 and VPS34 exist in different complexes for regulating a variety of cellular functions. The autophagy-specific complex consists of VPS34, p150, Beclin1 and Atg14L. The non-autophagy complex consists of VPS34, p150, Beclin1 and VPS38, which functions for retrograde trafficking of endosome-to-Golgi. AMPK phosphorylates T163/S165 in VPS34 and suppresses its non-autophagy activity. In parallel, AMPK phosphorylates S91/S94 in Beclin1, which increases proautophagy VPS34 kinase activity to promote autophagy induction, which is further enhanced by Atg14L [36]. Thus, AMPK activates autophagy through at least three layers of signaling pathways to ensure the activation of the ULK1 and Beclin1 complex: suppression of mTORC1, direct activation of ULK1, and activation of Beclin1 complex via phosphorylation. In the context of APAP-induced autophagy, it is possible that APAP may inhibit mTORC1 activity through increased generation of mitochondrial ROS and decreased cellular ATP levels to trigger AMPK activation. Increased mitochondrial ROS levels have been shown to suppress mTOR through the activation of LKB1–AMPK pathway [37], although other un-identified mechanisms may also be involved [38,39]. More importantly, we and others have demonstrated that pharmacological inhibition of mTOR or activation of AMPK attenuates APAP-induced liver injury [40–42]. Although knockout mice for AMPK, ULK1, mTOR, VPS34 or Beclin1 heterozygous mice are available, their roles in regulating necrosis in the liver remain elusive. The possible cellular events in regulating APAP-induced autophagy are described in Fig. 2.
Fig. 2.
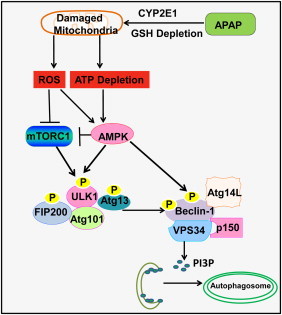
Possible role of AMPK–mTOR–ULK1–Beclin 1 signaling cascade in APAP-induced autophagy. APAP-induced damaged mitochondria increases mitochondria-mediated reactive oxygen species (ROS) and decreases cellular ATP levels. ROS may inactivate mTORC1 through AMPK and other not yet known mechanisms. mTORC1 negatively regulates autophagy through direct phosphorylation of S757 at ULK1 to inactivate ULK1 complex activity. ULK1 directly phosphorylates Beclin-1 and enhances VPS34 kinase activity to promote autophagy. TORC1-mediated phosphorylation of ULK1 also prevents ULK1 interaction with AMPK, which phosphorylates ULK1 at different sites to activate ULK1 and autophagy. AMPK positively regulates autophagy through three layers of regulation: (1) suppression of mTORC1 activity through phosphorylation of TSC2 and raptor; (2) phosphorylation of ULK1 at S317, S467, S555, T574, S637 and S777; (3) promotion of VPS34 kinase activity through phosphorylation of Beclin-1. Activated VPS34 increases the production of phosphatidylinositol 3-phosphate (PI3P), which promotes the biogenesis of autophagosomes.
Pharmacologic or genetic modulating autophagy has been widely used in many experimental models to determine the role of autophagy in liver diseases. Because of the essential role of autophagy in cell survival, mice with global knockout of the key autophagy genes results in prenatal death. Therefore, to study the role of autophagy in liver cell death has to rely on the using of liver-specific knockout mice. However, liver-specific knockout of either Atg7, Atg5 or VPS34 leads to severe hepatomegaly and cell death, suggesting that hepatic basal autophagy is a critical survival mechanism [43–45]. In addition, accumulation of p62 in the autophagy deficient mouse liver also leads to persistent activation of Nrf2, a key transcription factor for antioxidant genes. Because of the extensive adaptation mechanisms to the stress of impaired autophagy, the mice are resistant to other insults, e.g., APAP overdose [43]. As a result, these autophagy gene knockout mice are not suitable for experimental studies for liver injury [43].
It has been well known that liver displays a remarkable metabolic zonation, which generally is divided into three zones: periportal, intermediate and perivenous/or centrilobular zone [46]. Thus the liver forms functional gradients due to different activities of metabolic enzymes or oxygen concentrations as well as hormonal factors. For example, hepatocytes in the periportal zone have higher gluconeogenesis and higher O2 concentration, whereas hepatocytes in the perivenous zone have higher glycolytic and lower O2 concentration [47,48]. Moreover, the expression pattern of the P450 enzymes in the liver also displays a zonated pattern in normal rodent liver, with higher expression levels of CYP2E1, CYP1A2, CYP3A and CYP4A at the perivenous zone [49]. These P450 enzymes are important for APAP metabolism [1,50], and thus it is not surprising that APAP is mainly metabolized in the perivenous/centrilobular zone where APAP formed protein adducts are enriched, which results in mitochondrial damage and centrilobular necrosis [51]. A typical APAP-induced centrilobular necrosis in mouse liver is shown in Fig. 3A. Interestingly, using GFP–LC3 transgenic mice that constitutively express GFP–LC3 in the liver to monitor the formation of GFP–LC3 punctated autophagosome formation, we found that APAP increased GFP–LC3 positive autophagosomes, which were also enriched in the centrilobular area [42]. These findings are not surprising since APAP-induced mitochondrial damage is a key event in APAP-induced necrosis, and autophagy may be induced in that area to remove APAP-induced damaged mitochondria to promote cell survival and restrict the expansion of necrotic areas. Results from electron microscopy studies further revealed a striking and unique pattern of zonated changes in the centrilobular area, which we divided into four different zones (Fig. 3). Within the center adjacent to the central vein are the typical necrotic cells with swollen mitochondria, accumulated lipid droplets and disrupted plasma membrane (zone 1). In the areas next to the necrotic areas, we found distinctive mitochondrial structure changes where the mitochondria display ring-like or cup-like morphology, which we have termed mitochondria spheroids [28,52]. Similar mitochondrial structural changes are also observed in CCCP-treated mouse embryonic fibroblasts (MEF), which causes mitochondrial depolarization and fragmentation in these cells [52]. Morphologically, mitochondrial spheroids look like autophagosomes with lumen surrounded by mitochondrial membranes (Fig. 3C, zone 2). Mitochondrial spheroid lumen can also enwrap cytosolic contents, small pieces of endoplasmic reticulum, lipid droplets or even another mitochondrion. These mitochondrial spheroids are also positive for some lysosomal proteins and are acidic and seem to have limited degradation capacity [28]. However, it remains to be determined whether this formation of mitochondrial spheroid would represent another alternative pathway for the regulation of mitochondrial turn over and homeostasis.
Fig. 3.
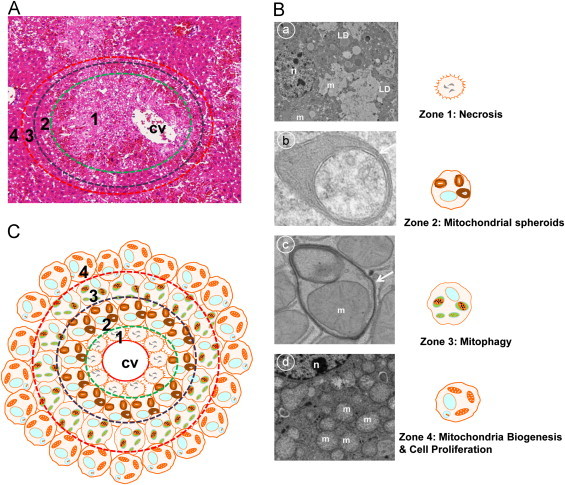
Distinctive zonated changes for necrosis, mitochondrial spheroids, mitophagy/autophagy and mitochondrial biogenesis/hepatocyte Proliferation in APAP-induced liver injury. (A) Typical histological changes of APAP-induced centrilobular necrosis. Male C57BL/6 mice were treated with APAP (500 mg kg, i.p.) for 6 h and liver tissues were processed for H & E staining. Necrotic areas were mainly detected around the central vein (green circled area) and adjacent areas appeared to be normal unaffected hepatocytes. (B) Distinctive zonated morphological changes detected by electron microscopy analysis in APAP-induced mouse liver. Four distinctive zonated morphological changes are: Zone 1-necrosis (panel a) where cells display swollen mitochondria, condensed nuclei and accumulated lipid droplets (LD); Zone 2-mitochondrial spheroids (panel b) where mitochondria undergo structural remodeling with squeezed matrix and form a lumen that can enwrap cytosol, endoplasmic reticulum and even another mitochondria (an arrow denotes the squeezed matrix of a mitochondrial spheroid); Zone 3-mitophagy (panel c) where double membrane autophagosomes envelop mitochondria (an arrow denotes a typical autophagosome); Zone 4-mitochondrial biogenesis and cell proliferation (panel d) where hepatocytes have an increased number of mitochondria and PCNA positive cells. n: nucleus; m: mitochondria; LD: lipid droplet.
The third zone next to zone 2 is the area where most hepatocytes contain typical double-membrane autophagosomes, and the majority of the autophagosomes have enclosed mitochondria, which we named zone 3 to represent the autophagy active area. Finally the cells at zone 4, which are in the outer areas surrounding the autophagy zone 3, have increased numbers of mitochondria indicating an increased mitochondrial biogenesis. We also found that most of the cells in zone 4 are positive for the hepatocyte proliferation marker proliferating cell nuclear antigen (PCNA). It is well known that liver has the high capacity to regenerate hepatocytes, which plays a critical role in the recovery phase of APAP-induced liver injury [1]. Liver regeneration/repair is an energy consuming process, and it is possible that the removal of damaged mitochondria at zone 2/zone 3, and mitochondrial biogenesis in hepatocytes in zone 4 will also be important for the restriction of cell injury and recovery from APAP-induced liver injury.
Why does APAP induce these zonated changes in the liver? As discussed above, liver has a gradient distribution of the P450 enzymes that are required for the metabolism of APAP [49]. In addition, centrilobular hepatocytes have the lower glutathione levels in the liver [53]. Therefore, the levels of APAP-AD and APAP-induced ROS and peroxynitrite as well as APAP-induced glutathione depletion also display a gradient pattern in that they are higher near the perivenous zone and lower near the periportal zone [54,55]. These gradient changes may eventually induce the specific zonated pathophysiological changes as we discussed above (Fig. 3C). The highest levels of these detrimental factors will lead to overwhelming mitochondrial damage and necrosis in the perivenous areas. When the levels of these detrimental factors are declining (with increasing distance from the perivenous areas), the cells may adapt and use autophagy/mitophagy to remove damaged mitochondria when the numbers and extent of damaged mitochondria are limited. In the areas where these detrimental factors are relatively low, there will be minimal or no mitochondrial damage. Thus, there would be no need to induce autophagy/mitophagy in these areas and instead, the mitochondria in these cells may proliferate to generate more ATP for cell proliferation and repair. It is most likely that different signaling pathways could be involved in regulating each specific zonated change. In addition to the signaling pathways that has been discussed in this manuscript (mTOR, AMPK and NO), more signaling pathways need to be further investigated to better understand the formation of these zonated changes. Moreover, it should be noted that these zonated changes induced by APAP in the mouse livers may also occur with other drugs such as thioacetamide and carbon tetrachloride, which also require metabolism and bioactivation by P450 enzymes and induce centrilobular necrosis in the liver [56,57]. However, the extent of the formation of mitochondrial spheroids and induction of mitophagy could be different because thioacetamide and carbon tetrachloride do not induce robust mitochondrial damage like APAP.
How is mitochondrial spheroid formation regulated? Mitochondria are dynamic organelles that constantly undergo fission and fusion. Fission is regulated by the dynamin-related protein 1 (Drp1), fission protein 1 (Fis1) and mitochondrial protein 18 kDa (MTP18). Drp1 does not have any transmembrane domains and requires interaction with Fis1, a protein anchored to the outer membrane of mitochondria. It has been suggested that Drp1 and Fis1 mainly regulate the outer mitochondrial membrane fission while MTP18 regulates the inner mitochondrial membrane fission [58,59]. Mitochondrial fusion is mainly regulated by mitofusin 1 (Mfn1), Mfn2 and optic atrophy 1 (OPA1), where Mfn1 and Mfn2 regulate the outer mitochondrial membrane fusion and OPA1 regulates the inner membrane fusion [60]. Once Parkin is translocated to mitochondria, it promotes the ubiquitination of Mfn1 and Mfn2 and their proteasomal degradation results in mitochondrial fragmentation [21,22]. We found that the formation of mitochondrial spheroids requires either Mfn1 or Mfn2 in cultured cells, and thus Parkin can negatively regulate the formation of mitochondrial spheroids by promoting the degradation of Mfn1 and Mfn2 [28]. Intriguingly, formation of mitochondrial spheroids is still detected in zone 2 next to the necrotic areas (zone 1) in APAP-treated mouse livers that expresses Parkin [61](Fig. 3). Moreover, no obvious changes in Mfn1 and Mfn2 levels were found in APAP-treated mouse livers by western blot analysis even though APAP treatment increased mitochondrial translocation of Parkin (Ding et al., unpublished observations). It is known that the E3 ligase function of Parkin is regulated by post-translational modifications such as phosphorylation, ubiquitination and S-nitrosylation [62]. S-nitrosylation of Parkin inhibits its E3 ligase activity and its protective functions [63]. APAP has been shown to increase levels of nitric oxide and protein nitration in mouse livers, especially in mitochondria [6,64]. Given the gradient and zonation pattern of cytochrome P450 enzymes in the liver, it is likely that the concentrations of NO and reactive nitric species induced by APAP could also display a gradient pattern where it could be enriched in the Zone 2 area next to the necrosis area. Therefore, it is possible that APAP may induce some of these post-translational modifications on Parkin resulting in inactivation of Parkin and formation of mitochondrial spheroids in Zone 2. The possible mechanisms for how APAP induces the formation of mitochondrial spheroids are summarized in Fig. 4. Further work is definitely needed to further elucidate the exact role of post-translational modifications of Parkin induced by APAP in mouse livers. We are currently investigating the role of Parkin-mediated mitophagy in APAP-induced liver injury using the Parkin KO mice.
Fig. 4.
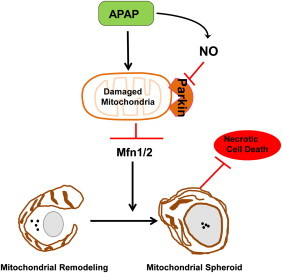
Parkin negatively regulates mitochondrial spheroid formation. Following mitochondria damage by APAP, mitochondria are depolarized and Parkin is translocated to the outer membrane of mitochondria. In most cases, Parkin promotes Mfn1 and Mfn2 (Mfn1/2) degradation resulting in mitochondrial fragmentation, and Parkin also promotes canonical selective mitophagy through mitochondrial ubiquitination. APAP increases production of nitric oxide (NO) and reactive nitrogen species in certain areas of the liver (such as Zone 2), which may lead to the s-nitrosylation/or other possible translational modifications and inactivation of Parkin to allow Mfn1/2 mediated formation of mitochondrial spheroids. The mitochondrial spheroid pathway could be a default pathway that serves as another alternative mechanism to regulate mitochondrial homeostasis when Parkin is inactivated. Mitochondrial spheroids can contain some lysosomal markers and are acidic and may undergo self-turnover and in turn protect against APAP-induced necrosis.
Summary and future perspectives
APAP induces distinctive cellular changes including centrilobular necrosis (Zone 1), mitochondrial spheroids (Zone 2), autophagy/mitophagy (Zone 3) and mitochondrial biogenesis and hepatocyte proliferation (Zone 4), which depends on the liver zonation. It seems that induction of autophagy/mitophagy may help to restrict the necrotic areas and promote liver regeneration and recovery for APAP-induced liver injury. Modulating autophagy/mitophagy pathways may thus provide a novel therapeutic avenue for treating APAP-induced liver injury. The recent observation that mitochondrial damage is also a critical feature in human overdose patients [65] supports the potential relevance of autophagy/mitophagy as a therapeutic target in the clinic. However, future studies will be needed to investigate the signaling pathways regulating autophagy/mitophagy downstream of mTOR and AMPK such as the ULK1 complex and VPS34–Beclin1 complex as well as the Pink1–Parkin–ubiquitination-mediated selective mitophagy pathway in APAP-induced liver injury in experimental animals and in humans to assess which pathway holds the most promise as therapeutic target.
Acknowledgments
We thank Ms. Barbara Fegley (KUMC Electron Microscopy Research Laboratory) for her excellent assistance for the EM studies. This study was supported in part by the National Institute of Health (NIH) funds R01 AA020518 (W.X.D) and AA12916 and DK070195 (to H.J.) and by Grants from the National Center for Research Resources (5P20RR021940-07) and the National Institute of General Medical Sciences (8 P20 GM103549-07). Jessica A Williams is a recipient of the Biomedical Research Training Program Fellowship from University of Kansas Medical Center.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Jaeschke H., Bajt M.L. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicological Sciences. 2006;89(1):31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 2.Bajt M.L. Apoptosis-inducing factor modulates mitochondrial oxidant stress in acetaminophen hepatotoxicity. Toxicological Sciences. 2011;122(2):598–605. doi: 10.1093/toxsci/kfr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaeschke H., McGill M.R., Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metabolism Reviews. 2012;44(1):88–106. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han D. Regulation of drug-induced liver injury by signal transduction pathways: critical role of mitochondria. Trends in Pharmacological Sciences. 2013;34(4):243–253. doi: 10.1016/j.tips.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hinson J.A. Acetaminophen-induced hepatotoxicity: role of metabolic activation, reactive oxygen/nitrogen species, and mitochondrial permeability transition. Drug Metabolism Reviews. 2004;36(3–4):805–822. doi: 10.1081/dmr-200033494. [DOI] [PubMed] [Google Scholar]
- 6.Saito C., Lemasters J.J., Jaeschke H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicology and Applied Pharmacology. 2010;246(1–2):8–17. doi: 10.1016/j.taap.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gunawan B.K. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131(1):165–178. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 8.Bourdi M. Protective role of c-Jun N-terminal kinase 2 in acetaminophen-induced liver injury. Biochemical and Biophysical Research Communications. 2008;374(1):6–10. doi: 10.1016/j.bbrc.2008.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramachandran A. The receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology. 2013 doi: 10.1002/hep.26547. [Epub ahead in print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 11.Shaid S. Ubiquitination and selective autophagy. Cell Death and Differentiation. 2013;20(1):21–30. doi: 10.1038/cdd.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johansen T., Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manley S., Williams J.A., Ding W.X. Role of p62/SQSTM1 in liver physiology and pathogenesis. Experimental Biology and Medicine (Maywood) 2013;238(5):525–538. doi: 10.1177/1535370213489446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanki T. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Developmental Cell. 2009;17(1):98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okamoto K., Kondo-Okamoto N., Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Developmental Cell. 2009;17(1):87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 16.Sandoval H. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454(7201):232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novak I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Reports. 2010;11(1):45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nature Cell Biology. 2012;14(2):177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 19.Ding W.X. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin–ubiquitin–p62-mediated mitochondrial priming. Journal of Biological Chemistry. 2010;285(36):27879–27890. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narendra D. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. Journal of Cell Biology. 2008;183(5):795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan N.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Human Molecular Genetics. 2011;20(9):1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshii S.R. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. Journal of Biological Chemistry. 2011;286(22):19630–19640. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geisler S. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature Cell Biology. 2010;12(2):119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 24.Narendra D. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6(8):1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Twig G., Hyde B., Shirihai O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochimica et Biophysica Acta. 2008;1777(9):1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mao K. The scaffold protein atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Developmental Cell. 2013;26(1):9–18. doi: 10.1016/j.devcel.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka A. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. Journal of Cell Biology. 2010;191(7):1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding W.X. Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. Journal of Biological Chemistry. 2012;287(50):42379–42388. doi: 10.1074/jbc.M112.413682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Current Opinion in Cell Biology. 2010;22(2):132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 30.Egan D. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7(6):643–644. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology. 2011;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Egan D.F. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gwinn D.M. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular Cell. 2008;30(2):214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inoki K., Zhu T., Guan K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 35.Russell R.C. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nature Cell Biology. 2013;15(7):741–750. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim J. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152(1–2):290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alexander A. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proceedings of the National Academy of Sciences. 2010;107(9):4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z.Y. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochemical and Biophysical Research Communications. 2011;414(1):5–8. doi: 10.1016/j.bbrc.2011.09.046. [DOI] [PubMed] [Google Scholar]
- 39.Gibson S.B. A matter of balance between life and death: targeting reactive oxygen species (ROS)-induced autophagy for cancer therapy. Autophagy. 2010;6(7):835–837. doi: 10.4161/auto.6.7.13335. [DOI] [PubMed] [Google Scholar]
- 40.Saberi B. Protein kinase C (PKC) participates in acetaminophen hepatotoxicity through c-jun-N-terminal kinase (JNK)-dependent and -independent signaling pathways. Hepatology. 2013 doi: 10.1002/hep.26625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ni H.M., Jaeschke H., Ding W.X. Targeting autophagy for drug-induced hepatotoxicity. Autophagy. 2012;8(4):709–710. doi: 10.4161/auto.19659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ni H.M. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55(1):222–232. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ni H.M. Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicological Sciences. 2012;127(2):438–450. doi: 10.1093/toxsci/kfs133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Komatsu M. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 45.Jaber N. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proceedings of the National Academy of Sciences. 2012;109(6):2003–2008. doi: 10.1073/pnas.1112848109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oinonen T. Zonation of cytochrome P450 enzyme expression in rat liver. Isozyme-specific regulation by pituitary dependent hormones. Biochemical Pharmacology. 1996;51(10):1379–1387. doi: 10.1016/0006-2952(96)00064-0. [DOI] [PubMed] [Google Scholar]
- 47.Jungermann K., Kietzmann T. Oxygen: modulator of metabolic zonation and disease of the liver. Hepatology. 2000;31(2):255–260. doi: 10.1002/hep.510310201. [DOI] [PubMed] [Google Scholar]
- 48.Burke Z.D., Tosh D. The Wnt/beta-catenin pathway: master regulator of liver zonation? Bioessays. 2006;28(11):1072–1077. doi: 10.1002/bies.20485. [DOI] [PubMed] [Google Scholar]
- 49.Lindros K.O. Zonation of cytochrome P450 expression, drug metabolism and toxicity in liver. General PharmacologyGen Pharmacol. 1997;28(2):191–196. doi: 10.1016/s0306-3623(96)00183-8. [DOI] [PubMed] [Google Scholar]
- 50.James L.P., Mayeux P.R., Hinson J.A. Acetaminophen-induced hepatotoxicity. Drug Metabolism and Disposition. 2003;31(12):1499–1506. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- 51.Jaeschke H. Mechanisms of hepatotoxicity. Toxicological Sciences. 2002;65(2):166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 52.Ding W.X. Electron microscopic analysis of a spherical mitochondrial structure. Journal of Biological Chemistry. 2012;287(50):42373–42378. doi: 10.1074/jbc.M112.413674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia-Ruiz C. Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. Journal of Clinical Investigation. 1994;94(1):193–201. doi: 10.1172/JCI117306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts D.W. Immunohistochemical localization and quantification of the 3-(cystein-S-yl)-acetaminophen protein adduct in acetaminophen hepatotoxicity. American Journal of Pathology. 1991;138(2):359–371. [PMC free article] [PubMed] [Google Scholar]
- 55.Knight T.R. Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicological Sciences. 2001;62(2):212–220. doi: 10.1093/toxsci/62.2.212. [DOI] [PubMed] [Google Scholar]
- 56.Chilakapati J. Saturation toxicokinetics of thioacetamide: role in initiation of liver injury. Drug Metabolism and Disposition. 2005;33(12):1877–1885. doi: 10.1124/dmd.105.005520. [DOI] [PubMed] [Google Scholar]
- 57.Manibusan M.K., Odin M., Eastmond D.A. Postulated carbon tetrachloride mode of action: a review. Journal of Environmental Science and Health: Part C, Environmental Carcinogenesis and Ecotoxicology Reviews. 2007;25(3):185–209. doi: 10.1080/10590500701569398. [DOI] [PubMed] [Google Scholar]
- 58.Liesa M., Palacin M., Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiological Reviews. 2009;89(3):799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 59.Detmer S.A., Chan D.C. Functions and dysfunctions of mitochondrial dynamics. Nature Reviews Molecular Cell Biology. 2007;8(11):870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 60.Chen H., Chan D.C. Physiological functions of mitochondrial fusion. Annals of the New York Academy of Sciences. 2010;1201:21–25. doi: 10.1111/j.1749-6632.2010.05615.x. [DOI] [PubMed] [Google Scholar]
- 61.Ding W.X., Yin X.M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biological Chemistry. 2012;393(7):547–564. doi: 10.1515/hsz-2012-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Walden H., Martinez-Torres R.J. Regulation of Parkin E3 ubiquitin ligase activity. Cellular and Molecular Life Sciences. 2012 doi: 10.1007/s00018-012-0978-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chung K.K. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004;304(5675):1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 64.Burke A.S., MacMillan-Crow L.A., Hinson J.A. Reactive nitrogen species in acetaminophen-induced mitochondrial damage and toxicity in mouse hepatocytes. Chemical Research in Toxicology. 2010;23(7):1286–1292. doi: 10.1021/tx1001755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McGill M.R. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. Journal of Clinical Investigation. 2012;122(4):1574–1583. doi: 10.1172/JCI59755. [DOI] [PMC free article] [PubMed] [Google Scholar]