

Abstract
Mitochondria are a major source of cellular oxidants and have been implicated in aging and associated pathologies, notably cardiovascular diseases. Vascular cell senescence is observed in experimental and human cardiovascular pathologies. Our previous data highlighted a role for angiotensin II in the induction of telomere-dependent and -independent premature senescence of human vascular smooth muscle cells and suggested this was due to production of superoxide by NADPH oxidase. However, since a role for mitochondrial oxidants was not ruled out we hypothesise that angiotensin II mediates senescence by mitochondrial superoxide generation and suggest that inhibition of superoxide may prevent vascular smooth muscle cell aging in vitro. Cellular senescence was induced using a stress-induced premature senescence protocol consisting of three successive once-daily exposure of cells to 1×10−8 mol/L angiotensin II and was dependent upon the type-1 angiotensin II receptor. Angiotensin stimulated NADPH-dependent superoxide production as estimated using lucigenin chemiluminescence in cell lysates and this was attenuated by the mitochondrial electron transport chain inhibitor, rotenone. Angiotensin also resulted in an increase in mitoSOX fluorescence indicating stimulation of mitochondrial superoxide. Significantly, the induction of senescence by angiotensin II was abrogated by rotenone and by the mitochondria-targeted superoxide dismutase mimetic, mitoTEMPO. These data suggest that mitochondrial superoxide is necessary for the induction of stress-induced premature senescence by angiotensin II and taken together with other data suggest that mitochondrial cross-talk with NADPH oxidases, via as yet unidentified signalling pathways, is likely to play a key role.
Keywords: Vascular smooth muscle cell, Angiotensin II, Mitochondria, Superoxide, Cell senescence, Stress-induced premature senescence
Graphical abstract
Highlights
-
•
Angiotensin II causes stress-induced premature senescence in hVSMC.
-
•
Mitochondrial superoxide is necessary for premature senescence.
-
•
Mitochondrial cross-talk with NADPH oxidases is implicated in this mechanism.
Introduction
Cell senescence prevents uncontrolled mitosis of transformed cells, but is also associated with mechanisms of age-related pathologies including cardiovascular disease [1]. Stress induced premature senescence (SIPS) is defined as the early senescence of cells resulting from repeated exposure to a variety of cellular stressors at sub-cytotoxic concentrations, e.g. H2O2, chemotherapeutic agents, ultraviolet and ionizing radiation [2]. Oxygen radicals and intermediates are implicated in the induction of SIPS with high concentrations of these oxidants acting through damage to DNA but there is also a role for physiological levels of H2O2 and in particular, mediating effects through cell signalling pathways.
SIPS has been studied in cultured skin fibroblast models [3] and endothelial cells [4] and to a limited extent in vascular smooth muscle cells (VSMC) [5,6]. Two main types of protocol have been used to study premature senescence or SIPS. Firstly, continuous stress and secondly, a series of stresses with recover periods in-between and following the final stress. Much lower concentrations are often used for the latter (SIPS) protocol. In the continuous model it is not possible to discriminate between immediate effects of stress and long term, irreversible effects on aging (e.g. p53 growth arrest by oxidative damage). With the repeat stress model the provision of complete growth media between stresses and following final stress allows cells to recover, thus avoiding interactions between acute effects and long term or aging effects; this paradigm has been adopted here to study angiotensin II (Ang II)-induced senescence in human VSMC.
In vivo, SIPS has been observed in chronic kidney disease patients [7] and in experimental models [8]. Notably, cells with a characteristic senescence phenotype have been observed in human arteries at sites prone to atherosclerosis [9] and their accumulation may account for the decline in tissue and organ function observed with aging in vivo.
Reports have suggested a correlation between dysfunctional or damaged mitochondria and the onset or accumulation of cellular senescence [10]. An increase in mitochondrial dysfunction has been observed in human tissue with advancing age [11], which correlates with a gradual accumulation of senescent cells. There is little evidence to determine whether mitochondria have a mechanistic role in promoting premature senescence, however, a very recent study showed that inhibition of mitochondrial superoxide promoted cell death and senescence of B16 mouse melanoma cells [12].
We and others have reported that Ang II induces senescence in human VSMC in vitro [5,13]. Ang II causes oxygen radical production in VSMC via NADPH oxidase [14,15] and oxidative DNA damage [5]. Both replicative senescence, by accelerated telomere attrition over a number of cell divisions and premature senescence have been identified as possible mechanisms for the effects of Ang II [5] but formal proof of induction of SIPS has not been demonstrated. Prevention of senescence may partly explain the beneficial effects, in terms of increasing lifespan and maintaining organ/tissue function in rodents, of drugs which inhibit Ang II synthesis and function [16]. We hypothesise that Ang II mediates senescence of VSMC by generation dependent upon mitochondrial function and that inhibition of mitochondrial may help delay or prevent VSMC aging in vitro.
Materials and methods
Reagents
All chemicals were supplied by Sigma unless otherwise noted. Mito-TEMPO was purchased from Enzo Life Sciences and mitoSOX from Invitrogen Ltd.
Thenoyltrifluoroacetone (TTFA) was supplied by Fisher Scientific. EXP3174 was a gift from Merck & Co Inc.
hVSMC culture and induction of SIPS
VSMC were cultured as previously described [5] and used between passage 2 and 9. Cells were cultured in RPMI 1640 media containing 10% foetal calf serum (FCS), 2.5% (v/v) smooth muscle cell growth supplement (TCS Cellworks), 100 μg/ml glutamine, 100 μg/ml penicillin/streptomycin and 20 mmol/L HEPES buffer and maintained in a 5% CO2 atmosphere. Near confluent cells were rendered quiescent in media containing 0.5% (v/v) FCS (without growth supplement) for 24 h prior to Ang II (1×10−8 or 1×10−7 mol/L) exposures.
In order to investigate induction of SIPS, the protocol of Toussaint et al. was adopted [3,2]. Sub-confluent cells were treated with successive Ang II for 2 h each day, for 3 days, followed by 24 h recovery periods after each treatment by replacing the culture media with fresh growth media. Treatment with tert-butylhydroperoxide (tert-BHP; 4×10−5 mol/L) was conducted in the same manner in parallel cultures as a positive control. The same schedule of media changes without the stress treatment was performed on control cells.
Senescence-associated β-galactosidase (SA-β-gal) staining
Senescence was determined by measurement of SA-β-gal activity [17] using the ‘Senescence Cell Staining kit’ (Sigma). Following treatments with peroxide or Ang II, hVSMC were re-plated (5×104 per well) in 12-well plates in media containing 10% FCS for 24 h prior to staining. Senescent cells were enumerated by light microscopy in five fields of view selected at random in each well.
To investigate the effect of antioxidants and mitochondrial inhibitors on senescence, VSMC were maintained in media containing 0.5% (v/v) foetal calf serum for 24 h. Cells were then incubated in media containing catalase (300 Units/ml for 3 h), N-acetylcysteine (NAC; 0.5×10−3 mol/L for 2 h), SOD (50 Units/ml for 3 h), rotenone (2×10−6 mol/L for 3 h), thenoyltrifluoroacetone (TTFA; 1×10−6 mol/L for 3 h) or the mitochondrial scavenger mito-TEMPO (25×10−9 mol/L for 4 h) prior to Ang II exposure at 1×10−8 mol/L for 24 h at 37 °C. Following Ang II treatment, cells were trypsinized, counted using a haemocytometer and re-plated at 5×104 cells per well in 12-well plates, in media containing 10% (v/v) FCS. Cells were then left to adhere for 24 h at 37 °C prior to fixation and staining for SA-β-gal.
Measurement of NADPH-dependent superoxide production
Quiescent hVSMC were stimulated with Ang II for 1 h then homogenized by sonication. The lucigenin chemiluminescence assay was used as previously described to determine NADPH-dependent superoxide production in cell lysates [5]. To assess whether Ang II-induced superoxide production was dependent upon mitochondrial activity, the effect of electron transport chain inhibitors for complex I (rotenone, 10×10−6 mol/L) and II (TTFA, 10×10−6 mol/L) was studied.
Detection of mitochondrial
MitoSOXTM red was used in live hVSMC. Near confluent cells were exposed to tert-BHP (50×10−6 mol/L) for 2 h or Ang II (1×10−7 mol/L) for 1 h at 37 °C. Following treatment, cells were loaded with 5×10−6 mol/L MitoSOXTM red in Hanks' balanced salt solution (HBSS) for 10 min at 37 °C. Cells were washed 3× in HBSS and finally 500 μl of HBSS was added to each well and fluorescence was measured using a Cary Eclipse Fluorescence Spectrophotometer plate reader (excitation: 510 nm; emission: 580 nm). Fluorescence measurements were calculated relative to untreated cells loaded with the probe.
Analysis of data
Comparison of means was performed using one way-ANOVA.
Results
Induction of SIPS in cultured hVSMC by tert-BHP
To our knowledge, the induction of SIPS under the defined protocol described by Toussaint et al. [2] has not been previously reported for hVSMC. In order to investigate this, hVSMC were exposed to mild, successive tert-BHP stresses for 2 h each over 3 days with 24 h recovery periods between each treatment followed by an extended period of recovery. Following tert-BHP, hVSMC displayed >2.5-fold increase in cells staining for SA-β-gal compared with controls (Fig. 1A). This is the first direct evidence, using the strict definition of SIPS induction, that hVSMC in culture undergo SIPS in response to peroxide stress.
Fig. 1.
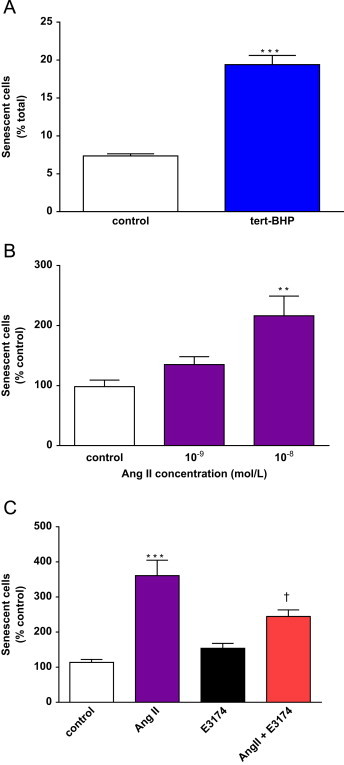
Tert-BHP and Ang II induce SIPS in hVSMC. A, Successive tert-BHP exposures induce SIPS in hVSMC. Sub-confluent cells were submitted to 3 stresses of 4×10−5 mol/L tert-BHP for 2 h, over 3 days. Senescence was determined by SA-β-gal activity on the third day after the final recovery period. Bars represent mean+SD; n=6 (***p<0.001 compared to the control). B, quiescent cells were submitted to 3, once daily stresses of Ang II for 2 h. Bars represent mean+SD; n=6–9 (**p<0.01 compared with control). C, Ang II-induced SIPS is mediated via AT1. Quiescent cells were pre-incubated with E3174 (1×10−5 mol/L for 1 h) prior to each Ang II (1×10−8 mol/L for 2 h) stress over the 3 days. Bars represent mean+SD; n=5 (⁎⁎⁎p<0.001 compared with control and †p<0.05 compared with Ang II alone).
Ang II stresses induce SIPS via angiotensin II receptor
Previous work from our group showed that Ang II accelerated the onset of hVSMC senescence via telomere-dependent and -independent mechanisms [5]. To determine whether Ang II induced SIPS, hVSMC were exposed to Ang II using the same regime as for tert-BHP, namely treatment followed by recovery and consistent with other reported studies on SIPS in other cell types [3,18]. A two-fold increase in SIPS was observed at an Ang II concentration of 1×10−8 mol/L (Fig. 1B). This is the first report that acute, repeated Ang II treatments cause senescence of hVSMC. Induction of senescence was reduced (p<0.05) by approximately 50% by pre-incubation of cells with the type-1 angiotensin II receptor (AT1) antagonist, EXP3174 (E3174) prior to each Ang II exposure (Fig. 1C) confirming a true receptor-mediated mechanism for induction of SIPS by Ang II.
Contribution of mitochondria to Ang II-induced superoxide production in hVSMC
Since Ang II initiates various cell signalling pathways in VSMC via increased ROS generation [14], which is also a key trigger of cellular senescence [19], it is therefore reasonable to hypothesize that is a significant mediator of SIPS in hVSMC. Previous work has shown that Ang II elevates superoxide generation via NADPH oxidase activity in VSMC [20,15] and that, in this model, superoxide production was not detected when Complex III of the electron transport chain was inhibited by antimycin A [5]. The addition of the mitochondrial complex I inhibitor rotenone almost fully suppressed NADPH-dependent superoxide production due to Ang II over a period of 40 min (Fig. 2). There was also a trend towards decreased Ang II-induced superoxide with TTFA but this did not reach statistical significance in this model (Fig. 2). These data implicated mitochondrial function as an important determinant of NADPH-dependent production following Ang II stimulation, however, a direct scavenging of by rotenone or a direct effect on NADPH oxidase activity in lysates cannot be ruled out from these data.
Fig. 2.
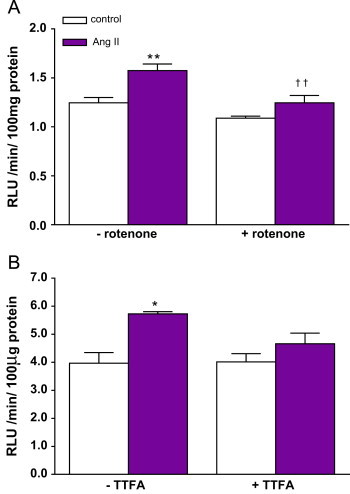
Ang II stimulated NADPH-dependent superoxide production is modulated by mitochondrial ETC inhibitors in hVSMC. Cells were pre-incubated with Ang II (1×10−7 mol/L) for 1 h then lysates were used for the measurement of NADPH-dependent superoxide production using lucigenin chemiluminescence. NADPH stimulated production was derived from the relative light units (RLU)/minute/100 μg of protein over a 40-min period. A, complex I inhibitor, rotenone significantly reduced Ang II-induced production. Bars represent mean+SD, n=3–4. ⁎⁎p<0.01 compared with control and ††p<0.01 compared with Ang II. B, Complex II inhibitor TTFA, did not affect Ang II-induced production. Bars represent mean+SD, n=2–4. ⁎p<0.05 compared with control.
The possible contribution of mitochondria to cellular following Ang II stimulation was investigated using mito-SOX as a superoxide reporter in live hVSMC (Fig. 3). Both tert-BHP and Ang II stimulated mitoSOX fluorescence by 1.5–2.0 fold compared to the control indicating that mitochondria within cells produce superoxide in response to Ang II.
Fig. 3.
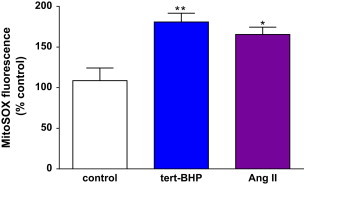
Ang II stimulated mitochondrial superoxide production in live hVSMC. Cells were pre-incubated with Ang II (1×10−7 mol/L) for 1 h then loaded with MitoSOX (5 µmol/L) for 10 min at 37 °C. Cells were washed to remove excess probe prior to measurement of fluorescence. Bars represent mean+SD, n=3–4. (⁎p<0.05 and **p<0.01 compared with control).
Ang II induced premature senescence of hVSMC involves oxidant generation and the mitochondrial electron transport chain
In non-phagocytic cells a major portion of cellular ROS is believed to be derived from the mitochondrial electron transport chain (mtETC). Our data on Ang II exposure suggested that superoxide was generated via the mtETC in addition to NADPH oxidase, as previously described in hVSMC [5]. Supporting our previous data, antioxidants catalase, NAC and SOD inhibited Ang II-induced premature senescence of VSMC (Fig. 4A–C) confirming the involvement of oxidants in senescence.
Fig. 4.
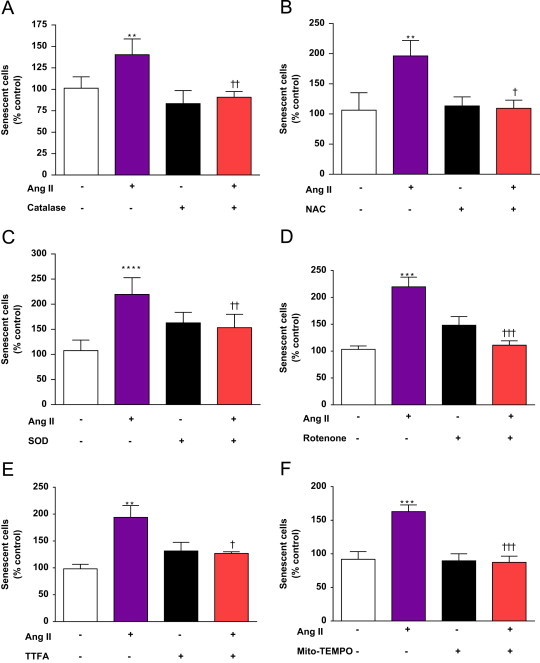
Ang II induction of premature senescence in hVSMC is dependent upon cellular oxidants, mtETC activity and mitochondrial superoxide. Quiescent cells were pre-incubated with catalase (A), NAC (B), SOD (C) or inhibitors of the mtETC, rotenone (D) or TTFA (E) or the mitochondrial scavenger mitoTEMPO (F) for 4 h prior to induction of senescence with 1×10−8 mol/L Ang II. Following treatment, senescence was assessed by staining for SA-β-gal activity. Bars represent mean+SD; n=3–5. ⁎⁎p<0.01, ⁎⁎⁎p<0.001, ⁎⁎⁎⁎p<0.0001 compared with control. †p<0.05, ††p<0.01, †††p<0.001 compared with Ang II alone.
To determine whether mitochondria were directly involved in the mechanism or contributed to the Ang II induced premature senescence of hVSMC, cells were pre-incubated with mitochondrial complex inhibitors. Ang II exposure for 24 h consistently caused a ~2-fold increase in senescent cells (Fig. 4). Pre-incubation with rotenone completely inhibited Ang II-induced senescence of hVSMC (Fig. 4D) as did incubation with the complex II inhibitor, TTFA (Fig. 4E).
Ang II-induced premature senescence is dependent upon superoxide generated by mitochondria
The data presented so far illustrate that Ang II induces NADPH-dependent in cell lysates and mt in living cells and that SIPS is dependent upon oxidant generation and mtETC activity. Further investigation into whether mitochondria stimulated by Ang II participate in the induction of senescence, was conducted using a mitochondrially-targeted oxygen radical scavenger, mito-TEMPO. Mito-TEMPO accumulates within mitochondria, where it scavenges and alkyl radicals specifically [21]. Incubation of hVSMC with a relatively low concentration of mito-TEMPO (25×10−9 mol/L) completely prevented Ang II-induced senescence, strongly implicating mitochondrial function and specifically in the rapid senescence response to Ang II.
Discussion
Here we show that mitochondria play a key role in Ang II-induced human VSMC senescence. Inhibition studies suggest that mitochondrial function, specifically mtETC activity, is important in the induction of premature senescence.
Our previous work showed that catalase inhibited Ang II-induced senescence [5] and this was not only confirmed here but extended with the application of SOD and NAC. Here, inhibition of and senescence by rotenone and the effectiveness of nanomolar concentrations of mitoTEMPO at blocking senescence induced by Ang II, implicate superoxide production by mitochondria in this mechanism. This is in agreement with the suggestion that mitoTEMPO mimicks the antioxidant effects of SOD2 in mitochondria [22] and further adds weight to the hypothesis that mitochondrial superoxide is a mediator of physiological and stress response pathways in human cells [23]. Complex I is proposed as the main source of in the mitochondrial electron transport chain, including via reverse electron transport following inhibition at complex II [24]. Therefore, the current data suggest that Complex I is an important source of superoxide generation by Ang II in hVSMC. The complex interactions between mitochondrial and NADPH oxidase generation are discussed below.
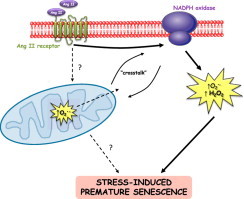
It is well described that Ang II stimulates superoxide production via activation of NADPH oxidases in vascular cells [25,26]. A mechanism for the observed mitochondrial dysfunction caused by Ang II in endothelial cells has been described involving upstream NADPH oxidase activity [27] and redox-dependent mitochondrial KATP channels which may be activated by either or H2O2 [28]. Nevertheless, there is also evidence in some cell models e.g. endothelial cells, that mitochondrial is upstream of NADPH oxidase (NOX) activity which leads to further production in the cytosolic phase [22]. In this endothelial cell model, in response to Ang II, SOD2 modulated NADPH oxidase activity in the cytosol, implying that mitochondrial superoxide was the signal for increased NOX activity. Moreover, mitoTEMPO effectively mimicked the activity of SOD2 in that it prevented Ang II-stimulated production in SOD2 depleted cells [22]. Furthermore, the ability of Ang II to mediate NADPH oxidase expression and activity in rabbit VSMC was eliminated by mtDNA depletion with ethidium bromide or inhibition of complex III by antimycin A [29]. In addition to suggesting a complex mitochondrial-NOX crosstalk [30], these findings support the contention that mitochondrial redox biology plays a key biological role in regulating important cellular processes [23]. In the current model (see Fig. 5), mitochondrial is vital for the senescence process but it is unknown whether the signal from mitochondria involves its dismutation to H2O2 and/or whether other intermediate signalling event(s) is/are required. In this respect, targeting catalase to mitochondria might be informative.
Fig. 5.
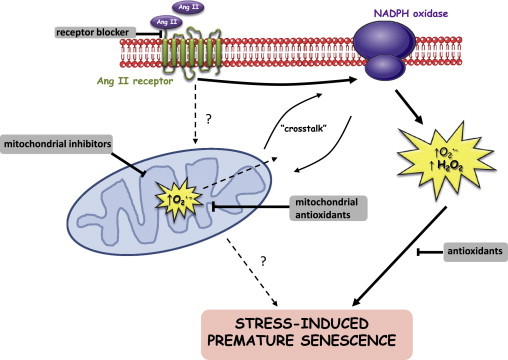
Working model for the mechanism of Ang II-induced SIPS in hVSMC. Ang II acts via the AT1 receptor to illicit superoxide production via a NADPH-linked process. Cytoplasmic oxidants, mitochondrial superoxide/mitochondrial function are necessary for the development of SIPS. This mechanism invokes NADPH oxidase-mitochondrial cross-talk via oxidant generation; the working hypothesis is that firstly NADPH oxidase is activated by Ang II which leads to stimulation of mitochondrial superoxide resulting in a feed-forward amplification of NOX activity and subsequent downstream induction of SIPS.
There are many implications of a mitochondrial pharmacology approach in biology and medicine. Mitochondrial function/signalling is a developing clinical target and mitochondria-targeted antoxidants are amongst several promising modalities [31]. Although it is some way from clinical fruition, the data presented here support the contention that this class(es) of compounds may be employed as potential therapies for targeting cells and tissues prone to aging and age-related disease with the aim of maintaining tissue/organ function and prolonging a healthy lifespan.
The present study provides the first report of SIPS in hVSMC using the specific experimental criteria developed by Toussaint et al. [2]. The current study demonstrates that short, successive exposures to either tert-BHP or Ang II, with periods of recovery between treatments, were sufficient to induce SIPS of hVSMC. Both of these mediators act via redox mechanisms, which have been intimately associated with induction of cellular senescence in several cell systems [3,13,5].
Data from the current and previous studies [5] suggest that Ang II acts via its cognate receptor, AT1, since the inhibition of the AT1 with EXP3174 prior to daily Ang II exposure blocked the senescence response in hVSMC. Complete inhibition of senescence was not attained with EXP3174 which may mean that this antagonist only partially prevents Ang II agonism under the experimental conditions employed, however an alternate mechanism for Ang II, for example via binding to the type-2 angiotensin II receptor, cannot be ruled out. Nevertheless, we favour the former explanation since in previous studies EXP3174 fully inhibited DNA damage and telomere attrition in addition to induction of senescence by Ang II [5].
Induction of senescence by Ang II was first reported in vitro in aortic VSMC and also in vivo in apoE deficient mice [13]. These in vivo analyses showed that Ang II infusion promoted staining for SA-β-gal activity within mouse aortas and co-staining for smooth muscle actin suggested that much of the senescence staining was associated with VSMC [13]. This evidence provides an important in vivo context for the observations on Ang II-induced senescence and provides a rationale for studying mechanisms in vitro. Subsequent workers [32] have highlighted the complexity of pathways which may be involved in signal transduction and may also include a significant contribution from the cytoskeleton-extra cellular matrix axis [33].
A body of work looking at the possible role of Ang II in aging in vivo has been reported [16] with the pleiotropic actions of Ang II implying several mechanisms may be involved. One initial experimental paradigm showed that lifelong feeding of ACE inhibitors to CF1 mice resulted in lifespan extension and reduced pathology usually associated with the normal aging process, at least for laboratory animals [34]. This and subsequent work by the same group has suggested that preservation of mitochondrial function is a key determinant of anti-Ang II treatments in rodent models of aging and is not dependent on blood pressure lowering [16]. It confirms that, perhaps inadvertently, these drugs are amongst the most effective antioxidant therapies currently used in man. In this respect, blockade of the renin-angiotensin system in humans is likely to affect several biological processes/pathways and provides an avenue for further exploration particularly to mitigate cardiovascular aging.
Conclusions
The data presented here demonstrate that Ang II induces true stress-induced premature senescence in human VSMC and suggest that mitochondrial is necessary for this premature aging response. Furthermore, the data confirm an Ang II receptor mediated process which is likely to involve mitochondria-NADPH oxidase crosstalk by as yet undefined signalling pathways.
Source of funding
This project was supported by a Grant from the British Heart Foundation PG/2001110. The authors are grateful to Merck & Co. Inc. for the gift of EXP3174.
Disclosures
Bryan Williams has received independent investigator-led grant support and honoraria from Merck for lectures and consultancy.
Acknowledgements
The authors wish to thank Dr Sergey I. Dikalov (Vanderbilt University) for helpful suggestions and Merck & Co Inc. for the kind gift of EXP3174. Bryan Williams is a National Institute for Health Research (NIHR) Senior Investigator and his research is supported by the NIHR University College London Hospitals Biomedical Research Centre.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Tchkonia T., Zhu Y., van Deursen J., Campisi J., Kirkland J.L. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. Journal of Clinical Investigation. 2013;123:966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Toussaint O., Medrano E.E., von Zglinicki T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Experimental Gerontology. 2000;35:927–945. doi: 10.1016/s0531-5565(00)00180-7. [DOI] [PubMed] [Google Scholar]
- 3.Dumont P., Burton M., Chen Q.M., Gonos E.S., Frippiat C., Mazarati J.B., Eliaers F., Remacle J., Toussaint O. Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radical Biology & Medicine. 2000;28:361–373. doi: 10.1016/s0891-5849(99)00249-x. [DOI] [PubMed] [Google Scholar]
- 4.Goligorsky M.S., Chen J., Patschan S. Stress-induced premature senescence of endothelial cells: a perilous state between recovery and point of no return. Current Opinion in Hematology. 2009;16:215–219. doi: 10.1097/MOH.0b013e32832a07bd. [DOI] [PubMed] [Google Scholar]
- 5.Herbert K.E., Mistry Y., Hastings R., Poolman T., Niklason L., Williams B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circulation Research. 2008;102:201–208. doi: 10.1161/CIRCRESAHA.107.158626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin-Pardillos A., Sosa C., Sorribas V. Arsenic increases Pi-mediated vascular calcification and induces premature senescence in vascular smooth muscle cells. Toxicological Sciences. 2013;131:641–653. doi: 10.1093/toxsci/kfs313. [DOI] [PubMed] [Google Scholar]
- 7.Jimenez R., Carracedo J., Santamaria R., Soriano S., Madueno J.A., Ramirez R., Rodriguez M., Martin-Malo A., Aljama P. Replicative senescence in patients with chronic kidney failure. Kidney International Supplements. 2005;99:S11–5. doi: 10.1111/j.1523-1755.2005.09903.x. [DOI] [PubMed] [Google Scholar]
- 8.Carracedo J., Buendia P., Merino A., Soriano S., Esquivias E., Martin-Malo A., Aljama P., Ramirez R. Cellular senescence determines endothelial cell damage induced by uremia. Experimental Gerontology. 2013;48:766–773. doi: 10.1016/j.exger.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Minamino T., Miyauchi H., Yoshida T., Ishida Y., Yoshida H., Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105:1541–1544. doi: 10.1161/01.cir.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- 10.Passos J.F., Saretzki G., von Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: Is there a connection? Nucleic Acids Research. 2007;35:7505–7513. doi: 10.1093/nar/gkm893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomez-Cabrera M.C., Sanchis-Gomar F., Garcia-Valles R., Pareja-Galeano H., Gambini J., Borras C., Vina J. Mitochondria as sources and targets of damage in cellular aging. Clinical Chemistry and Laboratory Medicine. 2012;50:1287–1295. doi: 10.1515/cclm-2011-0795. [DOI] [PubMed] [Google Scholar]
- 12.Nazarewicz R.R., Dikalova A., Bikineyeva A., Ivanov S., Kirilyuk I.A., Grigor'ev I.A., Dikalov S.I. Does scavenging of mitochondrial superoxide attenuate cancer prosurvival signaling pathways? Antioxidants & Redox Signaling. 2013;19:344–349. doi: 10.1089/ars.2013.5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kunieda T., Minamino T., Nishi J., Tateno K., Oyama T., Katsuno T., Miyauchi H., Orimo M., Okada S., Takamura M., Nagai T., Kaneko S., Komuro I. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation. 2006;114:953–960. doi: 10.1161/CIRCULATIONAHA.106.626606. [DOI] [PubMed] [Google Scholar]
- 14.Griendling K.K., Minieri C.A., Ollerenshaw J.D., Alexander R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circulation Research. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 15.Touyz R.M., Chen X., Tabet F., Yao G., He G., Quinn M.T., Pagano P.J., Schiffrin E.L. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circulation Research. 2002;90:1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 16.de Cavanagh E.M., Inserra F., Ferder L. Angiotensin II blockade: a strategy to slow ageing by protecting mitochondria? Cardiovascular Research. 2011;89:31–40. doi: 10.1093/cvr/cvq285. [DOI] [PubMed] [Google Scholar]
- 17.Dimri G.P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E.E., Linskens M., Rubelj I., Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proceedings of the National Academy of Sciences USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chainiaux F., Magalhaes J.P., Eliaers F., Remacle J., Toussaint O. UVB-induced premature senescence of human diploid skin fibroblasts. International Journal of Biochemistry & Cell Biology. 2002;34:1331–1339. doi: 10.1016/s1357-2725(02)00022-5. [DOI] [PubMed] [Google Scholar]
- 19.Ben-Porath I., Weinberg R.A. The signals and pathways activating cellular senescence. International Journal of Biochemistry & Cell Biology. 2005;37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 20.Touyz R.M., Schiffrin E.L. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. Journal of Hypertension. 2001;19:1245–1254. doi: 10.1097/00004872-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 21.Murphy M.P., Smith R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annual Review of Pharmacology and Toxicology. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 22.Dikalova A.E., Bikineyeva A.T., Budzyn K., Nazarewicz R.R., McCann L., Lewis W., Harrison D.G., Dikalov S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circulation Research. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamanaka R.B., Chandel N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends in Biochemical Sciences. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy M.P. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lassegue B., Sorescu D., Szocs K., Yin Q., Akers M., Zhang Y., Grant S.L. Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circulation Research. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 26.Mehta P.K., Griendling K.K. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. American Journal of Physiology—Cell Physiology. 2007;292:C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 27.Doughan A.K., Harrison D.G., Dikalov S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circulation Research. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 28.Queliconi B.B., Wojtovich A.P., Nadtochiy S.M., Kowaltowski A.J., Brookes P.S. Redox regulation of the mitochondrial K(ATP) channel in cardioprotection. Biochimica et Biophysica Acta. 2011;1813:1309–1315. doi: 10.1016/j.bbamcr.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wosniak J., Jr., Santos C.X., Kowaltowski A.J., Laurindo F.R. Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in nox isoform expression and activity in vascular smooth muscle cells. Antioxidants & Redox Signaling. 2009;11:1265–1278. doi: 10.1089/ars.2009.2392. [DOI] [PubMed] [Google Scholar]
- 30.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radical Biology & Medicine. 2011;51:1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith R.A., Hartley R.C., Cocheme H.M., Murphy M.P. Mitochondrial pharmacology. Trends in Pharmacological Sciences. 2012;33:341–352. doi: 10.1016/j.tips.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Min L.J., Mogi M., Iwai M., Horiuchi M. Signaling mechanisms of angiotensin II in regulating vascular senescence. Ageing Research Reviews. 2009;8:113–121. doi: 10.1016/j.arr.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 33.de Cavanagh E.M., Ferder M., Inserra F., Ferder L. Angiotensin II, mitochondria, cytoskeletal, and extracellular matrix connections: an integrating viewpoint. American Journal of Physiology. Heart and Circulatory Physiology. 2009;296:H550–8. doi: 10.1152/ajpheart.01176.2008. [DOI] [PubMed] [Google Scholar]
- 34.Ferder L., Inserra F., Romano L., Ercole L., Pszenny V. Effects of angiotensin-converting enzyme inhibition on mitochondrial number in the aging mouse. American Journal of Physiology. 1993;265:C15–8. doi: 10.1152/ajpcell.1993.265.1.C15. [DOI] [PubMed] [Google Scholar]