

Abstract
Adaptive mechanisms involving upregulation of cytoprotective genes under the control of transcription factors such as Nrf2 exist to protect cells from permanent damage and dysfunction under stress conditions. Here we explore of the hypothesis that Nrf2 activation by reactive oxygen and nitrogen species modulates cytotoxicity during hypoxia (H) with and without reoxygenation (H/R) in H9C2 cardiomyoblasts. Using MnTBap as a cell permeable superoxide dismutase (SOD) mimetic and peroxynitrite scavenger and L-NAME as an inhibitor of nitric oxide synthase (NOS), we have shown that MnTBap inhibited the cytotoxic effects of hypoxic stress with and without reoxygenation. However, L-NAME only afforded protection during H. Under reoxygenation, conditions, cytotoxicity was increased by the presence of L-NAME. Nrf2 activation was inhibited independently by MnTBap and L-NAME under H and H/R. The increased cytotoxicity and inhibition of Nrf2 activation by the presence of L-NAME during reoxygenation suggests that NOS activity plays an important role in cell survival at least in part via Nrf2-independent pathways. In contrast, O2−• scavenging by MnTBap prevented both toxicity and Nrf2 activation during H and H/R implying that toxicity is largely dependent on O2−•.To confirm the importance of Nrf2 for myoblast metabolism, Nrf2 knockdown with siRNA reduced cell survival by 50% during 4 h hypoxia with and without 2 h of reoxygenation and although cellular glutathione (GSH) was depleted during H and H/R, GSH loss was not exacerbated by Nrf2 knockdown. These data support distinctive roles for ROS and RNS during H and H/R for Nrf2 induction which are important for survival independently of GSH salvage.
Abbreviations: CREB, cAMP-responsive element-binding protein; HIF-1, hypoxia-inducible factor; KEAP1, Kelch-like ECH-associated protein 1; L-NAME, L-NG-nitroarginine methyl ester; MnTBap, manganese [III] tetrakis (4-benzoic acid) porphyrin; NO, nitric oxide; NFκB, nuclear factor kappa B; NOS, nitric oxide synthase; NOX, NADPH oxidase; RNS, reactive nitrogen species; ROS, reactive oxygen species; Nrf2, nuclear factor erythroid 2-related factor 2; DHE, dihydroethidium; DAF-2-DA, 4,5-diaminofluorescein diacetate
Keywords: Adaptive, MnTBap, L-NAME, RNS, ROS, Glutathione
Graphical abstract
ROS and RNS in cell survival and death during hypoxia and reoxygenation; the role of Nrf2. There is evidence of superoxide anion radical and nitric oxide production in both hypoxia and H/R. While MnTBap inhibited the toxicity of both stressors, L-NAME only protected against the toxicity of hypoxia implying that nitric oxide was required for survival during H/R. Both superoxide anion radical and nitric oxide elicited Nrf2 activation during H/R and to a lesser extent during hypoxia. Nrf2 knockdown prevented GSH induction and also reduced survival during H/R by 50% but only by 30% during hypoxia. RNS are essential for Nrf2 activation but may also exert cytoprotective effects during H/R by other means
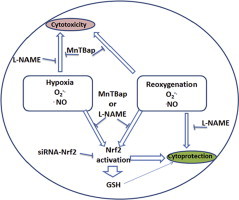
.
Highlights
-
•
Cardiomyoblast toxicity during hypoxia is dependent on O2−• and NO•.
-
•
Nrf2 activation is important for cardiomyoblast survival during hypoxia or hypoxia/reoxygenation, but, restoration of GSH is not required.
-
•
NOS activity is essential for the adaptation of cardiomyoblasts to hypoxia/reoxygenation but survival may be independent of Nrf2.
Introduction
Oxygen insufficiency, hypoxia, may arise in tissues during physiological and pathophysiological conditions such as exercise or a reduction in blood flow during ischemia. Adaptive mechanisms exist to protect cells from permanent damage and dysfunction due to hypoxia [1,2]. Effective sensing of oxygen availability allows cells to activate specific adaptive responses during hypoxia that confer protection [3]. Many of these responses are controlled by hypoxia-inducible factors (HIFs) whose activity is regulated by oxygen via HIF-prolyl hydroxylase and which mediate the expression of molecules such as haem oxygenase-1 and ferritin [4–6].When the oxygen supply is inadequate, cellular respiration shifts from aerobic fatty acid metabolism to anaerobic glycolysis thereby sustaining ATP production, albeit at a lower level [7,8].
Reactive oxygen (ROS) and/or nitrogen (RNS) species have been strongly implicated in ischaemic or hypoxic-toxicity, with and without reperfusion although there is a lack of agreement over their respective mechanisms of action [9–12]. There are a number of transcription factors whose activation is regulated either directly or indirectly by ROS/RNS; For example, ROS/RNS can act as potent electrophiles and key activators of nuclear factor erythroid 2-related factor 2 (Nrf2) [13,14]; a master regulator of the specific antioxidant phenotype [15,16]. During normoxia, Nrf2 is held in the cytoplasm and maintained at low levels by a cytoskeletal-associated inhibitory protein; Kelch-like ECH-associated protein 1 (KEAP1), which promotes rapid degradation of Nrf2 via KEAP1-dependent ubiquitin conjugation [13]. During oxidative stress, thiol oxidation occurs in the hinge region of KEAP1, resulting in a conformational change in KEAP1 with the loss of Nrf2 binding [17]. Then Nrf2 accumulates, undergoes modification by phosphorylation mediated by PKCδ and Akt and is translocated to the nucleus e.g. in epithelial cells [18]. cAMP-responsive element-binding protein (CREB)-dependent acetylation of Nrf2 promotes its binding to DNA [18]. With its cofactors, nuclear Nrf2 up-regulates cytoprotective genes through the transcriptional activation of genes binding to a cis-acting enhancer sequence of upstream of the antioxidant response element (ARE) [19]. Nrf2 is essential for transcriptional activation of several genes including gamma glutamyl cysteinyl ligase the rate limiting enzyme in glutathione synthesis and the subunits of the proteasome responsible for degradation of oxidatively damage proteins [20,21].
A change in expression and activation of Nrf2 has profound effects on the physiological response to oxygen insufficiency; for example, infarcted volume is reduced following focal cerebral ischaemia in the presence of the Nrf2 inducer, sulforaphane [22]. Moreover, Lee et al. [23] showed that neural cells lacking in Nrf2 (Nrf2−/− mice) were more susceptible to oxidative stress than control neurons from wild-type mice (Nrf2 +/+ wild mice). However, when the cells derived from Nrf2−/− mice were transfected with a functional Nrf2 construct, they became resistant to oxidative stress. Dhakshinamoorthy and Porter, reported that dominant negative-Nrf2 stable neuroblastoma cells were more prone to apoptosis induced by nitric oxide when they were Nrf2 silenced with siRNA [24] compared to Nrf2 expressing cells. This mechanistic controversy over ROS/RNS functions limits their potential as targets for addressing H and H/R toxicity. Therefore, we have developed a simple model to define the role for Nrf2, its induction via NOS and O2−• dependent pathways and effect in protecting cardiomyoblasts during H and H/R.
Experimental
Routine cell culture
ATCC H9C2 (1-2) rat-cardiomyoblasts were maintained routinely in Dulbecco′s modified Eagle′s medium (DMEM) up to passage 20 and supplemented with heat inactivated 10% fetal bovine serum (FBS), 4 mM l-glutamine and 200 U/ml penicillin and 200 µg/ml streptomycin at 37 °C in a humidified atmosphere of 5% CO2 and 95% air.
Induction of hypoxia (H) or hypoxia/reoxygenation (H/R)
Cells were maintained in preincubated hypoxic or normoxic phenol red-free DMEM which was supplemented with 25 mM HEPES (4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid), with penicillin/streptomycin as above (complete media). Estimates of O2 tension in tissue vary between 10 and 14% O2 and will fluctuate during physiological stress and pathophysiological conditions which reduce O2 delivery to tissues [25]. In normal myocardial perfusion in rat hearts, mean PO2 values were reported as 240 ±140 Torr and during local ischemia, the mean PO2 changed to 50±90 Torr which is equivalent to 6.7% oxygen [26]. For hypoxia or normoxia experiments, the medium was preincubated for 24 h in a hypoxic environment (2% O2+98% N2) and 10% O2+90% N2 and 21% O2+79% N2 for control experiments (BOC, UK). pH stability was assessed as described previously [27,28]. The experimental design is shown in Fig. 1.
Fig. 1.
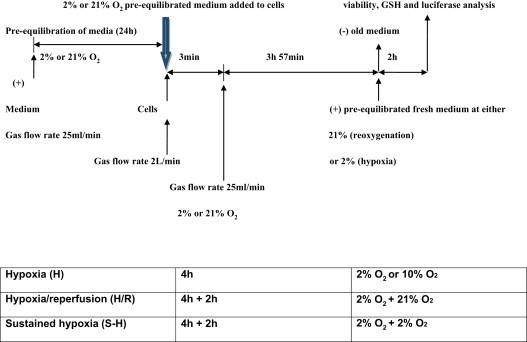
Schematic plan of experimental hypoxia and reoxygenation experiment. Media was pre-equilibrated at desired oxygen tension for 24 h prior to each experiment. At the start of each experiment, pre-equilibrated media was added to near confluent cells and the incubator was flushed with appropriate oxygen tension at a high flow rate for 3 min, then flow rate was returned to 25 ml/min for the remainder of the study period. After 4 h, for H/R experiments, 2% oxygen media was removed and pre-equilibrated 21% oxygen media added with 21% oxygen flushed into the incubator for the remaining 2 h. Cells that were to be retained in the same oxygen tension (either sustained hypoxia or normoxia) also underwent a change in media at the same time points but oxygen tension remained unchanged.
Measurement of superoxide/RNS generation during hypoxia (H) and hypoxia/reoxygenation (H/R)
For analysis of ROS and RNS, plates were sealed at 3 min prior to the end of each experiment and centrifuged at 400g for 3 min. For H/R, supernatants were gently removed and replaced with pre-equilibrated reoxygenated medium (21% O2+79% N2) and incubated further for 2 h reoxygenation. 45 min before completing each experiment fluorescent probe (20 µM DHE or 100 µM DAF-2-DA) was added into wells without incorporating air. The plate was then returned to the pre-equilibrated chamber and flushed with premixed normoxic gas at a higher flow rate (2000 ml/min) for 3 min to stabilize the experimental conditions. This was calculated in the basis of the volume of the incubating chamber and allowed for 10 exchanges of air. Fluorescence was determined at 37 °C in a preheated fluorescence reader (Molecular Devices). Cells were then lysed using 2% triton. Fluorescence in each well was calculated in arbitrary fluorescent units per 1 mg of total protein. Live imaging was achieved with a Carl Zeiss LSM 700 confocal microscope (Germany). The 488 nm argon laser line was used to excite DHE, which was measured by fluorescence emission using a band pass filter from 570 to 590 nm, with DAF-2-DA fluorescent probe monitored at Ex: 480–490 nm and Em: 510–520 nm. Illumination intensity was set up at a minimum (0.1–0.2% of laser output) to avoid photo-oxidation and the pinhole set to give the optical beam at 2 nm for optimum resolution. Each experiment was undertaken using by at least 3 independent chamber wells.
Cytotoxicity assays
Cell viability was assessed as the ratio of excluded propidium iodide (DNA staining due to permeable membrane) to Hoechst 33342 (DNA staining in all cells) and as caspase 3 cleavage by western blotting as described previously [28]. MTT assay was also employed to assess the loss of metabolic activity during hypoxia (H), sustained hypoxia (S-H) or hypoxia/reoxygenation (H/R). Cells were seeded at a density of 3×104 cells/well until they reached 80–90% confluence. 2 h prior to completion of S-H or H/R, dimethyl thiazolyl diphenyl tetrazolium salt (MTT) solution (100 µl of 5 mg/ml in 0.01 M PBS) was added to all wells. Control cells under normoxia also received the MTT solution prior to 1 h or 2 h as appropriate to the experimental set up. After completing hypoxia or reoxygenation, lysis buffer (100 µl, 20% w/v SDS in 50% DMF, dH2O (50%), pH 4.7 adjusted with 2.5% of glacial acetic acid) was added to each well and incubated for a further 16 h at 37 °C in a humidified atmosphere of 5% CO2 and 95% air [29] and then formazan production was assessed at 570 nm. Loss of metabolic activity was calculated by comparing results to normoxic cells which were considered as 100% metabolically active.
Transient transfection of plasmid DNA into H9C2 cardiomyoblasts
The luciferase reporter plasmid vector pGL 3 [nqo1/luc] was generated using the pGL3-promoter vector (Promega,UK) containing an ARE consensus sequence from the upstream of nqo1 promoter and the firefly luciferase reporter gene (luc), The pGL4.74[hRluc/TK] plasmid vector containing the thymidine kinase (TK) promoter upstream of hRluc for Renilla luciferase expression was employed as an internal control in co-transfected H9C2 cardiomyoblasts.
Expression of Renilla luciferase (Rluc) upon the activation of TK promoter in the pGL4.74 [TK/hRluc] plasmid provides an internal control value to which expression of the experimental luc gene with nqo1 promoter was normalized in the subsequent reporter gene assay [30]. Lipofectamine 2000 was diluted and gently mixed with 50 µl of Opti-MEM I reduced serum medium and incubated for 25 min [29]. After 25 min incubation, the diluted DNA samples (1:10) were combined with diluted Lipofectamine 2000 and incubated for a further 25 min at room temperature. 100 µl of DNA and Lipofectamine 2000 complex was then added to each well with 400 µl of medium and the content was gently mixed. After 6 h of transfection, 500 µl of culture medium containing 20% FBS and 8 mM l-glutamine was added to all transfected and non-transfected cell wells.
Luciferase reporter gene assay
To measure the luciferase reporter activity in transfected cells after exposure to hypoxia, a Dual Glo luciferase assay (Promega, UK) was performed according to the manufacturer′s protocol. Plates were centrifuged at 300g for 3 min to collect dead cells due to hypoxia treatment.
The ratio of luminescence of experimental reporter to control reporter was normalised to account for any cell loss. Hydrogen peroxide (100 µM) was employed over 6 h to activate transcription factors as a positive control.
Nrf2-RNA interference assay
Transient transfection with Nrf2-siRNA was performed using Lipofectamine RNAimax reagent according to a modified protocol and manufacturer′s instructions. H9C2 cells at approximately 50 to 70% confluence were transfected with siNrf2 or scrambled Nrf2-siRNA (scr/siRNA) (Invitrogen, UK). Cell culture medium was replaced at 6 h after transfection, and cells were then further incubated for 16–24 h before expose to hypoxia and/or reoxygenation or S-H. The knockdown efficiency was validated by western blotting [25].
Cellular glutathione
After 6 h incubation, cells were scraped from the plate and washed twice with PBS and sulfosalicylic acid (SSA; 3.33 µl of 100% made up in distilled water) was then added to the cell pellet. Following centrifugation at 6600g for 1.5 min, stock buffer (96.6 µl of 125 mM sodium phosphate, 6.3 mM disodium EDTA, pH 7.5) was then added to each tube and supernatants were collected carefully and stored immediately at −80 °C 80 °C prior to analysis of GSH by the GSR-DTNB (5,5'-dithiobis-(2-nitrobenzoic acid)) recycling assay within one month [27].
Statistical analysis
Results are presented as sample mean±SEM. Statistical analysis was performed using Graphpad Prism software (version 5) and tested by one-way ANOVA (nonparametric) using Tukey′s post-hoc test. All results are means of three independent experiments and P< (0.05)⁎ was considered as significantly different from controls.
Results
Initial studies were performed to determine the stability of the hypoxic chamber system. The dissolved O2 concentration in control medium (normoxia) was measured as 5.93±0.23 mg/l. After 24 h, the dissolved O2 concentration in the medium under 2% O2 was decreased by 26-fold (2%O2 0.23±0.04 mg/l) and at 10% O2 decreased by 3-fold (2.78±0.77 mg/l). Conversely, the pH of the medium was stable over a 24 h period during both normoxia (21%O2; pH 7.4±0.30), hypoxia (2%O2 pH 7.5±0.21) and at 10%O2 (pH 7.5±0.23).
The effect of oxygen tension on metabolic activity and viability of cardiomyoblasts
The overproduction of ROS/RNS during H and H/R may arise from and/or lead to mitochondrial dysfunction with associated cell death [9]. To test this hypothesis, we determined H and H/R-induced cell death and metabolic change by Hoechst and PI staining, caspase blots and MTT activity assays.
Analysis of necrotic death was undertaken by co-staining with Hoechst and PI. PI uptake was significant during 4 h H (2% O2 P<0.05) but not at 10% O2 compared to normoxia (Fig. 2A). After incubation at 2%, 10% and 21%O2 for 4 h, total cell lysates (20 µg) were resolved by SDS-PAGE and transferred to PVDF membranes for immunoblotting for procaspase 3 and cleaved caspase 3. The larger cleaved fragment (18 kDa) of procaspase-3 was detected in cardiomyoblasts exposed to 2% and 10% O2 for 4 h (Fig. 2B and C). However, no cleavage band was detected under normoxia confirming that apoptosis is increased by H only.
Fig. 2.
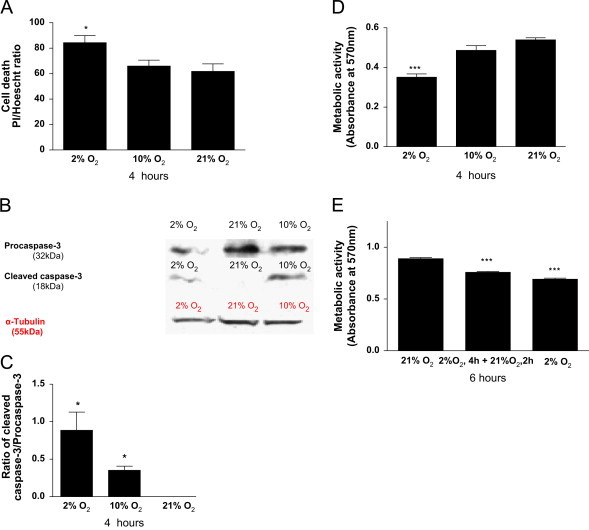
Hypoxia and hypoxia/reoxygenation induces cell death and a decrease in metabolic activity but not protein concentration. H9C2 cells at 70–80% confluence were incubated in 2% 10% or 21% O2 equilibrated, HEPES-buffered, phenol red-free DMEM and exposed to (4 h) hypoxia. (A); The ratio of propidium iodide (PI)-positive stained cells to total Hoechst stained cells was measured as an index of necrosis. ((B) and (C)); Procaspase 3 activation measured as cleavage relative to tubulin expression and determined by western blotting. (D); Metabolic activity determined by MTT assay. (E); After 4 h hypoxia (2% O2), the medium was replaced with reoxygenated medium (2 h; 21% O2) for control and H/R experiments, and with hypoxic medium (2 h; 2% O2) for sustained hypoxia experiments and MTT activity was determined. Data represent the mean ±S.E.M of three independent experiments conducted in triplicate. ⁎ represents P<0.05 and ⁎⁎ represents P<0.001 (one-way ANOVA) compared to controls, Tukey′s post-hoc test.
Hypoxia (2%O2 4 h) caused more than 20% loss in metabolic activity (P<0.001; Fig. 2D and E) whereas H/R (2%O2 4 h+21%O2 2 h), caused a 15% loss in metabolic activity compared to normoxia (21%O2 6 h)-maintained controls (P<0.001; Fig. 2E). These data show that the severity of H associates with metabolic inhibition but that the metabolic effects of H/R are less than those of S-H alone.
Hypoxia/reoxygenation induced O2−• generation and increased •NO production
To investigate whether H/R may increase O2−• production more than H alone in cardiomyoblasts, O2−• production was measured using DHE oxidation and inhibition by MnTBap. The specificity of DHE for discrete ROS such as the superoxide anion radical is dependent on the analytical method employed with HPLC analysis being the most specific method and although less-specific spectrophotometric analysis has been used here, this approach in combination with superoxide scavenging e.g. by SOD or the mimetic MnTBap, can infer O2−• involvement in a biological process. As shown in Fig. 3A and C, DHE fluorescence was significantly increased after 2 h of reoxygenation compared to hypoxic and normoxic controls and this was inhibitable by MnTBap, suggesting a significant increase in O2−• production during the reoxygenation period alone after S-H (P<0.001). The findings of DHE oxidation inhibition by MnTBap suggest production of superoxide anion during H and H/R.
Fig. 3.
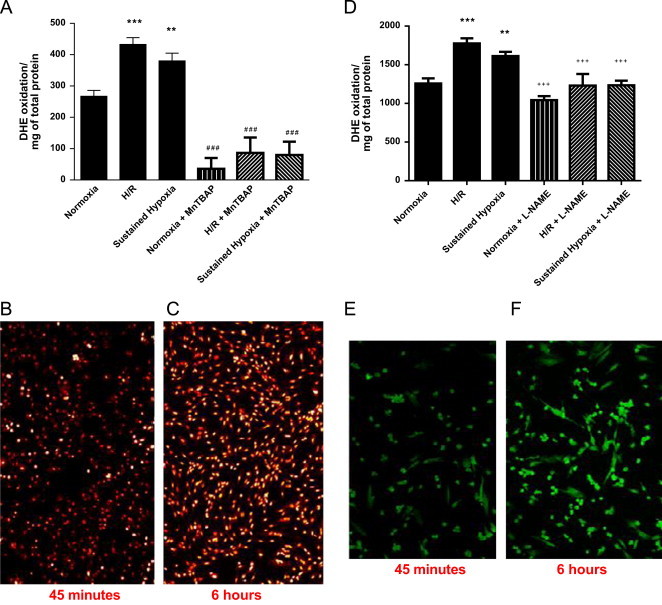
Effects of MnTBap and L-NAME on DHE and DAF2 oxidation during hypoxia/reperfusion. H9C2 cells at 70–80% confluence were incubated in hypoxic medium (2%O2; 4 h) or normoxic medium (21%O2; 4 h). After 4 h, culture medium was replaced with reoxygenated medium (21%O2; 2 h), for H/R and normoxia controls) or hypoxic medium (2%O2; 2 h) in the presence and absence of 50 µM MnTBap or 100 µM L-NAME. Prior to analysis (t-45 min), cells were treated with 20 µM DHE ((A)–(C)) or 100 µM DAF2-AM ((D)–(F)) and further incubated for 45 min to 6 h. The fluorescence was measured at 37 °C in a Spectramax GEMINI EM fluorescence reader at Ex: 488 nm, Em; 570 nm for DHE; and Ex: 480–490 nm and Em: 510–520 nm for DAF2. Fluorescent images of normoxia ((B) and (E)) and H/R ((C) and (F)) were captured using Zeiss 700 LCM confocal microscope. Data represent the mean±SEM fluorescence of three independent experiments conducted in triplicates. ⁎ represents P<0.05 or ⁎⁎ for P<0.01 or ⁎⁎⁎ for P<0.001 (one-way ANOVA) compared to controls and+denotes inhibitor significance relative to non-inhibitor treated cells at the same oxygen tension using Tukey′s post-hoc test.
It has been reported that an increase in myocardial •NO production occurs during hypoxia/reperfusion. To test this hypothesis in cardiomyoblasts, •NO generation during H and H/R was measured by quantitative oxidation of DAF-2-DA (a fluorescent probe oxidized by •NO) and inhibition by the NOS inhibitor L-NAME. As illustrated in Fig. 3D and F, there is a high level of basal oxidation of DAF-2 which is not inhibitable by L-NAME and is indicative of NOS-independent oxidation of the probe. Nevertheless, H/R significantly increased DAF fluorescence compared to normoxia (Fig. 3D; P<0.001) and this increase was prevented by the presence of L-NAME. In cells maintained in H, the increase in DAF fluorescence was also significant compared to normoxia (128.3±8.1% of normoxic control, P<0.01) and again could be prevented by the presence of L-NAME. These findings of DAF-2 oxidation inhibition by L-NAME implicate NOS formation during H and H/R.
ROS and RNS mediate the cytotoxicity of hypoxia
To identify whether O2−•, •NO and/or ONOO− production are implicated in H or H/R toxicity in cardiomyoblasts, the effects of 50 μM MnTBap or 100 μM L-NAME on metabolic activity were analysed.
While MnTBap and L-NAME have no effect on the metabolic activity of H9C2 cells under normoxic conditions, treatment of hypoxic cells with MnTBap during the reoxygenation period and in sustained H afforded protection against toxicity (Fig. 4A–C). In contrast, cytotoxicity was greater after inhibition of NOS with L-NAME during H/R with ~50% activity compared to cells treated with L-NAME in normoxia (P<0.05; Fig. 4B) however, L-NAME offered some protection (~12%; P<0.05) for cells maintained under S-H (P<0.001; Fig. 4C).
Fig. 4.
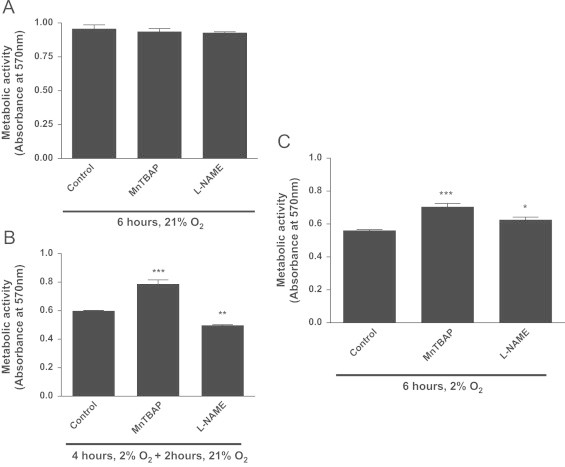
MnTBap and L-NAME differentially influence the toxicity of hypoxia and hypoxia/reoxygenation in rat cardiomyoblasts. H9C2 cells were exposed to hypoxia (2%O2; 4 h) or normoxic medium (21%O2; 4 h). After 4 h, culture medium was replaced with reoxygenated medium (21%O2 2 h) for H/R (B) and normoxia controls (A), or hypoxic medium (2%O2; 2 h) (C) in the presence and absence of 50 µM MnTBap or 100 µM L-NAME and then further incubated for 2 h in the presence of MTT reagent. Data represents the mean±S.E.M of three independent experiments conducted in triplicate. ⁎⁎⁎ represents P<0.001 and ⁎⁎ for P<0.01, ⁎ represents P<0.05 (one-way ANOVA), with Tukey′s post-hoc test.
These data suggest that O2−• production contributes in part to the toxicity during both H and reoxygenation. In contrast to their toxicity in S-H, •NO and/or related RNS are protective during reoxygenation. This suggests a complex picture of NOS/ O2−•-dependent survival mechanisms and death pathways which co-exist and where the nature and sites of reactive species production may be key factors in determining toxicity.
ROS and RNS mediate Nrf2 activation during hypoxia/reoxygenation (H/R)
A marked increase in the activation of Nrf2 during ischaemia-reperfusion has been reported in several cell types [17,23]. To confirm the activation of Nrf2 in H9C2 cells during H or H/R, cells at 90–95% confluence were co-transfected with pGL 3 [nqo1/luc] plasmid with the NQO1 promoter / pGL 4.4 [TK/hRluc] control plasmid for 24 h then incubated under normoxia, H/R or S-H for 6 h. The efficiency of knockdown is shown in supplementary figure 1.
In the luciferase reporter assay, activated Nrf2 binds with ARE region in NQO1-promoter to induce luciferase transcription/translation. Therefore, luciferase enzyme production and activity is proportional to Nrf2 activation. Nrf2 was significantly activated during S-H and H/R in H2C9 cells (Fig. 5A; p<0.01, and p<0.001, respectively).
Fig. 5.
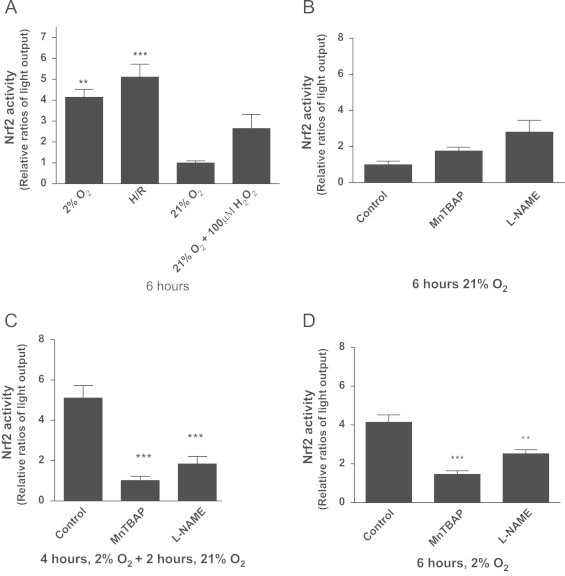
Hypoxia and hypoxia/reoxygenation increase Nrf2 activity in H9C2 cells in a superoxide and nitric oxide dependent manner. H9C2 cells grown to 90–95% confluence were transfected pGL3[nqo1/luc]/pGL 4.4 [hRluc/TK] plasmids (1.2 µg/ml): Lipofectamine 2000 complexes at 1:4 ratio for Nrf2 expression and cells were then incubated for further 24 h. Cells were then incubated under 2% oxygen with and without re-oxygenation or 21% oxygen (normoxia) over 6 h; 100 µM H2O2 was used as a positive control for Nrf2 activation after 4 h at 37 °C. Nrf2 activation was measured as the ratio of firefly to renila luciferase luminescence (A). The effects of MnTBap and L-NAME were investigated on Nrf2 activation in normoxia (B), H/R (C) and sustained hypoxia (D). Data represent the mean±S.E.M of three independent experiments conducted in triplicate. ⁎ represents P<0.05 (one-way ANOVA), Tukey′s post-hoc test.
To investigate if ROS and RNS produced during H and H/R were mediating Nrf2 activation, cells were cultured under different oxygen tensions in the presence of MnTBap or L-NAME. Inhibitors had no significant effect on Nrf2 activation under normoxic conditions (Fig. 5B). MnTBap reduced significantly the ARE-mediated luciferase gene expression in H/R and S-H, suggesting that O2−• is important in the activation of Nrf2 gene transcription under both conditions (Fig. 5C and Fig. 5D, respectively).
L-NAME also reduced ARE/NQO1 mediated luciferase gene expression during H/R and during S-H, (Fig. 5C and D).
Nrf2 activation supports cell survival during hypoxia and hypoxia/reoxygenation
Further investigation of the role for Nrf2 during H and H/R in cardiomyoblasts was undertaken following Nrf2 knockdown (KD). siRNA-treated Nrf2 cells exhibited a significant decrease (>40%) in metabolic activity during H/R and S-H in comparison to non-transfected controls and cells that had been transfected with scrambled si-RNA implying an important role for Nrf2 in survival during H and H/R (Fig. 6A). Nrf2-KD cells grown under normoxia showed little cytotoxicity after knockdown beyond 6 h (data not shown). Analysis of cellular glutathione confirmed a significant loss of glutathione from cells during H with or without reoxygenation which was not exacerbated by Nrf2 knockdown (Fig. 6B).
Fig. 6.
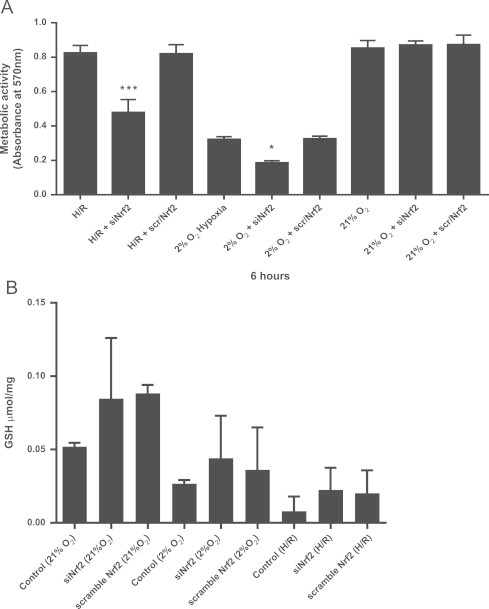
Nrf2 is a protective factor for cell survival during hypoxia/reperfusion and sustained hypoxia but does not contribute to the salvage of glutathione in H9C2 cells. H9C2 cells grown to 50–70% confluence were transiently transfected with Nrf2-siRNA or scramble (scr)-siRNA using Lipofectamine RNAimax and incubated for 16–24 h. Transfected cells were then incubated in hypoxic or normoxic medium for 4 h followed by further incubation for 2 h either at 2% or 21% O2. (A) Metabolic activity (MTT reducing activity) of cells was measured over last 2 h incubation in each experiment. (B) Cellular glutathione (GSH) was determined by the DTNB recycling assay after H/R or sustained hypoxia and is expressed as µmol/mg protein. Data are the mean±S.E.M of three independent experiments conducted in triplicate. Compared to controls in the absence of inhibitors, ⁎ represents P<0.05, ⁎⁎ represents P<0.01 and ⁎⁎⁎ represents P<0.001 (one-way ANOVA), Tukey′s post-hoc test.
These data show that Nrf2 activation, in S-H and to a greater extent by H/R, is associated with less cytotoxicity than when Nrf2 expression is knocked down but that the protective effects of Nrf2 appear to be independent of glutathione salvage after S-H or H/R. In S-H and H/R, ROS and RNS appear to play a significant role in Nrf2 induction.
In both environments, loss of Nrf2 impacted on cell survival but the effect of Nrf2 loss in S-H was the greatest. This suggests that other regulatory systems may be involved in the activation of Nrf2 or be activated independently of Nrf2 during H/R and that nitric oxide/RNS may be Involved in an alternative pathway of cytoprotection.
Discussion
Previously, we have shown that mitochondrial superoxide anion radicals mediate toxicity during H in cardiomyoblasts [28]. Here we have extended our studies to demonstrate that NOS activity also plays an important role in cytoprotection, particularly during reoxygenation. ROS/RNS are also important mediators of adaptive responses to stress, through reversibly modifying the redox state of cysteine residues in KEAP-1 [13]. When cysteines in KEAP-1 are reduced, KEAP1 binds to and negatively regulates Nrf2 by enhancing its rate of proteasomal degradation and altering its subcellular distribution. However, once KEAP-1 is oxidised, Nrf2 is stabilised and promotes induction of antioxidant gene expression [14]. The observation that the mole rat, which burrows deep and survives in hypoxic conditions, has constitutively activated Nrf2 supports the thesis that Nrf2 is a key regulator of survival during H [33]. Our data using L-NAME as an inhibitor of NOS, suggest that in addition to superoxide anion radical production, NOS activity is an important contributor to the transactivation activity of Nrf2 which in turn supports cardiomyoblast survival during H andH/R. Indeed, partial Nrf2 knockdown increases cytotoxicity of H and H/R. Nrf2-mediated antioxidant gene expression affords a highly specific and co-ordinated response to protect the cells against H or H/R-induced oxidative stress through induced expression of antioxidant genes; e.g. NQO1, MnSOD and the glutathione transferase (GST) pathway [19,34]. Here, MnTBap was able to inhibit Nrf2 activation during hypoxia and reoxygenation but as it can scavenge both peroxynitrite and O2−• [35] it is not possible to deduce from our present study which specific oxygen and nitrogen radical species mediate the Nrf2 activation and protective effects during H/R. Nevertheless, the protective effect of adding MnTBap but not L-NAME during the reoxygenation phase despite its capacity to prevent Nrf-2 activation highlights an additional important protective effect of scavenging O2−• during reoxygenation.
In support of a role for peroxynitrite in Nrf2 activation during H/R, Kang et al. [36] showed that during sulphur depletion, peroxynitrite plays an essential role in nuclear translocation of Nrf2 and ARE activation in rat hepatoma cells through the PI3-kinase pathway and that nitric oxide synthase is involved. Similarly, Li showed that peroxynitrite activates Nrf2 via PI3K/Akt signalling, enhances Nrf2-ARE binding, upregulates HO-1 expression and may confer an adaptive survival response against nitrosative stress in PC12 cells [37]. More recently, in HUVEC Mattart et al. showed that sublethal peroxynitrite concentrations could exert protective effects by modulating the balance between autophagy and apoptosis through Nrf2-dependent pathways [38].
The inhibition of NOS and the scavenging of •NO have been described previously by Kawahara et al. to result in a significant decrease in the survival rate of myocytes during 3 h ischaemia [39]. •NO served to balance ATP supply and demand during ischemia, and during ischemia/reperfusion [39]. A recent study in isolated ischemic cardiomyocytes, reported that the heat shock protein Hsp22 enhanced NFκB and STAT-dependent iNOS expression and reduced H2O2-mediated apoptosis by 60%; this effect was abolished by NOS inhibitors illustrating that NOS activation is an alternative approach for preemptive treatment of myocardial ischemia [40]. The difference in survival outcome that we observed here following L-NAME treatment of cardiomyoblasts during hypoxia (i.e. L-NAME protects) compared with H/R (i.e. L-NAME exacerbates) may be due to differential local concentrations of •NO and O2−• [41]. In addition, the activation of an alternative cytotoxic pathway during hypoxia and protective pathway during reoxygenation may also be important.
L-NAME exacerbated toxicity during reoxygenation, confirming the absolute requirement for NO production to achieve cytoprotection. In contrast the decrease in cytotoxicity in the presence of MnTBap despite a reduction in Nrf-2 suggests that the toxic effects of O2−• outweigh their beneficial effects through activation of adaptive pathways. It is likely that ROS/RNS may activate other cytoprotective transcription factor pathways, such as those driven by HIF-1 alpha [42].
Cellular redox imbalance is prevalent and complex in H and H/R; this may explain the limited success of redox-modulating interventions to improve outcome in H and H/R. Therapeutic use of •NO donors such as nitrosoprusside may offer benefits for cardioprotection through supporting the adaptive response to hypoxia and reoxygenation, in addition to their well- established benefits in vascular smooth muscle relaxation.
Conclusions
NOS activity is essential for the adaptation of cardiomyoblasts to H/R via Nrf2-dependent effects, however, during hypoxia O2−• with NO appears to contribute to toxicity which is further exacerbated in the absence of Nrf2.
Acknowledgments
RTK was funded by an Aston University postgraduate scholarship. IHKD was funded by an Aston University postgraduate scholarship and is now supported by The Dunhill Medical Trust [grant number: R92/1108]. HRG is funded under COST CM1001 and CM1203.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary materials
Supplementary material
References
- 1.Webster KA. Hypoxia: life on the Edge. Antioxidants & Redox Signaling. 2007;9:1303–1308. doi: 10.1089/ars.2007.1730. [DOI] [PubMed] [Google Scholar]
- 2.Lynn EG, Lu Z, Minerbi D, Sack MN. The regulation, control, and consequences of mitochondrial oxygen utilization and disposition in the heart and skeletal muscle during hypoxia. Antioxidants & Redox Signaling. 2007;9:1353–1362. doi: 10.1089/ars.2007.1700. [DOI] [PubMed] [Google Scholar]
- 3.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metabolism. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL, Shimoda LA, Prabhakar NR (2006) Regulation of gene expression by HIF-1. Novartis found symp 272, 2-8; discussion 8-14, 33-16. [PubMed]
- 5.Vangeison G, Carr D, Federoff HJ, Rempe DA. The good, the bad, and the cell type-specific roles of hypoxia inducible factor-1{alpha} in neurons and astrocytes. J Neurosci. 2008;28:1988–1993. doi: 10.1523/JNEUROSCI.5323-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. 2002;64:993–998. doi: 10.1016/s0006-2952(02)01168-1. [DOI] [PubMed] [Google Scholar]
- 7.Chen C-H, Liu Y-F, Lee S-D, Huang C-Y, Lee W-C, Tsai Y-L, Hou C-W, Chan Y-S, Kuo C-H. Altitude hypoxia increases glucose uptake in human heart. High Altitude Medicine & Biology. 2009;10:83–86. doi: 10.1089/ham.2008.1064. [DOI] [PubMed] [Google Scholar]
- 8.Malhotra R, Brosius FC., 3rd Glucose uptake and glycolysis reduce hypoxia-induced apoptosis in cultured neonatal rat cardiac myocytes. The Journal of Biological Chemistry. 1999;274:12567–12575. doi: 10.1074/jbc.274.18.12567. [DOI] [PubMed] [Google Scholar]
- 9.Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. The Journal of Neuroscience. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffman DL, Salter JD, Brookes PS. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: implications for hypoxic cell signaling. American Journal of Physiology. Heart and Circulatory Physiology. 2007;292:H101–108. doi: 10.1152/ajpheart.00699.2006. [DOI] [PubMed] [Google Scholar]
- 11.Lacza Z, Puskar M, Figueroa JP, Zhang J, Rajapakse N, Busija DW. Mitochondrial nitric oxide synthase is constitutively active and is functionally upregulated in hypoxia. Free Radical Biology & Medicine. 2001;31:1609–1615. doi: 10.1016/s0891-5849(01)00754-7. [DOI] [PubMed] [Google Scholar]
- 12.Levrand S, Vannay-Bouchiche C, Pesse B, Pacher P, Feihl F, Waeber B, Liaudet L. Peroxynitrite is a major trigger of cardiomyocyte apoptosis in vitro and in vivo. Free Radical Biology and Medicine. 2006;41:886. doi: 10.1016/j.freeradbiomed.2006.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMahon M, Itoh K, Yamamoto M, Hayes JD. KRAP1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven Gene expression. Journal of Biological Chemistry. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 14.Leonard MO, Kieran NE, Howell K, Burne MJ, Varadarajan R, Dhakshinamoorthy S, Porter A, Farrelly C, Rabb H, Taylor CT. Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia-reperfusion injury. The FASEB Journal. 2006;20:2624–2626. doi: 10.1096/fj.06-5097fje. [DOI] [PubMed] [Google Scholar]
- 15.Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Molecular Aspects of Medicine. 2011;32:234–246. doi: 10.1016/j.mam.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Li X, Zhang D, Hannink M, Beamer LJ. Crystal structure of the Kelch domain of human KEAP1. Journal of Biological Chemistry. 2004;279:54750–54758. doi: 10.1074/jbc.M410073200. [DOI] [PubMed] [Google Scholar]
- 17.Zhang DD. Mechanistic studies of the Nrf2-KEAP1 signaling pathway. Drug Metabolism Reviews. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 18.Kawai Y, Garduño L, Theodore M, Yang J, Arinze IJ. Acetylation–deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. Journal of Biological Chemistry. 2011;286:7629–7640. doi: 10.1074/jbc.M110.208173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochemical Journal. 2003;374:337–348. doi: 10.1042/BJ20030754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. Journal of Biological Chemistry. 1991;266:11632–11639. [PubMed] [Google Scholar]
- 21.Pickering AM, Linder RA, Zhang H, Forman HJ, Davies KJA. Nrf2-dependent induction of proteasome and Pa28αβ regulator are required for adaptation to oxidative stress. Journal of Biological Chemistry. 2012;287:10021–10031. doi: 10.1074/jbc.M111.277145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shah ZA LR, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Dore S. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience. 2007;147:53–59. doi: 10.1016/j.neuroscience.2007.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J-M, Shih AY, Murphy TH, Johnson JA. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. Journal of Biological Chemistry. 2003;278:37948–37956. doi: 10.1074/jbc.M305204200. [DOI] [PubMed] [Google Scholar]
- 24.Dhakshinamoorthy S, Porter AG. Nitric oxide-induced transcriptional up-regulation of protective genes by Nrf2 via the antioxidant response element counteracts apoptosis of neuroblastoma cells. Journal of Biological Chemistry. 2004;279:20096–20107. doi: 10.1074/jbc.M312492200. [DOI] [PubMed] [Google Scholar]
- 25.Zuurbier CJ, van Iterson M, Ince C. Functional heterogeneity of oxygen supply-consumption ratio in the heart. Cardiovascular Research. 1999;44:488–497. doi: 10.1016/s0008-6363(99)00231-x. [DOI] [PubMed] [Google Scholar]
- 26.Shukla HP, Mason RP, Bansal N, Antich PP. Regional myocardial oxygen tension: 19F MRI of sequestered perfluorocarbon. Magnetic resonance in medicine. Official Journal of the Society of Magnetic Resonance in Medicine/Society of Magnetic Resonance in Medicine. 1996;35:827–833. doi: 10.1002/mrm.1910350607. [DOI] [PubMed] [Google Scholar]
- 27.Grant MM, Griffiths HR. Cell passage-associated transient high oxygenation causes a transient decrease in cellular glutathione and affects T cell responses to apoptotic and mitogenic stimuli. Environmental Toxicology and Pharmacology. 2007;23:335–339. doi: 10.1016/j.etap.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 28.Kolamunne RT, Clare M, Griffiths HR. Mitochondrial superoxide anion radicals mediate induction of apoptosis in cardiac myoblasts exposed to chronic hypoxia. Archives of Biochemistry and Biophysics. 2011;505:256–265. doi: 10.1016/j.abb.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 29.Grant MM, Barber VS, Griffiths HR. The presence of ascorbate induces expression of brain derived neurotrophic factor in SH-SY5Y neuroblastoma cells after peroxide insult, which is associated with increased survival. Proteomics. 2005;5:534–540. doi: 10.1002/pmic.200300924. [DOI] [PubMed] [Google Scholar]
- 30.Dalby B, Cates S, Harris A, Ohki EC, Tilkins ML, Price PJ, Ciccarone VC. Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods. 2004;33:95–103. doi: 10.1016/j.ymeth.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 33.Schülke S, Dreidax D, Malik A, Burmester T, Nevo E, Band M, Avivi A, Hankeln T Living with stress: regulation of antioxidant defense genes in the subterranean, hypoxia-tolerant mole rat, Spalax. Gene 500, 199–206. [DOI] [PubMed]
- 34.Rushworth SA, MacEwan DJ, O′Connell MA. Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. The Journal of Immunology. 2008;181:6730–6737. doi: 10.4049/jimmunol.181.10.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Batinić-Haberlea I, Cuzzocrea S, Rebouças JS, Ferrer-Sueta G, Mazzon E, Di Paola R, Radi R, Spasojević I, Benov L, Salvemini D. Pure MnTBap selectively scavenges peroxynitrite over superoxide: comparison of pure and commercial MnTBap samples to MnTE-2-PyP in two models of oxidative stress injury, an SOD-specific Escherichia coli model and carrageenan-induced pleurisy. Free Radical Biology and Medicine. 2009;46:192–201. doi: 10.1016/j.freeradbiomed.2008.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang KW, Choi SH, Kim SG. Peroxynitrite activates NF-E2-related factor 2/antioxidant response element through the pathway of phosphatidylinositol 3-kinase: the role of nitric oxide synthase in rat glutathione S-transferase A2 induction. Nitric Oxide. 2002;7:244–253. doi: 10.1016/s1089-8603(02)00117-9. [DOI] [PubMed] [Google Scholar]
- 37.Li M-H, Cha Y-N, Surh Y-J. Peroxynitrite induces HO-1 expression via PI3K/Akt-dependent activation of NF-E2-related factor 2 in PC12 cells. Free Radical Biology and Medicine. 2006;41:1079–1091. doi: 10.1016/j.freeradbiomed.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 38.Mattart L, Calay D, Simon D, Roebroeck L, Caesens-Koenig L, Van Steenbrugge M, Tevel V, Michiels C, Arnould T, Boudjeltia KZ. The peroxynitrite donor 3-morpholinosydnonimine activates Nrf2 and the UPR leading to a cytoprotective response in endothelial cells. Cellular Signalling. 2011;24:199–213. doi: 10.1016/j.cellsig.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 39.Kawahara K, Hachiro T, Yokokawa T, Nakajima T, Yamauchi Y, Nakayama Y. Ischemia/reperfusion-induced death of cardiac myocytes: possible involvement of nitric oxide in the coordination of ATP supply and demand during ischemia. Journal of Molecular and Cellular Cardiology. 2006;40:35–46. doi: 10.1016/j.yjmcc.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 40.Chen L, Lizano P, Zhao X, Sui X, Dhar SK, Shen Y-T, Vatner DE, Vatner SF, Depre C. Preemptive conditioning of the swine heart by H11 kinase/Hsp22 provides cardiac protection through inducible nitric oxide synthase. American Journal of Physiology: Heart and Circulatory Physiology. 2012;300:H1303–H1310. doi: 10.1152/ajpheart.00979.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malhotra JD KR. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxidants & Redox Signaling. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 42.Kohl R, Zhou J, Brune B. Reactive oxygen species attenuate nitric-oxide-mediated hypoxia-inducible factor-1alpha stabilization. Free Radical Biology & Medicine. 2006;40:1430–1442. doi: 10.1016/j.freeradbiomed.2005.12.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material