

Abstract
Rasmussen encephalitis (RE) is a rare neurologic disorder of childhood characterized by unihemispheric inflammation, progressive neurologic deficits, and intractable focal epilepsy. The pathogenesis of RE is still enigmatic. Adenosine is a key endogenous signaling molecule with anticonvulsive and anti-inflammatory effects, and our previous work demonstrated that dysfunction of the adenosine kinase (ADK)–adenosine system and astrogliosis are the hallmarks of epilepsy. We hypothesized that the epileptogenic mechanisms underlying RE are related to changes in ADK expression and that those changes might be associated with the development of epilepsy in RE patients. Immunohistochemistry was used to examine the expression of ADK and glial fibrillary acidic protein in surgically resected human epileptic cortical specimens from RE patients (n = 12) and compared with control cortical tissues (n = 6). Adenosine kinase expression using Western blot and enzymatic activity for ADK were assessed in RE versus control samples. Focal astrogliosis and marked expression of ADK were observed in the lesions of RE. Significantly greater ADK expression in RE versus controls was demonstrated by Western blot, and greater enzymatic activity for ADK was demonstrated using an enzyme-coupled bioluminescent assay. These results suggest that upregulation of ADK is a common pathologic hallmark of RE and that ADK might be a target in the treatment of epilepsy associated with RE.
Key Words: Adenosine kinase, Astrogliosis, Epilepsy, Rasmussen encephalitis
INTRODUCTION
Rasmussen encephalitis (RE) is a very rare, chronic, progressive, inflammatory, neurologic disorder of uncertain etiology that mostly affects children. It is associated with hemispheric atrophy, focal epilepsy (epilepsia partialis continua), cognitive deterioration, and progressive neurologic deficits that result from progressive loss of function subserved by the involved cerebral hemisphere (1–3). The pathogenesis of this severely disabling inflammatory disease remains unknown. An intriguing feature of RE is the restriction of the inflammatory process to one brain hemisphere, setting it apart from other inflammatory diseases of the CNS. The factors responsible for the characteristic of asymmetry are still enigmatic. The neuropathologic hallmarks of RE consist of lymphocytic infiltrates (perivascular lymphocytic cuffing), microglial nodules, neuronal destruction, and gliosis of the affected hemisphere (4, 5). Although this constellation of histologic features is strongly suggestive of inflammatory disease of the CNS, no precise etiology of RE has been elucidated. Recent studies strongly support the hypothesis of an antigen-driven MHC class I–restricted, CD8-positive, T cell–mediated attack against neurons and astrocytes in the CNS dominating the pathogenesis in RE, in contrast to a random attraction of cells as part of a secondary immune response (6). Seizures are a prominent clinical feature of RE, and recent data suggest that the inflammation plays a direct role in the pathogenetic mechanisms of epileptogenesis.
Adenosine is an endogenous purine nucleoside that modulates a wide range of physiologic functions (7). Most notable among its many roles is its importance in controlling inflammation (8, 9) and inhibiting seizures (10–16). Adenosine is formed by dephosphorylation of adenosine monophosphate by ecto- and endo-5′-nucleotidase; through hydrolysis of S-adenylhomocysteine, it is removed by adenosine deaminase and adenosine kinase (ADK) (17). However, based on its low KM for adenosine, ADK is the primary route of adenosine metabolism in the brain. Therefore, minor changes in ADK activity translate rapidly into major changes in adenosine (17). Recently, we and others have provided considerable evidence that dysfunctional adenosine homeostasis as one of the astrocytic bases for epilepsy (18–21). Adenosine kinase, the main adenosine-removing enzyme, has been highlighted as a diagnostic marker to predict epileptogenesis as well as a potential target for antiepileptogenesis or disease modification (13, 19–22). Our previous results demonstrated that astrogliosis via disruption of adenosine homeostasis per se and in the absence of any other overt pathology associated with the emergence of spontaneous recurrent subclinical seizures may be a first step in epileptogenesis (20, 21).
In addition, in vitro experiments in human astrocyte cultures showed that ADK expression was increased by several proinflammatory molecules (e.g. interleukin-1β and lipopolysaccharide), which indicates the existence of an additional layer of modulatory cross talk between the astrocyte-based adenosine cycle and inflammation (18).
Based on the data that adenosine signaling plays a crucial role in anticonvulsion and anti-inflammation, we hypothesized that the epileptogenic mechanisms underlying RE, a chronic progressive inflammatory neurologic disorder, are related to changes in ADK expression and that those changes might be associated with the development of epilepsy in patients with RE.
MATERIALS AND METHODS
Patients and Diagnosis
The local Ethics Committee (Beijing Sanbo Hospital, Capital Medical University, Beijing, China) approved all studies, and clinical investigations were conducted according to the Declaration of Helsinki. Informed consent was obtained from all participants or their parents or legal guardians. Twelve childhood-onset RE patients diagnosed between January 2004 and August 2011 according to the typical clinical, magnetic resonance imaging (MRI), and neuropathologic findings (as proposed in Bien et al[2]) were enrolled in this study. Presurgical evaluation in Beijing Sanbo Hospital included MRI (spin-echo T1-weighted axial and T2-weighted axial; coronal sequences; and fluid-attenuated inversion recovery images with 5-mm-thick axial, sagittal, and coronal sections), fluorodeoxyglucose positron emission tomography, scalp video-electroencephalography, intracranial electroencephalographic monitoring, seizure semiology analysis, as well as neuropsychologic testing were peformed. There were six male and six female patients, with a mean age of seizure onset of 4.14 ± 1.96 years (range, 3.0–14.5 years) and a mean age at surgery of 7.30 ± 3.15 years (range, 3.0–14.5 years). Epilepsy was the first manifestation of the condition in all patients. None of the patients had a relevant perinatal history before the onset of the disease. Clinical details of the RE patients are summarized in the Table. Brains samples were obtained from all 12 RE patients during neurosurgical operations (functional hemispherectomies or anatomical hemispherectomies). Brain tissue was paraffin embedded. In 6 of the patients, parts of the brain samples were cryoprotected for Western blot analysis. In addition, we studied control patient brain samples obtained at autopsy from 6 children without a history of seizures or other neurologic diseases. All autopsies were performed within 12 hours after death.
TABLE.
Clinical Details of Patients With Rasmussen Encephalitis
Tissue Preparation
Formalin-fixed, paraffin-embedded tissue samples (i.e. 1 representative paraffin block per case containing the complete lesion or the largest part of the lesion resected at surgery) were sectioned at 4 μm and mounted on precoated glass slides (Star Frost, Waldemar Knittel GmbH, Braunschweig, Germany). Sections of all specimens were processed for hematoxylin and eosin (H&E) stain and for immunohistochemistry for CD8, CD3, the neuronal marker neuronal nuclear protein (NeuN), the glial marker glial fibrillary acidic protein (GFAP), and ADK. Brain tissue samples from RE patients (n = 3) and autopsy controls (n = 3) were snap frozen in liquid nitrogen and stored at -80°C until use for Western blot analysis.
Immunohistochemistry
Antibodies to GFAP (polyclonal rabbit, 1:4000; DAKO, Glostrup, Denmark), NeuN (mouse clone MAB377, IgG1, 1:2000; Chemicon, Temecula, CA), CD8 (mouse-IgG, clone C8/144B, 1:50; DakoCytomation, Glostrup, Denmark), CD3 (rabbit-IgG, clone SP7, 1:500; Lab Vision, Fremont, CA), and ADK (polyclonal rabbit, 1:500; provided by Professor Detlev Boison, R.S. Dow Neurobiology Laboratories, Legacy Research Institute, Portland, OR) (11, 13, 18) were used in the routine immunohistochemical analysis of RE human specimens. Double label immunocytochemistry was performed as described previously (13, 18). After incubation with primary antibodies, sections were incubated for 2 hours at room temperature with AlexaFluor 568 and AlexaFluor 488 (anti-rabbit IgG or anti-mouse IgG; 1:200; Molecular Probes, Eugene, OR). Images were visualized using a Leica microscope under Ex/Em wavelength of 500/550 nm (green), collected using an Optronics DEI-750 three-chip camera equipped with a BQ 8000 sVGA frame grabber and analyzed using Bioquant software (Nashville, TN).
Western Blot Analysis
Preparation and analysis of Western blots were performed as previously described (11, 18). Freshly frozen histologically normal autopsy cerebral cortex and RE samples (n = 3 per group) were homogenized in lysis buffer containing 10 mmol/L Tris (pH 8.0), 150 mmol/L NaCl, 10% glycerol, 1% NP-40, Na-orthovanadate (10.4 mg/mL), 5 mmol/L EDTA (pH 8.0), 5 mmol/L NaF, and protease inhibitor cocktail (Boehringer Mannheim, Heidesheim am Rhein, Germany). Protein content was determined using the bicinchoninic acid method. For electrophoresis, equal amounts of proteins (30 μg per lane) were separated by sodium dodecylsulfate–polyacrylamide gel electrophoretic analysis. Separated proteins were transferred to nitrocellulose paper for 1.5 hours using a semidry electroblotting system (Transblot SD; BioRad, Hercules, CA). Blots were incubated overnight in Tris-buffered saline ([TTBS] 20 mmol/L Tris, 150 mmol/L NaCl, 0.1% Tween, pH 7.5)/5% nonfat dry milk, containing the primary antibody (1:5000). After several washes in TTBS, the membranes were incubated in TTBS/5% nonfat dry milk/1% bovine serum albumin, containing the goat anti-rabbit or goat anti-mouse antibodies coupled to horseradish peroxidase (1:2500; Dako) for 1 hour. After washes in TTBS, immunoreactivity was visualized using Lumi-light PLUS Western blotting substrate (Roche Diagnostics, Mannheim, Germany) and digitized using a luminescent image analyzer (LAS-3000; Fuji Film, Japan). Expression of β-actin (monoclonal mouse, 1:50,000; Sigma, St. Louis, MO) was used as reference. Adenosine kinase levels were normalized to internal standards and reported relative to control.
ADK Enzyme Activity Analysis
The ADK enzyme activity was evaluated in 3 specimens removed from RE patients and 3 specimens from controls. Evaluation of the enzymatic activity for ADK was performed as previously described (15, 22). Briefly, tissues were homogenized individually at 4°C in 200 μL of 50 mmol/L NaCl, 20 mmol/L Na2HPO4, and 5 mmol/L EDTA, pH 6.5. The homogenates were then centrifuged at 100,000 × g for 10 minutes. The protein contents of the supernatants were quantified using the bicinchoninic acid method; 75 μL of each sample was applied to a Micro Bio-Spin 6 column (catalog no. 732–6221; BioRad) and equilibrated with 500 μL of the adenosine 5′-triphosphate assay mix dilution buffer ([FL-AAB] Sigma, Buchs, Switzerland). Each sample was centrifuged at 1,000 × g for 4 minutes and diluted with FL-AAB to reach a final protein concentration of 1.0 mg/mL, and 5 μL of each sample was used for ADK activity determinations with an enzyme-linked bioluminescent assay according to the previously described method. Adenosine kinase activity values were normalized to endogenous lactate dehydrogenase activity, used as an internal standard and determined with a commercial assay kit according to the manufacturer’s protocol (Roche Applied Science, Mannheim, Germany).
Statistical Analysis
Statistical analyses were performed with SPSS for Windows (SPSS 11.5, SPSS Inc., Chicago, IL) using 2-tailed Student t test; p < 0.05 was considered significant.
RESULTS
Clinical and Neuropathologic Features
All 12 RE patients had refractory epilepsy and underwent surgical resection for intractable seizures (Table). Eleven of 12 patients in the study cohort were in the acute stage of RE and 1 patient was in a residual stage, according to Bien’s stage-wise course of RE (23). The seizures types of patients included simple partial seizure, epilepsia partialis continua, complex partial seizure, and secondary generalized tonic-clonic seizure. Eleven of 12 patients had epilepsia partialis continua, which appeared 0.78 ± 0.39 years after the first seizure onset. All RE patients exhibited a focal motor deficit, which appeared 0.82 ± 0.38 years after the first seizure onset. The duration from the seizure to first operation was 3.36 ± 2.67 years (range, 0.67–10 years) (Table).
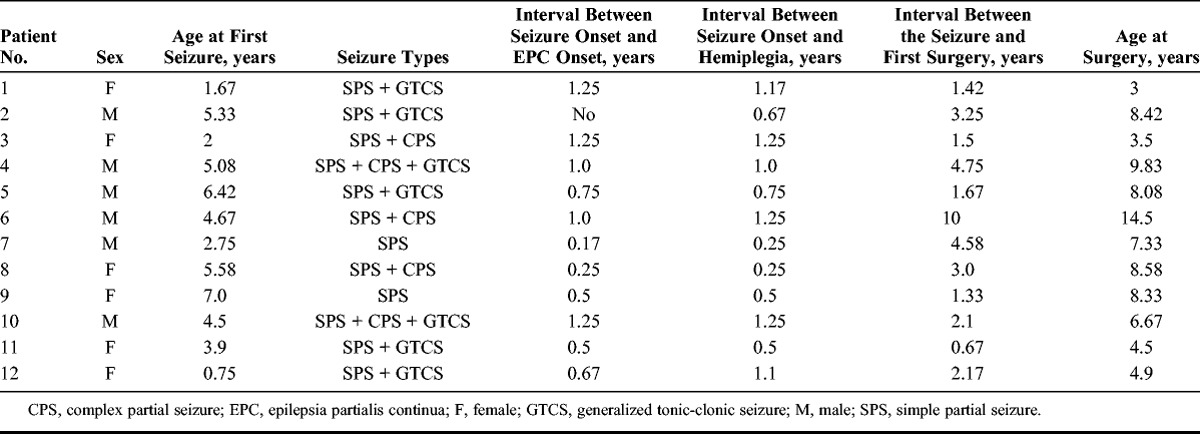
Microscopically, all 12 cases demonstrated the features of inflammatory pathology of RE with varying severity. These findings included perivascular lymphocyte cuffing (Fig. 1A, E) and foci of parenchymal lymphocyte infiltrates (Fig. 1D, F) with microglial nodules (Fig. 1B); double labeling revealed that most of the infiltrating lymphocytes were CD8-positive, CD3-positive T lymphocytes (Fig. 1F, inset). There was also diffuse microglial activation, neuron loss, reactive astrogliosis, and occasional foci of neuronophagia (Fig. 1B, C, D, F). Some cases showed a band of remote infarct with extensive gliosis in the middle layers of the cortical gray matter, loss of neurons, and a prominent branching vascular pattern.
FIGURE 1.
Neuropathologic findings in surgically resected brain tissue from Rasmussen encephalitis patients. (A) Perivascular lymphocytic cuffing in the cerebral cortex (arrows, H&E stain). (B) Formation of microglial nodules and diffuse microglial activation (arrows, H&E stain). (C) Neuronal loss and astrogliosis (H&E stain). (D) Cortical neuron with neuronophagia (arrows, NeuN immunostaining). (E) Perivascular lymphocytic cuffing in the cerebral cortex (arrows, CD8 immunostaining). (F) Parenchymal lymphocytic in the cerebral cortex (arrows, CD8 immunostaining). Inset in (G): colocalization of CD3 and CD8. Scale bar = 25 μm. Original magnification: (G) inset 2,000×.
Overexpression of ADK Concomitant With Astrogliosis Within RE Lesions
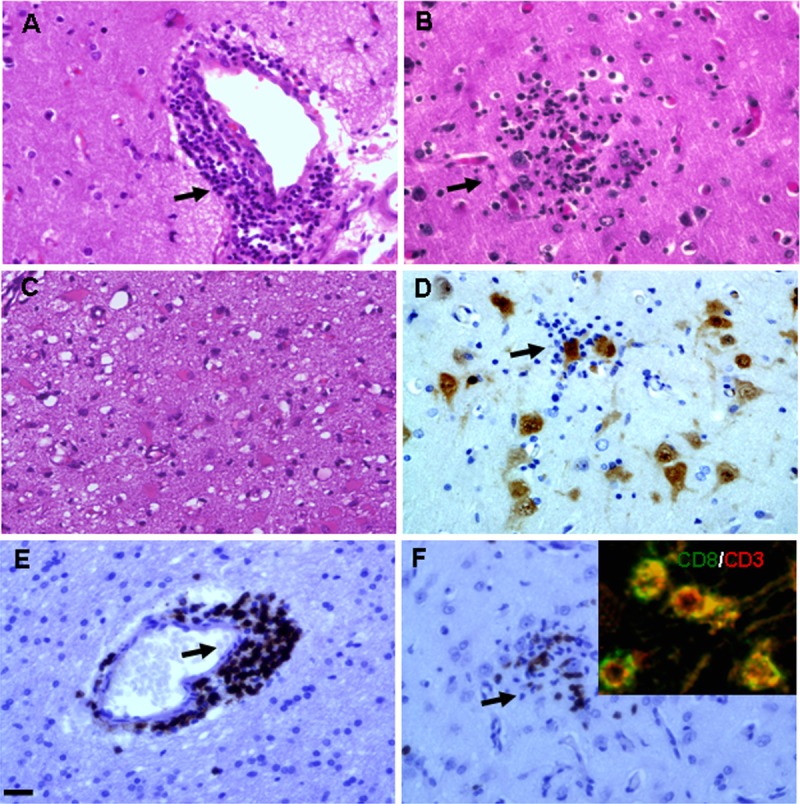
In control (autopsy) white matter, ADK immunoreactivity was present in sparse glial cells with only weak staining (Fig. 2D3); control cortical gray matter also displayed weak astroglial staining (Fig. 2D1, D2).
FIGURE 2.
Adenosine kinase, GFAP, and NeuN immunostaining in brain tissue of patients with RE. (A1–A3) NeuN staining demonstrates neuron loss in the lesion cortex (A1), occasional foci of neuronophagia (arrows [A2]), and marked cell loss in the perivascular area (arrows [A3]). (B1–B3) GFAP-positive reactive astrocytes within the lesion area (B1, B2) and marked reactive astrogliosis in the perivascular area (arrows [B3]). (C1–C3) Concomitant with reactive astrogliosis, there is cytoplasmic localization of ADK immunoreactivity in cells with typical astroglial morphology within the lesion (C1, C2) and prominent in the perivascular area (arrows [C3]). (D1–D3) There is weak immunostaining for ADK in control cortical gray matter (D1, D2) and sparse glial cells in control white matter (D3). Scale bars = (A1–D1) 100 μm; (A2–D3) 25 μm.
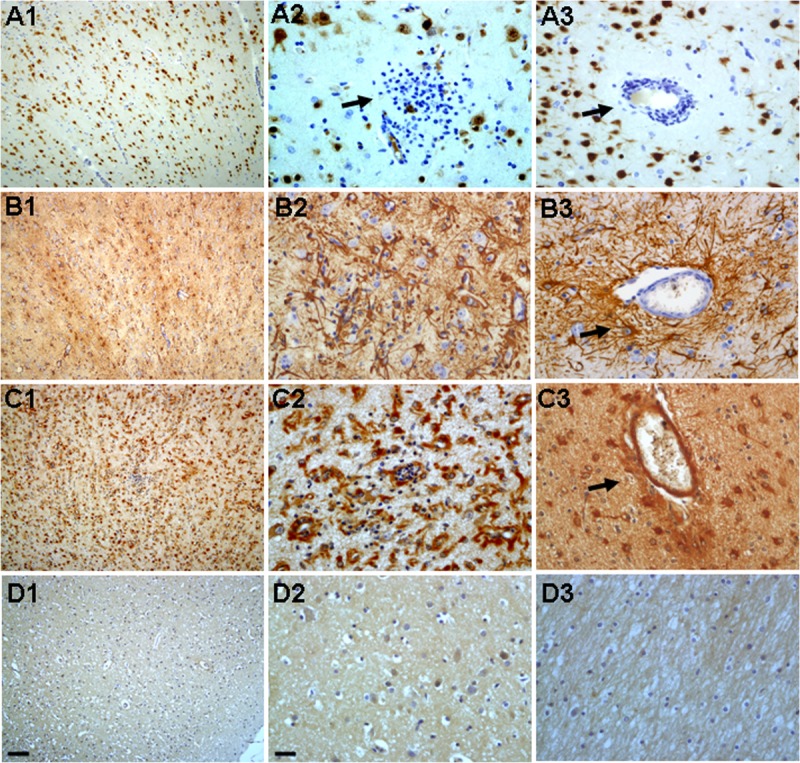
In RE specimens, neuronal loss was observed in the lesion area (Fig. 2A1, A2, A3). Occasional foci of neuronophagia (Fig. 2A2) and marked perivascular cell loss (Fig. 2A3) were seen. There were GFAP-positive reactive astrocytes within lesions (Fig. 2B1, B2, B3) and marked reactive astrogliosis in perivascular areas (Fig. 2B3), as well as bandlike subpial gliosis (Chaslin gliosis) (Fig. 3A, B). Concomitant with reactive astrogliosis, ADK immunoreactivity was observed in cells with typical astroglial morphology (Fig. 2C1, C2, C3) within lesions; they were prominent in perivascular areas (Fig. 2C3) and in areas with bandlike subpial gliosis region (Fig. 3C); ADK was remarkably observed in the astroglial component, with a predominant cytoplasmic localization (Figs. 2C2, C3; 3C). Double labeling confirmed ADK expression in GFAP-positive reactive astrocytes within lesions (Fig. 4A4, B4, C4, D4) and perivascular areas (Fig. 4A1, B1, C1, D1, A2, B2, C2, D2, A3, B3, C3, D3).
FIGURE 3.
Adenosine kinase (ADK) immunoreactivity in bandlike subpial gliosis (Chaslin gliosis). (A–C) Chaslin gliosis in the cortical gray matter ([A] arrow, H&E stain), GFAP immunoreactivity ([B] arrow) and ADK immunoreactivity ([C] arrow) in the astroglial component within the bandlike subpial gliosis. Scale bar = 25 μm.
FIGURE 4.
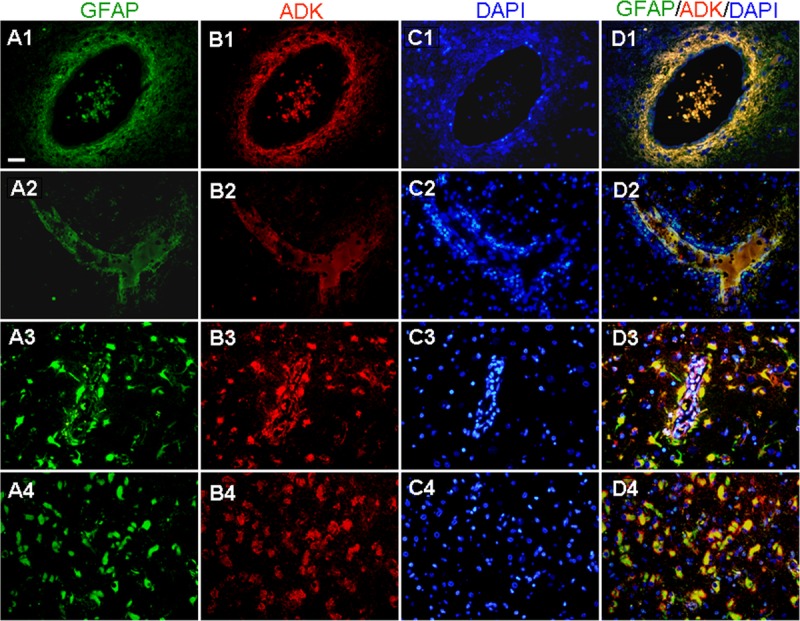
Double labeling for ADK and GFAP in lesion areas in patients with RE. (A–D) Colocalization of ADK and GFAP confirms ADK expression in GFAP-positive reactive astrocytes within lesion areas (A4, B4, C4, D4) and prominent colocalization in perivascular areas, as shown in cross sections (A1–D1) and longitudinal sections (A2–D2, A3–D3). Scale bar = 25 μm for all panels.
Western blot analysis was also performed to quantify the total amount of ADK in total homogenates of control autopsy cortex and surgical cortex from RE patients (Fig. 5A). There was significantly greater ADK expression (p < 0.05) in RE versus control specimens (Fig. 5B).
FIGURE 5.
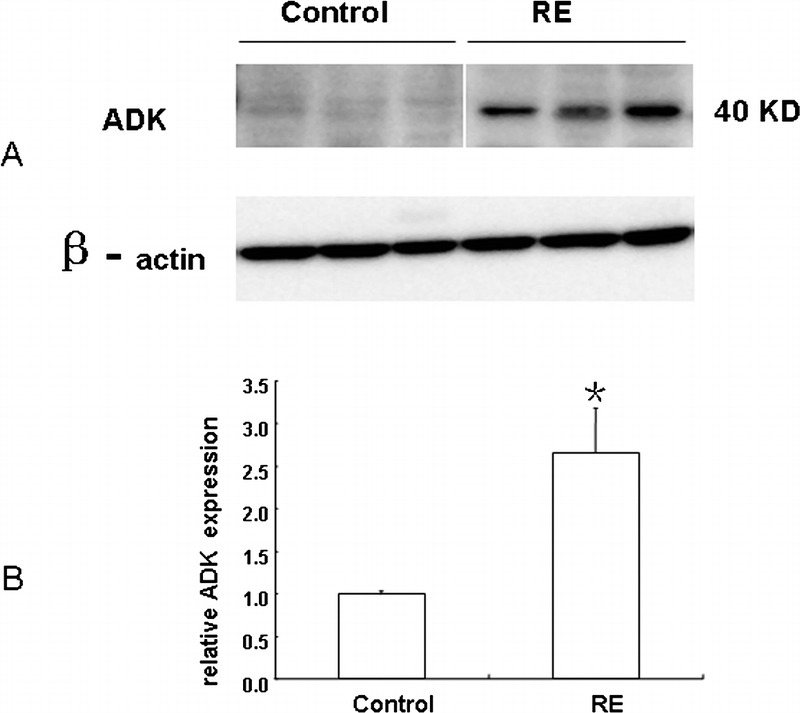
Western blot analysis of adenosine kinase (ADK) in RE and control specimens. (A) Representative immunoblots of ADK (∼40 kDa) in total homogenates of RE and control specimens. Beta-actin immunoreactivity was used to normalize for equal protein loading. (B) Quantitative analysis of ADK expression levels. Data are mean ± SEM (n = 3 per group). * p < 0.05 vs control.
ADK Activity Within RE Lesions
To study the possible relationship between increased ADK immunoreactivity observed in RE tissue with enhanced adenosine metabolism, the enzymatic activity of ADK was evaluated in homogenates derived from RE specimens and controls. Enzyme activity of ADK was determined by performing an enzyme-coupled bioluminescent assay. Rasmussen encephalitis samples from epileptic patients displayed significantly greater ADK activity versus the ADK activity detected in control samples (p < 0.05) (Fig. 6).
FIGURE 6.
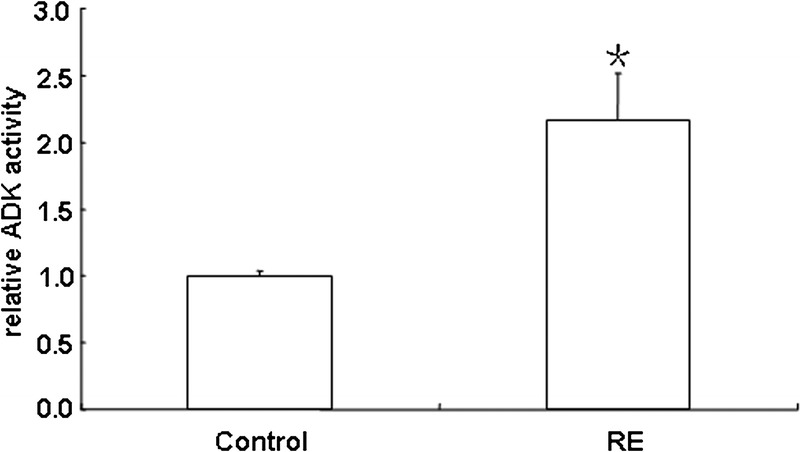
Adenosine kinase enzyme activity measured using an enzyme-coupled bioluminescent assay. The results were normalized to the ADK activity found in control samples and expressed as mean ± SEM of RE (n = 3), control cortex (n = 3); * p < 0.05 vs control.
DISCUSSION
Since the original description of 3 pediatric patients with the title “Focal seizures due to chronic localized encephalitis” by Rasmussen in 1958 (1), most researchers and clinicians have used the term Rasmussen encephalitis (RE) or Rasmussen syndrome for this condition (24). Rasmussen encephalitis is a rare progressive inflammatory disorder of uncertain etiology that characteristically occurs in children who present with refractory seizures, cognitive deterioration, and progressive hemiparesis, resulting from dysfunction of 1 cerebral hemisphere. Radiologically, characteristic MRI features are areas of cortical hyperintense T2/fluid-attenuated inversion recovery signal and progressive atrophy in the affected cerebral hemisphere (25, 26). Rasmussen encephalitis is a pathophysiologically fascinating condition because both seizures and inflammation are seen only in 1 cerebral hemisphere. The neuropathologic hallmarks of RE include inflammation (perivascular lymphocytic cuffing, microglial nodules, leptomeningeal infiltrates), neuronal loss, and astrogliosis (4, 5, 27). The inflammation and astrogliosis have a crucial impact on the seizure induction. Recent data suggest that the inflammation has a direct role in the pathogenetic mechanisms of epileptogenesis (28–30).
Adenosine is an endogenous neuromodulator with important roles on inhibition of inflammation (8, 9) and seizures (10–16). Adenosine is a powerful inhibitory substance released during seizures and implicated in seizure arrest, postictal refractoriness, and suppression of epileptogenesis (14). We and others have recently provided evidence of adenosine dysfunction in astrogliosis, including a decrease in the density of A1Rs (31), upregulation of the major adenosine-removing enzyme ADK (11, 13, 16, 18, 32, 33), and reduced adenosine “tone” in epileptic foci, which can promote further seizures (14). In particular, ADK has been shown to be a critical molecular link between astrogliosis and neuronal dysfunction of epilepsy (13, 15, 18, 32).
Extracellular levels of adenosine are regulated largely by an astrocyte-based adenosine cycle, and astrocytic ADK is the major adenosine-removing enzyme (34). Because ADK activity is the principal factor controlling adenosine metabolism in the brain, minor changes in ADK activity translate rapidly into major changes in adenosine (17). Upregulation of ADK in astrogliosis leads to reducing the tone of ambient adenosine, leading to insufficient activation of adenosine receptors (19). Overexpression of ADK per se might be sufficient to trigger electrographic seizures. The concept is supported by the following findings: i) transgenic mice overexpressing ADK display increased sensitivity to brain injury and seizures; conversely, after pharmacologic induction of an otherwise epileptogenesis-precipitating acute brain injury, transgenic mice with reduced forebrain ADK are resistant to subsequent epileptogenesis; ii) wild-type mice overexpressing the virus-induced ADK in astrocytes of the CA3 area of the hippocampal formation displayed electrographic seizures (35); iii) intrahippocampal implants of stem cells engineered by biallelic genetic disruption of the adenosine kinase gene (Adk-/-) prevent epileptogenesis in a limbic mouse model induced by injection of kainate into the amygdale (13); iv) a ketogenic diet suppresses seizures in transgenic mice overexpressing ADK mice and A1R+/– mice through inhibition of ADK expression (thus increasing ambient levels of adenosine), and thereby increasing activation of adenosine A1 receptors (11); and v) ADK is overexpressed in human temporal lobe epilepsy tissue and glial tumor tissue, as well as the peritumoral region infiltrated by glia, suggesting that reduced adenosine could play a role in the development of epilepsy in patients with glial tumors (18, 22) and in seizure foci of patients with temporal lobe epilepsy (18). Basal adenosine is reduced in epileptic versus control human hippocampus, consistent with ADK contributing to epileptogenesis (14, 18, 22). Thus, ADK has been highlighted as a target for diagnostic and therapeutic intervention.
In the present study, we assessed the expression of the adenosine-metabolizing enzyme ADK in the focal astrogliotic lesion of RE. To our knowledge, this is the first study to describe the cellular distribution and expression of ADK in RE brain. Immunohistochemical analysis showed ADK expression in RE astrocytes with both nuclear and cytoplasmic labeling; however, expression was predominant in the cytoplasm. The cellular distribution and expression of ADK in RE are similar to the findings of ADK expression in brain astroglial tumors (22). Recent findings suggested that ADK exists in 2 isoforms: ADK-long and ADK-short isoforms (36). It has been demonstrated that ADK-long is mainly localized in the nucleus and has an essential role in methylation reactions, possibly being involved in epigenetic controlling mechanisms. On the other hand, ADK-short is localized in the cytoplasm and regulates extracellular adenosine concentrations (22, 36, 37). Therefore, the latter is believed to be more involved in the regulation of neuronal excitability. Accordingly, both experimental and human studies demonstrated that overexpression of ADK in mice resulted in a decrease in the adenosinergic tone and subsequently increased seizure activity (11, 13, 15, 18, 21). Quantification of ADK protein by Western blot analysis proved that ADK increased significantly in RE compared with control. To study the possible relationship between increased ADK immunoreactivity observed in RE tissue with enhanced adenosine metabolism, the enzymatic activity of ADK was also evaluated in homogenates derived from RE specimens and controls. Higher expression of ADK activity was found in RE versus the controls. Thus, the dysregulation of ADK in RE-affected cortex supports the role of this enzyme in the epileptogenesis of RE.
Adenosine kinase expression was more markedly increased in perivascular areas concomitant with reactive astrogliosis, neuron loss, and inflammation (i.e. CD8-positive, CD3-positive T lymphocytes). In addition, ADK expression was prominent in the bandlike subpial gliosis (Chaslin gliosis) region. These findings suggest that i) the perivascular region of the affected cortex may be relevant to seizure generation and propagation and contribute to the epileptogenicity of the region, and ii) increased expression of ADK is inflammation associated. A recent study has demonstrated that inflammation-associated molecules such as interleukin-1β and lipopolysaccharide could play a role in the upregulation of ADK levels in astrocytes, providing a potential additional layer of modulatory cross talk between the astrocyte-based adenosine cycle and inflammation (18).
As an endogenous anticonvulsant of the brain (12), adenosine also modulates important functions of the brain immune system (8, 38–40). It is well known that inflammatory responses, microglial activation, and changes in the blood-brain barrier contribute to epileptogenesis (41, 42). It is also well documented that extracellular adenosine has potent anti-inflammatory properties (9), that is, increases of extracellular adenosine levels turn off local inflammatory responses to decrease cellular damage in the surrounding tissue (9). Increased expression of ADK in the involved cerebral hemisphere in RE patients may decrease extracellular adenosine levels, which may explain the natural history of RE, that is, deterioration of cortical inflammation, lower seizure threshold, and development of pharmacoresistant focal epilepsy and epilepsia partialis continua over time.
We acknowledge limitations to the interpretation of these results. Because we analyzed a relatively small cohort of patients, we cannot exclude the influence of clinical variables, including gender. Moreover, the expression patterns and regulation of adenosine receptors (A1, A2A, A2B, and A3) deserve further investigation. Consequently, adenosine-augmenting therapeutic strategies might combine antiproliferative effects with the well-known anticonvulsive effects (22), as well as anti-inflammatory effects of adenosine (9).
In conclusion, increased expression of the major adenosine-removing enzyme ADK in RE patient plays an important role in the epileptogensis of RE. Our findings suggest that overexpression of ADK is a common pathologic hallmark of RE. Adenosine-augmenting therapeutic strategies with anticonvulsant and anti-inflammatory effects might be the ideal treatment for RE.
ACKNOWLEDGMENTS
The authors thank Professor Detlev Boison (R.S. Dow Neurobiology Laboratories, Legacy Research Institute, Portland, OR) for his kind and generous gift of ADK antibody. The authors confirm that they have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Footnotes
Send correspondence and reprint requests to: Tianfu Li, MD, PhD, Department of Neurology, Epilepsy Center, Beijing Sanbo Brain Hospital, Capital Medical University, Xianshan Yikesong 50, Haidian District, Beijing, 100093 China; E-mail: tianfuli88@hotmail.com
This project was supported by grants from the National Natural Science Foundation of China (81171228), Beijing Key Laboratory of Epilepsy Research, and Construction project of National Clinical Key Specialties (SG2011-02-1-6).
None of the authors has any conflict of interest to disclose.
REFERENCES
- 1. Rasmussen T, Olszewski J, Lloydsmith D. Focal seizures due to chronic localized encephalitis. Neurology 1958; 8: 435– 45 [DOI] [PubMed] [Google Scholar]
- 2. Bien CG, Granata T, Antozzi C, et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: A European consensus statement. Brain 2005; 128: 454– 71 [DOI] [PubMed] [Google Scholar]
- 3. Takei H, Wilfong A, Malphrus A, et al. Dual pathology in Rasmussen’s encephalitis: A study of seven cases and review of the literature. Neuropathology 2010; 30: 381– 91 [DOI] [PubMed] [Google Scholar]
- 4. Farrell MA, Droogan O, Secor DL, et al. Chronic encephalitis associated with epilepsy: Immunohistochemical and ultrastructural studies. Acta Neuropathol 1995; 89: 313– 21 [DOI] [PubMed] [Google Scholar]
- 5. Rogers SW, Andrews PI, Gahring LC, et al. Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science 1994; 265: 648– 51 [DOI] [PubMed] [Google Scholar]
- 6. Schwab N, Bien CG, Waschbisch A, et al. CD8+ T-cell clones dominate brain infiltrates in Rasmussen encephalitis and persist in the periphery. Brain 2009; 132: 1236– 46 [DOI] [PubMed] [Google Scholar]
- 7. Fredholm BB, IJzerman AP, Jacobson KA, et al. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol Rev 2011; 63: 1– 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mills JH, Kim DG, Krenz A, et al. A2A adenosine receptor signaling in lymphocytes and the central nervous system regulates inflammation during experimental autoimmune encephalomyelitis. J Immunol 2012; 188: 5713– 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blackburn MR, Vance CO, Morschl E, et al. Adenosine receptors and inflammation. Handb Exp Pharmacol 2009; 193: 215– 69 [DOI] [PubMed] [Google Scholar]
- 10. Wilz A, Pritchard EM, Li T, et al. Silk polymer-based adenosine release: Therapeutic potential for epilepsy. Biomaterials 2008; 29: 3609– 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Masino SA, Li T, Theofilas P, et al. A ketogenic diet suppresses seizures in mice through adenosine A(1) receptors. J Clin Invest 2011; 121: 2679– 83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li T, Steinbeck JA, Lusardi T, et al. Suppression of kindling epileptogenesis by adenosine releasing stem cell–derived brain implants. Brain 2007; 130: 1276– 88 [DOI] [PubMed] [Google Scholar]
- 13. Li T, Ren G, Lusardi T, et al. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest 2008; 118: 571– 82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. During MJ, Spencer DD. Adenosine: A potential mediator of seizure arrest and postictal refractoriness. Ann Neurol 1992; 32: 618– 24 [DOI] [PubMed] [Google Scholar]
- 15. Gouder N, Scheurer L, Fritschy JM, et al. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci 2004; 24: 692– 701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fedele DE, Gouder N, Guttinger M, et al. Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain 2005; 128: 2383– 95 [DOI] [PubMed] [Google Scholar]
- 17. Boison D. Adenosine kinase, epilepsy and stroke: Mechanisms and therapies. Trends Pharmacol Sci 2006; 27: 652– 58 [DOI] [PubMed] [Google Scholar]
- 18. Aronica E, Zurolo E, Iyer A, et al. Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia 2011; 52: 1645– 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boison D. The adenosine kinase hypothesis of epileptogenesis. Prog Neurobiol 2008; 84: 249– 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boison D. Adenosine dysfunction in epilepsy. Glia 2012; 60: 1234– 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li T, Lytle N, Lan JQ, et al. Local disruption of glial adenosine homeostasis in mice associates with focal electrographic seizures: A first step in epileptogenesis? Glia 2012; 60: 83– 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Groot M, Iyer A, Zurolo E, et al. Overexpression of ADK in human astrocytic tumors and peritumoral tissue is related to tumor-associated epilepsy. Epilepsia 2012; 53: 58– 66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bien CG, Widman G, Urbach H, et al. The natural history of Rasmussen’s encephalitis. Brain 2002; 125: 1751– 59 [DOI] [PubMed] [Google Scholar]
- 24. Piatt JJ, Hwang PA, Armstrong DC, et al. Chronic focal encephalitis (Rasmussen syndrome): Six cases. Epilepsia 1988; 29: 268– 79 [DOI] [PubMed] [Google Scholar]
- 25. Bien CG, Urbach H, Deckert M, et al. Diagnosis and staging of Rasmussen’s encephalitis by serial MRI and histopathology. Neurology 2002; 58: 250– 57 [DOI] [PubMed] [Google Scholar]
- 26. Yacubian EM, Marie SK, Valerio RM, et al. Neuroimaging findings in Rasmussen’s syndrome. J Neuroimaging 1997; 7: 16– 22 [DOI] [PubMed] [Google Scholar]
- 27. Pardo CA, Vining EP, Guo L, et al. The pathology of Rasmussen syndrome: Stages of cortical involvement and neuropathological studies in 45 hemispherectomies. Epilepsia 2004; 45: 516– 26 [DOI] [PubMed] [Google Scholar]
- 28. Vezzani A, French J, Bartfai T, et al. The role of inflammation in epilepsy. Nat Rev Neurol 2011; 7: 31– 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nabbout R, Vezzani A, Dulac O, et al. Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol 2011; 10: 99– 108 [DOI] [PubMed] [Google Scholar]
- 30. Devinsky O, Vezzani A, Najjar S, et al. Glia and epilepsy: Excitability and inflammation. Trends Neurosci 2013; 36: 174– 84 [DOI] [PubMed] [Google Scholar]
- 31. Glass M, Faull RL, Bullock JY, et al. Loss of A1 adenosine receptors in human temporal lobe epilepsy. Brain Res 1996; 710: 56– 68 [DOI] [PubMed] [Google Scholar]
- 32. Li T, Lan JQ, Boison D. Uncoupling of astrogliosis from epileptogenesis in adenosine kinase (ADK) transgenic mice. Neuron Glia Biol 2008; 4: 91– 99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li T, Quan LJ, Fredholm BB, et al. Adenosine dysfunction in astrogliosis: Cause for seizure generation? Neuron Glia Biol 2007; 3: 353– 66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boison D, Chen JF, Fredholm BB. Adenosine signaling and function in glial cells. Cell Death Differ 2010; 17: 1071– 82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Theofilas P, Brar S, Stewart KA, et al. Adenosine kinase as a target for therapeutic antisense strategies in epilepsy. Epilepsia 2011; 52: 589– 601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cui XA, Singh B, Park J, et al. Subcellular localization of adenosine kinase in mammalian cells: The long isoform of AdK is localized in the nucleus. Biochem Biophys Res Commun 2009; 388: 46– 50 [DOI] [PubMed] [Google Scholar]
- 37. Boison D. Adenosine as a modulator of brain activity. Drug News Perspect 2007; 20: 607– 11 [DOI] [PubMed] [Google Scholar]
- 38. Hasko G, Pacher P, Vizi ES, et al. Adenosine receptor signaling in the brain immune system. Trends Pharmacol Sci 2005; 26: 511– 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wei W, Du C, Lv J, et al. Blocking A2B adenosine receptor alleviates pathogenesis of experimental autoimmune encephalomyelitis via inhibition of IL-6 production and Th17 differentiation. J Immunol 2013; 190: 138– 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mills JH, Alabanza LM, Mahamed DA, et al. Extracellular adenosine signaling induces CX3CL1 expression in the brain to promote experimental autoimmune encephalomyelitis. J Neuroinflammation 2012; 9: 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ravizza T, Gagliardi B, Noe F, et al. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis 2008; 29: 142– 60 [DOI] [PubMed] [Google Scholar]
- 42. Uva L, Librizzi L, Marchi N, et al. Acute induction of epileptiform discharges by pilocarpine in the in vitro isolated guinea-pig brain requires enhancement of blood-brain barrier permeability. Neuroscience 2008; 151: 303– 12 [DOI] [PMC free article] [PubMed] [Google Scholar]