

Abstract
Homeostatic synaptic plasticity allows neural circuits to function stably despite fluctuations to their inputs. Previous work has shown that excitatory synaptic strength increases globally when neuronal inputs are chronically silenced. A recent paper by Kim and Tsien describes a new type of synapse-specific homeostatic plasticity in which input silencing causes simultaneous strengthening and weakening of different populations of excitatory synapses within a hippocampal network. These seemingly antagonistic homeostatic adaptations maintain synaptic gain and preserve overall network stability by limiting harmful reverberatory bursting, which may underlie epileptic seizures.
Synaptic transmission can undergo rapid, long-lasting changes as a result of neural activity. This plasticity provides neural networks with the flexibility to be extensively modified by experience to store neural information. However, the unchecked persistence of this type of plasticity can result in loss of network stability and thus impair normal network function. Fortunately, neural networks are able to maintain a stable output in the face of perturbations by using a multitude of additional plasticity mechanisms. One of these mechanisms is known as homeostatic plasticity. In this form of plasticity, neurons adapt their synaptic strength and intrinsic excitability in response to long-term changes in activity levels. Among the most well characterized homeostatic plasticity mechanisms is synaptic scaling, in which the strength of all synapses onto a postsynaptic neuron are adjusted or “scaled” up or down together in response to activity-dependent changes of network function. The interplay between rapid forms of synaptic plasticity and homeostatic plasticity allows a network to remain sufficiently flexible so that it can accommodate experience-dependent changes, while still retaining a level of network stability.
One classic method of inducing homeostatic plasticity is by altering neural activity through the use of pharmacological agents to block action potential firing.This manipulation typically results in homeostatic increases in synaptic function, such as synaptic scaling in neocortical neurons (Turrigiano et al. 1998). However, recent work carried out in the hippocampus has shown that synaptic changes induced by activity blockade can also be nonuniform, meaning that they extend beyond simple synaptic scaling (Echegoyen et al. 2007; Thiagarajan et al. 2005).
A recent paper published in Neuron by Kim and Tsien (2008) expands on these findings and shows that in the hippocampus homeostasis can be both cell type and synapse specific, allowing for the simultaneous strengthening and weakening of different classes of inputs to the same cell. As a result of these different changes, the hippocampus is able to enhance throughput of activity, yet prevent the runaway reverberatory activity that may result from synaptic scaling. Therefore individual neurons are not only regulating their own firing rate, but they are also regulating the activity of the entire hippocampal network.
To examine the underlying mechanisms of homeostatic plasticity, the authors induced inactivity within a neuronal network by treating organotypic hippocampal slices derived from 6- to 7-day-old rats with tetrodotoxin (TTX), a drug that blocks action potential firing leading to overall network silencing. To measure changes in synaptic function, the authors recorded miniature glutamatergic excitatory postsynaptic currents (mEPSCs). These represent the postsynaptic response to the spontaneous release of an individual neurotransmitter-containing vesicle. Following TTX treatment, the authors observed an increase in mEPSC amplitude at CA3-to-CA1 synapses and an increase in mEPSC frequency at mossy fiber (MF)-to-CA3 synapses, both consistent with previous reports (Echegoyen et al. 2007; Maffei et al. 2008; Turrigiano et al. 1998). However, when the authors looked at the effect of activity deprivation on another class of synapses received by CA3 neurons, they encountered an unexpected result (Fig. 1).
FIG. 1.
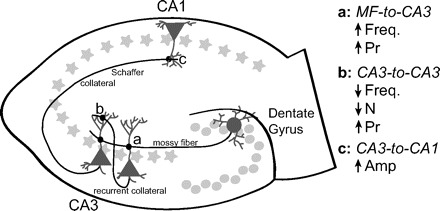
Homeostatic adaptations at tetrodotoxin (TTX)-treated hippocampal synapses maintain network stability. The hippocampal circuit is primarily viewed as a relatively simplistic feedforward excitatory pathway. Information from the entorhinal cortex travels along the perforant pathway to dentate gyrus granule (DG) cells, which send mossy fibers (MFs) to CA3 cells. From here, information flows from CA3 cells to CA1 cells via Schaffer collaterals. Following chronic TTX application to suppress spiking two types of hippocampal throughput synapses, MF-to-CA3 and CA3-to-CA1, show an increase in activity. Such increases in activity following activity deprivation are an expected homeostatic adaptation. At MF-to-CA3 synapses, the frequency (Freq) of miniature excitatory postsynaptic currents (mEPSCs) and the probability of presynaptic neurotransmitter release (Pr) both increase. mEPSC amplitude (Amp) is up-regulated at CA3-to-CA1 synapses. On the other hand, CA3-to-CA3 synapses show a decrease in activity, an unexpected homeostatic adaptation to inactivity. At these synapses, the increase in probability of presynaptic transmitter release is counterbalanced by a decrease in the number of active postsynaptic sites (N) responding to transmitter release. This synaptic silencing is primarily responsible for the decrease in mEPSC frequency that is also observed at these synapses. All of these adaptations could function in concert to make the network more stable by controlling reverberatory bursting, thereby guarding the network against harmful runaway excitation underlying epileptogenic activity.
CA3 pyramidal cells receive inputs from two sources. Granule cells arising from the dentate gyrus (DG) send their inputs to CA3 pyramidal cells by way of MFs. The other input to CA3 cells arises from fellow CA3 cells. To investigate the effect of activity deprivation on these inputs separately, MF transmission was blocked to isolate CA3 collaterals. This manipulation uncovered a decrease in mEPSC frequency in the CA3-to-CA3 recurrent collaterals, meaning that when the hippocampal network was deprived of activity, these recurrent synapses actually weakened.
Despite this observed decrease in mEPSC frequency at recurrent CA3 collaterals, the overall mEPSC frequency recorded from CA3 neurons did not decrease when both recurrent CA3 and MF inputs were active. The authors concluded that the frequency of mEPSCs from MF inputs is up-regulated, counterbalancing the decrease in mEPSC frequency observed at the CA3-to-CA3 collaterals, thus allowing the overall mEPSC frequency to remain stable under the activity-deprived conditions induced by TTX. To account for the strengthening of the MF-to-CA3 synapses, the authors proposed that the ability of the presynaptic MF terminals to release excitatory transmitter had been increased in response to inactivity. To test this hypothesis, the authors used a paired-pulse protocol in which two stimuli are given in rapid succession. If the probability of transmitter release has increased at a given synapse, the response of the postsynaptic neuron (measured as EPSC amplitude) to the second pulse should be smaller than that to the first. This effect is believed to be due to the depletion of all (or most) of the readily releasable transmitter pool at the activated presynaptic release sites. The authors observed this paired-pulse depression in the MF-to-CA3 synapses, consistent with the theory that the probability of transmitter release had increased at this synapse.
They also investigated presynaptic properties of recurrent CA3-to-CA3 synapses to determine the mechanism of the unconventional decrease in mEPSC frequency. Surprisingly, the authors observed paired-pulse depression at CA3 recurrent synapses after TTX treatment, indicating that the probability of release increased at this synapse as well. This increase in release probability is not readily reconcilable with the observed decrease in mEPSC frequency at CA3 recurrent collateral synapses. Therefore the authors postulated that although the overall presynaptic release probability increased, the number of postsynaptic active sites responding to this increase in release probability had decreased, down-regulating overall synaptic drive at these synapses and leading to fewer mEPSCs. In this case, there would be no difference between the frequency of mEPSCs in TTX-treated and control conditions, assuming that mEPSCs measured in TTX were derived exclusively from active postsynaptic terminals. To test this prediction, the authors recorded asynchronously evoked EPSCs (aEPSCs) by substituting calcium in the extracellular medium with strontium. This particular element is not as effective as calcium in inducing neurotransmitter release, resulting in asynchronous release of synaptic vesicles in response to presynaptic stimulation. Since aEPSCs recorded in strontium resemble mEPSCs recorded in the same cell, it is believed that aEPSCs arise from the same set of synapses responsible for generating mEPSCs (Bekkers and Clements 1999). Thus recording aEPSCs in strontium is akin to recording mEPSCs from active synapses only. When asynchronous EPSCs from TTX-treated slices were compared with controls, the difference in mEPSC frequency ceased to exist, consistent with the idea that this discrepancy was caused by a decrease in the number of active CA3 synapses.
Kim and Tsien (2008) showed that in CA3-to-CA3 recurrent collaterals, postsynaptic silencing overrides the presynaptic increase in release probability, leading to a significant decrease in mEPSC frequency at these synapses. To gain a better understanding of how the simultaneous decrease and increase of mEPSC frequency at different classes of synapses onto the same CA3 cells might function in the overall neuronal network, the authors induced epileptiform activity. Seizure-like activity was induced in CA3 neurons by pharmacologically blocking all inhibitory currents, resulting in spontaneous bursts of action potentials and prolonged depolarization of CA3 neurons.
When inhibitory inputs were blocked in TTX-treated organotypic hippocampal slices, overall burst frequency increased whereas the duration of individual bursts decreased. The first of these effects—an increase in burst frequency—is mediated by MF input. The authors established this by showing that the increase in burst frequency in TTX-treated slices was abolished in the presence of MF antagonists. The decrease in burst width persisted in inhibitory and MF input-deprived CA3 neurons in TTX. Under these conditions, CA3-to-CA3 recurrent connections are the only synapses active and are therefore the cause of the truncated bursts. However, hippocampal circuitry is not exactly cut and dry. Not all projections are feedforward; in fact, it has been shown that reciprocal excitatory synapses exist between CA1/CA3 pyramidal and dentate gyrus granule cells (Bausch and McNamara 2000). It is assumed that stimulating MF inputs mimics input received from dentate gyrus granule cells, derived in turn from the entorhinal cortex. It would be interesting to repeat this experiment in an expanded slice that includes the entorhinal cortex to see whether the increase in burst frequency mediated by MF inputs is similar when the entorhinal cortex is directly stimulated or whether this effect may in fact be mediated through back-projections by CA1 or CA3 cells.
The experiments clearly demonstrate that the properties of burst length and frequency in CA3 neurons are under homeostatic control. Due to previous work showing that increases in presynaptic activity caused by activity blockade are also accompanied by elevation of a specific Ca2+/calmodulin-dependent protein kinase (bCaMKII) (Thiagarajan et al. 2005), Kim and Tsien investigated the potential role of CaMK in the truncated bursts of CA3 neurons. The authors showed that inhibiting CaMK at MF inputs eliminates the increased burst frequency seen when these cells are treated with TTX. However, inhibition of CaMK did not eliminate the decrease in burst width (shown to be mediated by CA3 recurrent collaterals) observed in TTX-treated slices. This suggests that, in addition to being regulated by two separate synapse types, burst frequency and burst width are regulated by disparate molecular mechanisms as well.
Following chronic inactivity, the striking differences in induced firing of CA3 pyramidal and the DG cells that excite MF revealed that synapse-specific homeostatic adaptations had an overall effect on the hippocampal network. Although the intrinsic firing rate of CA3 pyramidal cells decreased, the firing rate of DG cells increased dramatically. These changes corresponded to shifts in the resting membrane potential of each cell type. DG cells rest at more depolarized potentials following TTX treatment, an effect that functions to scale up the input of the MF synapses to CA3 cells, contributing to an increased burst frequency. The resting membrane potential of CA3 pyramidal cells shifted in the opposite direction to become more hyperpolarized, creating slightly more unfavorable firing conditions. Therefore the increase in CA3 burst rate is due to a stronger presynaptic component: i.e., more active DG cells, which function to increase MF output onto CA3 neurons. This effect is counterbalanced by the hyperpolarization of the resting membrane potential of CA3 pyramidal cells, making it harder for these cells to sustain bursting activity. Shifts to the resting membrane potential of CA3 cells help to prevent the reverberatory bursting in these cells believed to underlie epileptogenic activity in the hippocampus.
Interestingly, this shift in resting membrane potential may be partly due to the developmental stage of the animal tested. Kim and Tsien made organotypic hippocampal slices from 6- to 7-day-old rats. These slices were cultured and treated with TTX for 3 to 4 days, starting as early as 18 days in vitro, and physiology was performed after 21 to 25 days in vitro. A separate study by Bausch and colleagues (2006) did not report a change in resting membrane potential of DG cells in organotypic hippocampal slices derived from 11-day-old animals that were treated with TTX for 4 days. However, the physiology experiments on these animals were performed at 40–60 days in vitro. Hippocampi from animals used by Kim and Tsien were younger and spent about half as much time in culture than those used by Bausch et al. (2006). Although it has been shown that synaptic transmission and dendritic morphology are maintained in organotypic slices, compared with acute slices (De Simoni et al. 2003), it is difficult to ascertain the contribution of age to the shift in resting membrane potential. Further experiments are needed to determine the exact role of developmental stage and time spent in culture to synapse-specific plasticity mechanisms.
It is also important to consider that the “overall neuronal network” referred to here (Fig. 1) is an organotypic hippocampal slice. Although in vivo systems are the acknowledged gold standard of any sort of circuitry experiment, they are not always experimentally tractable. Organotypic slices preserve the connections from dentate gyrus granule cells to CA3 pyramidal neurons, on to CA1 neurons. However, it has been shown that a massive increase in synaptic connectivity occurs during the time these slices are in culture, leading to significant increases in excitatory activity compared with acute slices (Bausch and McNamara 2000; De Simoni et al. 2003). Furthermore, the increases in synaptic connectivity and overall network excitability observed in organotypic hippocampal slices mimic morphological and physiological alterations seen in human limbic epilepsy and animal models (Bausch and McNamara 2000). This means that organotypic hippocampal slices provide a snapshot of a diseased state and that conclusions drawn about homeostatic adaptations observed in this preparation are likely limited to responses of an epileptic brain to inactivity. Further experimentation involving activity deprivation in vivo could confirm whether such synapse-specific adaptation can also be induced in a more nearly “normal” state.
It has been shown that disparate brain areas and cell types make use of divergent mechanisms to maintain homeostasis. In cultured visual cortical pyramidal neurons, mEPSC amplitudes scale up in response to activity deprivation (Turrigiano et al. 1998). This effect was also observed in the intact visual cortex (Desai et al. 2002). In hippocampal granule cells mEPSC amplitude, frequency, and kinetics were all increased in response to TTX treatment (Bausch et al. 2006). Kim and Tsien showed that the increase observed in mEPSC amplitude in CA1 pyramidal hippocampal neurons is complemented by a decrease in mEPSC frequency at recurrent CA3 synapses, thereby demonstrating that homeostatic processes must not necessarily be neuronwide. The authors demonstrated that the synaptic strengths of outputs and inputs from and to the same population of cells in the hippocampal network are regulated independently at the level of individual synapses. The idea that synaptic strengths can be regulated according to synapse type in the hippocampus adds to our fundamental understanding of homeostatic mechanisms. It will be interesting to see whether such adaptations exist outside of the hippocampus and whether they are inducible by other disease paradigms. Kim and Tsien's work has shown that disparate and seemingly contradictory homeostatic adaptations in the hippocampus are integrated at the single-neuron level to regulate overall network activity, controlling potentially harmful reverberatory bursting believed to underlie epileptic seizures. Their research adds one more piece to the puzzle of how networks maintain their stability in response to particular disease states.
Acknowledgments
I thank Dr. C. Aizenman and members of the Aizenman lab and Dr. S. Cruikshank, Dr. J. Sanes, Dr. A. Heyland, and D. Valenzuela for many careful readings and insightful discussions of this manuscript.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- Bausch 2006.Bausch SB, He S, Petrova Y, Wang XM, McNamara JO. Plasticity of both excitatory and inhibitory synapses is associated with seizures induced by removal of chronic blockade of activity in cultured hippocampus. J Neurophysiol 96: 2151–2167, 2006 [DOI] [PubMed] [Google Scholar]
- Baush 2000.Baush SB, McNamara JO. Synaptic connections from multiple subfields contribute to granule cell hyperexcitability in hippocampal slice cultures. J Neurophysiol 84: 2918–2932, 2000 [DOI] [PubMed] [Google Scholar]
- Bekkers 1999.Bekkers JM, Clements JD. Quantal amplitude and quantal variance of strontium-induced asynchronous EPSCs in rat dentate granule neurons. J Physiol 516: 227–248, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai 2002.Desai NS, Cudmore RH, Nelson SB, Turrigiano GG. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci 5: 783–789, 2002 [DOI] [PubMed] [Google Scholar]
- De Simoni 2003.De Simoni A, Griesinger CB, Edwards FA. Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J Physiol 550: 135–147, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echegoyen 2007.Echegoyen J, Neu A, Graber KD, Soltesz I. Homeostatic plasticity studied using in vivo hippocampal activity-blockade: synaptic scaling, intrinsic plasticity and age-dependence. PLoS ONE 2: e700, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim 2008.Kim J, Tsien RW. Synapse-specific adaptations to inactivity in hippocampal circuits achieve homeostatic gain control while dampening network reverberation. Neuron 58: 925–937, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei 2008.Maffei A, Turrigiano GG. Multiple modes of network homeostasis in visual cortical layers 2/3. J Neurosci 28: 4377–4384, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan 2005.Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47: 725–737, 2005 [DOI] [PubMed] [Google Scholar]
- Turrigiano 1998.Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391: 892–896, 1998 [DOI] [PubMed] [Google Scholar]