

Abstract
Congenital dyserythropoietic anemia type II is an autosomally recessive form of hereditary anemia caused by SEC23B gene mutations. Patients exhibit characteristic phenotypes including multinucleate erythroblasts, erythrocytes with hypoglycosylated membrane proteins and an apparent double plasma membrane. Despite ubiquitous expression of SEC23B, the effects of mutations in this gene are confined to the erythroid lineage and the basis of this erythroid specificity remains to be defined. In addition, little is known regarding the stage at which the disparate phenotypes of this disease manifest during erythropoiesis. We employ an in vitro culture system to monitor the appearance of the defining phenotypes associated with congenital dyserythropoietic anemia type II during terminal differentiation of erythroblasts derived from small volumes of patient peripheral blood. Membrane protein hypoglycosylation was detected by the basophilic stage, preceding the onset of multinuclearity in orthochromatic erythroblasts that occurs coincident with the loss of secretory pathway proteins including SEC23A during erythropoiesis. Endoplasmic reticulum remnants were observed in nascent reticulocytes of both diseased and healthy donor cultures but were lost upon further maturation of normal reticulocytes, implicating a defect of ER clearance during reticulocyte maturation in congenital dyserythropoietic anemia type II. We also demonstrate distinct isoform and species-specific expression profiles of SEC23 during terminal erythroid differentiation and identify a prolonged expression of SEC23A in murine erythropoiesis compared to humans. We propose that SEC23A is able to compensate for the absence of SEC23B in mouse erythroblasts, providing a basis for the absence of phenotype within the erythroid lineage of a recently described SEC23B knockout mouse.
Introduction
Congenital dyserythropoietic anemia type II (CDAII) is a form of hereditary anemia characterized by the presence of bi- or multinuclear erythroblasts in the bone marrow. Clinically, patients present with typical symptoms associated with hemolytic anemia of variable degree, erythroid hyperplasia, splenomegaly, gallstones and iron overload.1–3 Other characteristic features of CDAII include erythrocytes with hypoglycosylated membrane proteins including band 3, and the glucose transporter Glut14–7 and a “double plasma membrane” due to residual endoplasmic reticulum, that stains positively for endoplasmic reticulum (ER) protein markers GRP78, calreticulin and protein disulfide isomerase (PDI).8 The stage at which the characteristic features of CDAII observed in patients’ erythrocytes manifest during erythropoiesis is currently unknown.
In 2009, the SEC23B gene, encoding the COPII coat component, was identified as the causative gene for CDAII by different approaches.9,10 Since then approximately 60 different causative mutations have been described along the gene including missense, nonsense, deletion and splice site mutations;11 the missense mutations affecting highly conserved residues in multiple domains of SEC23B.9–17 The defect is transmitted as an autosomal recessive trait; in almost all patients, mutations are detected at homozygous or compound heterozygote level. The coexistence of two severe nonsense mutations has never been described suggesting that this condition is most likely lethal.
SEC23 exists as two isoforms (SEC23A and SEC23B) encoded by two different genes, which form the first tier of the COPII coat complex alongside SEC24 (which exists as four isoforms). The multimeric COPII coat is comprised of five key subunits including the small GTPase SAR1, the SEC23–SEC24 inner coat complex and the SEC13–SEC31 outer coat complex. The COPII coat assembles at specialized exit sites on the ER where GTP bound active SAR1 inserts into ER membranes and recruits the inner SEC23–SEC24 coat via its interaction with SEC23. SEC24 interacts with the cytoplasmic domain of cargo proteins and the cargo bound SAR1-SEC23–SEC24 complex recruits SEC13–SEC31 heterotetramers to assemble a protein coat.18 Coat assembly on the ER membrane sculpts the membrane into a vesicle for cargo transport from the ER to the cis Golgi.19–22
The two SEC23 isoforms SEC23A and SEC23B share 85% amino acid sequence identity and are ubiquitously expressed,23 albeit at different levels. This differential expression, along with the existence of multiple isoforms of the other coat proteins such as SAR1 and SEC24, is thought to provide a combinatorial diversity to cargo selection and transport that is tissue specific.24 Mutations in genes encoding several COPII components cause human disease.25–27 These diseases generally occur in cells with a specific requirement for protein transport and delivery. The confinement of the effects of SEC23B mutations to the erythroid lineage, therefore, implies an essential role for this COPII coat component in normal development of the erythrocyte. Intriguingly, a SEC23B knockout mouse model did not reproduce the human disease phenotype suggesting species differences in the roles or regulation of the SEC23 proteins between humans and mice.28
In this study, we use an in vitro erythroid cell culture system to expand and differentiate erythroblasts derived from peripheral blood mononuclear cells of 3 CDAII patients to monitor the appearance of characteristic phenotypes associated with this disease during erythropoiesis. This enabled us to establish a temporal profile by which the phenotypes observed in CDAII patient erythroblasts and erythrocytes in vivo manifest during terminal erythroid differentiation. By comparing the expression of SEC23 protein isoforms during terminal differentiation in both human and mouse erythroblasts, we demonstrate erythroblast stage and species specific differences in expression of the respective SEC23 isoforms, which potentially explains the species and lineage specific phenotype observed in the presence of SEC23B mutations in human CDAII patients.
Methods
Antibodies
Mouse monoclonal antibodies used were BRIC170 (band 3; IGBRL, Bristol, UK) and PDI (Assay Designs). Rabbit polyclonal antibodies to RhAG, Glut1, GPA, CD47,29,30 SEC24C,31 SEC24D32 and SEC31A31 were available in house. Anti-mouse band 3 was from Prof. Carsten Wagner (University of Zurich), anti GM130 (BD Transduction), anti-GAPDH (Santa Cruz) and anti-GRP78 (Sigma). Initial antibodies against SEC23A and SEC23B were from Prof. Randy Schekman (University of California). Novel isoform specific purified polyclonal antibodies were generated for SEC23A, SEC23B, SEC24A and SEC24B. Synthetic peptide sequences used are listed in Online Supplementary Methods. Secondary antibodies were Alexa-488™ or Alexa594™ conjugated goat anti-mouse or anti-rabbit (Life Technologies) for immunofluorescence and HRP-conjugated rabbit anti-mouse and swine anti-rabbit (DAKO) for immunoblotting.
Donor and patient blood
Waste blood from anonymous platelet apheresis donors (NHSBT, Filton) or patient samples were provided with written informed consent for research use given in accordance with the Declaration of Helsinki. Research was reviewed and approved by Southmead and Bristol Research Ethics Committee Centre reference 08/H0102/26 and 12/SW/0199, respectively. Peripheral blood samples (25 mL) were obtained from 5 unrelated CDAII patients with known SEC23B gene mutations and 3 of the samples were successfully cultured. Clinical data are summarized in the Online Supplementary Table S1 and have already been reported10 for 4 patients. Online Supplementary Figure S1 shows as an example the characterization of the erythrocytes of 2 of the CDAII patients.
Isolation and expansion of human peripheral blood mononuclear cells
Erythroblasts derived from peripheral blood mononuclear cells were isolated, expanded and differentiated as previously described29,33,34 for initial experiments and then subsequently modified according to Bell et al.35
Preparation of erythrocyte ghosts and erythroblast lysates
Erythrocyte ghosts and erythroblast lysates were prepared from donor and patient cells as previously described.30,36 Samples were snap frozen in liquid nitrogen and stored at −80°C until use. Immunoblotting using tomato lectin is described in the Online Supplementary Methods.
Culture of mouse primary fetal liver erythroblasts
Primary fetal liver erythroblasts isolated from C57BL/6 Day E12–13 mice were isolated and cultured as previously described.37 The immortalized erythroid cell line II–3 was generated from fetal liver cultures as previously described.38
Cytospins and immunofluorescence
Erythrocytes or erythroblasts were cytospun and stained with May Grünwald Giemsa stains or processed for immunofluorescence as previously described.34 Reticulocyte imaging is described in the Online Supplementary Methods.
Transmission electron microscopy
Electron microscopy was conducted on cultured erythroblasts or erythrocytes as described in the Online Supplementary Methods.
Results
Mapping the temporal onset of the CDAII Multinuclear Phenotype using in vitro culture
To determine the stage at which the characteristic phenotypes associated with CDAII manifest during erythropoiesis, erythroblasts derived from peripheral blood mononuclear cells (PBMCs) of healthy donors and from patients with CDAII arising from mutations in the SEC23B gene were expanded and synchronously differentiated. Figure 1A shows representative differentiation courses from 3 patients with CDAII alongside a healthy donor control. No obvious morphological disruption or differences in proerythroblasts or basophilic erythroblasts were observed between 3 CDAII patients and 4 control samples up to 72 h of differentiation. In all CDAII patient cultures, multinuclear erythroblasts were most evident from the 72–96 h time point onwards at the late polychromatic and orthochromatic erythroblast stages (see Online Supplementary Figure S2 for confocal imaging examples). The prevalence of multinuclear cells was 2–3 fold in patient cells compared to control cells at the equivalent stage (Figure 1B).
Figure 1.

Multinuclear orthochromatic erythroblasts are observed from in vitro culture of CDAII patient-derived erythroblasts. (A) 5×105 cells were removed from differentiation culture of control and 3 independent CDAII patient-derived erythroblasts at 24-h intervals. Cells were cytospun and stained with May Grünwald Giemsa dyes, representative images are shown. White arrows highlight multinuclear erythroblasts. Scale bar 20 μm. (B) Histogram shows percentage of bi/multinuclear erythroblasts according to erythroblast cell stage. Data were obtained by counting at least 150 cells per cytospin. Values for multinuclear erythroblasts are expressed as a percentage of total nucleated erythroblasts. ProE/Baso: pro- and basophilic erythroblasts; Poly: polychromatic; Ortho: orthochromatic erythroblasts, respectively. The mean and standard deviation are shown for 4 control cultures.
Defective glycosylation precedes the onset of multinuclearity during CDAII erythropoiesis
Erythrocyte membrane protein hypoglycosylation is a defining characteristic of CDAII (Online Supplementary Figure S1). To determine the stage at which hypoglycosylation is first evident during erythropoiesis, proerythroblasts from 2 CDAII patients (R190X/S603L and R14W/R554X) and from healthy donors were differentiated and lysates collected at 24-h intervals. Immunoblots were conducted to assess glycosylation of the erythroid specific membrane protein band 3, hypoglycosylation of which is used as a diagnostic marker of CDAII. Figure 2 shows there is hypoglycosylation of band 3 from the onset of its synthesis in basophilic erythroblasts (T24–T48) that was maintained throughout terminal erythroid differentiation. Tomato lectin detects the presence of polylactosamines (complex glycosylation) and can be used to detect gross differences in glycosylation between control and CDAII erythrocytes39 (see Online Supplementary Figure S3A). Reduced tomato lectin binding to proteins from CDAII patient erythroblast lysates relative to control was detected within 24–48 h of terminal differentiation in 2 patients and this difference persisted throughout terminal differentiation (see Figure 2B for R190X/S603L throughout terminal differentiation and Online Supplementary Figure S3B for R14W/R554X and R190X/S603L again).
Figure 2.
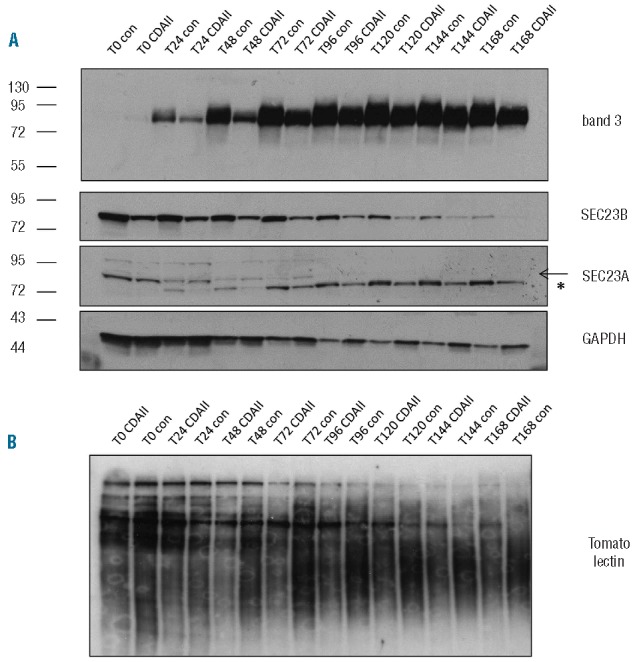
Impaired glycosylation of erythroid membrane proteins is evident from the onset of terminal differentiation. (A) 5×105 differentiating erythroblasts derived from the peripheral blood of healthy donor control or CDAII patient (R190X/S603L) were removed from culture at 24-h intervals, cells were lysed, proteins separated by SDS PAGE and immunoblotted with antibodies to band 3 (BRIC170) GAPDH, SEC23A and SEC23B. *Antibody cross reactivity with unrelated protein. Note that the GAPDH loading control indicates higher total protein levels in CDAII samples compared to control at the later stages of differentiation, reinforcing the observed reduction in SEC23B expression in the CDAII samples. (B) The equivalent of 2×104 cells was loaded per lane of control or CDAII (R190X/S603L) were separated by SDS PAGE and immunoblotted using tomato lectin to detect differences in glycosylation.
Loss of ER occurs during enucleation and after additional reticulocyte maturation
Another key feature of CDAII erythrocytes is the appearance of an apparent double plasma membrane as observed by electron microscopy, which is comprised of residual ER illustrated by its labeling by antibodies to ER proteins such as PDI.8 Intriguingly, no obvious double membrane structures could be observed in our late stage CDAII cultured erythroblasts by light or electron microscopy (data not shown) although the double membrane was evident in mature erythrocytes taken from CDAII patient peripheral blood samples (Online Supplementary Figure S1).
We recently reported that the bulk of ER proteins are lost with the extruded nucleus during enucleation. However, some ER remnants persist in the nascent reticulocyte that may be lost later during reticulocyte maturation.35 To establish whether the residual ER in erythrocytes from CDAII patients (see Online Supplementary Figure S1) originates from a failure of ER to partition correctly during enucleation, enucleating orthochromatic erythroblasts from CDAII patient cultures were labeled with antibodies specific for band 3 and PDI and examined using confocal microscopy. Online Supplementary Figure S4A (top and middle panels) shows PDI partitioning with the extruding nucleus in control patient samples, which is comparable to that in CDAII orthochromatic erythroblasts (bottom panel).
Post enucleation, nascent reticulocytes generated from both CDAII patient and healthy donor erythroblasts retained variable amounts of punctate PDI staining (Online Supplementary Figure S4B). To determine whether PDI is further lost upon reticulocyte maturation, control and the R14W/R554X patient reticulocytes were allowed to mature for an additional three days in culture. Figure 3A shows that PDI labeling was lost in a proportion of reticulocytes from both healthy donor and CDAII patient cultures. This is consistent with our observation that CDAII patient peripheral blood samples contain a majority of erythrocytes with no PDI immunoreactivity (82–87% for splenectomized patients and 95–97% in unsplenectomized CDAII patient peripheral blood samples; Online Supplementary Figure S1D). These ER remnants were largely lost in healthy donor reticulocytes that have passed through a leukofilter, a process which isolates mainly mature reticulocytes34,40 (Figure 3B). Leukofiltration of CDAII patient reticulocytes was not possible due to the minimal material available, however, the presence of residual ER in a proportion of erythrocytes is already an established feature of this disease8 (Online Supplementary Figure S1).
Figure 3.
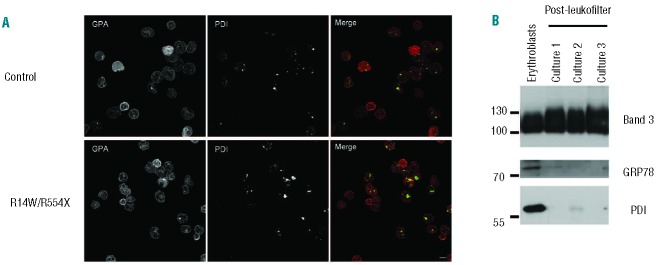
PDI is lost in normal mature reticulocytes and in a proportion of CDAII reticulocytes. (A) Control and CDAII patient reticulocytes were separated from erythroblasts and extruded nuclei, using a 3 μm polycarbonate filter and then allowed to mature in culture for an additional 72 h in differentiation medium containing 10% AB serum. The reticulocytes were then processed for immunofluorescence, using a rabbit glycophorin A antibody to label plasma membrane and a monoclonal PDI antibody to image ER remnants, which were detected using suitable secondary antibodies. Multiple sections were taken and the cell projection view is shown. Scale bar is 5 μm. (B) Three independent cultures of 300–450 million in vitro cultured reticulocytes that had been cultured for at least 24-h were filtered using a Leukofilter to isolate mature reticulocytes. 1×106 orthochromatic erythroblasts (from T120) and mature reticulocytes from the 3 cultures were subjected to SDS PAGE and immunoblotted using band 3 (Bric170), GRP78 and PDI antibodies.
SEC23 Isoform expression during erythropoiesis
The reason why mutations within the ubiquitously expressed SEC23B gene are confined specifically to the erythroid lineage in CDAII patients is unknown. One possibility is that a specific role exists for SEC23B within erythroid cells that is not conserved in other cell types. Alternatively, reduced expression of the functionally redundant SEC23A during erythropoiesis could account for erythroid specific phenotypes observed in the presence of mutated SEC23B, in a manner analogous to the tissue specificity observed in Cranio Lenticulo Sutural Dysplasia25,26 where mutations in SEC23A coincide with relatively low levels of SEC23B expression in calvarial osteoblasts.
To investigate this, we analyzed the protein expression profile of the two SEC23 isoforms during normal human terminal erythroid differentiation from proerythroblasts to reticulocytes using SEC23 isoform specific antibodies generated and characterized in house (see Online Supplementary Figure S5). Figure 4A shows that although the expression of both SEC23 isoforms decreases during human terminal erythroid differentiation, there is a distinctive expression profile for each isoform during this process. This is consistent with the available human mRNA expression profile during in vitro erythropoiesis41 (Online Supplementary Figure S6). SEC23B expression remains detectable throughout the process of erythropoiesis albeit reduced in the later stages of differentiation relative to that in pro/basophilic erythroblasts (Figure 4A). This is consistent with the detection of SEC23B by proteomics in erythrocytes.10,42 Strikingly, a rapid drop in expression of SEC23A was observed during erythropoiesis, which was evident from the onset of terminal differentiation, with levels decreasing below the level of antibody detection after 72–96 h of differentiation. This time point corresponds to the late polychromatic/early orthochromatic erythroblast stage in our culture model34 (Figure 1A), a point at which other proteins that comprise the secretory pathway, e.g. the cis-Golgi matrix protein GM130, are also lost (Figure 4B). Expression of other COPII coat proteins known to interact with SEC23, including multiple isoforms of SEC24 (SEC24A-D) and SEC31A, was also differentially lost during terminal erythroid differentiation (Figure 4B).
Figure 4.
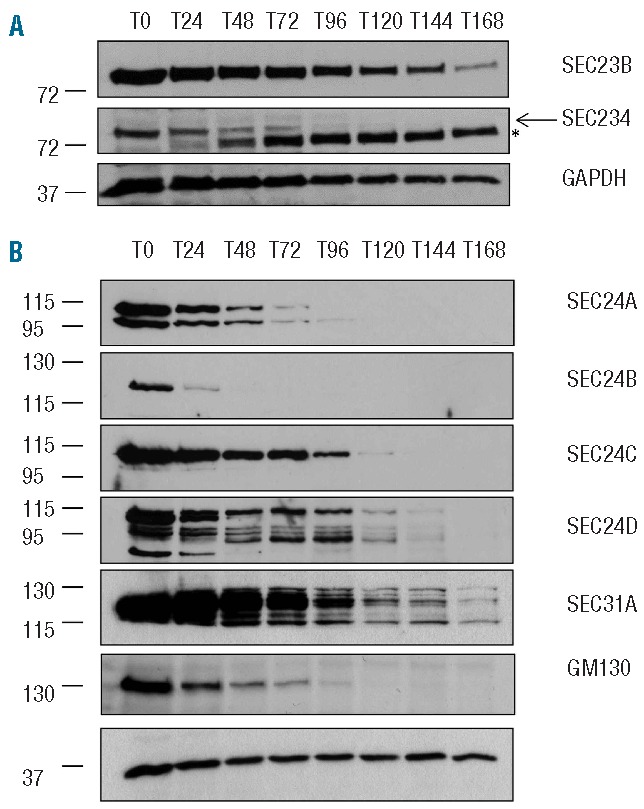
SEC23A is lost prior to SEC23B during normal human terminal erythroid differentiation. 1×106 differentiating erythroblasts were removed from culture at 24 h intervals, cells were lysed, proteins separated by SDS PAGE and immunoblotted with (A) rabbit antibodies to SEC23B, SEC23A, and GAPDH. Arrow indicates SEC23A band; *antibody cross reactivity with unrelated protein. (B) Rabbit antibodies to SEC24A, SEC24B, SEC24C, SEC24D, SEC31A, GM130 and GAPDH. SEC24 antibodies detect multiple splice forms.
Immunoblotting of cell lysates from the (R190X/S603L) CDAII patient culture, with antibodies to SEC23A and SEC23B shows that the temporal expression profiles of both SEC23 isoforms remain unaltered between control and CDAII erythroblast samples throughout differentiation, with SEC23A expression again dropping to below detectable levels after 72–96 h of differentiation and expression of mutant SEC23B maintained throughout terminal differentiation (see Figure 2A and Online Supplementary Figure S3C for R14W/R554X). SEC23A protein expression levels are not up-regulated in CDAII erythroblasts to compensate for SEC23B. In contrast, SEC23B expression levels are reduced to ~50% in proerythroblasts and there is further reduction (to <30%) during subsequent differentiation in the CDAII sample (R190X/S603L). This reflects the S603L protein expression as R190X truncation mutant would not be detected using our antibody. A similar reduction in expression of R14W/R554X patient full length SEC23B was observed reflecting expression of the R14W allele (Online Supplementary Figure S3). Expression of the truncated R554X product was not detected (data not shown).
SEC23A expression is maintained throughout murine erythropoiesis
A recently published SEC23B knockout mouse model28 failed to recapitulate the erythroid phenotypes observed in CDAII patients with SEC23B mutations. To determine whether different expression profiles of the SEC23 isoforms in human and mouse erythropoiesis could underlie the absence of phenotype within the SEC23B knockout mouse, we studied the expression profile of these isoforms in murine erythroblasts. Importantly, the corresponding regions of human and mouse protein sequences to which our in-house SEC23 isoform specific antibodies are raised, are completely conserved for SEC23A and differ minimally for SEC23B (3 of 24 amino acid residues). Therefore, using these antibodies we investigated the expression profile of SEC23A and B in murine erythroblasts using an immortalized erythroblast cell line38 and primary fetal liver erythroblasts. The immortalized erythroblasts were synchronously differentiated and cell lysates collected every 24 h over the 5-day period of murine erythroblast differentiation (see Figure 5A and Online Supplementary Figure S7 for cytospins and flow cytometry data). Figure 5 shows a gradual decrease in the expression of SEC23B similar to that observed in human erythropoiesis, with SEC23B still detectable in late stage orthochromatic erythroblasts and reticulocytes. Unlike in humans, SEC23A expression is maintained throughout murine erythroid differentiation, with only a small reduction in protein levels evident during erythropoiesis. Similar results were obtained using primary cell erythroblasts isolated from fetal liver of wild-type mice (C57BL/6) (Figure 5B), by using a second independent murine erythroblast cell line and erythroblasts derived from wild-type mouse bone marrow (data not shown). These data are consistent with the available murine bone marrow mRNA expression data43 (Online Supplementary Figure S6).
Figure 5.
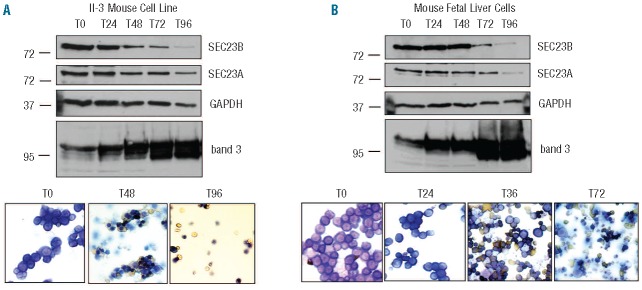
SEC23A expression is maintained during murine terminal erythroid differentiation. 1×106 differentiating erythroblasts were removed from culture of (A) immortalized erythroblast cell line (II–3) or (B) primary fetal liver-derived mouse erythroblasts. Cytospins were prepared and stained with Giemsa dye and neutral benzidine. Cells were lysed, proteins separated by SDS PAGE and immunoblotted with rabbit antibodies to SEC23B, SEC23A, GAPDH and band 3.
Discussion
Although the genetic basis for the human CDAII phenotype is now firmly established, detailed studies of the phenotypes arising from these mutations and their appearance during erythrocyte development are limited. Similarly, the question of why impairment of SEC23B expression or function results in an erythroid specific phenotype remains to be resolved. The scope of such investigations is however limited by the requirement to study the lineage specific effects of such mutations within a primary erythroid cell context, our current inability to fully mimic the bone marrow microenvironment and by the inherent technical difficulties of culturing dyserythropoietic cells.
In this study, we have successfully characterized the onset of key erythroid specific phenotypes associated with mutations in SEC23B. Furthermore, we report for the first time protein expression profiles of multiple COPII core coat component proteins during human erythropoiesis; importantly demonstrating a loss or reduction of both SEC23A and SEC23B during this process. We show that despite reducing levels, SEC23B is expressed throughout erythropoiesis, whereas SEC23A expression is lost by the early orthochromatic stage (approx 72–96 h of differentiation). Our findings are consistent with the increased ratio of SEC23B:SEC23A transcript observed previously in differentiated CD34+ cells.9 Thus analogous to Cranio Lenticulo Sutural Dysplasia,25,26 CDAII likely arises specifically during erythropoiesis because of the abundant SEC23B and rapidly reducing SEC23A expression profile during this process.
Membrane protein hypoglycosylation precedes the appearance of multinuclear erythroblasts in CDAII patient erythroblast culture
Our results demonstrate for the first time that a glycosylation defect is detectable early (basophilic stage) during the process of terminal erythroid differentiation. It remains unknown why a glycosylation defect manifests specifically in erythroblasts at this stage where both SEC23B and SEC23A are present but not in other cell types. However, it is notable that at this stage during erythropoiesis the expression of SEC23A, although not lost, is reduced relative to undifferentiated erythroblasts, resulting in an increased SEC23B/SEC23A protein ratio that could explain a dose-dependent glycosylation defect in the presence of mutated SEC23B protein. During the early stages of terminal differentiation, there is a significant secretory pathway burden whereby expression of erythroid specific proteins increases dramatically and must be delivered to the plasma membrane.29,34 Thus this work suggests that there is an important role for SEC23B in general COPII mediated trafficking or in the transport of cargo with function specific to the erythroid cellular context that cannot be completely compensated by the decreasing levels of SEC23A.
It is also notable that the appearance of multinuclear cells in culture of CDAII patient derived erythroblasts is coincident with the loss of SEC23A expression at the early orthochromatic erythroblast stage of erythropoiesis. This observation is consistent with earlier reports from EM studies of bone marrow aspirates in which early erythroblasts appear unaffected but are bi/multinucleate at the later developmental stages.1,44,45 An attractive hypothesis is that loss of compensation for the mutated SEC23B through the functional redundancy of SEC23A at this stage of differentiation, could account for the appearance of these multinuclear cells at this stage. However, we cannot currently discount a role for mutant SEC23B protein in the multinuclear phenotype. SEC23B was identified in a proteomic screen of midbody components in CHO cells,46 however, there is no direct evidence that it plays an active role in assembly or deconstruction of the midbody. It is nevertheless possible that SEC23B is required for transport of a specific cargo important for efficient cytokinesis in erythroblasts, which needs to be down-regulated during cell cycle arrest in differentiation. An alternative explanation is that the multinuclear phenotype is secondary to the aberrant glycosylation of specific proteins required for some unknown aspect of cell division leading to defects in this tightly controlled process. In support of this, Fukuda et al.47 found that the in vitro treatment of cultured erythroblasts with α mannosidase inhibitors resulted in the appearance of erythroblast multinuclearity and the α mannosidase II knockout mouse exhibits a dyserythropoietic phenotype.48
Clearance of ER proteins occurs both during and post erythroblast enucleation
The ER origin of ‘double membranes’ in the erythrocytes of CDAII patients was established by immunoblotting of erythrocyte ghost proteins and immunoelectron microscopy of these inner membranes with antibodies to ER markers.8 Within the small number of representative CDAII patient blood samples that we used in the current studies, we have found this ranges between 13–18% for splenectomized patients and 3–5% in unsplenectomized patient samples. It is likely that this discrepancy can be accounted for by splenic sequestration/clearance of PDI positive erythrocytes from the circulation. However, further studies incorporating additional CDAII patient peripheral blood samples are needed to confirm this.
We have shown that the bulk of the ER partitions with the nucleus during enucleation in normal and CDAII erythroblasts but some remaining ER remnants are detectable in reticulocytes from both cell sources after the enucleation process. Importantly, we have also demonstrated that the residual ER remnants are normally lost upon additional maturation in culture of healthy donor reticulocytes (see Figure 3B). This corroborates our recent suggestion that clearance of the residual ER membrane continues post erythroblast enucleation.35 This ER clearance nevertheless appears to occur effectively in a proportion of in vitro cultured CDAII patient reticulocytes and this is consistent with presence of PDI negative erythrocytes observed in these patients. These observations now raise the possibility that reticulocyte maturation is a stage at which defective SEC23B function can result in erythrocyte ER protein retention, potentially explaining both the residual ER observed in a proportion of CDAII patients’ erythrocytes and the prolonged expression of SEC23B in erythropoiesis. Further work is necessary to determine whether there is a delay or an actual disruption of reticulocyte maturation in a proportion of CDAII patients’ reticulocytes. Reticulocyte maturation involves both autophagy and exosome release40 and interestingly, mutations in yeast Sec23, Sec24 and Sec16 cause defective autophagy, which is independent of their role in ER to Golgi transport.49 It is possible that SEC23B deficiency or the presence of mutant SEC23B in human erythroblasts disturbs an aspect of autophagy or ER clearance either directly or indirectly. However, a gross disruption of autophagy in CDAII patients is unlikely since the removal of other cellular organelles by autophagy (e.g. mitochondria) is unaffected.
It should be noted that previous reports have documented the appearance of ‘double membranes’ in EM images of nucleated erythroblasts taken from bone marrow aspirates of CDAII patients.1,44,50,51 We saw no evidence of these structures in our in vitro cultured erythroblasts from the specific CDAII patient’s peripheral blood samples we obtained. One possible explanation is that erythroblasts exhibiting this aspect of the disease phenotype do not persist outside of the protective microenvironment of the bone marrow. Hence the possibility remains that all the CDAII reticulocytes produced by in vitro culture will eventually lose their ER remnants or that CDAII mutations cause disruption of ER earlier in erythropoiesis in a subset of cells in a process not recapitulated here under in vitro culture conditions.
Recently, the absence of an erythroid phenotype in SEC23B knockout mice was reported28 in contrast to the human erythroid specific CDAII phenotype. Our observation here of perdurance of SEC23A during murine erythropoiesis could explain the absence of erythroid phenotype in these mice,28 with SEC23A able to complement the SEC23B deficiency in the knockout model. Thus it is possible that in mice, SEC23A may completely compensate for loss of SEC23B throughout erythropoiesis or alternatively trafficking in mouse erythroblasts could be more SEC23A orientated from the start due to SEC23A possessing similar functional properties to SEC23B in mice. The possibility that expression of a mutated SEC23B, allied to the absence of wild-type expression, as observed in CDAII patients is required to recapitulate the disease phenotypes also remains. Further dissection of the regulation pathways and species differences involved may provide further insight into the precise roles played by SEC23 isoforms in erythroid cells of both species.
Conclusion
This study widens our understanding of the different functions that the SEC23B isoform of the COPII coat plays in the specialized development of the erythrocyte. By culturing erythroblasts derived from CDAII patients we have begun to establish the temporal order in which the characteristic phenotypes observed in patient erythrocytes manifest during terminal erythroid differentiation. We have shown that: 1) the loss of SEC23A in early orthochromatic erythroblasts is coincident with the appearance of multinuclear cells in CDAII patient erythroblasts cultured in vitro; 2) impaired glycosylation of membrane proteins precedes the loss of detectable SEC23A suggesting that it is most likely due to the reduction in SEC23A observed during terminal differentiation and/or that SEC23A cannot fully compensate for reduced/defective SEC23B in erythroblasts; 3) the clearance of ER remnants continues post enucleation during normal reticulocyte maturation suggesting that retention of ER remnants may result from a delay in reticulocyte maturation or defective ER clearance; and finally, 4) expression of SEC23 isoforms during erythropoiesis differs between mice and humans. This striking species difference offers one possible explanation for the absence of phenotype in erythroid cells of a recently described SEC23B knockout mouse.28
Acknowledgments
TJS work was funded initially funded by a BBSRC Case PhD NHSBT Studentship and is currently supported by a Wellcome Trust project grant (N. 094277) awarded to AMT. SP is funded by an NHSBT R&D project grant (09-25-02-01) awarded to AMT. This work was funded in part by National Institute for Health Research program grant funding (RP-PG-0310-1004-AMT) and the UK Medical Research Council and Biotechnology and Biological Sciences Research Council (DJS and AB, respectively), and by an Italian Ministry of Health program grant funding (RF-2010 n.2303934). The authors thank Prof. Randy Schekman (University of California at Berkeley) for providing initial SEC23 antibodies and the HA-tagged human SEC24 constructs.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Wickramasinghe SN. Congenital dyserythropoietic anaemias: clinical features, haematological morphology and new biochemical data. Blood Rev. 1998;12(3):178–200 [DOI] [PubMed] [Google Scholar]
- 2.Khoriaty R, Vasievich MP, Ginsburg D. The COPII pathway and hematologic disease. Blood. 2012;120(1):31–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iolascon A, Esposito MR, Russo R. Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: from morphology to molecular approach. Haematologica. 2012;97(12):1786–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anselstetter V, Horstmann HJ, Heimpel H. Congenital dyserythropoietic anaemia, types I and II: aberrant pattern of erythrocyte membrane proteins in CDA II, as revealed by two-dimensional polyacrylamide gel electrophoresis. Br J Haematol. 1977;35(2):209–15 [DOI] [PubMed] [Google Scholar]
- 5.Baines AJ, Banga JP, Gratzer WB, Linch DC, Huehns ER. Red cell membrane protein anomalies in congenital dyserythropoietic anaemia, type II (HEMP AS). Br J Haematol. 1982;50(4):563–74 [DOI] [PubMed] [Google Scholar]
- 6.Scartezzini P, Forni GL, Baldi M, Izzo C, Sansone G. Decreased glycosylation of band 3 and band 4.5 glycoproteins of erythrocyte membrane in congenital dyserythropoietic anaemia type II. Br J Haematol. 1982;51(4): 569–76 [DOI] [PubMed] [Google Scholar]
- 7.Mawby WJ, Tanner MJ, Anstee DJ, Clamp JR. Incomplete glycosylation of erythrocyte membrane proteins in congenital dyserythropoietic anaemia type II (CDA II). Br J Haematol. 1983;55(2):357–68 [DOI] [PubMed] [Google Scholar]
- 8.Alloisio N, Texier P, Denoroy L, Berger C, Miraglia del Giudice E, Perrotta S, et al. The cisternae decorating the red blood cell membrane in congenital dyserythropoietic anemia (type II) originate from the endoplasmic reticulum. Blood. 1996;87(10):4433–9 [PubMed] [Google Scholar]
- 9.Schwarz K, Iolascon A, Verissimo F, Trede NS, Horsley W, Chen W, et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet. 2009;41(8):936–40 [DOI] [PubMed] [Google Scholar]
- 10.Bianchi P, Fermo E, Vercellati C, Boschetti C, Barcellini W, Iurlo A, et al. Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum Mutat. 2009;30(9):1292–8 [DOI] [PubMed] [Google Scholar]
- 11.Iolascon A, Russo R, Esposito MR, Asci R, Piscopo C, Perrotta S, et al. Molecular analysis of 42 patients with congenital dyserythropoietic anemia type II: new mutations in the SEC23B gene and a search for a genotype-phenotype relationship. Haematologica. 2010;95(5):708–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russo R, Esposito MR, Asci R, Gambale A, Perrotta S, Ramenghi U, et al. Mutational spectrum in congenital dyserythropoietic anemia type II: identification of 19 novel variants in SEC23B gene. Am J Hematol. 2010;85(12):915–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Punzo F, Bertoli-Avella AM, Scianguetta S, Della Ragione F, Casale M, Ronzoni L, et al. Congenital dyserythropoietic anemia type II: molecular analysis and expression of the SEC23B gene. Orphanet J Rare Dis. 2011;6:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russo R, Esposito MR, Iolascon A. Inherited hematological disorders due to defects in coat protein (COP)II complex. Am J Hematol. 2013;88(2):135–40 [DOI] [PubMed] [Google Scholar]
- 15.Fermo E, Bianchi P, Notarangelo LD, Binda S, Vercellati C, Marcello AP, et al. CDAII presenting as hydrops foetalis: molecular characterization of two cases. Blood Cells Mol Dis. 2010;45(1):20–2 [DOI] [PubMed] [Google Scholar]
- 16.Liu G, Niu S, Dong A, Cai H, Anderson GJ, Han B, et al. A Chinese family carrying novel mutations in SEC23B and HFE2, the genes responsible for congenital dyserythropoietic anaemia II (CDA II) and primary iron overload, respectively. Br J Haematol. 2012; 158(1):143–5 [DOI] [PubMed] [Google Scholar]
- 17.Amir A, Dgany O, Krasnov T, Resnitzky P, Mor-Cohen R, Bennett M, et al. E109K is a SEC23B founder mutation among Israeli Moroccan Jewish patients with congenital dyserythropoietic anemia type II. Acta Haematol. 2011;125(4):202–7 [DOI] [PubMed] [Google Scholar]
- 18.Fromme JC, Orci L, Schekman R. Coordination of COPII vesicle trafficking by Sec23. Trends Cell Biol. 2008;18(7):330–6 [DOI] [PubMed] [Google Scholar]
- 19.Stagg SM, LaPointe P, Razvi A, Gurkan C, Potter CS, Carragher B, et al. Structural basis for cargo regulation of COPII coat assembly. Cell. 2008;134(3):474–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gurkan C, Stagg SM, Lapointe P, Balch WE. The COPII cage: unifying principles of vesicle coat assembly. Nat Rev Mol Cell Biol. 2006;7(10):727–38 [DOI] [PubMed] [Google Scholar]
- 21.Russell C, Stagg SM. New insights into the structural mechanisms of the COPII coat. Traffic. 2010;11(3):303–10 [DOI] [PubMed] [Google Scholar]
- 22.Hughes H, Stephens DJ. Assembly, organization, and function of the COPII coat. Histochem Cell Biol. 2008;129(2):129–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paccaud JP, Reith W, Carpentier JL, Ravazzola M, Amherdt M, Schekman R, et al. Cloning and functional characterization of mammalian homologues of the COPII component Sec23. Mol Biol Cell. 1996;7(10): 1535–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wendeler MW, Paccaud JP, Hauri HP. Role of Sec24 isoforms in selective export of membrane proteins from the endoplasmic reticulum. EMBO Rep. 2007;8(3):258–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C, Hur DJ, et al. Cranio-lenticulosutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet. 2006;38(10):1192–7 [DOI] [PubMed] [Google Scholar]
- 26.Fromme JC, Ravazzola M, Hamamoto S, Al-Balwi M, Eyaid W, Boyadjiev SA, et al. The genetic basis of a craniofacial disease provides insight into COPII coat assembly. Dev Cell. 2007;13(5):623–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, et al. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet. 2003;34(1):29–31 [DOI] [PubMed] [Google Scholar]
- 28.Tao J, Zhu M, Wang H, Afelik S, Vasievich MP, Chen XW, et al. SEC23B is required for the maintenance of murine professional secretory tissues. Proc Natl Acad Sci USA. 2012;109(29):E2001–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Akker E, Satchwell TJ, Pellegrin S, Flatt JF, Maigre M, Daniels G, et al. Investigating the key membrane protein changes during in vitro erythropoiesis of protein 4.2 (−) cells (mutations Chartres 1 and 2). Haematologica. 2010;95(8):1278–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruce LJ, Beckmann R, Ribeiro ML, Peters LL, Chasis JA, Delaunay J, et al. A band 3-based macrocomplex of integral and peripheral proteins in the RBC membrane. Blood. 2003;101(10):4180–8 [DOI] [PubMed] [Google Scholar]
- 31.Townley AK, Feng Y, Schmidt K, Carter DA, Porter R, Verkade P, et al. Efficient coupling of Sec23–Sec24 to Sec13–Sec31 drives COPII-dependent collagen secretion and is essential for normal craniofacial development. J Cell Sci. 2008;121(Pt 18):3025–34 [DOI] [PubMed] [Google Scholar]
- 32.Palmer KJ, Konkel JE, Stephens DJ. PCTAIRE protein kinases interact directly with the COPII complex and modulate secretory cargo transport. J Cell Sci. 2005;118(Pt 17):3839–47 [DOI] [PubMed] [Google Scholar]
- 33.van den Akker E, Satchwell TJ, Pellegrin S, Daniels G, Toye AM. The majority of the in vitro erythroid expansion potential resides in CD34(−) cells, outweighing the contribution of CD34(+) cells and significantly increasing the erythroblast yield from peripheral blood samples. Haematologica. 2010;95(9):1594–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Satchwell TJ, Bell AJ, Pellegrin S, Kupzig S, Ridgwell K, Daniels G, et al. Critical band 3 multiprotein complex interactions establish early during human erythropoiesis. Blood. 2011;118(1):182–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bell AJ, Satchwell TJ, Heesom KJ, Hawley BR, Kupzig S, Hazell M, et al. Protein distribution during human erythroblast enucleation in vitro. PLOS One. 2013;8(4):e60300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van den Akker E, Satchwell TJ, Williamson RC, Toye AM. Band 3 multiprotein complexes in the red cell membrane; of mice and men. Blood Cells Mol Dis. 2010;45(1):1–8 [DOI] [PubMed] [Google Scholar]
- 37.Schmidt U, van den Akker E, Parren-van Amelsvoort M, Litos G, de Bruijn M, Gutierrez L, et al. Btk is required for an efficient response to erythropoietin and for SCF-controlled protection against TRAIL in erythroid progenitors. J Exp Med. 2004;199 (6):785–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.von Lindern M, Deiner EM, Dolznig H, Parren-Van Amelsvoort M, Hayman MJ, Mullner EW, et al. Leukemic transformation of normal murine erythroid progenitors: v-and c-ErbB act through signaling pathways activated by the EpoR and c-Kit in stress erythropoiesis. Oncogene. 2001;20(28):3651–64 [DOI] [PubMed] [Google Scholar]
- 39.Denecke J, Kranz C, Nimtz M, Conradt HS, Brune T, Heimpel H, et al. Characterization of the N-glycosylation phenotype of erythrocyte membrane proteins in congenital dyserythropoietic anemia type II (CDA II/HEMPAS). Glycoconj J. 2008;25(4):375–82 [DOI] [PubMed] [Google Scholar]
- 40.Griffiths RE, Kupzig S, Cogan N, Mankelow TJ, Betin VM, Trakarnsanga K, et al. Maturing reticulocytes internalize plasma membrane in glycophorin A-containing vesicles that fuse with autophagosomes before exocytosis. Blood. 2012;119(26): 6296–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merryweather-Clarke AT, Atzberger A, Soneji S, Gray N, Clark K, Waugh C, et al. Global gene expression analysis of human erythroid progenitors. Blood. 2011;117(13):e96–108 [DOI] [PubMed] [Google Scholar]
- 42.Roux-Dalvai F, Gonzalez de Peredo A, Simo C, Guerrier L, Bouyssie D, Zanella A, et al. Extensive analysis of the cytoplasmic proteome of human erythrocytes using the peptide ligand library technology and advanced mass spectrometry. Mol Cell Proteomics. 2008;7(11):2254–69 [DOI] [PubMed] [Google Scholar]
- 43.Kingsley PD, Greenfest-Allen E, Frame JM, Bushnell TP, Malik J, McGrath KE, et al. Ontogeny of erythroid gene expression. Blood. 2013;121(6):e5–e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vermylen C, Scheiff JM, Rodhain J, Ninane J, Cornu G. A variant of the congenital dyserythropoietic anaemia type II with structural abnormalities in the granulocytic series. Eur J Pediatr. 1986;145(3):232–5 [DOI] [PubMed] [Google Scholar]
- 45.Heimpel H, Kellermann K, Neuschwander N, Hogel J, Schwarz K. The morphological diagnosis of congenital dyserythropoietic anemia: results of a quantitative analysis of peripheral blood and bone marrow cells. Haematologica. 2010;95(6):1034–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skop AR, Liu H, Yates J, 3rd, Meyer BJ, Heald R. Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science. 2004;305(5680):61–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fukuda MN. Congenital dyserythropoietic anaemia type II (HEMPAS) and its molecular basis. Baillieres Clin Haematol. 1993;6(2): 493–511 [DOI] [PubMed] [Google Scholar]
- 48.Chui D, Oh-Eda M, Liao YF, Panneerselvam K, Lal A, Marek KW, et al. Alpha-mannosidase-II deficiency results in dyserythropoiesis and unveils an alternate pathway in oligosaccharide biosynthesis. Cell. 1997;90(1):157–67 [DOI] [PubMed] [Google Scholar]
- 49.Ishihara N, Hamasaki M, Yokota S, Suzuki K, Kamada Y, Kihara A, et al. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell. 2001;12 (11):3690–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wickramasinghe SN, Wood WG. Advances in the understanding of the congenital dyserythropoietic anaemias. Br J Haematol. 2005;131(4):431–46 [DOI] [PubMed] [Google Scholar]
- 51.Wong KY, Hug G, Lampkin BC. Congenital dyserythropoietic anemia type II: ultrastructural and radioautographic studies of blood and bone marrow. Blood. 1972;39(1):23–30 [PubMed] [Google Scholar]