

Abstract
Mouse cytochrome P450 2A5 (CYP2A5) is upregulated in various liver diseases and a putative common feature for all of these conditions is altered cellular redox status. Nuclear factor erythroid 2-like 2 (Nrf2) is a transcription factor that is post-translationally regulated by oxidative stress and controls the transcription of protective target genes. In the present study, we have characterized the regulation of CYP2A5 by Nrf2 and evaluated gene expression, protein content and activity of anti-oxidant enzymes in the Nrf2+/+ and Nrf2−/− mice model of non-alcoholic fatty liver (NAFLD). After eight weeks of feeding on a high-fat diet, livers from Nrf2−/− mice showed a substantial increase in macro and microvesicular steatosis and a massive increase in the number of neutrophil polymorphs, compared to livers from wild-type mice treated similarly. Livers of Nrf2−/− mice on the high-fat diet exhibited more oxidative stress than their wild-type counterparts as assessed by a significant depletion of reduced glutathione that was coupled with increases in malondialdehyde. Furthermore, results in Nrf2-deficient mice showed that CYP2A5 expression was significantly attenuated in the absence of Nrf2, as was found with the conventional target genes of Nrf2. The treatment of wild-type mice with high-fat diet leaded to nuclear accumulation of Nrf2, and co-immunoprecipitation experiments showed that Nrf2 was bound to Cyp2a5. These findings suggest that the high-fat diet induced alteration in cellular redox status and induction of CYP2A5 was modulated through the redox-sensitive transcription Nrf2.
Abbreviations: ARE, Antioxidant response element; Nrf2, Nuclear factor erythroid 2-like 2; KO, Knockout; WT, Wild-type; P450, Cytochrome P450; NASH, Nonalcoholic steatohepatitis; NAFLD, Nonalcoholic fatty liver disease; co-IP, Co-immunoprecipitation
Keywords: Coumarin 7-hydroxylase, Oxidative stress, glutathione S-transferases, High-fat diet, Knockout mice
Graphical abstract
Highlights
-
•
CYP2A5 up-regulation in response to NAFLD was Nrf2 dependent.
-
•
NAFLD induces oxidant stress.
-
•
A protective role for Nrf2 against hepatic damage by NAFLD was demonstrated.
-
•
NAFLD induces translocation of Nrf2 from the cytoplasm to the nucleus.
-
•
Nrf2 binding to CYP2a5 was shown.
Introduction
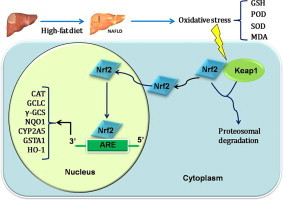
Nonalcoholic fatty liver disease (NAFLD) is composed of a spectrum of pathologies ranging from simple fatty liver (steatosis) to the more severe nonalcoholic steatohepatitis (NASH). The proposed mechanism for progression of NAFLD involves a two-hit theory where lipid accumulation in hepatocytes (the“first hit”) is followed by a “second hit,” including insulin resistance, oxidative stress, and cytokine production [1]. The capacity of a cell to manage oxidative stress is primarily mediated through antioxidant response elements or electrophile response elements (AREs), which are largely under the control of the transcription factor nuclear factor erythroid 2-like 2 (Nrf2) [2,3]. Nrf2 is primarily a cytosolic protein due to proteasomal degradation regulated by Keap1 [4]. When a cell is undergoing oxidative stress, Nrf2 escapes Keap1-mediated degradation, translocates to the nucleus, actives a battery of redox-homeostasis regulatory genes such as γ-glutamyl cysteine ligase (γ-GCS), and phase II genes such as glutathione S-transferases (GST) and NAD(P)H:quinone oxidoreductase1 (NQO1) [5]. Activation of Nrf2 plays a crucial role in regulating the cytoprotective responses to a variety of drugs, toxicants, and cellular stresses.
CYP2A5 belongs to the group of P450 enzymes involved in the metabolism of xenobiotics, such as drugs and environmental toxicants [6]. Murine CYP2A5 and its human orthologue CYP2A6 are the main catalysts of coumarin and nicotine metabolism but have also been shown to participate in metabolic activation of procarcinogens, such as aflatoxin B1 and nitrosamines [7,8]. A number of studies have indicated high expression of CYP2A5 in mouse hepatomas and preneoplastic liver lesions; therefore, CYP2A5 has been suggested to be a useful marker for hepatocarcinogenesis [9–12]. Additionally, CYP2A5 has been found to be up-regulated in several types of hepatic infections of viral, bacterial, and parasitic origin, which are associated with an increased risk of hepatocarcinogenesis [13–16]. The mechanistic basis behind these elevated expression levels has remained enigmatic. A possible explanation for these apparently unrelated effects on CYP2A5 expression could be involvement of a common indirect mechanism.
We previously found that mice fed with high-fat diet resulted in an increase in expression of Nrf2 and CYP2A5 in liver. We hypothesized that feeding a high-fat diet would cause an elevation of ROS, which increases Nrf2 levels, and the elevated Nrf2 up-regulates CYP2A5. To test this hypothesis, we have characterized a high-fat diet induced model of NAFLD and evaluated gene expression and protein activity of anti-oxidant enzymes in Nrf2+/+ and Nrf2−/− mice.
Materials and methods
Chemicals and antibodies
All reagents were obtained from Sigma Chemical Company (St. Louis, MO, USA) unless otherwise indicated. Rabbit IgG (sc-2027) and rabbit polyclonal anti-Nrf2 (sc-13032), were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Polyclonal chicken Cyp2a5 antiserum was a kind gift from Dr. H. Raunio (University of Kuopio, Kuopio, Finland).
Animal treatment
Six to eight-week-old, ICR male wild-type (Nrf2+/+) and Nrf2 knockout mice (Nrf2−/−) were purchased from the Animal Experiment Center of General Hospital of Nanjing Military Area Command [Certification no. SCXK (JUN) 2007-012]. They were divided into four groups of three mice in each group and housed in filter-top polycarbonate cages containing wood chip bedding and maintained in a 12-h light/dark cycle with free access to standard mouse chow and tap water. After a one-week adaptive period, the four groups of mice were treated for eight consecutive weeks. The Nrf2+/+ and Nrf2−/− control groups were fed with the standard diet. The Nrf2+/+ and Nrf2−/− high-fat model groups were fed with a high-fat diet (68.5% standard diet, 15% lard, 1% cholesterol, 0.5% bile and 15% dextrin) which induces NAFLD (data not shown). The mice in all groups were sacrificed by CO2 overdose. At the time of death, blood was collected from the thoracic aorta. The serum was separated from the cellular elements by centrifugation. The livers were removed, rinsed in ice-cold saline, and divided for various assays, as outlined below. All the experimental procedures were approved by, and conducted in accordance with, the Institutional Animal Use and Care Committee of the Northeast Agricultural University.
Liver histological examination
Fresh liver tissue pieces from four groups of mice were fixed in 10% neutral formalin, embedded in paraffin, sliced and stained with H&E. The slides were examined by a pathologist to detect the presence of fat, necrosis, fibrosis and inflammation. The pathologist scored them from 0 to 4 using the standards proposed by Dixon for assessing changes in fat and inflammation [17]. The sum of the fat, necrosis, inflammation and fibrosis scores was termed the total histological score. The presence of NAFLD in mice was diagnosed using standard criteria [18].
Biochemistry analysis
Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), triglycerides (TG), glucose (GLU), total protein (TP), high-density lipoprotein (HDL), low-density lipoprotein (LDL) and total cholesterol (TC) were determined using a Beckman CX4 automatic biochemical analyzer (Beckman Colter, Inc., USA).
Measurement of reduced glutathione levels
Liver homogenate was mixed with trichloroacetic acid (TCA) to a final concentration of 5%, and the mixture was incubated at 4 °C for 30 min to extract glutathione (GSH). The TCA extracts (10 μl) were added to 200 μl of methanol containing 1 mg/ml o-phthaldehyde and then were incubated for 15 min at 37 °C in the dark. Fluorescence was measured at 350/420 nm (excitation/emission). The concentration of GSH was determined from a GSH standard curve [19].
Lipid peroxidation in livers
Lipid peroxidation in liver was monitored by quantifying thiobarbituric acid reactive substances (TBARS). About 25 mg of liver was homogenized in 2 fold volumes of ice-cold PBS, and TBARS were determined via the OXltek TBARS kit (ZeptoMetrix, Buffalo, NY). Sample homogenates, as well as malondialdehyde (MDA) standards, were incubated with sodium dodecyl sulfate and thiobarbituric acid at 95 °C for 1 h. After incubation, samples were cooled on ice and centrifuged at 1800g for 15 min. Supernatants (200 μl) were transferred to a 96-well plate and quantified at 532 nm. MDA equivalents were expressed as nmol of MDA equivalents per gram liver.
Preparation of hepatic microsomes.
Hepatic microsomes were prepared by placing liver aliquots in 0.15 M KCl and homogenized in a polytron homogenizer for 10 strokes. The homogenate was centrifuged at 9000g for 20 min, and then the resulting supernatant fraction was centrifuged further at 105,000g for 60 min. The resulting pellets (microsomes) were re-suspended in 50 mM sodium phosphate buffer (pH 7.4). All procedures were carried out on ice.
Real-time qPCR
Differences in gene expression levels were quantitated using Brilliant SYBR Green QPCR Master Mix (Stratagene, La Jolla, CA) and Mx3005P Q-PCR system (Stratagene). Analyses were done by the comparative Ct (ΔΔCt) method, wih fluorescence correction by the passive reference dye ROX and subsequent normalization to reference gene GAPDH. Sequences for primers used in quantitation of gene expression are listed in Table 1.
Table 1.
Primer sequences used in mRNA quantitation by reverse transciption-polymerase chain reaction.
| Gene | Forward primers | Reverse primers |
|---|---|---|
| Cyp2a5 | 5′–GGACAAAGAGTTCCTGTCACTGCTTC-3′ | 5′–GTGTTCCACTTTCTTGGTTATGAAGTCC-3′ |
| Gclc | 5′–TTTGAGAACTCTGCCTATGTGGT-3′ | 5′–ATAAAACATCCCCTGCAAGACA-3′ |
| r-GCS | 5′–ATGCGGTGGTGCTACTGATTG-3′ | 5′–CATCTCGGAAGTACACCACAG-3′ |
| Catalase | 5′–ACATGGTCTGGGACTTCTGG-3′ | 5′–CAAGTTTTTGATGCCCTGGT-3′ |
| Ngo1 | 5′–CGCCTCGAGGCCTCTGAATACTTTCAACAA-3′ | 5′–GCGAAGCTTTCGGAGAGATCCTTAGGGCTG-3′ |
| Gsta1 | 5′–CCCCTTTCCCTCTGCTGAAG-3′ | 5′–TGCAGCTTCACTGAATCTTGAAAG-3′ |
| Ho-1 | 5′–CACGCATATACCCGCTACCT-3′ | 5′–CCAGAGTGTTCATTCGAGCA-3′ |
| GAPDH | 5′–GCACCACCAACTGCTTAGCCCCCCTG-3′ | 5′–CACAAACATGGGGGCATCGGCAGAAG-3′ |
Isolation of nuclear and cytoplasmic proteins from mouse liver
Nuclear and cytoplasmic extracts were prepared with nuclear and cytoplasmic protein extraction kit according to their protocols (Beyotime Institute of Biotechnology, Beijing, China). Protein concentrations were determined with the BCA Assay Kit from Beyotime Biotechnology. Briefly, approximately 0.2 g of mouse liver was homogenated with 2 ml of ice cold hypotonic buffer A and buffer B (20:1). The homogenate was incubated at 4 °C for 15 min and centrifuged at 12,000g for 10 min at 4 °C. The supernatant was obtained as cytoplasmic extract and the residue was added with 0.2 ml of hypertonic buffer C. The pellet suspension was shaken gently for 30 min at 4 °C and then centrifuged at 12,000g 10 min at 4 °C. The supernatant was obtained as nuclei extract. PMSF and leupeptin were added into buffer A and buffer C before use.
Immunoprecipitation
Liver fractions were extracted as described above. Sufficient amount of Nrf2 antibody or Cyp2a5 antibody or mouse IgG negative control was added to 200 μg protein and gently rotated at 4 °C overnight. The immunocomplex was captured by adding 25 μl protein A+G agarose beads (Beyotime, Beijing, China) and gently rotating at 4 °C for 3 h. Then the mixture was centrifuged at 2500g for 5 min at 4 °C and the supernatant was discarded. The precipitate was washed for three times with ice-cold RIPA buffer, resuspended in 3×sample buffer and boiled for 5 min to dissociate the immunocomplex from the beads. The supernatant was collected by centrifugation and subjected to SDS-PAGE as descriped followed by western blotting with Cyp2a5 or Nrf2 antibody.
Western blot
Protein levels were determined by Western blotting. Solubilized sample suspensions were boiled for 5 min, and proteins (20 μg of liver microsomal proteins for detection of CYP2A5 proteins; 15 μg of cytosolic or nuclear protein for detection of Nrf2 protein) were loaded and separated using SDS-polyacrylamide gel electrophoresis (12%), electrophoretically transferred to nitrocellulose/polyvinylidene difluoride membranes (Pierce Biotechnology, Rockford, IL/Bio-Rad Laboratories, Hercules, CA), and blocked for 1 h in phosphate-buffered saline containing Tween 20 (0.1%) and nonfat milk (5%). Blots were incubated with CYP2A5 (1:1000 dilution), or Nrf2 (1:500 dilution) antibody for 3 h. Membranes were then incubated for 1 h with horseradish peroxidase-conjugated goat anti-rabbit (1:5000 dilution) or goat anti-mouse (1:5000 dilution) antibody. After further washing with phosphate-buffered saline, blots were incubated in commercial chemoluminescence reagents (Amersham Biosciences). Band intensities were measured using Quantity One software (Bio-Rad, Hercules, CA).
Data analysis
Data was analyzed by the SAS 13.0 (The SAS Institute, Cary, NC). Differences in all of the measured endpoints between the control and the treatment groups were compared by ANOVA and GLM Model of the SAS.
Results
Serological profiles
Experiments were performed to determine the effect of eight weeks high-fat diet on hepatocellular integrity in Nrf2+/+ mice and in the Nrf2−/− mice (Table 2). The mice (Nrf2+/+ or Nrf2−/−) with high-fat diet had significantly higher TC, LDL, GLU, ALT, AST (P<0.01), and ALP levels (P<0.05) than did the mice in the control group. However, the TP levels were significantly lower in the mice with high-fat diet than mice in the control (P<0.01). Mice in the Nrf2−/−control group exhibited significantly different levels of TC, HDL, TP, ALT and ALP (P<0.05 or P<0.01) than the mice in the Nrf2+/+ control group. Similarly, the mice in the Nrf2−/− high-fat model group exhibited significantly different levels of TG, TC, HDL, LDL, TP, ALT, AST and ALP (P<0.05 or P<0.01) compared to the mice in the Nrf2+/+ high-fat group.
Table 2.
Serum characteristics of WT and KO mice with or without NAFLD.
| Parameters | Controls(Nrf2+/+) | NAFLD(Nrf2+/+) | Controls(Nrf2−/−) | NAFLD(Nrf2−/−) |
|---|---|---|---|---|
| TG (mmo/L) | 0.91±0.14 | 0.78±0.15 | 1.06±0.11 | 1.23±0.23† |
| TC (mmo/L) | 2.39±0.39 | 11.33±1.39⁎⁎ | 5.30±0.42†† | 7.81±0.23⁎⁎† |
| HDL (mmo/L) | 1.29±0.20 | 1.44±0.10 | 2.75±0.51† | 2.81±0.18†† |
| LDL (mmo/L) | 0.26±0.6 | 2.12±0.28⁎⁎ | 0.2±0.04 | 0.88±0.05⁎⁎†† |
| TP (g/L) | 1.90±0.10 | 1.01±0.19⁎⁎ | 1.56±0.05†† | 1.04±0.07⁎⁎ |
| GLU (mmo/L) | 4.90±0.40 | 8.53±0.46⁎⁎ | 5.16±0.15 | 8.57±0.30⁎⁎ |
| ALT (IU/L) | 71.33±10.21 | 135.67±10.97⁎⁎ | 45.67±8.08† | 165.33±14.98⁎⁎† |
| AST (IU/L) | 238.33±15.89 | 439±44.16⁎⁎ | 223.67±13.32 | 336.33±13.65⁎⁎† |
| ALP (IU/L) | 101.33±10.02 | 192.33±19.13⁎ | 205±7.21†† | 430.67±22.03⁎⁎†† |
The data are expressed as the mean ±SD (n=3 per treatment group). †Statistically different from wild type on the same diet; ⁎statistical difference caused by the high-fat diet within each genotype. ⁎P<0.05, ⁎⁎P<0.01; †P<0.05, ††P<0.01. ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase; TG, triglyerides; TC, total cholesterol; TP, total protein; HDL, high-density lipoprotein; LDL, low-density lipoprotein; NAFLD, nonalcoholic fatty liver disease.
Histological profile
All of the sections in the experimental groups exhibited diffuse hepatic steatosis (Fig. 1) under a light microscope, whereas no fatty liver was observed in the control group. The relative sizes of the hepatic cell nuclei were uneven. Hepatic steatosis (mostly microvesicular and macrovesicular mixed steatosis) was most obvious around the portal area and was accompanied by liver cell necrosis and inflammatory cell infiltration. The lobular and portal areas exhibited considerably more inflammatory cell infiltration in the experimental group than in the control group. The total histological scores of the livers in the model-group mice reached grade 2 or 3. Livers from WT-high-fat mice showed a minimal degree of microvesicular steatosis. Most significantly, livers from Nrf2 KO-high-fat mice exhibited severe macrovesicular steatosis and mild microvesicular steatosis, indicating that diffuse hepatic steatosis with moderate inflammation (NAFLD) had developed.
Fig. 1.
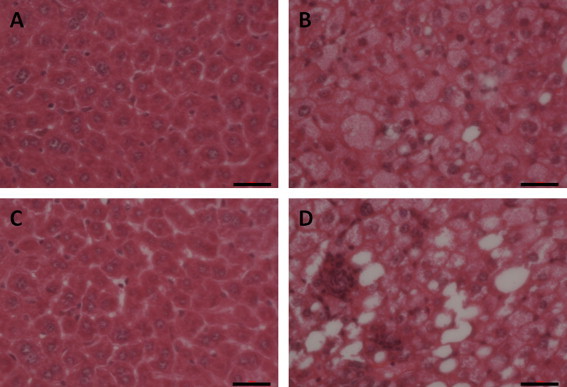
NAFLD models in WT and KO mice were established by feeding with high-fat diet for eight consecutive weeks. Fresh sections were stained with H&E to demonstrate lipid accumulation. The sections were examined by light microscopy, and the liver images are displayed at 200× original magnification: (A) Nrf2+/+ mice on control diet; (B) Nrf2+/+ mice on high-fat diet; (C) Nrf2−/− mice on control diet; (D) Nrf2−/− mice on high-fat diet. The Nrf2+/+ and Nrf2−/− high-fat model groups liver sections exhibited severe hepatosteatosis consisting of mixed microvesicular and macrovesicular fat accumulation. The horizontal scale bar represents 100 μm.
Hepatic antioxidant capability and lipid peroxidation
The levels of oxidative liver damage were assessed by GSH, SOD, POD and MDA levels (Fig. 2). In WT and KO mice fed with high-fat diet, hepatic GSH and SOD levels were significantly lower (P<0.05) than the mice in control group, while, WT and KO mice fed with high-fat diet had higher (P<0.05) POD and MDA concentrations than mice fed with normal diet. Compared with WT mice, KO mice MDA levels increased by 49% when fed with high-fat diet (P<0.05). GSH, SOD and POD levels in WT and KO mice were not significantly changed when fed with high-fat diet, while fed with normal diet, SOD, POD and MDA levels in KO mice were significantly higher (P<0.05 or P<0.01) than in WT mice, but GSH was significantly lower (P<0.01).
Fig. 2.
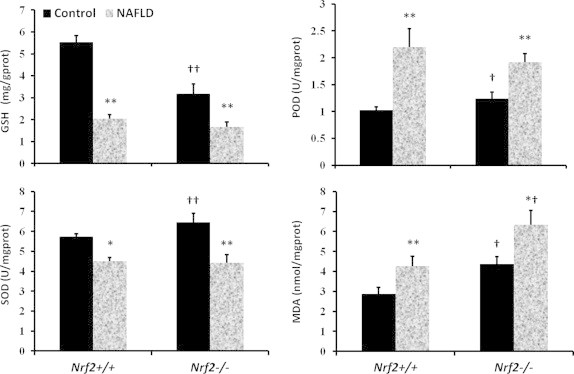
NAFLD increased liver antioxidant and lipid peroxidation capacities in Nrf2−/− mice. Glutathione (GSH) and Superoxide dismutase (SOD) concentrations were quantified by colorimetric method to indicate the antioxidant capability of livers from mice expressing various amount of Nrf2. Peroxidase (POD) and Malondialdehyde (MDA) levels were quantified as a measure of lipid peroxidation in mouse livers. The data are expressed as the mean ±SD (n=3 per treatment group). †Statistically different from wild type on the same diet; ⁎statistical difference caused by the high-fat diet within each genotype.⁎P<0.05, ⁎⁎P<0.01; †P<0.05, ††P<0.01.
Effects of the high-fat diet on the mRNA expression of Nrf2 target genes
Messenger RNA expression of several Nrf2 target genes is shown in Fig. 3. The expression of γ-GCS, Nqo1, GSTA1and HO-1 mRNA was induced by the high-fat diet in Nrf2+/+ and Nrf2−/−mice. However, no induction of CAT and GCLC was observed in KO mice fed with the high-fat diet. Furthermore, in KO mice fed with high-fat diet all of the Nrf2 target genes expression were significantly lower (P<0.05 or P<0.01) than the WT mice fed with high-fat diet.
Fig. 3.
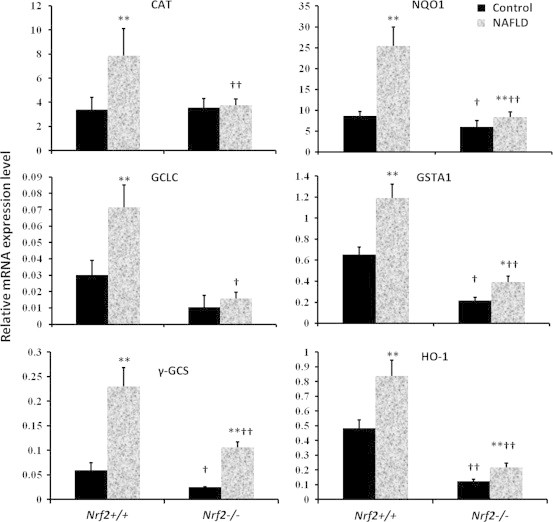
NAFLD reduced mRNA expression of Nrf2 target genes in Nrf2−/−mice. CAT, catalase; GClC, glutamate cysteine ligase catalytic; γ-GCS, γ-glutamylcysteine synthetase; Nqo1, NAD(P)H:quinine oxidoreductase 1; GSTA1, glutathione S-transferase alpha 1 gene; HO-1, heme oxygense-1. The mRNA expressions of various genes were quantified by RT-PCR, using mRNA GAPDH as an internal control. The data are expressed as the mean±SD (n=3 per treatment group). †Statistically different from wild type on the same diet; ⁎statistical difference caused by the high-fat diet within each genotype.⁎P<0.05, ⁎⁎P<0.01; †P<0.05, ††P<0.01.
Effect of high-fat diet on cellular distribution of Nrf2
Cellular localization of Nrf2 protein was determined by Western blotting. As shown in Fig. 4, a significant decrease in Nrf2 protein levels in the cytoplasm was observed after treatment with high-fat diet. However, Nrf2 protein levels in the nucleus were higher in high-fat diet group mice livers than that in control mice livers. These results suggested that the high-fat diet induced a translocation of Nrf2 from the cytoplasm to the nucleus.
Fig. 4.
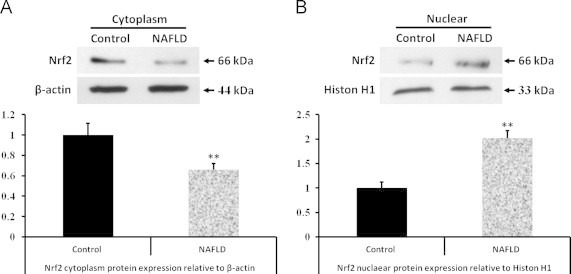
NAFLD induces a translocation of Nrf2 from cytoplasma to nuclear in WT mice liver. (A) Western blot analysis of cytosolic proteins from wild-type control and high-fat diet group mice livers, and quantitated protein band intensities presented as β-actin normalized mean values (n=3)±SD. (B) Western blot analysis of nuclear proteins from wild-type control and high-fat diet group mice livers, and quantitated protein band intensities presented as Histon H1 normalized mean values (n=3) ±SDs. Mean difference is significant from control group at ⁎⁎, P<0.01.
Effects of high-fat diet on Cyp2a5 expression in Nrf2+/+ and Nrf2−/− mice
To evaluate the role of Nrf2 in high-fat diet induced CYP2A5, levels and activities of CYP2A5 in livers from WT and KO mice were assayed (Fig. 5). Expression of CYP2A5 was increased by 23% in WT mice fed with high-fat diet, but CYP2A5 expression was increased by 13% in KO mice fed with the high-fat diet compared with the mice in the control group (Fig. 5B). CYP2A5 mRNA induction by high-fat diet was comparable (73% vs. 20%) in WT mice and Nrf2−/− mice (Fig. 5C). CYP2A5 activity was increased by 35% in WT mice fed with the high-fat diet but 16% in KO mice (Fig. 5D). Furthermore, induction of CYP2A5 by high-fat diet in Nrf2−/− Mice was lower compared to the WT mice. These results suggested that Nrf2 plays a modulatory role in the high-fat diet induction of CYP2A5.
Fig. 5.
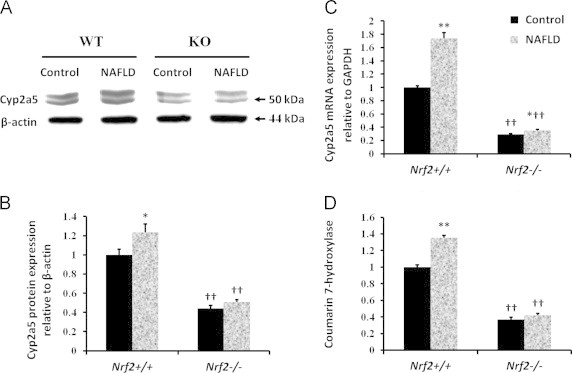
CYP2A5 induction by NAFLD requires Nrf2. (A) Western blot analysis of microsomal proteins extracted from Nrf2+/+ and Nrf2−/− livers. (B) Protein blot densities were quantified by Quantity One 1-D Analysis Software. Cyp2a5 protein was normalized to the loading control β-actin. (C) The mRNA expression of Cyp2a5 was quantified by RT-PCR, using mRNA GAPDH as an internal control. (D) COH activities are in pmol of product formed/min per mg of microsomal protein. All values represent the mean±SD (n=3 per treatment group). †Statistically different from wild type on the same diet; ⁎statistical difference caused by the high-fat diet within each genotype.⁎P<0.05, ⁎⁎P<0.01; †P<0.05, ††P<0.01.
The interaction between Nrf2 and Cyp2a5
As shown in Fig. 6, Cyp2a5 protein was detected in the protein complex from the imunoprecipitation of cell lysates using an Nrf2 antibody (Fig. 6A), and Nrf2 protein also was detected in the imunoprecipitations of Cyp2a5 (Fig. 6B). Furthermore, in the high-fat diet group both Nrf2 and Cyp2a5 protein expression were higher significantly (P<0.05 or P<0.01) than in control group mice. There was no co-immunoprecipitation of Nrf2 with Cyp2a5 in the absence of antibody.
Fig. 6.
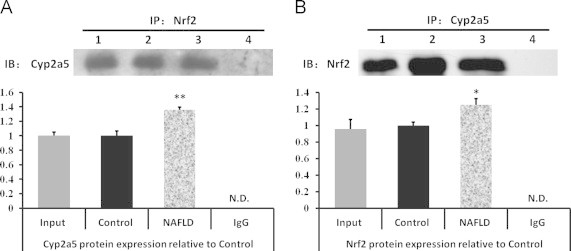
Nrf2 protein expresses along with Cyp2a5 protein interactively in WT mice and NAFLD induces the two protein expression. (A) Co-immunoprecipitated with anti-Nrf2 and Western blot with anti-Cpy2a5 showing expression of Cyp2a5. (B) Co-immunoprecipitated with anti-Cpy2a5 and Western blot with anti-Nrf2 showing expression of Nrf2. Relative amount of the co-immunoprecipitation complex Nrf2 and Cyp2a5 valuated by the ratio of band optical density. All values represent the mean±SD (n=3) against the control levels. Mean differences are significant from control group at P<0.05(⁎) and P<0.01(⁎⁎). (1) liver extracts (input) were used as positive control; (2) immunoprecipitates from wild-type control group mice livers; (3) immunoprecipitates from high-fat diet group mice livers; (4) immunoprecipitates with IgG used as negative control; IP, co-immunoprecipitation; IB: immunoblot.
Discussion
In this study we have shown the expected effects of high-fat diet on hepatic steatosis, and induced hepatocyte necrosis and inflammatory cell infiltration was more severe in the Nrf2−/− mice compared to the wild type Intact Nrf2/ARE signaling has been demonstrated to effectively prevent hepatotoxicity in mice against compounds such as acetaminophen [22], ethanol [23], and pyrazole [24] and this can now be extended to the high fat diets. These data are consistent with previous studies which have shown hepatic mitochondrial dysfunction, hypoxia and increased oxidative and nitrative stress in response to high fat diets [25], An important mechanism by which Nrf2 helps to maintain cellular redox status is through regulating glutathione synthesis [26]. In the current study, the Nrf2 signaling pathway was activated by the high-fat diet as indicated by increased nuclear accumulation of Nrf2 proteins and the induction of Nrf2-target genes (Figs. 3 and 4). Reduced glutathione (GSH) represents a major intracellular antioxidant that protects against oxidative stress and the Nrf2−/− mice showed lower levels of GSH and several other ARE regulated genes (Figs. 2 and 3) [27,28]. The impairment in antioxidant defenses was associated with increased levels in the Nrf2−/− mice under both basal conditions and consumption of the high fat diet.
Interestingly, Cyp2a5 is upregulated in various pathophysiological liver diseases and induced by structurally variable hepatotoxic chemicals [20]. A putative common feature for all of these conditions is altered cellular redox status. Nrf2 is a transcription factor that is post-translationally regulated by oxidative stress and controls transcription of numerous protective target genes. We hypothesized that the magnitude of Cyp2a5 expression is tightly connected with the hepatic protein level of Nrf2 in NAFLD model mice, which could explain high CYP2A5 levels in pathophysiological conditions involving oxidative stress. Our data suggested that Nrf2−/− mice suffer oxidative stress on the high-fat diet, as evidenced by increases in hepatic POD and MDA. It seems plausible that the inability of Nrf2−/− mice to up-regulate their antioxidant defenses when placed on an high-fat diet results in lipid peroxidation and that this in turn leads to increased inflammatory cell infiltration and a strongly pronounced inflammatory response. Presumably the increased lipid burden coupled with an inability to maintain redox homeostasis facilitates the development of NAFLD in the livers of mutant animals on the high-fat diet. It has been reported that lipid peroxidation represents an important event in the etiology of NAFLD [21], but further work is still required to determine its precise contribution to the rapid onset of NAFLD in Nrf2−/− mice on the high-fat diet.
Xenobiotic metabolizing P450s may produce reactive metabolites and sometimes ROS via enzymatic uncoupling. For example, induction of CYP2E1 by ethanol is one of the central pathways by which ethanol generates a state of oxidative stress in hepatocytes [29]. Our current findings clearly show that Cyp2a5 is modulated by Nrf2. Furthermore, our finding that Nrf2is bound to Cyp2a5 in the liver might suggest some yet unidentified redox regulatory function for CYP2A5. The putative role of CYP2A5 in antioxidant defense is unclear at this moment. Interestingly, observations by Abu-Bakar et al. [30], proposed a role for CYP2A5 in oxidative metabolism of cellular bilirubin in conditions of excess production. Thus, CYP2A5 could participate in controlling the cellular levels of this potentially toxic compound with antioxidant properties.
Nrf2 regulates primarily an adaptive genetic survival program that may, however, be harnessed by cancerous cells to promote carcinogenesis and avoid apoptosis. In fact, increased Nrf2 activation and upregulation of several target genes has been detected in various cancer cell lines and tumors [31]. This might explain why elevated CYP2A5 expression is often detected in hepatocellular carcinomas [9–12]. Detection of elevated expression of CYP2A5 in liver tumors has resulted in the idea that this P450 enzyme, capable of activating some hepatic carcinogens, could promote carcinogenesis. However, regulation by Nrf2 suggests that upregulation of CYP2A5 could also be a consequence than a cause of tumor formation. Elevated expression of the orthologous human isoform CYP2A6 has been associated with chronic inflammation, and elevated CYP2A6 colocalizes with the inflamed areas in human hepatocellular carcinomas [8,32,33]. A relevant question is whether regulatory similarities between mouse CYP2A5 and human CYP2A6 exist in relation to Nrf2-mediated signaling. Interestingly, a positive correlation between hepatic CYP2A6 activity and environmental cadmium exposure was found in an epidemiological study on a healthy Thai population [34], supporting possible involvement of Nrf2 also in regulation of CYP2A6. In conclusion, our current results suggest that expression of CYP2A5 is modulated by Nrf2 in response to a high fat diet.
Acknowledgments
We thank Dr. H. Raunio (University of Kuopio, Kuopio, Finland) for generously providing antibodies against Cyp2a5. This work was supported by the Natural Science Foundation of China, Grant no. 31172369.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Bellentani S., Bedogni G., Miglioli L., Tiribelli C. The epidemiology of fatty liver. European Journal of Gastroenterology and Hepatology. 2004;16:1087–1093. doi: 10.1097/00042737-200411000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., Yamamoto M., Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochemical and Biophysical Research Communications. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 3.Jaiswal A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radical Biology and Medicine. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 4.Zhang D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metabolism Reviews. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 5.Copple I.M., Goldring C.E., Kitteringham N.R., Park B.K. The Nrf2-Keap1 defence pathway: role in protection against drug-induced toxicity. Toxicology. 2008;246:24–33. doi: 10.1016/j.tox.2007.10.029. [DOI] [PubMed] [Google Scholar]
- 6.Jana S., Paliwal J. Molecular mechanisms of cytochrome p450 induction: potential for drug–drug interactions. Current Protein and Peptide Science. 2007;8:619–628. doi: 10.2174/138920307783018668. [DOI] [PubMed] [Google Scholar]
- 7.Pelkonen P., Lang M.A., Negishi M., Wild C.P., Juvonen R.O. Interaction of aflatoxin B1 with cytochrome P450 2A5 and its mutants: correlation with metabolic activation and toxicity. Chemical Research in Toxicology. 1997;10:85–90. doi: 10.1021/tx960078m. [DOI] [PubMed] [Google Scholar]
- 8.Raunio H., Juvonen R., Pasanen M., Pelkonen O., Paakko P., Soini Y. Cytochrome P4502A6 (CYP2A6) expression in human hepatocellular carcinoma. Hepatology. 1998;27:427–432. doi: 10.1002/hep.510270217. [DOI] [PubMed] [Google Scholar]
- 9.Kobliakov V., Kulikova L., Samoilov D., Lang M.A. High expression of cytochrome P450 2a-5 (Coumarin 7-Hydroxylase) in mouse hepatomas. Molecular Carcinogenesis. 1993;7:276–280. doi: 10.1002/mc.2940070411. [DOI] [PubMed] [Google Scholar]
- 10.Jounaidi Y., Bonfils C., Perin F., Negishi M., Lange R. Overexpression of a cytochrome P-450 of the 2a family (Cyp2a-5) in chemically induced hepatomas from female mice. European Journal of Biochemistry. 1994;219:791–798. doi: 10.1111/j.1432-1033.1994.tb18559.x. [DOI] [PubMed] [Google Scholar]
- 11.Takagi H., Aida K., Sohara N., Mori M., Merlino G., Negishi M. Steroid hormone-dependent overexpression of cytochromes P450 2A in liver tumors of TGF alpha transgenic male mice. Journal of Gastroenterology. 1997;32:708–711. doi: 10.1007/BF02934127. [DOI] [PubMed] [Google Scholar]
- 12.Wastl U.M., Rossmanith W., Lang M.A., Camus-Randon A.M., Grasl-Kraupp B., Bursch W., Schulte-Hermann R. Expression of cytochrome P450 2A5 in preneoplastic and neoplastic mouse liver lesions. Molecular Carcinogenesis. 1998;22:229–234. doi: 10.1002/(sici)1098-2744(199808)22:4<229::aid-mc4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 13.Kirby G.M., Chemin I., Montesano R., Chisari F.V., Lang M.A., Wild C.P. Induction of specific cytochrome p450s involved in aflatoxin B1metabolism in hepatitis B virus transgenic mice. Molecular Carcinogenesis. 1994;11:74–80. doi: 10.1002/mc.2940110204. [DOI] [PubMed] [Google Scholar]
- 14.Sipowicz M.A., Chomarat P., Diwan B.A., Anver M.A., Awasthi Y.C., Ward J.M., Rice J.M., Kasprzak K.S., Wild C.P., Anderson L.M. Increased oxidative dna damage and hepatocyte overexpression of specific cytochrome P450 Isoforms in hepatitis of mice infected with helicobacter hepaticus. American Journal of Pathology. 1997;151:933–941. [PMC free article] [PubMed] [Google Scholar]
- 15.Montero R., Gentile G.J., Frederick L., McMannis J., Murphy T., Silva G., Blankespoor H., Gentile J.M. Induced expression of CYP2A5 in inflamed trematode-infested mouse liver. Mutagenesis. 1999;14:217–220. doi: 10.1093/mutage/14.2.217. [DOI] [PubMed] [Google Scholar]
- 16.De-Oliveira A.C., Da-Matta A.C., Paumgartten F.J. Plasmodium berghei (ANKA): infection induces CYP2A5 and 2E1 while depressing other cyp isoforms in the mouse liver. Experimental Parasitology. 2006;113:256–261. doi: 10.1016/j.exppara.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 17.Dixon J.B., Bhathal P.S., Hughes N.R., O'Brien P.E. Nonalcoholic fatty liver disease: improvement in liver histological analysis with weight loss. Hepatology. 2004;39:1647–1654. doi: 10.1002/hep.20251. [DOI] [PubMed] [Google Scholar]
- 18.Kleiner D.E., Brunt E.M., Natta M.V. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 19.Lu Y., Cederbaum A.I. Cisplatin-induced hepatotoxicity is enhanced by elevated expression of cytochrome P450 2E1. Toxicological Sciences. 2006;89:515–523. doi: 10.1093/toxsci/kfj031. [DOI] [PubMed] [Google Scholar]
- 20.H. Raunio, J. Hakkola, O. Pelkonen The CYP2A subfamily, in: C. Ioannides (Ed.), Cytochromes p450s Role in the Metabolism and Toxicity of Drugs and Other Xenobiotics, 2008, pp. 150–177.
- 21.Ikura Y., Ohsawa M., Suekane T., Fukushima H., Itabe H., Jomura H., Nishiguchi S., Inoue T., Naruko T., Ehara S., Kawada N., Arakawa T., Ueda M. Localization of oxidized phosphatidylcholine in nonalcoholic fatty liver disease: impact on disease progression. Hepatology. 2006;43:506–514. doi: 10.1002/hep.21070. [DOI] [PubMed] [Google Scholar]
- 22.Enomoto A., Itoh K., Nagayoshi E., Haruta J., Kimura T., O'Connor T., Harada T., Yamamoto M. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of are-regulated drug metabolizing enzymes and antioxidant genes. Toxicological Sciences. 2001;59:169–177. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- 23.Lamle J., Marhenke S., Borlak J., von Wasielewski R., Eriksson C.J., Geffers R., Manns M.P., Yamamoto M., Vogel A. Nuclear factor-eythroid 2-related factor 2 prevents alcohol-induced fulminant liver injury. Gastroenterology. 2008;134:1159–1168. doi: 10.1053/j.gastro.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 24.Lu Y., Gong P., Cederbaum A.I. Pyrazole induced oxidative liver injury independent of CYP2E1/2A5 induction due to Nrf2 deficiency. Toxicology. 2008;252:9–16. doi: 10.1016/j.tox.2008.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mantena S.K., Vaughn D.P., Andringa K.K., Eccleston H.B., King A.L., Abrams G.A., Doeller J.E., Kraus D.W., Darley-Usmar V.M., Bailey S.M. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochemical Journal. 2009;417:183–193. doi: 10.1042/BJ20080868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reddy N.M., Kleeberger S.R., Yamamoto M., Kensler T.W., Scollick C., Biswal S., Reddy S.P. Genetic dissection of the Nrf2-dependent redox signalingregulated transcriptional programs of cell proliferation and cytoprotection. Physiological Genomics. 2007;32:74–81. doi: 10.1152/physiolgenomics.00126.2007. [DOI] [PubMed] [Google Scholar]
- 27.Forman H.J., Zhang H., Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Molecular Aspects of Medicine. 2009;30:1–12. doi: 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan L., Kaplowitz N. Glutathione in liver diseases and hepatotoxicity. Molecular Aspects of Medicine. 2009;30:29–41. doi: 10.1016/j.mam.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 29.Lu Y., Zhang X.H., Cederbaum A.I. Ethanol induction of CYP2A5: role of CYP2E1-ROS-Nrf2 pathway. Toxicological Sciences. 2012;128:427–438. doi: 10.1093/toxsci/kfs164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abu-Bakar A., Moore M.R., Lang M.A. Evidence for induced microsomal bilirubin degradation by cytochrome P450 2A5. Biochemical Pharmacology. 2005;70:1527–1535. doi: 10.1016/j.bcp.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 31.Lau A., Villeneuve N.F., Sun Z., Wong P.K., Zhang D.D. Dual roles of Nrf2 in cancer. Pharmacological Research. 2008;58:262–270. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirby G.M., Batist G., Alpert L., Lamoureux E., Cameron R.G., Alaoui-Jamali M.A. Overexpression of cytochrome P-450 isoforms involved in aflatoxin B1 bioactivation in human liver with cirrhosis and hepatitis. Toxicologic Pathology. 1996;24:458–467. doi: 10.1177/019262339602400408. [DOI] [PubMed] [Google Scholar]
- 33.Satarug S., Lang M., Yongvanit P., Sithithaworn P., Mairiang E., Mairiang P., Pelkonen P., Bartsch H., Haswell-Elkins M. Induction of cytochrome P450 2A6 expression in humans by the carcinogenic parasite infection. Opisthorchiasis Viverrini. Cancer Epidemiology Biomarkers PreV. 1996;5:795–800. [PubMed] [Google Scholar]
- 34.Satarug S., Nishijo M., Ujjin P., Vanavanitkun Y., Baker J.R., Moore M.R. Evidence for concurrent effects of exposure to environmental cadmium and lead on hepatic CYP2A6 phenotype and renal function biomarkers in nonsmokers. Environmental Health Perspectives. 2004;112:1512–1518. doi: 10.1289/ehp.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]