

Abstract
Hepcidin, the liver-produced peptide hormone, is a principal regulator of iron homeostasis. Abnormal hepcidin production has emerged as a causative factor in several common iron disorders. Hepcidin insufficiency results in iron overload in hereditary hemochromatosis and iron-loading anemias, whereas hepcidin excess causes or contributes to the development of iron-restricted anemias in inflammatory diseases, infections, some cancers and chronic kidney disease. Not surprisingly, hepcidin and related pathways have become the target for the development of novel therapeutics for iron disorders. In this review, we will summarize the strategies and development programs that have been devised for agonizing or antagonizing hepcidin and its receptor ferroportin.
Systemic iron homeostasis and the iron hormone hepcidin
Iron plays an essential role in cellular metabolism and erythropoiesis. In a healthy adult human, plasma iron is strictly maintained in the range of 10–30 μM. Long-term deviations from this range cause clinically important iron-related disorders.1 Low plasma iron (hypoferremia) restricts the amount of iron available to erythrocyte precursors eventually resulting in anemia. Cellular processes requiring ferroproteins may also be compromised in a hypoferremic state. Non-hematologic manifestations of iron deficiency can include changes in nails, tongue, and esophagus as well as deficits in muscular function.2 At the other extreme, when plasma iron concentrations exceed the iron-binding capacity of transferrin, iron will complex with organic anions such as citrate or albumin3 (commonly referred to as non-transferrin-bound iron or NTBI). High concentrations of iron transferrin and the presence of NTBI in circulation result in iron accumulation in parenchymal cells. Excessive intracellular iron catalyzes the generation of reactive oxygen species that can cause extensive damage to cells and tissues, with resulting dysfunction of the liver, heart or endocrine glands.3
To meet the iron demands of the organism while avoiding iron toxicity, systemic iron balance is tightly regulated by the peptide hormone hepcidin (HAMP),1 produced primarily in hepatocytes. Hepcidin controls plasma iron concentrations by regulating the delivery of iron to plasma through the iron exporting protein ferroportin.4 Ferroportin (SLC40A1, Solute carrier family 40, member 1) is the sole known cellular iron exporter in vertebrates.5 It is mainly expressed in cells processing large amounts of iron: enterocytes in the duodenum involved in dietary iron absorption, macrophages of the spleen and liver that recycle senescent erythrocytes, hepatocytes involved in iron storage, and placental syncytiotrophoblast that transfers iron from the mother to the fetus. Hepcidin binding triggers rapid ubiquitination of ferroportin, resulting in endocytosis of the ligand-receptor complex and their ultimate proteolysis.6,7 Hepcidin-induced degradation of ferroportin decreases the delivery of iron from iron exporting cells into plasma, resulting in hypoferremia. Because of the central role hepcidin plays in the maintenance of iron homeostasis, dysregulation of hepcidin production or of its interaction with ferroportin results in a spectrum of iron disorders.
Regulation of hepcidin production
Multiple new therapeutic approaches targeting hepcidin are based on manipulating the mechanisms regulating hepcidin production. A brief overview of the main pathways regulating hepcidin production is provided.
Hepcidin regulation by iron availability
Similarly to other hormones that are regulated by their substrates, hepcidin production is homeostatically regulated by iron. Hepcidin transcription, and consequently its synthesis and secretion, is induced in response to increases in plasma iron or cellular iron stores, and this generates a negative feedback loop as hepcidin restricts the flows of iron into the plasma and blocks further dietary iron absorption. Mutations in the proteins involved in iron sensing or signal transduction can lead to hepcidin deficiency and the development of iron overload in humans and mice. Our current understanding of the pathways involved in hepcidin regulation by iron is shown in Figure 1A.
Figure 1.
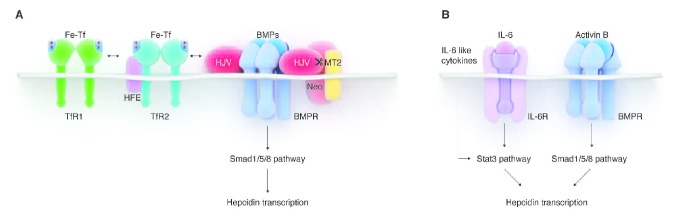
Pathways regulating hepcidin expression. (A) Hepcidin regulation by iron. Binding of holo-transferrin (Fe-Tf) to TfR1 displaces HFE from the complex with TfR1. HFE then interacts with TfR2, which is itself stabilized by the binding of Fe-Tf. The HFE/TfR2 is thought to form a complex with hemojuvelin (HJV), a BMP co-receptor. The BMP pathway is consequently stimulated, resulting in the phosphorylation of Smad1/5/8 and an increase in hepcidin transcription. Additional proteins (TMPRSS6/matriptase-2 (MT2) and neogenin) mediate the cleavage of membrane HJV and thus modulate hepcidin transcription. (B) Hepcidin regulation by inflammation. During inflammation, IL-6 and other cytokines (e.g. oncostatin M, IL-22) activate the Stat3 pathway to promote transcription of hepcidin. Activin B acting via BMP receptors and the Smad1/5/8 pathway was also proposed to stimulate hepcidin expression during inflammation.
The bone morphogenetic protein receptors (BMPR) and their SMAD signaling pathway mediate the hepcidin transcriptional response to iron levels. ALK2 and ALK3 have recently been identified as the specific BMP type I receptors involved in hepcidin regulation8 as mice with liver-specific deletion of Alk2 or Alk3 exhibited decreased hepcidin expression and iron overload. These mice also failed to increase hepcidin rapidly in response to iron-dextran injection.
BMP ligands and co-receptors also participate in hepcidin regulation by iron. Multiple BMP ligands can activate hepcidin transcription in vitro, but so far only BMP6 was shown to be an important non-redundant regulator of hepcidin transcription in vivo in mice.9,10 BMP6 knockout mice have profoundly decreased hepcidin expression and develop severe iron overload. BMP6−/− mice did not increase hepcidin in response to acute dietary iron challenge, and had a blunted hepcidin response to chronic iron loading.11 However, in humans, no iron-related role of BMP6 has so far been demonstrated. The glycosylphosphatidylinositol (GPI)-linked protein hemojuvelin (HJV) is the BMP co-receptor that specifically regulates hepcidin expression.12 Humans with HJV mutations develop the most severe form of genetic iron overload, juvenile hemochromatosis, due to profound hepcidin deficiency.13Hjv knockout mice are also severely iron-loaded, fail to increase hepcidin after acute iron loading, and show severe impairment in response to chronic iron loading.11
The BMP pathway signaling to hepcidin is modulated by plasma iron concentrations.14 In blood plasma, iron is normally bound to transferrin. It has been proposed that transferrin receptors 1 and 2 (TfR1 and TfR2) and their interacting partner HFE serve as sensors of holo-transferrin concentrations. Increasing concentrations of holo-transferrin displace HFE from TfR1 and promote HFE interaction with TfR2.15 HFE and TfR2 were shown to interact with HJV, forming a membrane-associated protein complex, at least when over-expressed in hepatoma cells.16 It has, therefore, been thought that HFE and TfR2 promote hepcidin expression by enhancing SMAD activation.17 However, there is evidence that HFE and TfR2 can also regulate hepcidin independently of each other,18 although the specific signaling mechanisms are not understood. Mutations in HFE and TfR2 in humans lead to relative hepcidin deficiency and hereditary hemochromatosis.1,19
Hepcidin production is further modulated by proteins that posttranslationally regulate the levels of membrane-associated HJV. Transmembrane serine protease TMPRSS6, also known as matriptase-2, cleaves membrane HJV,20 possibly in an iron-dependent manner as TMPRSS6 protein levels were increased in iron deficient rats.21 Inactivation of TMPRSS6 in humans and mice leads to inappropriately high hepcidin levels and consequent iron restriction and anemia.22 A multifunctional membrane protein neogenin was also shown to interact with TMPRSS6 and HJV, and it facilitated cleavage of HJV by TMPRSS6 in vitro.23 The specific involvement of these proteins in iron sensing is still uncertain.
Hepcidin transcription is also controlled by intracellular iron concentrations via a mechanism that is distinct from that related to extracellular iron.11 The specific pathways or cell types involved in the sensing of intracellular iron are not known. One of the mediators may be BMP6, whose expression increases with liver iron loading.24
Hepcidin regulation by erythropoietic signals
Increased erythropoietic activity, such as after hemorrhage or hemolysis, suppresses hepcidin expression.25 Intact bone marrow is necessary for this effect,26 and it is thought that precursors in the bone marrow secrete an ‘erythroid factor’ which suppresses hepcidin in proportion to the erythropoietic activity. Lower hepcidin makes more iron available for erythropoiesis. Exuberant erythropoietic activity that fails to produce mature erythrocytes, such as in anemias with ineffective erythropoiesis, leads to pathological suppression of hepcidin and consequent iron overload.27,28 GDF-15 and TWSG1 were proposed as candidate suppressors of hepcidin in beta(β)-thalassemia and other iron-loading anemias.29,30 The question as to whether the same pathways mediate both physiological and pathological suppression of hepcidin in response to increased erythropoietic activity remains unresolved.
Hepcidin regulation by inflammatory stimuli
Hepcidin increase caused by inflammation may have evolved as a host response to infection where hepcidin-triggered hypoferremia could slow the growth of microorganisms. A protective role of hepcidin in infections, however, has not been formally demonstrated. Nevertheless, hepcidin regulation by inflammation has been extensively explored and elevated hepcidin is thought to contribute to the development of anemia in a variety of inflammatory conditions.1 Hepcidin transcription is increased by interleukin-6 (IL-6)31 (Figure 1B). The binding of IL-6 to its receptor triggers a signal transduction cascade that results in the phosphorylation of the transcriptional factor STAT3 by the signaling kinase JAK2. Phosphorylated STAT3 in turns binds to the cognate motifs in the hepcidin promoter and increases hepcidin transcription.32 Other cytokines that activate the STAT pathway can also increase hepcidin transcription, including IL-22 and oncostatin M.33,34 The BMP pathway synergistically stimulates hepcidin expression during inflammation.33,35 Additionally, activin B, a member of the TGF-β superfamily, has been suggested to mediate hepcidin increase after LPS injection even in IL-6 knockout mice,36 by activating the Smad1/5/8 pathway.
The role of hepcidin in iron disorders
Hepcidin deficiency in iron overload disorders
Hereditary hemochromatoses are a group of genetic disorders of iron homeostasis in which hepcidin is deficient, due to deleterious mutations in the genes encoding either regulators of hepcidin or hepcidin itself.1 Low hepcidin allows unchecked ferroportin activity giving rise to dietary iron hyperabsorption and rapid iron release from macrophages (Figure 2B). As transferrin iron-binding capacity is exceeded by the large iron flux, non-transferrin-bound iron (NTBI) appears in the circulation. NTBI is taken up by the liver and other parenchyma, eventually resulting in iron overload and associated morbidity. The severity of iron overload correlates with the degree of hepcidin deficiency. In juvenile hemochromatosis, mutations in the genes encoding hepcidin37 or hemojuvelin13 result in almost complete loss of hepcidin synthesis, with little or no hepcidin detectable in plasma. In the adult form of the disease caused by mutations in HFE and TfR2, hepcidin response to iron loading is partially preserved,11,19 thus iron overload develops later in life and tends to be less severe.
Figure 2.
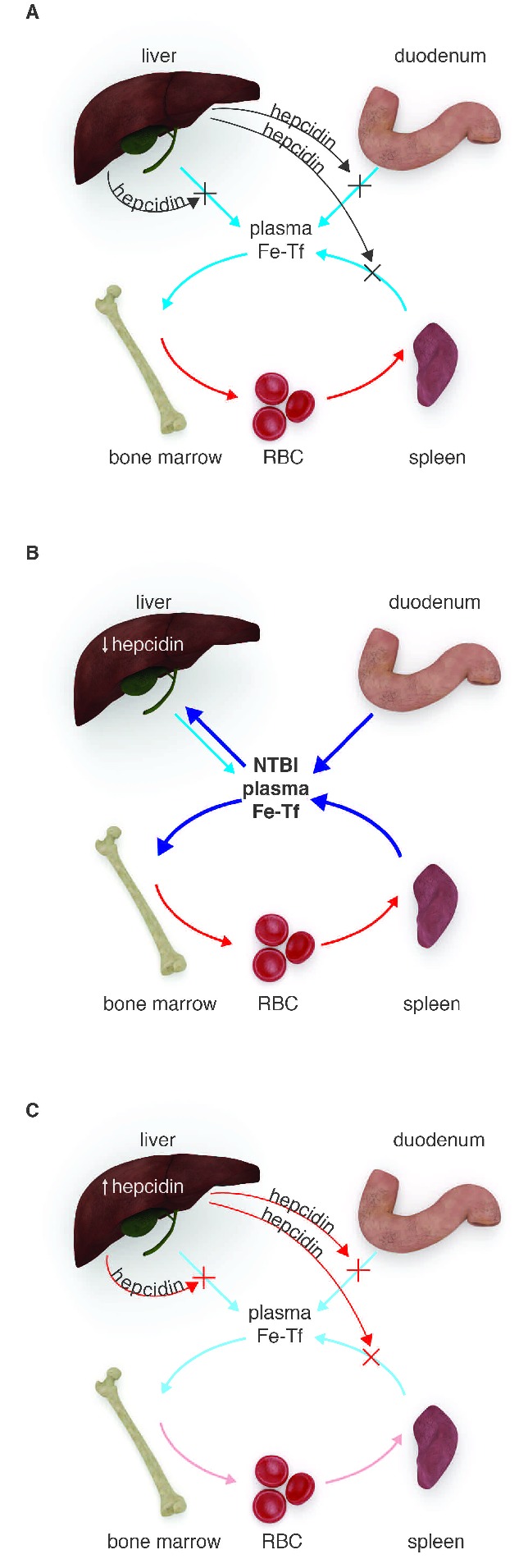
The role of hepcidin in the pathogenesis of iron disorders. (A) Normal iron homeostasis: hepcidin modulates iron flows into plasma from splenic and liver macrophages, hepatocytes and the duodenum to maintain relatively constant plasma iron concentrations. Most of the plasma iron is utilized by erythroid precursors in the bone marrow to synthesize hemoglobin. This iron is eventually recycled from old red blood cells by erythrophagocytosing macrophages. (B) Iron overload: hepcidin insufficiency causes iron overload in hereditary hemochromatosis and iron-loading anemias. In hereditary hemochromatosis, hepcidin production is low because of inactivating mutations in the genes encoding hepcidin or its regulators. In iron-loading anemias, hepcidin is suppressed by the high erythropoietic drive, via as yet poorly understood mechanisms. In either disease, low hepcidin allows excessive iron absorption and rapid release of recycled iron from macrophages. Plasma iron levels increase, non-transferrin-bound iron (NTBI) accumulates, and excess iron is deposited in vital organs where it causes organ damage. (C) Iron-restricted anemias: hepcidin is elevated in inflammatory disorders, certain cancers and chronic kidney disease because proinflammatory cytokines stimulate hepcidin transcription. In kidney diseases, decreased renal clearance of hepcidin may also contribute. In iron-refractory iron deficiency anemia, hepcidin is increased because of the mutations in TMPRSS6, a negative regulator of hepcidin. In all of these conditions, elevated hepcidin inhibits iron efflux from macrophages, hepatocytes and the duodenum. As a result, hypoferremia develops and erythropoiesis becomes iron-limited, eventually manifesting as anemia.
Another form of iron overload associated with hepcidin deficiency includes iron-loading anemias such as β-thalassemia and congenital dyserythropoietic anemias.38 These diseases are characterized by ineffective erythropoiesis with extreme expansion of the erythroid precursor pool and premature death of these cells before they develop into functional erythrocytes. It is thought that precursors release one or more suppressive factors which act on the liver to decrease hepcidin production. In non-transfused patients, similarly to hereditary hemochromatosis, low hepcidin leads to excessive iron absorption, high concentrations of NTBI and development of iron overload (Figure 2B). Despite iron loading, hepcidin remains inappropriately suppressed in these patients because the exuberant erythroid activity opposes the effects of iron on hepcidin production.27 Even in transfused patients, where expanded erythropoiesis is partially ameliorated, hepcidin is lower than would be expected for the degree of iron overload.28,39 Furthermore, hepcidin levels fluctuate depending on the phase of the transfusion cycle30 suggesting that, as the effect of transfusion wears off and hepcidin is lowered, dietary iron absorption could contribute to iron loading even in transfused patients.
Hepcidin excess in iron-restrictive disorders
By causing ferroportin degradation, excessive plasma hepcidin inhibits iron absorption in the duodenum and restricts iron export from macrophages, thereby decreasing the iron available for hemoglobin production (Figure 2C). Accordingly, transgenic mice over-expressing hepcidin developed anemia with iron-restricted erythropoiesis.40 Increased hepcidin is seen in diverse human disorders associated with iron restriction and anemia1,41 but the underlying causes are varied. In inflammatory diseases and infections, proinflammatory cytokines such as IL-6 stimulate hepcidin production. BMP-2 was shown to be a major stimulus of hepcidin production in multiple myeloma.35 In rare hepatic adenomas, autonomous hepcidin production by tumors was observed.42 In the genetic disorder iron-refractory iron deficiency anemia (IRIDA), loss-of-function mutations in TMPRSS6, the negative regulator of hepcidin transcription, lead to hepcidin overproduction.22 In chronic kidney disease, impaired renal clearance of hepcidin from plasma may contribute to increased hepcidin levels.43,44 The specific mechanisms by which hepcidin excess and the resulting iron restriction affect the size of erythrocytes, their hemoglobin content and the production rate are not yet known. Remarkably, whereas in non-inflammatory syndromes of hepcidin excess (IRIDA,22 hepcidin-producing adenomas,42 transgenic hepcidin mice40) erythrocytes are characteristically small, during inflammatory iron restriction erythrocyte numbers are decreased but their size and hemoglobin content are relatively preserved.45 These differences and early experimental evidence suggest that inflammatory cytokines, including interferon-γ and IL-6, may contribute to anemia through hepcidin-independent effects on hematopoietic precursor differentiation and erythrocyte maturation.46,47 Nevertheless, decreased erythropoietic response to endogenous and exogenous erythropoietin appears to be characteristic of both inflammatory and non-inflammatory forms of iron-restricted erythropoiesis.
Hepcidin-based therapeutics for iron disorders
Proof of principle in mouse genetic models
Increasing evidence indicates that hepcidin deficiency and excess play a pathogenic role in iron disorders, thus hepcidin agonists and antagonists would be expected to improve the treatment of these common conditions. Genetic studies in animal models provided the proof of principle that hepcidin could be an effective therapeutic target. Transgenic overexpression of hepcidin in HFE−/− mice, a model of the most common form of human hereditary hemochromatosis, prevented the liver iron overload normally seen in these mutants.48 In β-thalassemia intermedia mouse models, moderate transgenic hepcidin expression not only decreased iron loading of the liver, but also had beneficial effects on erythropoiesis.49 The lifespan of red cells was prolonged, hemoglobin levels increased and splenomegaly was diminished, apparently because mild iron restriction in these mice decreased formation of insoluble membrane-bound globins and reactive oxygen species.
Conversely, in mouse models of anemia of inflammation (AI), hepcidin deficiency was demonstrated to ameliorate the phenotype. When anemia was induced by heat-killed Brucella abortus or by turpentine-induced sterile abscesses, hepcidin knockout mice demonstrated milder anemia with faster recovery than wild-type mice.50–52 Similarly, in mice with liver-specific deletion of the BMP receptor Alk3, a critical regulator of hepcidin, chronic turpentine injections failed to decrease hemoglobin.53
Hepcidin agonists
Current treatment options for patients with iron overload diseases are relatively limited. Hereditary hemochromatosis is typically treated with phlebotomies. This is an effective and inexpensive treatment for many patients, but not all patients tolerate phlebotomy.54 Iron-loading anemias such as β-thalassemia are treated by iron chelation but this treatment can have serious side effects.55 As hepcidin deficiency causes iron overload in both of these disorders, agents that can mimic hepcidin function or potentiate its endogenous production would be expected to prevent systemic accumulation of iron. Both of these types of approaches are under development (Table 1). The hepcidin peptide itself does not have desirable pharmacological properties as the synthesis of bioactive hepcidin is difficult and costly and the peptide has a relatively short half-life in circulation (several minutes).56
Table 1.
Hepcidin-targeting approaches currently under development

Class 1: hepcidin mimics
Minihepcidins are peptide-based hepcidin agonists which were rationally designed based on the region of hepcidin that interacts with ferroportin.57 Mutagenesis studies and biomolecular modeling indicated that the first 9 amino acids of the hepcidin N-terminus were important for hepcidin activity. Synthetic N-terminal peptides were further engineered to increase their bioavailability. Unnatural amino acids were introduced to increase resistance to proteolysis and fatty acids were conjugated to prolong the half-life in circulation and potentially increase oral absorption. Some of the minihepcidins that were developed were at least as potent as the full-length hepcidin, and had a longer duration of action.58 To confirm the results of the principle studies, a minihepcidin (PR65) was tested in hepcidin knockout mice, a model of severe hemochromatosis. In a ‘preventive mode’, iron-depleted hepcidin knockouts were given subcutaneous injections of either PR65 or solvent for two weeks while they were maintained on an iron-loading chow. Unlike the solvent injections, PR65 completely prevented iron loading of the liver, decreased heart iron levels, and normalized iron retention in the spleen and duodenum.58 At high doses, PR65 also caused profound iron restriction and anemia. This would be expected as the main side effect of the therapy, and titration to effect may be required to avoid excessive hypoferremia and iron restriction. In a ‘treatment mode’, hepcidin knockout mice with pre-existing iron overload were injected with PR65 for two weeks. This caused partial redistribution of iron from the liver to the spleen, but the effect was more moderate.58 Minihepcidins could thus be useful for the prevention of iron overload, or may be used in combination with phlebotomy or chelation for the treatment of existing iron overload.
Class 2: stimulators of hepcidin production
Targeting TMPRSS6: Nai et al.59 demonstrated that homozygous inactivation of TMPRSS6 in thalassemic th3/+ mice increased hepcidin levels, ameliorated iron overload, and improved ineffective erythropoiesis. The study provided the proof of principle that targeting of Tmprss6, a negative regulator of hepcidin production, may be a useful approach for the therapy of iron overload disorders. Recently, two studies harnessed RNA-based technologies to decrease the levels of Tmprss6 and demonstrated their effectiveness in increasing hepcidin transcription and reverting iron overload in mouse models.60,61 Guo et al. utilized anti-sense oligonucleotides (ASOs) against Tmprss6 mRNA.60 ASOs are single-stranded nucleic acids containing modified bases that protect ASOs from degradation by exonucleases. These ASOs trigger the degradation of their target mRNA based on the RNaseH mechanism. Schmidt et al. used small interfering RNA (siRNA) against Tmprss6.61 Unlike ASOs, siRNAs are double-stranded nucleic acids designed to inhibit the expression of its target genes through the RNA interference pathway. Tmprss6 siRNA was packaged in lipid nanoparticles that promotes their delivery to the liver, the main site of TMPRSS6 expression and activity. Both studies evaluated the effect of Tmprss6 knockdown in HFE−/− mice and thalassemic th3/+ mice after six weeks. ASOs were injected twice a week (100 mg/kg/wk) and siRNA every two weeks (1 mg/kg). The effects of the two approaches on endogenous hepcidin expression, iron and hematologic parameters were remarkably similar. With both approaches, hepcidin mRNA increased 2–3 fold in HFE−/− and th3/+ mice compared to control injections.60,61 In HFE−/− mice, serum iron and liver iron concentrations were reduced compared to the vehicle control group, and spleen iron was increased. A mild reduction in hemoglobin was also observed indicating some iron restriction. In the mouse model of thalassemia intermedia (th3/+), 6-week treatment decreased liver iron but also improved anemia and ineffective erythropoiesis, with reduced spleen size and improved maturation of erythroid precursors, reproducing the effect of transgenic hepcidin overexpression in th3+/− mice.49 Targeting TMPRSS6 in humans may be a promising approach to treat iron overload disorders associated with hepcidin insufficiency.
BMP agonists: a study by Corradini et al.62 highlighted the potential of BMP6 agonists for the management of iron overload disorders. As mentioned earlier, BMP6 appears to be the major endogenous BMP regulating hepcidin transcription, at least in mice. In BMP6 knockout mice, no other significant abnormalities were noted except that hepatic hepcidin mRNA was virtually undetectable and iron overload phenotype was seen in the liver and other organs.9,10 Corradini et al. treated HFE–/– mice with exogenous BMP6 twice daily for ten days. This resulted in increased hepcidin mRNA expression, reduced serum iron, and the expected iron retention in the spleen and duodenum.62 No decrease in the iron content of the liver, heart or pancreas was observed within ten days, probably because the mice were already iron over-loaded and longer dosing would be necessary to observe any effects. Similar to other approaches that increased hepcidin activity, erythropoiesis was mildly iron-restricted. The main challenge with therapeutic application of BMP agonists in iron disorders will be off-target effects such as bone formation. For example, 2-week injections of BMP6 in mice resulted in peritoneal calcifications.
A small-scale chemical screen in zebrafish embryos identified the isoflavone genistein as an enhancer of hepcidin transcription.63 In a cellular system in vitro, genistein promoted hepcidin expression via both Stat3 and BMP-dependent pathways, but not via estrogen receptors, known targets of genistein. This is the first demonstration of a small molecule acting to promote hepcidin activity, and its potential for the treatment of iron overload remains to be tested in mouse models.
Hepcidin antagonists
Elevated serum hepcidin is thought to contribute to the development of anemia associated with inflammation, chronic kidney disease, some cancers and IRIDA. Treatment options for these diseases include erythropoiesis-stimulating agents (ESAs) with or without high-dose intravenous iron, but erythropoietin resistance is commonly observed as a consequence of inflammation.64,65 The long-term effects of high doses of ESAs and IV iron are not known but concerns have arisen about the potential side effects of ESAs66,67 most of which appear to be dose-related. More specific interventions are needed, and hepcidin antagonists may offer an alternative and/or allow safer dosing of erythropoietic drugs.
Multiple strategies to counter the effect of hepcidin in iron-restrictive disorders have been described (Table 1). Decreased hepcidin production could be achieved by targeting the hepcidin regulatory pathways (iron-related, inflammatory or erythropoietic pathway) or by targeting hepcidin mRNA using anti-gene therapies. The hepcidin peptide itself can be neutralized with pharmacological agents. In addition, the hepcidin receptor ferroportin could be targeted in different ways: by interfering with the hepcidin binding site on ferroportin, by preventing hepcidin-mediated ferroportin endocytosis, or by stimulating de novo ferroportin production.
Class 1: decreasing hepcidin production
Targeting the BMP pathway: as the BMP pathway plays a key role in stimulating hepcidin transcription, sequestration of BMP ligands could decrease hepcidin expression. Heparin, a glycosaminoglycan widely used as an anticoagulant, has long been known to bind BMPs.68 Poli and colleagues demonstrated that heparin inhibited hepcidin expression in hepatic cell lines as well as in mice.69 Daily injections in mice for seven days (50 mg/kg/d) decreased hepcidin mRNA expression and SMAD phosphorylation, increased serum iron and reduced spleen iron concentration. In 5 patients treated with low molecular weight heparin to prevent deep vein thrombosis, serum hepcidin concentration decreased by 80–85% within 2–5 days after the start of the treatment. Concurrently, increased serum iron levels and transferrin saturation were noted in all 5 patients. Although the safety profile of heparin is well understood, its anticoagulant activity hinders its wider application to iron-restricted disorders. To address this, Poli and colleagues recently prepared non-anticoagulant heparins70 that retained BMP-modulating activity and potently suppressed hepcidin in vitro and in mice in vivo, also in the presence of an inflammatory stimulus. Interestingly, heparin is also known to have anti-inflammatory activity69 and this could further contribute to its usefulness for the treatment of iron-restricted disorders.
Soluble hemojuvelin-Fc fusion protein (sHJV.Fc) is another agent aimed at preventing the interaction of BMPs with their receptors. sHJV is a soluble fragment of the membrane-linked BMP co-receptor hemojuvelin, and the soluble and membrane form have opposing effects on hepcidin expression.71 sHJV is known to bind BMP6 and other BMPs, and this results in decreased Smad signaling in the cell and decreased hepcidin expression.72,73 sHJV.Fc was also shown to ameliorate anemia of inflammation in a rat model.74 Rats were injected with Group A Streptococcal Peptidoglycan-Polysaccharide (PG-APS), and three weeks later they were injected with sHJV.Fc (20 mg/kg) twice a week for an additional four weeks. sHJV.Fc treatment resulted in higher hemoglobin, increased ferroportin levels in the spleen and higher serum iron, although by this point hepcidin mRNA was not significantly decreased. Interestingly, sHJV.Fc was shown to be a broad-spectrum BMP antagonist75 that blunted BMP2- and BMP6-mediated induction of a host of genes in Hep3B cells. This raises the question of the possible off-target effects of sHJV-Fc and other BMP-targeting therapeutics.
Inhibition of BMP type I receptor signaling by small-molecule inhibitors was also effective in alleviating iron-restricted anemias. LDN-193189 is a derivative of dorsomorphin, a small-molecule inhibitor of the BMP pathway, and it specifically antagonizes BMP receptor isotypes ALK2 and ALK3.76 Theurl et al. used the rat PG-APS model of anemia of inflammation, and starting at three weeks after PG-APS injection administered LDN-193189 every other day for four weeks. This regimen decreased hepcidin, increased serum iron and effectively reversed anemia.74 Similarly, in a chronic turpentine model of anemia of inflammation in mice, daily injections of LDN-193189 administered concurrently with turpentine injections77 prevented the development of anemia. LDN-193189 improved hemoglobin even when administered to mice after the turpentine-induced anemia was already established.77 In a rat model of anemia of chronic kidney disease caused by adenine treatment, Sun and colleagues administered LDN-193189 for five weeks.78 Although overall hemoglobin levels did not improve, hepcidin mRNA was decreased, and this was accompanied by improved mobilization of iron from the spleen, increased serum iron and increased hemoglobin content of reticulocytes.
Inhibiting the BMP pathways clearly results in decreased hepcidin levels in animal models and even in humans. However, as BMP pathways have pleiotropic effects, the main challenge of the BMP-targeted agents will be to achieve specificity for hepcidin over other BMP-regulated processes.
RNA-based targeting of hepcidin regulators is also under development (hemojuvelin antisense oligonucleotides79 and TfR2 siRNA).80 TfR2 siRNA administration potently decreased hepcidin mRNA in the rat model of anemia of inflammation (PG-APS) and resulted in the correction of anemia.80 Given that clinical consequences of HJV or TfR2 mutations in human patients are limited to iron overload, these anti-gene approaches may offer reasonable specificity for hepcidin suppression, subject to the general challenges associated with anti-gene technologies.
Targeting the inflammatory pathway: during inflammation, hepcidin expression is highly induced by the IL-6/Stat3 and possibly other pathways. Therefore, anti-cytokine agents that are used to treat inflammatory diseases may have an added benefit of reducing hepcidin expression. Neutralizing monoclonal antibodies against IL-6 or IL-6 receptor have been shown to decrease hepcidin production in animal models and even humans. Multicentric Castleman’s disease is a rare lymphoproliferative disorder that is commonly associated with overproduction of IL-6 and anemia. Treatment of Castleman’s disease patients with anti-IL6R resulted in a rapid and prolonged reduction of serum hepcidin and the correction of anemia.81 The same drug rapidly resolved anemia in monkeys with collagen-induced arthritis.82 Anti-IL-6 antibody treatment in patients with renal cell carcinoma, multiple myeloma or Castleman’s disease also effectively decreased hepcidin and increased hemoglobin.83,84 Anti-TNFα antibody in patients with rheumatoid arthritis similarly caused a sustained decrease in hepcidin.85 Although TNFα by itself suppresses hepcidin in vitro,31 the observed opposite effects of the anti-TNFα treatment in patients are likely a consequence of a general decrease in inflammation and IL-6 levels. Treatment with a small molecule STAT3 inhibitor (AG490) also decreased hepcidin and increased serum iron in healthy mice.86 Although anti-cytokine therapies are effective in lowering hepcidin production, their side effects include increased risk of infection due to impaired host defense,87,88 thus these types of therapies may be confined to the treatment of severe inflammatory diseases.
Targeting the erythropoietic pathway: ESAs are another example of pharmacological strategies that are not directly targeted at hepcidin but whose beneficial effects in anemia may include a suppression of hepcidin production to increase iron availability. Large pharmacological doses of erythropoietin (Epo) can overcome the resistance to erythropoietin seen in anemia of inflammation,89 and this may be in part due to hepcidin suppression, mediated indirectly by inducing the production of one or more unknown erythroid regulators. A single injection of Epo in human volunteers results in a rapid suppression of serum hepcidin.90 In clinical trials in CKD patients either receiving hemodialysis or not, prolyl hydroxylase inhibitors which stabilize hypoxia-inducible factor and increase Epo production were effective in lowering hepcidin and correcting hemoglobin even without intravenous (IV) iron supplementation.91,92 However, the use of ESAs in patients with chronic kidney disease or cancer has been associated with serious adverse effects.66,67 A US Food and Drug Administration (FDA) black box warning states that “ESAs increase the risk of death, myocardial infarction, stroke, venous thromboembolism, thrombosis of vascular access and tumor progression or recurrence”. An early candidate for prolyl hydroxylase inhibitors also led to fatal hepatic necrosis in one patient and abnormal elevations of liver enzymes in other patients.93 Thus, the use of ESAs as an approach to lower hepcidin in disorders where it is elevated may be subject to safety concerns.
Hepcidin anti-gene therapies: programs targeting hepcidin mRNA itself using siRNAs or antisense oligonucleotides have been announced.79,80 As hepcidin mRNA is highly expressed, particularly during inflammation, decreasing an abundant target with anti-gene approaches may present a technical challenge. At the same time, because these agents preferentially accumulate in the liver, the primarily hepatic expression of hepcidin makes it a suitable target for these technologies.
Class 2: neutralizing hepcidin peptide
A number of strategies have been developed that neutralize hepcidin activity by directly binding to the hepcidin peptide. These include monoclonal antibodies (mAbs) as well as engineered protein and RNA-based binders.
Monoclonal antibodies: in a mouse model of anemia of inflammation caused by heat-killed Brucella abortus, injection of an anti-hepcidin mAb helped overcome erythropoietin resistance.94 When the mAb was administered together with an erythropoietic agent the development of anemia was prevented, even though each agent was ineffective by itself. A fully humanized mAb against hepcidin (LY2787106) is currently in phase I human trials for cancer-related anemia.95
Anticalins: anticalins are derivatives of small extracellular proteins lipocalins that naturally bind diverse biological ligands. Anticalins are engineered to bind and antagonize specific targets by mutagenizing the lipocalins’ binding pocket and screening phage display libraries.96 Anticalin PRS-080 specifically binds human hepcidin with sub-nanomolar affinity. In cynomolgus monkeys, PRS-080 administration resulted in effective mobilization of iron and hyperferremia.97 As a recently developed therapeutic platform, relatively little is known about anticalins’ general safety and tolerability. The first clinical trial with PRS-080 is programmed to start in 2013.97
Spiegelmers: spiegelmers are RNA-like oligonucleotides with L-stereochemistry that confers resistance to nucleases and high stability in circulation. Spiegelmers for specific targets are selected by screening of large combinatorial libraries. Anti-hepcidin spiegelmer NOX-H94 was tested in cynomolgus monkeys in which anemia was caused by daily injection of IL-6 for a week. Concurrent administration of NOX-H94 blunted the development of anemia.98 In the phase I trial in humans volunteers, NOX-H94 was safe and well tolerated, and after a single NOX-H94 dose, serum iron and transferrin saturation increased in a dose-dependent manner.99 In experimental human endotoxemia caused by an injection of LPS, treatment with NOX–H94 30 min after LPS greatly delayed the onset of hypoferremia.100 NOX-H94 is currently in phase II clinical trials to examine the efficacy in patients with anemia of cancer.101
The main challenge for the efficacy of hepcidin binding molecules may be the high rate of hepcidin production. Xiao et al.56 estimated hepcidin production in macaque monkeys at 7.6 nmole/kg/h (20.5 μg/kg/h). Scaled to humans, the estimated hepcidin production in a 75 kg man would be 12 mg/day (20.5 μg/kg/h × 75kg × 24h × 12/37*) (*a conversion factor to estimate human equivalent dose based on the differences in body surface area).102 In addition, hepcidin binders may reduce the natural clearance of hepcidin and thus result in further hepcidin accumulation. This aspect at least could be addressed by developing agents with shorter half-life, or in case of antibodies, those that can be recycled after delivering hepcidin for degradation to macrophages.103 Furthermore, even a partial or intermittent blocking of hepcidin activity may be sufficient to increase iron delivery to the bone marrow and improve erythropoiesis. Clinical trials testing the efficacy of hepcidin binders in chronic conditions will help answer these questions.
Class 3: interfering with hepcidin binding to ferroportin
Agents that interfere with the binding of hepcidin to ferroportin have been reported recently. A high-throughput screening of small molecules identified a thiol-reactive compound fursultiamine as a hepcidin antagonist.104In vitro, fursultiamine acted by blocking ferroportin residue Cys326-SH that is necessary for hepcidin binding.105 However, because of its short half-life in vivo, fursultiamine did not consistently reverse the effect of hepcidin on serum iron in mice. Although not tested, non-specific reactivity with other proteins containing an active thiol form would also be a concern.
Anti-ferroportin antibodies have been designed that bind to an extracellular loop of ferroportin adjacent to the hepcidin-binding site. These antibodies prevent hepcidin-ferroportin interaction, while maintaining the iron-exporting function of ferroportin.106 A phase I trial in human volunteers107 has recently been completed that evaluated the effect on serum iron although no results have been reported yet.
Class 4 and 5: targeting ferroportin endocytosis or production
Because hepcidin acts by causing ferroportin internalization, preventing ferroportin endocytosis would be expected to antagonize hepcidin activity. Similarly, stimulating ferroportin synthesis could potentiate iron delivery to plasma. High-throughput screening studies have been reported that were designed to target ferroportin endocytosis or synthesis104,108 and these await confirmation in cellular and animal studies.
Conclusion
Within only 12 years since the first publication on hepcidin, a plethora of hepcidin-targeted therapeutics is under development for different iron disorders. This is not unexpected considering the important role hepcidin plays in the pathogenesis of iron disorders. Several hepcidin-targeted therapies have already entered human clinical trials. It remains to be seen how the efficacy of hepcidin-based approaches will compare to established treatment regimens in different diseases, and whether combinations of established and hepcidin-based approaches offer any advantages.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Ganz T, Nemeth E. Hepcidin and disorders of iron metabolism. Annu Rev Med. 2011;62:347–60 [DOI] [PubMed] [Google Scholar]
- 2.Andrews NC. Iron Deficiency and Related Disorders. In: Greer JP, Foerster J, eds. Wintrobe’s Clinical Hematology. 12th ed 2009. p. 810–34 [Google Scholar]
- 3.Brissot P, Ropert M, Le LC, Loreal O. Non-transferrin bound iron: a key role in iron overload and iron toxicity. Biochim Biophys Acta. 2012;1820(3):403–10 [DOI] [PubMed] [Google Scholar]
- 4.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–3 [DOI] [PubMed] [Google Scholar]
- 5.Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1(3): 191–200 [DOI] [PubMed] [Google Scholar]
- 6.Qiao B, Sugianto P, Fung E, Del-Castillo-Rueda A, Moran-Jimenez MJ, Ganz T, et al. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012;15(6):918–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Preza GC, Pinon R, Ganz T, Nemeth E. Cellular catabolism of the iron-regulatory peptide hormone hepcidin. PLOS One. 2013;8(3):e58934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinbicker AU, Bartnikas TB, Lohmeyer LK, Leyton P, Mayeur C, Kao SM, et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood. 2011;118(15):4224–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009; 41(4):478–81 [DOI] [PubMed] [Google Scholar]
- 10.Andriopoulos B, Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009;41(4):482–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramos E, Kautz L, Rodriguez R, Hansen M, Gabayan V, Ginzburg Y, et al. Evidence for distinct pathways of hepcidin regulation by acute and chronic iron loading in mice. Hepatology. 2011;53(4):1333–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38(5):531–9 [DOI] [PubMed] [Google Scholar]
- 13.Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36 (1):77–82 [DOI] [PubMed] [Google Scholar]
- 14.Corradini E, Meynard D, Wu Q, Chen S, Ventura P, Pietrangelo A, et al. Serum and liver iron differently regulate the bone morphogenetic protein 6 (BMP6)-SMAD signaling pathway in mice. Hepatology. 2011;54(1):273–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281(39):28494–8 [DOI] [PubMed] [Google Scholar]
- 16.D’Alessio F, Hentze MW, Muckenthaler MU. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J Hepatol. 2012;57(5):1052–60 [DOI] [PubMed] [Google Scholar]
- 17.Corradini E, Rozier M, Meynard D, Odhiambo A, Lin HY, Feng Q, et al. Iron regulation of hepcidin despite attenuated Smad1,5,8 signaling in mice without transferrin receptor 2 or Hfe. Gastroenterology. 2011;141(5):1907–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt PJ, Fleming MD. Transgenic HFE-dependent induction of hepcidin in mice does not require transferrin receptor-2. Am J Hematol. 2012;87(6):588–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piperno A, Girelli D, Nemeth E, Trombini P, Bozzini C, Poggiali E, et al. Blunted hepcidin response to oral iron challenge in HFE-related hemochromatosis. Blood. 2007;110(12):4096–100 [DOI] [PubMed] [Google Scholar]
- 20.Silvestri L, Pagani A, Nai A, De Domenico I, Kaplan J, Camaschella C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8(6):502–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang AS, Anderson SA, Wang J, Yang F, DeMaster K, Ahmed R, et al. Suppression of hepatic hepcidin expression in response to acute iron deprivation is associated with an increase of matriptase-2 protein. Blood. 2011;117(5):1687–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finberg KE, Heeney MM, Campagna DR, Aydinok Y, Pearson HA, Hartman KR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat Genet. 2008;40(5):569–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Enns CA, Ahmed R, Zhang AS. Neogenin interacts with matriptase-2 to facilitate hemojuvelin cleavage. J Biol Chem. 2012;287(42):35104–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kautz L, Meynard D, Monnier A, Darnaud V, Bouvet R, Wang RH, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood. 2008;112(4):1503–9 [DOI] [PubMed] [Google Scholar]
- 25.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110(7):1037–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108(12):3730–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Origa R, Galanello R, Ganz T, Giagu N, Maccioni L, Faa G, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007;92(5):583–8 [DOI] [PubMed] [Google Scholar]
- 28.Kearney SL, Nemeth E, Neufeld EJ, Thapa D, Ganz T, Weinstein DA, et al. Urinary hepcidin in congenital chronic anemias. Pediatr Blood Cancer. 2007;48(1):57–63 [DOI] [PubMed] [Google Scholar]
- 29.Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13(9):1096–101 [DOI] [PubMed] [Google Scholar]
- 30.Tanno T, Porayette P, Sripichai O, Noh SJ, Byrnes C, Bhupatiraju A, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood. 2009;114(1):181–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113(9): 1271–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verga Falzacappa MV, Vujic SM, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood. 2007;109(1):353–8 [DOI] [PubMed] [Google Scholar]
- 33.Armitage AE, Eddowes LA, Gileadi U, Cole S, Spottiswoode N, Selvakumar TA, et al. Hepcidin regulation by innate immune and infectious stimuli. Blood. 2011;118(15): 4129–39 [DOI] [PubMed] [Google Scholar]
- 34.Chung B, Verdier F, Matak P, Deschemin JC, Mayeux P, Vaulont S. Oncostatin M is a potent inducer of hepcidin, the iron regulatory hormone. FASEB J. 2010;24(6):2093–103 [DOI] [PubMed] [Google Scholar]
- 35.Maes K, Nemeth E, Roodman GD, Huston A, Esteve F, Freytes C, et al. In anemia of multiple myeloma, hepcidin is induced by increased bone morphogenetic protein 2. Blood. 2010;116(18):3635–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Besson-Fournier C, Latour C, Kautz L, Bertrand J, Ganz T, Roth MP, et al. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood. 2012;120(2): 431–9 [DOI] [PubMed] [Google Scholar]
- 37.Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33(1):21–2 [DOI] [PubMed] [Google Scholar]
- 38.Nemeth E, Ganz T. Hepcidin and iron-loading anemias. Haematologica. 2006;91(6): 727–32 [PubMed] [Google Scholar]
- 39.Pasricha SR, Frazer DM, Bowden DK, Anderson GJ. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with beta-thalassemia major: a longitudinal study. Blood. 2013;122(1):124–33 [DOI] [PubMed] [Google Scholar]
- 40.Roy CN, Mak HH, Akpan I, Losyev G, Zurakowski D, Andrews NC. Hepcidin antimicrobial peptide transgenic mice exhibit features of the anemia of inflammation. Blood. 2007;109(9):4038–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ganz T, Olbina G, Girelli D, Nemeth E, Westerman M. Immunoassay for human serum hepcidin. Blood. 2008;112(10):4292–7 [DOI] [PubMed] [Google Scholar]
- 42.Weinstein DA, Roy CN, Fleming MD, Loda MF, Wolfsdorf JI, Andrews NC. Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood. 2002;100(10):3776–81 [DOI] [PubMed] [Google Scholar]
- 43.Zaritsky J, Young B, Wang HJ, Westerman M, Olbina G, Nemeth E, et al. Hepcidin--a potential novel biomarker for iron status in chronic kidney disease. Clin J Am Soc Nephrol. 2009;4(6):1051–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashby DR, Gale DP, Busbridge M, Murphy KG, Duncan ND, Cairns TD, et al. Plasma hepcidin levels are elevated but responsive to erythropoietin therapy in renal disease. Kidney Int. 2009;75(9):976–81 [DOI] [PubMed] [Google Scholar]
- 45.Wians FH, Jr, Urban JE, Keffer JH, Kroft SH. Discriminating between iron deficiency anemia and anemia of chronic disease using traditional indices of iron status vs transferrin receptor concentration. Am J Clin Pathol. 2001;115(1):112–8 [DOI] [PubMed] [Google Scholar]
- 46.Libregts SF, Gutierrez L, de Bruin AM, Wensveen FM, Papadopoulos P, van IW, et al. Chronic IFN-gamma production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF-1/PU.1 axis. Blood. 2011; 118(9):2578–88 [DOI] [PubMed] [Google Scholar]
- 47.Prince OD, Langdon JM, Layman AJ, Prince IC, Sabogal M, Mak HH, et al. Late stage erythroid precursor production is impaired in mice with chronic inflammation. Haematologica. 2012;97(11):1648–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nicolas G, Viatte L, Lou DQ, Bennoun M, Beaumont C, Kahn A, et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet. 2003;34(1):97–101 [DOI] [PubMed] [Google Scholar]
- 49.Gardenghi S, Ramos P, Marongiu MF, Melchiori L, Breda L, Guy E, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J Clin Invest. 2010;120(12):4466–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim A, Fung E, Parikh S, Gabayan V, Nemeth E, Ganz T. Detailed characterization of the mouse model of anemia of inflammation caused by heatkilled brucella abortus reveals multiple causes of anemia and partial dependence on hepcidin [abstract]. Am J Hematol. 2013; 5(88):E137 [Google Scholar]
- 51.Gardenghi S, Casu C, Renaud TM, Meloni A, Cooke KS, Sasu BJ, et al. Investigating the role of cytokines and hepcidin in anemia of inflammation [Abstract]. Am J Hematol. 2013; 5(88):E124 [Google Scholar]
- 52.Langdon J, Yates S, Femnou L, McCranor B, Xue QL, Vaulont S, et al. Hepcidin-dependent and hepcidin-independent regulation of erythropoiesis in a mouse model of anemia of chronic inflammation [abstract]. Am J Hematol. 2013; 5(88):E139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mayeur C, Steinbicker AU, Kolodziej SA, Bloch KD. In a mouse model of chronic inflammation, liver-specific deletion of ALK3 (a BMP type I receptor) prevents the development of anemia of inflammation [abstract]. Am J Hematol 2013; 5(88):E29 [Google Scholar]
- 54.Barton JC. Chelation therapy for iron overload. Curr Gastroenterol Rep. 2007;9(1):74–82 [DOI] [PubMed] [Google Scholar]
- 55.Porter JB. Optimizing iron chelation strategies in beta-thalassaemia major. Blood Rev. 2009;23 Suppl 1:S3–S7 [DOI] [PubMed] [Google Scholar]
- 56.Xiao JJ, Krzyzanski W, Wang YM, Li H, Rose MJ, Ma M, et al. Pharmacokinetics of anti-hepcidin monoclonal antibody Ab 12B9m and hepcidin in cynomolgus monkeys. AAPS J 2010;12(4):646–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Preza GC, Ruchala P, Pinon R, Ramos E, Qiao B, Peralta MA, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest 2011;121(12):4880–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramos E, Ruchala P, Goodnough JB, Kautz L, Preza GC, Nemeth E, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood 2012; 120(18):3829–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nai A, Pagani A, Mandelli G, Lidonnici MR, Silvestri L, Ferrari G, et al. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood 2012;119(21):5021–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo S, Casu C, Gardenghi S, Booten S, Aghajan M, Peralta R, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest 2013;123(4):1531–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmidt PJ, Toudjarska I, Sendamarai AK, Racie T, Milstein S, Bettencourt BR, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(−/−) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood 2013;121(7):1200–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Corradini E, Schmidt PJ, Meynard D, Garuti C, Montosi G, Chen S, et al. BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in Hfe knockout mice. Gastroenterology 2010;139(5):1721–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhen AW, Nguyen NH, Gibert Y, Motola S, Buckett P, Wessling-Resnick M, et al. The Small Molecule Genistein Increases Hepcidin Expression in Human Hepatocytes. Hepatology. 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Macdougall IC, Cooper AC. Erythropoietin resistance: the role of inflammation and pro-inflammatory cytokines. Nephrol Dial Transplant. 2002;17 (Suppl 11):39–43 [DOI] [PubMed] [Google Scholar]
- 65.Horl WH. Iron therapy for renal anemia: how much needed, how much harmful? Pediatr Nephrol. 2007;22(4):480–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steinbrook R. Erythropoietin, the FDA, and oncology. N Engl J Med. 2007;356(24):2448–51 [DOI] [PubMed] [Google Scholar]
- 67.Bennett CL, Spiegel DM, Macdougall IC, Norris L, Qureshi ZP, Sartor O, et al. A review of safety, efficacy, and utilization of erythropoietin, darbepoetin, and peginesatide for patients with cancer or chronic kidney disease: a report from the Southern Network on Adverse Reactions (SONAR). Semin Thromb Hemost. 2012;38(8):783–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, et al. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242(4885): 1528–34 [DOI] [PubMed] [Google Scholar]
- 69.Poli M, Girelli D, Campostrini N, Maccarinelli F, Finazzi D, Luscieti S, et al. Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood. 2011; 117(3):997–1004 [DOI] [PubMed] [Google Scholar]
- 70.Poli M, Girelli D, Naggi A, Campostrini N, Asperti M, Finazzi D, et al. and others. Identification of heparins without anticoagulant activity which inhibit hepcidin in vivo [abstract]. Am J Hematol. 2013; 5(88):E40 [Google Scholar]
- 71.Lin L, Goldberg YP, Ganz T. Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood. 2005;106(8):2884–9 [DOI] [PubMed] [Google Scholar]
- 72.Wu Q, Sun CC, Lin HY, Babitt JL. Repulsive guidance molecule (RGM) family proteins exhibit differential binding kinetics for bone morphogenetic proteins (BMPs). PLoS One. 2012;7(9):e46307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, Lin HY. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J Clin Invest. 2007;117(7):1933–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Theurl I, Schroll A, Sonnweber T, Nairz M, Theurl M, Willenbacher W, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood. 2011;118(18): 4977–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nili M, Shinde U, Rotwein P. Soluble repulsive guidance molecule c/hemojuvelin is a broad spectrum bone morphogenetic protein (BMP) antagonist and inhibits both BMP2- and BMP6-mediated signaling and gene expression. J Biol Chem. 2010;285(32): 24783–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cuny GD, Yu PB, Laha JK, Xing X, Liu JF, Lai CS, et al. Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg Med Chem Lett. 2008;18(15):4388–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Steinbicker AU, Sachidanandan C, Vonner AJ, Yusuf RZ, Deng DY, Lai CS, et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood. 2011;117(18): 4915–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun CC, Vaja V, Chen S, Theurl I, Stepanek A, Brown DE, et al. A hepcidin lowering agent mobilizes iron for incorporation into red blood cells in an adenine-induced kidney disease model of anemia in rats. Nephrol Dial Transplant. 2013;28(7):1733–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Isis Pharmaceuticals Xenon licenses anti-sense drug XEN701 from Isis and initiates preclinical toxicology studies. Updated 2013. Available from: http://ir.isispharm.com/phoenix.zhtml?c=222170&p=irol-newsArticle&ID=1828284&high-light=
- 80.Akinc A, Chan-Daniels A, Sehgal A, Foster D, Bettencourt BR, Hettinger J, et al. Targeting the hepcidin pathway with RNAi therapeutics for the treatment of anemia. Blood. 2011;21(118):688 [Google Scholar]
- 81.Song SN, Tomosugi N, Kawabata H, Ishikawa T, Nishikawa T, Yoshizaki K. Down-regulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multicentric Castleman disease. Blood. 2010;116(18): 3627–34 [DOI] [PubMed] [Google Scholar]
- 82.Hashizume M, Uchiyama Y, Horai N, Tomosugi N, Mihara M. Tocilizumab, a humanized anti-interleukin-6 receptor antibody, improved anemia in monkey arthritis by suppressing IL-6-induced hepcidin production. Rheumatol Int. 2010;30(7):917–23 [DOI] [PubMed] [Google Scholar]
- 83.Schipperus M, Rijnbeek B, Reddy M, Qin X, Cornfield MJ. CNTO328 (Anti-IL-6 mAb) Treatment Is Associated with An Increase in Hemoglobin (Hb) and Decrease in Hepcidin Levels in Renal Cell Carcinoma (RCC) [abstract]. Blood. 2009;22(114): 4045 [Google Scholar]
- 84.Kurzrock R, Voorhees PM, Casper C, Furman RR, Fayad L, Lonial S, et al. A Phase I, Open-Label Study of Siltuximab, an Anti-IL-6 Monoclonal Antibody, in Patients with B-cell Non-Hodgkin Lymphoma, Multiple Myeloma, or Castleman Disease. Clin Cancer Res. 2013;19(13):3659–70 [DOI] [PubMed] [Google Scholar]
- 85.Doyle MK, Rahman MU, Frederick B, Birbara CA, de Vries D, Toedter G, et al. Effects of subcutaneous and intravenous golimumab on inflammatory biomarkers in patients with rheumatoid arthritis: results of a phase 1, randomized, open-label trial. Rheumatology (Oxford) 2013;52(7):1214–9 [DOI] [PubMed] [Google Scholar]
- 86.Zhang SP, Wang Z, Wang LX, Liu SJ. AG490: an inhibitor of hepcidin expression in vivo. World J Gastroenterol. 2011;17(45): 5032–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lang VR, Englbrecht M, Rech J, Nusslein H, Manger K, Schuch F, et al. Risk of infections in rheumatoid arthritis patients treated with tocilizumab. Rheumatology (Oxford) 2012;51(5):852–7 [DOI] [PubMed] [Google Scholar]
- 88.van de Vosse E, van Agtmael MA. Targets of anticytokine therapy and the risk of infections in humans and mice. Curr Opin Rheumatol. 2007;19(6):626–35 [DOI] [PubMed] [Google Scholar]
- 89.Elliott J, Mishler D, Agarwal R. Hyporesponsiveness to erythropoietin: causes and management. Adv Chronic Kidney Dis. 2009;16(2):94–100 [DOI] [PubMed] [Google Scholar]
- 90.Ashby DR, Gale DP, Busbridge M, Murphy KG, Duncan ND, Cairns TD, et al. Erythropoietin administration in humans causes a marked and prolonged reduction in circulating hepcidin. Haematologica. 2010;95(3):505–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Besarab A, Chernyavskaya EN, Motylev I, Evgeny S, Yampolskiy AF, Kumbar LM, et al. FG-4592, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor, corrects anemia without iron supplementation in incident dialysis patients [abstract]. J Am Soc Nephrol. 2012;(23):21A [Google Scholar]
- 92.Besarab A, Hulter HN, Klaus S, Lee TT, Lilienfeld DE, Neff TB, et al. FG-4592, a novel oral HIF prolyl hydroxylase inhibitor, elevates hemoglobin in anemic stage 3/4 CKD Patients [abstract]. J Am Soc Nephrol. 2010;(21):95A [Google Scholar]
- 93.Macdougall IC. New anemia therapies: translating novel strategies from bench to bedside. Am J Kidney Dis. 2012;59(3):444–51 [DOI] [PubMed] [Google Scholar]
- 94.Sasu BJ, Cooke KS, Arvedson TL, Plewa C, Ellison AR, Sheng J, et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood. 2010;115 (17):3616–24 [DOI] [PubMed] [Google Scholar]
- 95.Eli Lilly and Company A Phase 1 Study of LY2787106 in Cancer and Anemia. Updated 2013. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01340976
- 96.Skerra A. Alternative binding proteins: anticalins - harnessing the structural plasticity of the lipocalin ligand pocket to engineer novel binding activities. FEBS J. 2008; 275(11):2677–83 [DOI] [PubMed] [Google Scholar]
- 97.Hohlbaum A, Gille H, Christian J, Allersdorfer A, Jaworski J, Burrows J, et al. Iron mobilization and pharmacodynamic marker measurements in non-human primates following administration of PRS-080, a novel and highly specific anti-hepcidin therapeutic [abstract]. Am J Hematol. 2013; 5(88):E41 [Google Scholar]
- 98.Schwoebel F, van Eijk LT, Zboralski D, Sell S, Buchner K, Maasch C, et al. The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood. 2013;121 (12):2311–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Riecke K, Zollner S, Boyce M, van Hecke B, Vauleon S, Summo L, et al. Single and repeated dose first-in-human study with the anti-hepcidin spiegelmer NOX-H94 [abstract]. Am J Hematol 2013;5(88):E42 [Google Scholar]
- 100.van Eijk L, Swinkels DW, John A, Schwobel F, Fliegert F, Summo L, et al. Randomized double blind placebo controlled PK/PD study on the effects of a single intravenous dose of the anti-hepcidin spiegelmer NOX-H94 on serum iron during experimental human endotoxemia [abstract]. Am J Hematol 2013; 5(88):E225 [Google Scholar]
- 101.Noxxon Pharma AG Efficacy of NOX-H94 on Anemia of Chronic Disease in Patients With Cancer. Updated 2013. Available from: http://clinicaltrials.gov/show/NCT01691040
- 102.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. The FASEB J. 2008;22(3):659–61 [DOI] [PubMed] [Google Scholar]
- 103.Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S, et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol. 2010;28(11):1203–7 [DOI] [PubMed] [Google Scholar]
- 104.Fung E, Sugianto P, Hsu J, Damoiseaux R, Ganz T, Nemeth E. High-throughput screening of small molecules identifies hepcidin antagonists. Mol Pharmacol. 2013; 83(3):681–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fernandes A, Preza GC, Phung Y, De Domenico I, Kaplan J, Ganz T, et al. The molecular basis of hepcidin-resistant hereditary hemochromatosis. Blood. 2009; 114(2):437–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leung DDM, Luan P, Menetta JV, Tang Y, Witcher D, inventors; Eli Lilly and Company, assignee. Anti-Ferroportin 1 Monoclonal Antibodies and Uses Thereof. Patent US 8183346 B2 2012
- 107.Eli Lilly and Company A First Human Study of a Ferroportin Antibody. Updated 2011. Available from: http://www.clinical-trials.gov/ct2/show/NCT01330953
- 108.Guida C, Altamura S, Klein F, Boutros M, Hentze MW, Muckenthaler MU. Identification of novel regulators of ferro-portin expression [abstract]. Am J Hematol 2013;5(88):E128 [Google Scholar]