

Abstract
In order to identify novel somatic mutations associated with classic BCR/ABL1-negative myeloproliferative neoplasms, we performed high-coverage genome sequencing of DNA from peripheral blood granulocytes and cultured skin fibroblasts from a patient with MPL W515K-positive primary myelofibrosis. The primary myelofibrosis genome had a low somatic mutation rate, consistent with that observed in similar hematopoietic tumor genomes. Interfacing of whole-genome DNA sequence data with RNA expression data identified three somatic mutations of potential functional significance: i) a nonsense mutation in CARD6, implicated in modulation of NF-kappaB activation; ii) a 19-base pair deletion involving a potential regulatory region in the 5′-untranslated region of BRD2, implicated in transcriptional regulation and cell cycle control; and iii) a non-synonymous point mutation in KIAA0355, an uncharacterized protein. Additional mutations in three genes (CAP2, SOX30, and MFRP) were also evident, albeit with no support for expression at the RNA level. Re-sequencing of these six genes in 178 patients with polycythemia vera, essential thrombocythemia, and myelofibrosis did not identify recurrent somatic mutations in these genes. Finally, we describe methods for reducing false-positive variant calls in the analysis of hematologic malignancies with a low somatic mutation rate. This trial is registered with ClinicalTrials.gov (NCT01108159).
Introduction
The classic BCR/ABL1-negative myeloproliferative neoplasms include polycythemia vera, essential thrombocythemia, and primary myelofibrosis. Myeloproliferative neoplasms are derived from hematopoietic stem or early progenitor cells and are characterized early in the disease by increased production of mature myeloid cells.1 Primary myelofibrosis is a myeloproliferative neoplasm subtype typically characterized by megakaryocyte proliferation and atypical morphology, extensive bone marrow fibrosis, and splenomegaly secondary to extramedullary hematopoiesis.
The genetics underlying the pathogenesis of primary myelofibrosis and other classic BCR/ABL1-negative myeloproliferative neoplasms is an active area of investigation, particularly since the discovery of the JAK2 V617F mutation in 2005. This gain-of-function mutation in the JAK2 tyrosine kinase gene results in activation of the JAK-STAT pathway and is found in 50–60% of patients with primary myelofibrosis.2–5 The activating mutation(s) W515L/K in MPL, the gene encoding the thrombopoietin receptor, also result in constitutive JAK-STAT pathway signaling, and are found in approximately 5–10% of primary myelofibrosis patients.6,7 Further emphasizing the importance of these molecular abnormalities, expression of mutated JAK2 and MPL in mouse models recapitulates the phenotype observed in myeloproliferative neoplasm patients.6,8 Despite these findings, there are several lines of evidence that suggest these mutations in JAK2 and MPL may not be the initiating lesion in these neoplasms.9–12 In addition, recurrent, somatic mutations in TET2, CBL, SH2B3, ASXL1, DNMT3A, IDH1/2, IKZF1, EZH2, SRSF2, and TP53 have also been implicated in myeloproliferative neoplasm initiation and/or progression, generally in a small minority of cases (reviewed by Tefferi et al.13 and Abdel-Wahab et al.14).
In order to identify novel somatic mutations associated with primary myelofibrosis, we performed whole-genome sequencing of matched neoplastic and germ-line specimens from a patient with MPL W515K-positive primary myelofibrosis. We used high-coverage sequencing, two independent sequencing technologies, and multiple analysis approaches on a case with a high percentage of neoplastic cells to maximize the detection of somatic variants. Genome sequencing permitted evaluation of somatic variants outside of exons, and examination of coding regions that are difficult to capture with exome selection technologies. Using this approach, we have calculated the mutation rate for the exome and estimated the mutation rate for non-repetitive regions of the genome, finding comparable low mutation rates in each. Based on the identification of somatic mutations and expression pattern, we identified three candidate genes potentially involved in myeloproliferative neoplasm pathogenesis in this patient: CARD6, BRD2, and KIAA0355. Our specimen preparation and data analysis approaches in this case highlight effective strategies for reducing false-positive candidate somatic variants, a common obstacle in genome interpretation for hematologic malignancies with a low somatic mutation rate.
Methods
Samples for genome sequencing
Patient specimens were collected under protocols approved by the Stanford University Administrative Panel for the Protection of Human Subjects and the Stanford Cancer Institute Scientific Review Committee. The procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 2008. The participant was counseled and gave consent for genome sequencing and other described studies. Peripheral blood specimens were either drawn into PAXgene Blood RNA (PreAnalytiX GmbH, Hombrechtikon, Switzerland) or sodium heparin tubes. Following isolation of the buffy coat, granulocytes and peripheral blood mononuclear cells were isolated using Ficoll-Paque PREMIUM (GE Healthcare Biosciences, Pittsburgh, PA, USA) according to the manufacturer’s protocol. A 200-cell count differential performed on the cytospin from the isolated granulocyte fraction demonstrated 95% granulocytes. As a series of control populations of non-hematopoietic cells, multiple independent fibroblast cultures were initiated by mechanical and enzymatic disaggregation of a 4 mm skin punch biopsy and inoculation into culture flasks. These primary cultures were sub-cultured and then combined to provide approximately 5–10 million purified fibroblasts, representing the patient’s germ-line DNA complement. DNA was extracted using the Gentra Puregene Cell Kit (Qiagen, Valencia, CA, USA) or DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s protocol. RNA was extracted using the PAXgene Blood RNA Kit (Qiagen) or RNeasy Mini Kit (Qiagen).
Genome and RNA sequencing
Genome sequencing of DNA obtained from the patient’s peripheral blood granulocytes and from purified cultured fibroblasts was performed in parallel at Illumina (San Diego, CA, USA) and Complete Genomics (Mountain View, CA, USA). To obtain the Illumina sequence data, paired-end 100-bp reads were generated using the Illumina HiSeq 2000 sequencer, and alignment and variant calling were performed using CASAVA version 1.8 and the HugeSeq pipeline15 version 1.0, which uses BWA for alignment and GATK and SAMtool MPileup for variant calling. In parallel, a full set of ~35 bp paired-end reads were generated by the ligation-based nanoball sequencing at Complete Genomics. Alignment and variant calling of the Complete Genomics data were performed using the Complete Genomics internal pipeline and CGA Tools 1.5.0. Illumina paired-end 100-bp RNA sequence reads (49.3 million gross reads) were aligned with Tophat216/Bowtie217 and analyzed by Cufflinks.18 Further details about the analysis, including variant filtering approaches, are provided in the Online Supplementary Methods.
Sanger DNA and RNA sequencing of candidate mutations
PCR amplification and sequencing primers were designed with the Primer3Plus software. For RNA confirmations, reverse transcriptase PCR was performed with the SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Invitrogen, Grand Island, NY, USA). Following agarose gel electrophoresis, the specificity of any observed band was confirmed by Sanger sequencing.
Candidate gene sequencing in additional myeloproliferative neoplasm samples
Primers for re-sequencing of the coding regions of CARD6, KIAA0355, SOX30, CAP2, and MFRP as well as the exons of BRD2 were designed using Design Studio for a TruSeq Custom Amplicon kit (Illumina). Paired-end 150-bp reads from 178 myeloproliferative neoplasm patients’ peripheral blood samples were generated using the Illumina MiSeq, with protocols and analysis as described in the Online Supplementary Methods. Patients presented with: myelofibrosis (primary, post-polycythemia vera and post-essential thrombocythemia myelofibrosis) n=96; polycythemia vera, n=42; essential thrombocythemia, n=40.
Results
Case Report
An asymptomatic 51-year old man was found to have abnormal blood counts on a routine health care check in 2008. A complete blood count showed a white blood cell count of 5.1 × 109/L, hemoglobin 11.4 g/dL, and platelet count 147 × 109/L. The differential revealed 67% segmented neutrophils, 4% bands, 1% metamyelocytes, 4% myelocytes, 2% promyelocytes, 17% lymphocytes, 4% monocytes, 1% eosinophils, and 2 nucleated red blood cells/100 white blood cells. The smear showed teardrop red blood cells and bizarre platelets. His physical examination was notable for palpable splenomegaly of 5 cm below the left costal margin. The peripheral blood smear, bone marrow aspirate and biopsy (Online Supplementary Results and Online Supplementary Figure S1), and other laboratory findings were consistent with a diagnosis of primary myelofibrosis. Chromosome analysis detected no abnormalities. PCR testing for the JAK2 V617F mutation was negative; however, the MPL W515K mutation was detected in the peripheral blood. The patient has not required therapy since diagnosis and maintains a low-risk DIPSS Plus Prognostic Score with stable blood counts.
Genome sequencing analysis
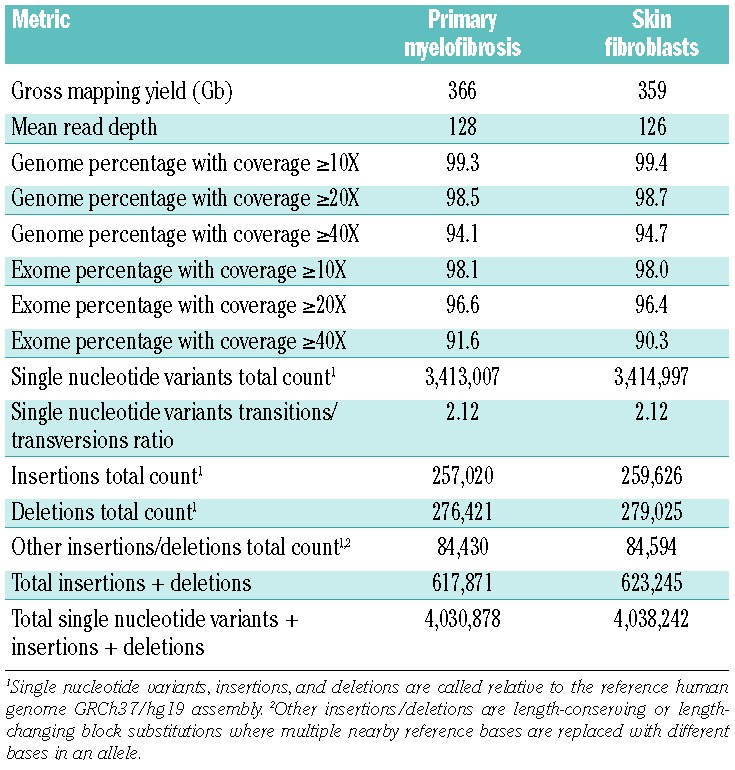
Using the Illumina chemistry, two independent libraries derived from the purified granulocytes were sequenced to an average depth of 45X and 43X for a total of 88X average depth, and the skin fibroblast specimen was sequenced to 47X (see Table 1 for a summary of Illumina genome data). Using Complete Genomics, the average depth was 128X for the purified granulocyte specimen and 126X for the skin fibroblast specimen (see Table 2 for a summary of Complete Genomics genome data). The coverage data, concordance with single nucleotide polymorphism arrays, and number of variants indicate the granulocyte and skin fibroblast genomes were sufficiently covered to allow detection of the majority of small somatic mutations occurring in the mappable regions of the human genome. Further discussion of the sequencing metrics is provided in the Online Supplementary Results and Discussion.
Table 1.
Illumina genome sequencing summary data.
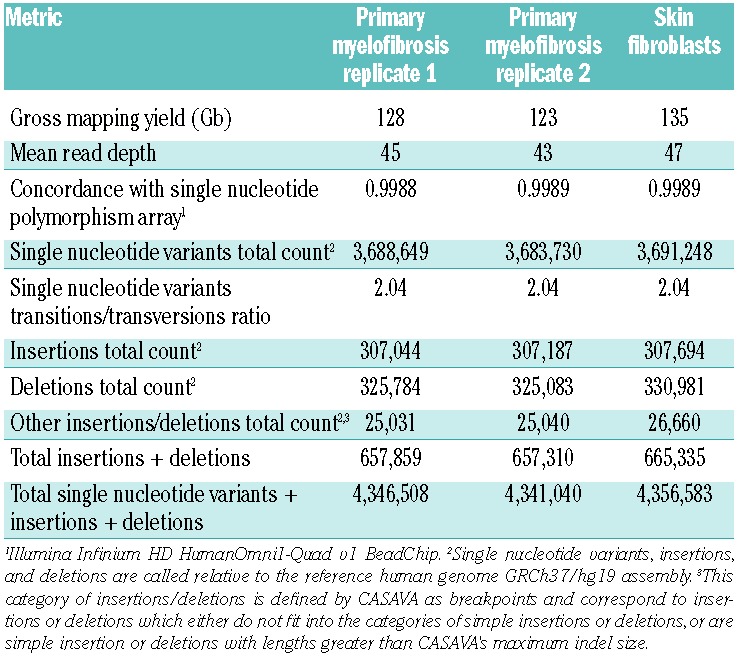
Table 2.
Complete Genomics genome sequencing summary data.
Analysis of the Illumina sequencing data did not identify large-scale copy number or structural variants involving genes in this specimen. In an attempt to comprehensively identify small somatic mutations in the specimen, we used four combinations of aligners and variant callers. The Complete Genomics sequence read data were aligned and variants called with the Complete Genomics internal pipeline; due to the complex sequence read structure of the Complete Genomics data, alternate high performance aligners and variant callers were not available. The Illumina read data were aligned and variants called with CASAVA as well as aligned with BWA and variants called by GATK and SAMTools MPileup. Focusing on coding regions, untranslated regions, and the invariant two bases of the donor or acceptor splice sites, the total number of somatic variant candidates derived from any of the four pipelines was 984. The 4-property Venn diagram in Figure 1 illustrates the number of candidate variant calls that were unique to or were shared among the various analyses.
Figure 1.
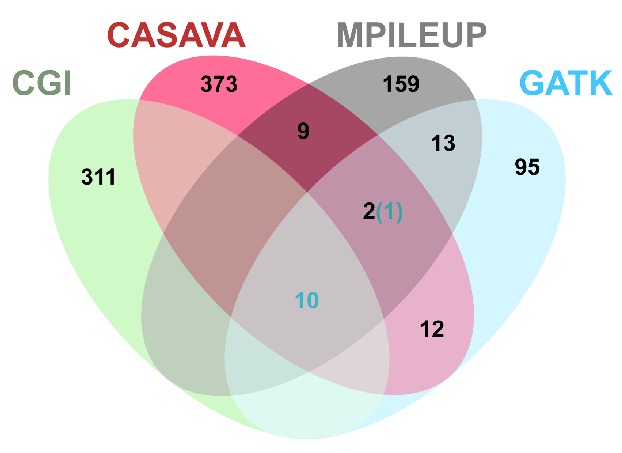
Four-property Venn Diagram of candidate somatic variants within exons and splice sites called by different analysis approaches. Venn Diagram illustrating the number of true (blue numbers) and false (black numbers) somatic variant calls within the coding regions, untranslated regions, and invariant two bases of the donor or acceptor splice sites that were unique to or shared among the various analysis approaches. Data are provided for four analysis approaches described in the Methods: GATK, MPileup, and CASAVA for the Illumina data and Complete Genomics analysis pipeline for the Complete Genomics data. Four-property Venn Diagram created by Gliffy Online (www.gliffy.com).
Bioinformatics approaches to low mutation rate tumor samples
Sequence analysis of genetic variation in tumor samples can present a variety of challenges. While tumors with a high mutation burden can pose problems due to the sheer number of genetic variation, such samples provide a strong signal-to-noise ratio in that the number of true mutations is large relative to a constant background of sequencing inaccuracies. Although tumors with a low mutation burden present an opportunity of more complete characterization of mutational events, these present a challenge in that even rare sequencing inaccuracies can lead to a low signal-to-noise ratio. We took several specific steps to maximize the detection of smaller mutation sets in the tumor sample while avoiding false positives that would have been dominant in standard analysis of a minimally mutated tumor population.
Use of cultured skin fibroblasts, in addition to allowing variants to be classified as germ-line versus somatic, enables detection of sequencing and mapping errors that result in false-positive candidate variant calls. Re-sequencing of the cultured skin fibroblast specimens at regions containing somatic variants did not detect the presence of sequencing reads containing the somatic variant at a rate above the error rate of the platform (Table 3), indicating that the cultured fibroblasts do not contain significant white blood cell contamination. Consequently, the finding of the same variant in sequence reads derived from the cultured fibroblasts and granulocytes indicates that the variant is likely either germ-line (and missed by the initial filtering of germ-line variants) or a sequencing/mapping error caused by repetitive or non-unique regions of the genome that are challenging to accurately examine using current analysis approaches. Excluding sequence variants found in even a single read in the skin fibroblast sample provided a marked improvement in the signal-to-noise ratio for true somatic variant detection. Indeed, all 11 confirmed somatic variants identified in this study (Table 4) were not present in reads from the skin fibroblast specimen. In contrast, direct use of the skin punch biopsy without culturing can result in contamination of the skin specimen with neoplastic cells from the blood,19,20 and a strict exclusion of variants found in the skin biopsy would be less appropriate under such circumstances.
Table 3.
Re-sequencing read counts at somatic mutations demonstrate no appreciable contamination of cultured skin fibroblasts by primary myelofibrosis cells.
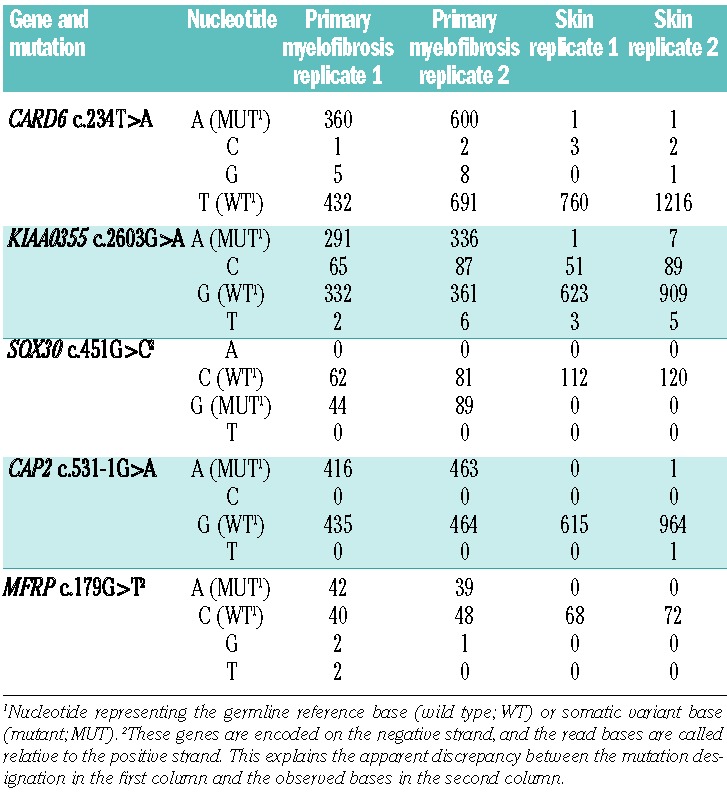
Table 4.
Confirmed somatic variants involving coding regions, untranslated regions (UTRs) and invariant two bases of donor/acceptor splice sites
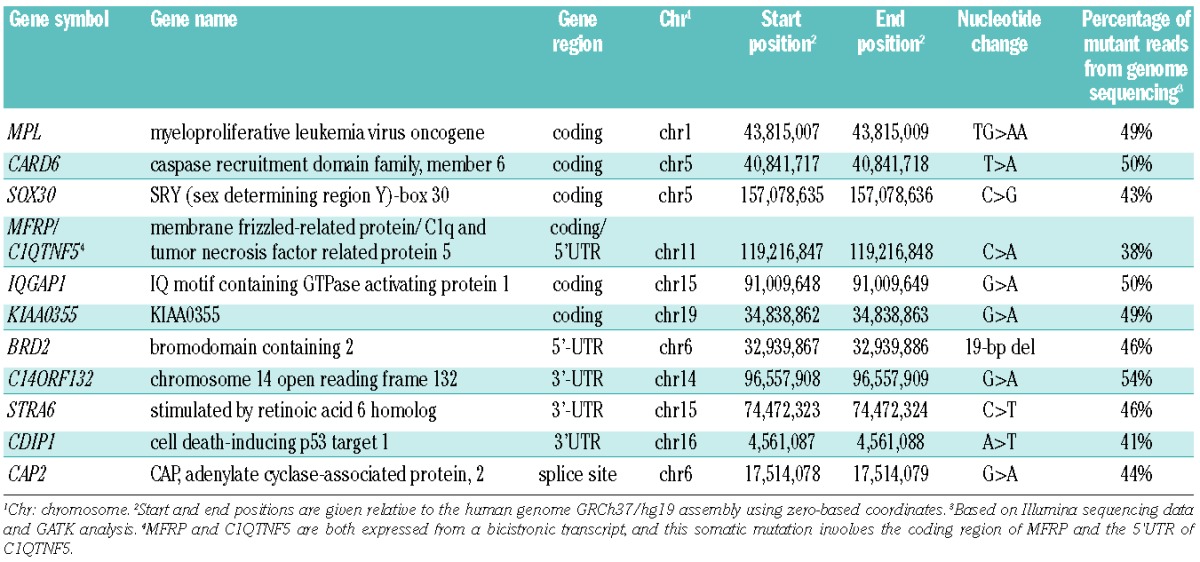
Application of two parallel sequencing chemistries (Illumina and Complete Genomics), use of multiple analysis methods for the Illumina sequencing data, and independent curation of each of the four resulting data sets were used to enhance detection of somatic mutations in this case. It is noteworthy that examination of common candidate variant calls between the Illumina and Complete Genomics data enriched for true somatic variants. In this study, 10 of 11 confirmed somatic variants within exons were identified by this approach (Figure 1). We recognize that, in most cases, using more than one sequencing chemistry is not practical for reasons of cost, but enrichment for true somatic variants can also be achieved by using multiple variant callers on Illumina data. As an example, there are 12 variants within exons that are jointly called by CASAVA, GATK and MPileup: 11 of these are confirmed somatic variants and one is a false-positive call.
To confirm identification of somatic variants in the neoplastic population and absence of the mutation in non-neoplastic cells (i.e. cultured skin fibroblasts in this study), we employed an orthogonal technology, bidirectional Sanger sequencing. Finally, application of other filters or approaches (e.g. base conservation, predicted effect of the mutation) was intentionally avoided due concerns about removing true somatic variants. For this case and other low mutation burden cases we have examined, the application of filtering based on cultured skin fibroblasts generates a manageable data set for manual curation.
Characterization of somatic variants
Following curation and, subsequently, PCR and Sanger confirmation, we identified 11 somatic variants among exons, untranslated regions, or splice sites in this primary myelofibrosis case (Table 4) corresponding to a mutation rate of 0.13 mutations per megabase (Mb) in these regions. This is similar to the estimated mutation rate in the non-repetitive portion of the patient’s genome of 0.15 single nucleotide variants per Mb (~210 variants in 1.4 Gb). We subsequently performed further genetic characterization of the 6 variants that resulted in non-synonymous amino acid changes, alteration of the invariant two bases of the donor/acceptor splice site sequence, or changes to putative regulatory regions (Table 5).
Table 5.
Somatic variants, RNA expression and recurrence data
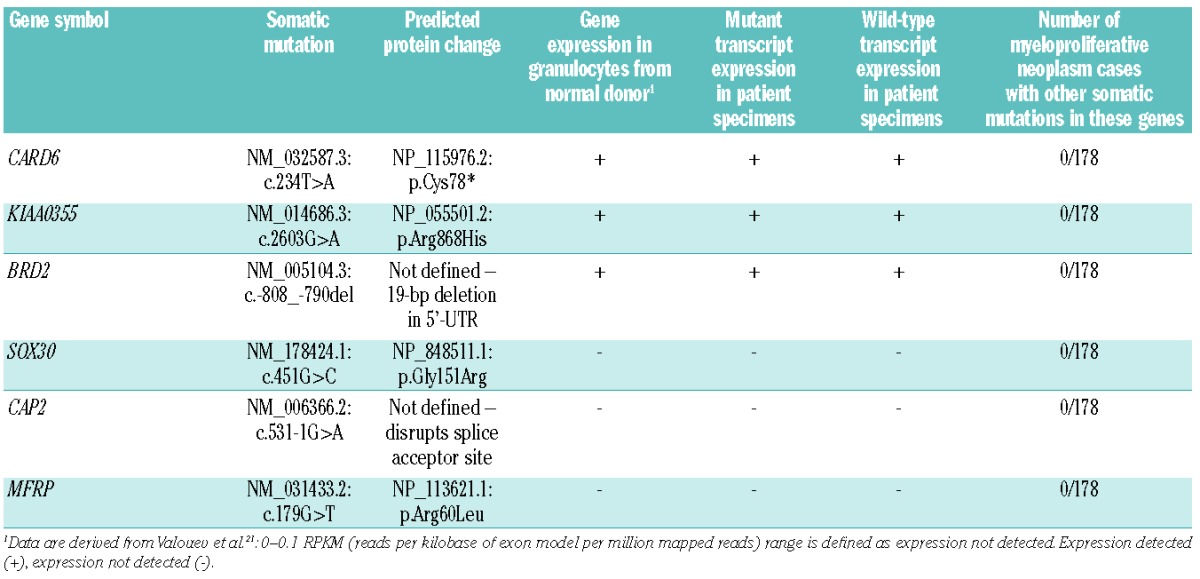
To evaluate which genes bearing somatic mutations might be most relevant to the pathogenesis of this patient’s disease, we examined expression of RNA transcripts using high-throughput RNA sequencing and reverse transcriptase PCR. Wild-type and mutant transcripts of CARD6 (6.3 fragments per kilobase of transcript per million fragments sequenced FPKM, a normalized measure of RNA expression levels derived from high-throughput RNA sequencing data in which larger numbers indicate higher expression levels18), BRD2 (2.8 FPKM), and KIAA0355 (1.1 FPKM) were detected in the primary myelofibrosis patient’s granulocyte and whole blood specimens by reverse transcriptase PCR, while expression of SOX30 (0.83 FPKM), CAP2 (0.60 FPKM), or MFRP (0 FPKM), was not detected in either patient specimen by reverse transcriptase PCR. The primary myelofibrosis patient’s reverse transcriptase PCR analyses are consistent with prior analysis on granulocytes derived from a normal donor,21 which found BRD2, CARD6, and KIAA0355 are expressed while SOX30, CAP2, and MFRP were not detected. These data indicate that mutations in BRD2, CARD6, and KIAA0355 are primary candidates as pathogenic contributors to this patient’s myeloproliferative neoplasm.
To search for other examples of mutations in the genes identified by analysis of this patient’s tumor, we re-sequenced the coding regions and donor/acceptor splice site sequences of BRD2, CARD6, KIAA0355, SOX30, CAP2, and MFRP in 178 myeloproliferative neoplasm specimens from other patients: myelofibrosis, n=96 (including 3 patients with MPL W515 mutations); polycythemia vera, n=42; and essential thrombocythemia, n=40. Since the BRD2 mutation was observed in the 5′ untranslated region, we also sequenced the 5′ and 3′ untranslated regions of BRD2 in these specimens. We did not identify any somatic mutations in these 6 genes in the 178 myeloproliferative neoplasm specimens.
Discussion
We performed high-coverage genome sequencing of granulocytes and cultured skin fibroblast samples from a patient with MPL W515K-positive primary myelofibrosis in an experimental design permitting comparison of Complete Genomics and Illumina sequencing methodologies, and several popular data analysis pipeline components. The average and median percentage of reads with mutant alleles for the 11 somatic variants in the exons or splice sites was 46% (range 38–54%, Table 4), indicating that the granulocyte specimen consisted predominantly of neoplastic granulocytes bearing heterozygous mutations. Deeper re-sequencing data of the cultured skin fibroblasts at regions containing somatic mutations in the granulocyte sample indicated that the skin fibroblast germ-line control did not contain detectable contamination by neoplastic cells (Table 3). These optimal specimens were sequenced using two independent sequencing chemistries at high coverage and were analyzed with multiple analysis methods to identify somatic variants in a very comprehensive manner. Our approach indicates that using cultured skin fibroblasts as a pure germ-line control allows detection of somatic variants in the neoplastic cells while permitting strict filtering and exclusion of false-positive mutations that dominate the standard analysis of tumors with a low mutation burden.
The 11 somatic variants we detected among 82 Mb of exons, untranslated regions, and splice sites corresponded to a mutation rate of 0.13 somatic variants per Mb in these regions. This is within the range reported for other myeloid malignancies including acute myeloid leukemia20,22 and myelodysplastic syndromes,23 and approximately 10- to over 50-fold below mean or median somatic mutation rates observed by The Cancer Genome Atlas Research Network for a variety of solid tumors.24–28 Likewise, the estimated non-repetitive genome mutation rate is also relatively low, 0.15 somatic variants per Mb.
In order to identify those somatic variants that were most likely to be relevant to the pathogenesis of primary myelofibrosis in this patient, we focused on 6 somatic variants resulting in non-synonymous amino acid changes, alteration of the invariant two bases of the donor/acceptor splice site sequence, or changes to characterized regulatory regions. Three of these genes, SOX30, MFRP, and CAP2, do not have detectable expression in granulocytes derived from a normal donor, do not have detectable expression by reverse transcriptase PCR in whole blood or purified granulocytes derived from the primary myelofibrosis patient, and have high-throughput RNA sequencing expression levels that are consistent with either very low expression or undetectable expression (<1 FPKM). Collectively, these expression data suggest that the somatic mutations in SOX30, MFRP, and CAP2 are less likely to significantly contribute to the pathogenesis of the myeloproliferative neoplasm in this case. For MFRP and SOX30, the known function and tissue expression of these genes further suggests they are less likely to be important for myeloproliferative neoplasm biology. SOX30 encodes a DNA-binding transcription factor that likely plays a role in gonadal differentiation and development.29SOX30 expression in humans appears to be limited to the testes and ovary, and its expression was not previously detected in leukocytes.29,30MFRP encodes a protein that is likely to be important for eye development.31 Its expression has been reported to be limited to the human eye and brain, and expression of MFRP was not detected in leukocytes.32,33 Finally, the function and tissue distribution of CAP2, which encodes an adenylate cyclase-associated protein, has not been characterized in humans.
The remaining three genes, KIAA0355, BRD2, and CARD6, are expressed in granulocytes from a normal donor, and both wild-type and mutant transcripts of these three genes were detected in whole blood or purified granulocytes derived from the primary myelofibrosis patient. KIAA0355 encodes a protein that has been conserved among vertebrates. It has not been functionally characterized, and no functional domains have been predicted for this protein. The somatic missense mutation in KIAA0355 results in the substitution of an evolutionarily conserved arginine residue at position 868 with a histidine residue, a non-conservative change.
BRD2 encodes a mitogen-activated kinase implicated in signal transduction and conserved among eukaryotes.34,35BRD2 has been reported to promote transactivation of promoters of cell cycle regulatory genes through E2F transcription factors.36 Denis and co-workers37 suggested that BRD2 functions as a scaffolding protein that facilitates access of transcriptional control proteins to chromatin at regulatory regions of cell cycle regulatory genes. Both upregulation and downregulation of BRD2 result in a marked phenotype in model systems. Homozygous null mouse mutants are embryonic lethal;38 in contrast, transgenic mice with overexpression of BRD2 restricted to the B-cell lineage results in the sporadic development of B-cell lymphoma.39 Likewise, BRD2 kinase activity is markedly increased in acute and chronic lymphocytic leukemias.34 The somatic mutation observed in the primary myelofibrosis case described in this manuscript was a 19-bp deletion in the 5′ untranslated region of BRD2. Analysis of previously published ribosome profiling data from HeLa cells40 suggests that this 5′ untranslated region contains either upstream translated regions or regions with unidentified heavy complexes that protect short (~25–35 nucleotides) sequences (Online Supplementary Figure S2). An attractive working hypothesis would be that this deletion could dysregulate synthesis of the BRD2 protein, leading in turn to derangement of the cell cycle and promotion of neoplastic transformation. It is of potential clinical interest that BRD2 is a member of the BET family of bromodomain proteins and is inhibited by small molecular inhibitors of BET bromodomains.41,42
CARD6, caspase recruitment domain family, member 6, encodes a protein whose exact function remains unclear. Caspase recruitment domain (CARD) proteins are generally involved in signal transduction pathways important for apoptosis, inflammation, immune function, and NF-κB activation. In vitro studies indicate that CARD6 is a micro-tubule-associated protein that positively modulates NF-κB activation.43 However, CARD6 homozygous deletion mice do not demonstrate evident defects in apoptosis, or in innate or adaptive immune pathways.44 The mutation detected in this primary myelofibrosis case results in a premature stop codon after the first 77 amino acids. Although this transcript might have been expected to undergo nonsense-mediated decay, we observed approximately equal expression of wild-type and mutant transcripts, as was also observed for the other genes. If the mutant protein was in fact expressed, this mutation would be predicted to result in the truncation of the protein near the end of the CARD domain (amino acids 3–94), leaving all but the very end of this protein-protein interaction domain completely intact. It has been demonstrated that a CARD6 mutant containing only the CARD domain is capable of interacting with the full-length CARD6,43 so it is possible that a 77 amino acid version could bind to and disrupt CARD6 targets and other CARD-containing proteins.
Although we did not detect recurrent somatic mutations in KIAA0355, BRD2, and CARD6, these somatic mutations could be functionally significant in this case. CARD6 and BRD2 have been reported to function in pathways or complexes that when disrupted are known to promote neoplastic transformation. As has been demonstrated for other hematologic neoplasms (e.g. chronic lymphocytic leukemia45), even though somatic mutations in these genes are not commonly recurrent, deregulation of these pathways via mutation or other genetic modification of other pathway members could be relevant to the pathogenesis of a significant fraction of primary myelofibrosis cases. It is also possible that non-coding changes present in this genome could contribute to the development of primary myelofibrosis in this patient, or that despite our attempts to maximize variant detection we did not detect relevant somatic mutations in coding regions. There is still a minor fraction of the non-repetitive genome and a major fraction of the repetitive genome that has not been fully examined by current technologies.
We cannot exclude that the somatic variants KIAA0355, BRD2, and CARD6 are passenger mutations that were accumulated in the hematopoietic stem or progenitor cell prior to transformation or in the neoplastic cell prior to the expansion of the current clonal population. Indeed, recent work in acute myeloid leukemia suggests that the majority of somatic mutations observed in acute myeloid leukemia genomes are stochastic events that occurred in the hematopoietic stem or progenitor cell prior to the initial transforming mutation, and most of these mutations do not influence pathogenesis.20 Murine models expressing MPL W515L recapitulate a phenotype consistent with a myeloproliferative neoplasm, specifically myelofibrosis,6 so other mutations may not be required for the development of a myeloproliferative neoplasm phenotype in mice. It is possible that the MPL W515L/K mutation alone is sufficient to generate a myeloproliferative neoplasm/myelofibrosis phenotype, as is true for BCR/ABL1 in chronic myelogenous leukemia.
More definitive classification of these somatic mutations as drivers of neoplasia will likely require the interrogation of mammalian model systems. In addition, examination of somatic variants identified in this study (and their associated pathways) in the context of additional genome and exome data will help clarify the importance of these mutations, genes, and biological pathways. As genome and exome sequencing is more commonly applied in clinical practice, it will be important to evaluate common and rare somatic mutations to determine their relevance to the practice of precision medicine.
In this study, we have utilized whole-genome sequencing to establish the low mutational complexity of MPL W515K-positive primary myelofibrosis, one of the first primary myelofibrosis genomes reported to date. Our comprehensive approach using high-coverage sequencing, two independent sequencing technologies, cultured skin fibroblasts, and multiple analysis approaches on a primary myelofibrosis specimen with a high percentage of neoplastic cells optimizes the detection of somatic variants. We estimate the exome mutation rate at 0.13 mutations/Mb and non-repetitive genome mutation rate at 0.15 mutations/Mb. In addition, the presence of somatic mutations and expression analysis of three genes, CARD6, BRD2, and KIAA0355, suggests these genes and associated pathways are potentially relevant to primary myelofibrosis pathogenesis, at least in rare cases. Finally, we describe bioinformatics approaches that improve somatic variant detection in hematologic malignancies. These data and approaches will enable further clarification of genes and biological pathways important for myeloproliferative neoplasm pathogenesis.
Acknowledgments
The authors would like to thank the Charles and Ann Johnson Foundation for funding support. We express gratitude to the Stanford Hematology Division Tissue Bank for collecting and processing samples. We would like to thank Anton Valouev for assistance with the structural and copy number variant analyses, and Norm Cyr for assistance with photography and figure layouts.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed Lyon: IARC Press, 2008 [Google Scholar]
- 2.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–61 [DOI] [PubMed] [Google Scholar]
- 3.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–90 [DOI] [PubMed] [Google Scholar]
- 4.Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106(6): 2162–8 [DOI] [PubMed] [Google Scholar]
- 5.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–97 [DOI] [PubMed] [Google Scholar]
- 6.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108(10):3472–6 [DOI] [PubMed] [Google Scholar]
- 8.Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111(8):3931–40 [DOI] [PubMed] [Google Scholar]
- 9.Campbell PJ, Baxter EJ, Beer PA, Scott LM, Bench AJ, Huntly BJ, et al. Mutation of JAK2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations, and role in leukemic transformation. Blood. 2006;108(10):3548–55 [DOI] [PubMed] [Google Scholar]
- 10.Pardanani A, Lasho TL, Finke C, Mesa RA, Hogan WJ, Ketterling RP, et al. Extending Jak2V617F and MplW515 mutation analysis to single hematopoietic colonies and B and T lymphocytes. Stem Cells. 2007;25(9):2358–62 [DOI] [PubMed] [Google Scholar]
- 11.Nussenzveig RH, Swierczek SI, Jelinek J, Gaikwad A, Liu E, Verstovsek S, et al. Polycythemia vera is not initiated by JAK2V617F mutation. Exp Hematol. 2007;35 (1):32–8 [DOI] [PubMed] [Google Scholar]
- 12.Theocharides A, Boissinot M, Girodon F, Garand R, Teo SS, Lippert E, et al. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood. 2007;110(1):375–9 [DOI] [PubMed] [Google Scholar]
- 13.Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol. 2011;29(5):573–82 [DOI] [PubMed] [Google Scholar]
- 14.Abdel-Wahab O, Pardanani A, Bernard OA, Finazzi G, Crispino JD, Gisslinger H, et al. Unraveling the genetic underpinnings of myeloproliferative neoplasms and understanding their effect on disease course and response to therapy: Proceedings from the 6th international post-ASH symposium. Am J Hematol. 2012;87(5):562–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lam HY, Pan C, Clark MJ, Lacroute P, Chen R, Haraksingh R, et al. Detecting and annotating genetic variations using the HugeSeq pipeline. Nat Biotechnol. 2012;30(3):226–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. Determinants of nucleosome organization in primary human cells. Nature. 2011;474(7352):516–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The Cancer Genome Atlas Research Network Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med. 2013; 368(22):2059–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–9 [DOI] [PubMed] [Google Scholar]
- 24.The Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455 (7216):1061–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353): 609–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The Cancer Genome Atlas Research Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The Cancer Genome Atlas Research Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Osaki E, Nishina Y, Inazawa J, Copeland NG, Gilbert DJ, Jenkins NA, et al. Identification of a novel Sry-related gene and its germ cell-specific expression. Nucleic Acids Res. 1999;27(12):2503–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Assou S, Anahory T, Pantesco V, Le Carrour T, Pellestor F, Klein B, et al. The human cumulus--oocyte complex gene-expression profile. Hum Reprod. 2006;21(7):1705–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sundin OH, Dharmaraj S, Bhutto IA, Hasegawa T, McLeod DS, Merges CA, et al. Developmental basis of nanophthalmos: MFRP Is required for both prenatal ocular growth and postnatal emmetropization. Ophthalmic Genet. 2008;29(1):1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayyagari R, Mandal MN, Karoukis AJ, Chen L, McLaren NC, Lichter M, et al. Late-onset macular degeneration and long anterior lens zonules result from a CTRP5 gene mutation. Invest Ophthalmol Vis Sci. 2005;46(9):3363–71 [DOI] [PubMed] [Google Scholar]
- 33.Katoh M. Molecular cloning and characterization of MFRP, a novel gene encoding a membrane-type Frizzled-related protein. Biochem Biophys Res Commun. 2001;282 (1):116–23 [DOI] [PubMed] [Google Scholar]
- 34.Denis GV, Green MR. A novel, mitogen-activated nuclear kinase is related to a Drosophila developmental regulator. Genes Dev. 1996;10(3):261–71 [DOI] [PubMed] [Google Scholar]
- 35.Thorpe KL, Abdulla S, Kaufman J, Trowsdale J, Beck S. Phylogeny and structure of the RING3 gene. Immunogenetics. 1996;44(5):391–6 [DOI] [PubMed] [Google Scholar]
- 36.Denis GV, Vaziri C, Guo N, Faller DV. RING3 kinase transactivates promoters of cell cycle regulatory genes through E2F. Cell Growth Differ. 2000;11(8):417–24 [PMC free article] [PubMed] [Google Scholar]
- 37.Denis GV, McComb ME, Faller DV, Sinha A, Romesser PB, Costello CE. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J Proteome Res. 2006;5(3):502–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shang E, Wang X, Wen D, Greenberg DA, Wolgemuth DJ. Double bromodomain-containing gene Brd2 is essential for embryonic development in mouse. Dev Dyn. 2009;238(4):908–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greenwald RJ, Tumang JR, Sinha A, Currier N, Cardiff RD, Rothstein TL, et al. E mu-BRD2 transgenic mice develop B-cell lymphoma and leukemia. Blood. 2004;103(4): 1475–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stadler M, Fire A. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA. 2011;17(12):2063–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370): 529–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dufner A, Pownall S, Mak TW. Caspase recruitment domain protein 6 is a micro-tubule-interacting protein that positively modulates NF-kappaB activation. Proc Natl Acad Sci USA. 2006;103(4):988–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dufner A, Duncan GS, Wakeham A, Elford AR, Hall HT, Ohashi PS, et al. CARD6 is interferon inducible but not involved in nucleotide-binding oligomerization domain protein signaling leading to NF-kappaB activation. Mol Cell Biol. 2008;28(5):1541–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365 (26):2497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]