

Summary
The development of efficient recovery processes is essential to reduce the cost of polyhydroxyalkanoates (PHAs) production. In this work, a programmed self‐disruptive Pseudomonas putida BXHL strain, derived from the prototype medium‐chain‐length PHA producer bacterium P. putida KT2440, was constructed as a proof of concept for exploring the possibility to control and facilitate the release of PHA granules to the extracellular medium. The new autolytic cell disruption system is based on two simultaneous strategies: the coordinated action of two proteins from the pneumococcal bacteriophage EJ‐1, an endolysin (Ejl) and a holin (Ejh), and the mutation of the tolB gene, which exhibits alterations in outer membrane integrity that induce lysis hypersensitivity. The ejl and ejh coding genes were expressed under a XylS/Pm monocopy expression system inserted into the chromosome of the tolB mutant strain, in the presence of 3‐methylbenzoate as inducer molecule. Our results demonstrate that the intracellular presence of PHA granules confers resistance to cell envelope. Conditions to control the cell autolysis in P. putida BXHL in terms of optimal fermentation, PHA content and PHA recovery have been set up by exploring the sensitivity to detergents, chelating agents and wet biomass solubility in organic solvents such as ethyl acetate.
Introduction
Human overpopulation combined with the current lifestyle urges the rational, efficient and sustainable use of natural resources to produce environmentally friendly plastic materials such as polyhydroxyalkanoic acids (PHAs), whose production/degradation cycle reduces undesirable wastes and emissions (Gavrilescu and Chisti, 2005). PHAs are optically active biopolyoxoesters composed of (R) 3‐hydroxy fatty acids, which represent a complex class of storage polyesters. They are synthesized by some Archaea and a wide range of Gram‐positive and Gram‐negative bacteria in aerobic and anaerobic environments (Madison and Huisman, 1999). These biopolymers are accumulated as inclusions (PHA granules) in the bacterial cytoplasm in response to inorganic nutrient limitations, generally, when the microbes are cultured in the presence of an excess carbon source (Madison and Huisman, 1999). At present, PHAs are classified in two major classes: short‐chain‐length PHAs (scl‐PHAs) with C4‐C5 monomers and medium‐chain‐length PHAs (mcl‐PHAs) with C6‐C14 monomers. Mcl‐PHAs are mainly produced by Pseudomonas species (revised in Prieto et al., 2007). Because of structural differences, the physical properties of mcl‐PHAs are generally quite different from the archetypal polyhydroxybutyrate (PHB) and other scl‐PHAs (Gagnon et al., 1992).
Using currently available technology, large‐scale production of PHA is suitable with expenditures almost evenly divided between carbon source, fermentation process and separation process (30% each) (Sun et al., 2007; Elbahloul and Steinbüchel, 2009). Because PHAs accumulate intracellularly, the development of an efficient recovery process is indispensable to reduce the total cost of PHAs production (Prieto, 2007). At present, different separation processes have been described, like filtration, froth flotation (van Hee et al., 2006) and continuous centrifugation (Gorenflo et al., 2001). Recovery procedures for mcl‐PHA mainly resemble those developed for PHB (Ramsay et al., 1994). PHB recovery using hypochlorite, SDS in an in situ extraction process (Thakor et al., 2005), or an enzyme cocktail (de Koning et al., 1997; Kellerhals et al., 1999) have been reported. The disadvantages related to applications of hypochlorite and detergents are the severe reduction in polymer molecular weight and the requirement of extensive washing steps to get rid of detergent residuals respectively. Furthermore, the use of acetone, ethylacetate or hexane extraction of PHAs from dried biomass in combination with subsequent precipitation of the extracted polymer using non‐solvents of PHAs, such as methanol, has been described (Williams et al., 1999; Furrer et al., 2007; Elbahloul and Steinbüchel, 2009).
Most bacteriophages accomplish lysis with a tandem, late transcriptional, two gene products: a holin, a small membrane protein that oligomerizes in the membrane to form non‐specific lesions or ‘holes’ and a specific endolysin. At a ‘programmed’ time, the holes cause a permeabilization of the membrane that facilitates the action of the active endolysins, murein hydrolases that degrade the bacterial cell wall. As endolysins coding genes do not harbour secretory signal sequence they accumulate in a fully folded and active state in the cytoplasm during the vegetative cycle until they reach the peptidoglycan, hydrolyse it and lyse the cells (Wang et al., 2000). The utilization of phage lysis genes to disrupt recombinant cells that produce PHB has been reported in Escherichia coli (Fidler and Dennis, 1992; Resch et al., 1998; Yu et al., 2000) and in Bacillus megaterium (Hori et al., 2002), but such technologies have not been tested yet in bacteria producing mcl‐PHAs.
In this work, a novel self‐disruption extraction system was tested as a proof of concept in Pseudomonas putida KT2440, a paradigmatic strain in environmental microbiology (Nelson et al., 2002), which produces mcl‐PHAs (Huijberts et al., 1992; de Eugenio et al., 2010). This technology was based on a controlled autolysis that utilizes the endolysin Ejl and the holin Ejh from the pneumococcal bacteriophage EJ‐1 (Díaz et al., 1996). To improve the efficiency of the lytic system we have tested the system in different P. putida tol‐pal mutant strains, which exhibit alterations in outer membrane integrity that induce lysis hypersensitivity (Llamas et al., 2000). The combination of different lytic facilitators renders the cells more susceptible to become extractable by simplified procedures.
Results
Construction of a stable P. putida KT2440 strain for controlling the expression of the ejh‐ejl lytic cassette
The ejh‐ejl cassette isolated from the EJ‐1 phage genome was previously cloned in the shuttle plasmid pNM185 rendering the recombinant plasmid pEDF12 (Table 1), as a multicopy XylS/Pm expression system, where transcription of Pm promoter is induced by the xylS gene product after its activation by effector molecules such as 3‐methylbenzoate (3MB) (Díaz et al., 1996). The simultaneous expression of the lytic cassette producing the Ejh holin and the Ejl endolysin reduced the viability of E. coli and P. putida cells. Moreover, the presence of nucleic acids into the culture medium suggested that cell lysis occurred (Díaz et al., 1996). To investigate if this inducible lytic system could be applied into the PHA production process by facilitating the release of the PHA granules in a controlled downstream process, we first studied the effect of the expression of ejh‐ejl genes on the growth profile of the strain P. putida KT2440 (pEDF12), in optimal PHA production conditions. P. putida KT2440 (pEDF12) and P. putida KT2440 (pNM185) cells were cultured in 200 ml of M63 0.1N with 15 mM octanoic acid medium to promote the PHA production in one step fermentation system (see Experimental procedures). At different times, 40 ml aliquots were taken and the effect of the ejh‐ejl cassette expression after the addition of 3MB inducer was studied (Fig. 1). In all aliquots tested, a growth inhibition was detected as soon as the inducer was added, indicating that the lytic system had been induced. However, a decrease in the turbidity was also observed in the absence of the inducer, when culture reached an optical density at 600 nm (OD600) ∼3. These results indicated that the gene expression system is not tightly controlled and some basal expression of the ejh and ejl genes caused a partial non‐programmed cell lysis. Another disadvantage of this multicopy system is that P. putida KT2440 (pEDF12) strain did not reach the same OD600 level, related to the biomass, as the wild‐type strain (Fig. 1).
Table 1.
Bacterial strains, plasmids and oligonucleotides used in this study.
| Strain | Relevant genotype | Reference |
|---|---|---|
| E. coli | ||
| CC118λpir | Δ(ara‐leu) araD, ΔlacX74, galE, galK, phoA20, thi‐1, rpsE, rpoB, argE (Am), recA1 Rfr, Spr, λpir | Herrero et al. (1990) |
| P. putida | ||
| KT2440 | hsdR1 | Nakazawa (2002) |
| KTHL | P. putida KT2440 with ejh and ejl genes in the chromosome, Kmr | This study |
| AX | P. putida KT2440 tolA::xylE (TolA shortened to 94 amino acids) | Llamas et al. (2000) |
| BX | P. putida KT2440 tolB::xylE (TolB shortened to 29 amino acids) | Llamas et al. (2000) |
| QX | P. putida KT2440 tolQ::xylE (TolQ shortened to 29 amino acids) | Llamas et al. (2000) |
| RX | P. putida KT2440 tolR::xylE (TolR completely removed) | Llamas et al. (2000) |
| AXHL | P. putida KT2440 tolA::xylE with ejh and ejl genes in the chromosome, Kmr | This study |
| BXHL | P. putida KT2440 tolB::xylE with ejh and ejl genes in the chromosome, Kmr | This study |
| QXHL | P. putida KT2440 tolQ::xylE with ejh and ejl genes in the chromosome, Kmr | This study |
| RXHL | P. putida KT2440 tolR::xylE with ejh and ejl genes in the chromosome, Kmr | This study |
| Plasmids | Description | Reference |
| pNM185 | xylS/Pm,Apr, Kmr | Mermod et al. (1986) |
| pEDF12 | pNM185 derivative with ejh and ejl genes | Díaz et al. (1996) |
| pCNB1 | pUTmini‐Tn5, xylS/Pm, Apr, Kmr | de Lorenzo et al. (1993) |
| pUC18Not | pUC18 with NotI sites flanking the polylinker, Apr | Herrero et al. (1990) |
| pUCHL | pUC18Not with ejh and ejl genes | This study |
| pCNBHL | pCNB1 with ejh and ejl genes | This study |
| Primersa | Sequence 5′→3′ | Reference |
| V08 | CTAGTCTAGAGGCCAACAACATTACCATAAT AGAA | This study |
| V09 | CGCGGATCCGTCTTTCTATTTTGTCGTAATCA AGCCG | This study |
Engineered endonuclease sites on the oligonucleotides are shown underlined.
Figure 1.
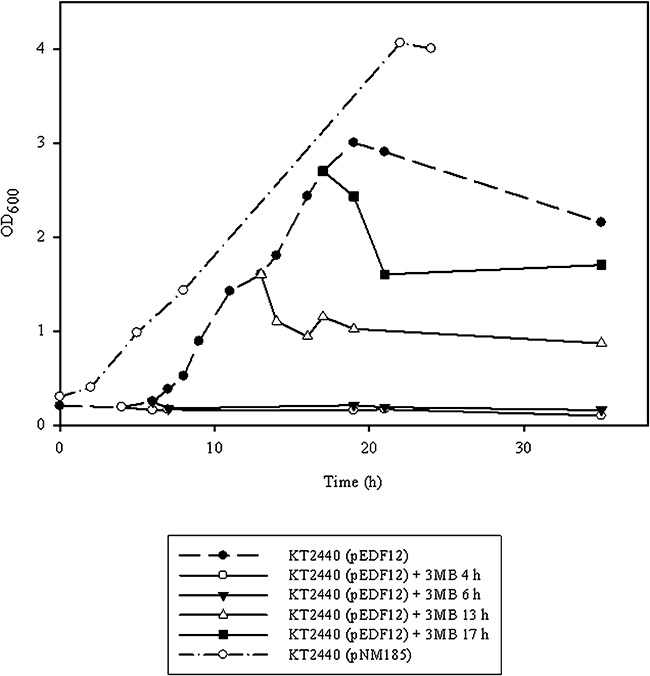
Profiles of cell lysis in P. putida strains caused by expression of the ejh‐ejl genes cloned in a multicopy plasmid under control of the 3MB inducible promoter. Strains were grown under one phase PHA production conditions and the bacterial growth was followed by measuring OD600 of cell cultures (see Experimental procedures). The effect of the ejh‐ejl cassette expression after the addition of the inducer (3MB) was studied in P. putida KT2440 (pEDF12) at different times. 3MB was added at the indicated times during the incubation (4, 6, 13 and 17 h). The growth curve of the control strain P. putida KT2440 (pNM185) without the lytic cassette is also shown.
The above results demonstrated the feasibility of the cell disruption strategy under PHA production conditions but also revealed the need for strain optimization to improve the process yield. To fully control the effect of the lytic system by reducing the XylS‐independent Pm activity, we constructed a monocopy expression system by transferring a xylS/Pm::ejh‐ejl cassette onto the chromosome of the wild type P. putida KT2440 strain, generating the strain P. putida KTHL (see Experimental procedures and Fig. 2 for details). We tested the ability of this strain for growing and producing PHA in one step fermentation system in minimal medium M63 0.1N containing 15 mM octanoic acid in the absence of the 3MB inducer. Table 2 compares the growth parameters and PHA production of P. putida KTHL versus P. putida KT2440 demonstrating that both strains were similar in terms of biomass production, PHA production and cell viability. However, the addition of 3MB to the medium during the fermentation reduced by one order of magnitude the viability of the KTHL cells but did not affect the survival of the control strain (Table 2). It is worth mentioning that when 3MB was added at different times along the fermentation process, the turbidity was not altered in P. putida KTHL. The lytic effect was exclusively detected when the inducer was added at the beginning of the culture (data not shown). In these growth conditions, biomass of induced P. putida KTHL cells was affected although the PHA production yield is similar to that of the wild‐type strain (Table 2).
Figure 2.
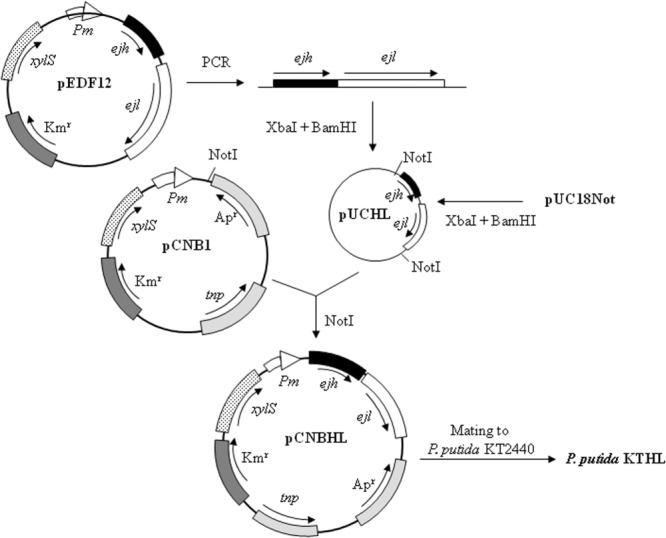
Schematic representation of the subcloning of ejh‐ejl cassette in the chromosome of P. putida KT2440 under transcriptional control of the positively regulated Pm promoter. Plasmids are drawn with the relevant elements and restriction sites indicated, as well as the direction of transcription of the genes. White arrows represent the Pm promoter of the meta‐pathway operon of the TOL plasmid. The ejh and ejl genes are shown as black and white blocks boxes respectively. xylS gene, encoding the cognate regulator of Pm, is indicated as a dotted block. Dark grey blocks indicate the antibiotic resistance (KmR indicates the gene conferring kanamycine resistance). The regions of the plasmids encoding the Tn5 transposase gene and the beta‐lactamase gene are also indicated.
Table 2.
Growth parameters and PHA production of P. putida KTHL versus P. putida KT2440 cultured in one phase fermentation system.
| P. putida strains | OD600a (24 h) | Total PHA content (g l−1) | Biomass (g l−1) | Viability (10−7 cells ml−1) |
|---|---|---|---|---|
| KT2440 | 6.7 ± 0.21 | 0.9 ± 0.07 | 1.45 ± 0.01 | 2.31 ± 1.04 |
| KT2440 + 3MB | 6.5 ± 0.49 | 0.86 ± 0.03 | 1.38 ± 0.05 | 2.09 ± 0.28 |
| KTHL | 6.43 ± 0.02 | 1 ± 0.05 | 1.51 ± 0.06 | 2.65 ± 0.59 |
| KTHL + 3MB | 4.13 ± 0.05 | 0.89 ± 0.03 | 0.95 ± 0.04 | 0.21 ± 0.06 |
Cells were grown in nitrogen limited mineral medium plus 15 mM octanoic acid (one phase) with and without inducer 3MB (5 mM) for 24 h (see Experimental procedures).
Two steps culture fermentation system
As an alternative culture approach, we established a two steps culture fermentation system where cells are initially cultured in a rich medium, and afterwards transferred to the PHA production medium (see Experimental procedures). Table 3 shows the growth parameters concerning P. putida KT2440 and KTHL strains cultured in a two steps system. When 3MB was added to the culture medium at the beginning of the second step, the viability of the KTHL cells was significantly reduced (Fig. 3) but biomass and PHA yield reached similar levels to those obtained with the wild‐type strain or with non‐induced KTHL strain (Table 3). Therefore, the two steps fermentation strategy was selected as the optimal method for producing PHA in P. putida KTHL strain.
Table 3.
Growth parameters and PHA production of P. putida KTHL and BXHL versus P. putida KT2440, cultured under two phases fermentation system.
| P. putida strains | OD600 (24 h) | Total PHA content (g l−1) | Biomass (g l−1) | Viability (10−9 cells ml−1) | Recovery of PHA extracted from wet biomass with ethyl acetate (g l−1) |
|---|---|---|---|---|---|
| KT2440 | 7.52 ± 0.13 | 0.84 ± 0.05 | 2.38 ± 0.04 | 2.33 ± 0.04 | 0 |
| KT2440 + 3MB | 7.66 ± 0.37 | 0.78 ± 0.04 | 2.26 ± 0.02 | 3.16 ± 1.19 | 0 |
| KTHL | 7.71 ± 0.03 | 0.88 ± 0.03 | 2.45 ± 0.04 | 3.33 ± 0.82 | 0 |
| KTHL + 3MB | 6.53 ± 0.18 | 0.79 ± 0.15 | 2.28 ± 0.14 | 0.51 ± 0.02 | 0.13 ± 0.01 |
| AX | Nd | 1.02 | 2.49 | Nd | Nd |
| BX | Nd | 0.82 | 2.56 | Nd | Nd |
| QX | Nd | 1.03 | 2.55 | Nd | Nd |
| RX | Nd | 0.87 | 2.45 | Nd | Nd |
| AXHL | 4.63 ± 0.62 | 0.43 | 1.38 | Nd | Nd |
| BXHL | 7.83 ± 0.36 | 0.86 ± 0.09 | 2.00 ± 0.01 | 1.05 ± 0.03 | 0.1 ± 0.07 |
| BXHL + 3MB | 7.1 ± 0.67 | 0.68 ± 0.01 | 1.52 ± 0.1 | 0.1 ± 0.4 | 0.28 ± 0.08 |
| QXHL | 0.78 ± 0.02 | 0 | 0.57 ± 0.13 | Nd | Nd |
| RXHL | 2.7 ± 0.11 | 0.07 ± 0.003 | 1.30 ± 0.09 | Nd | Nd |
3MB inducer was added at 5 mM to the PHA production medium (see Experimental procedures).
Nd, not determined.
Figure 3.
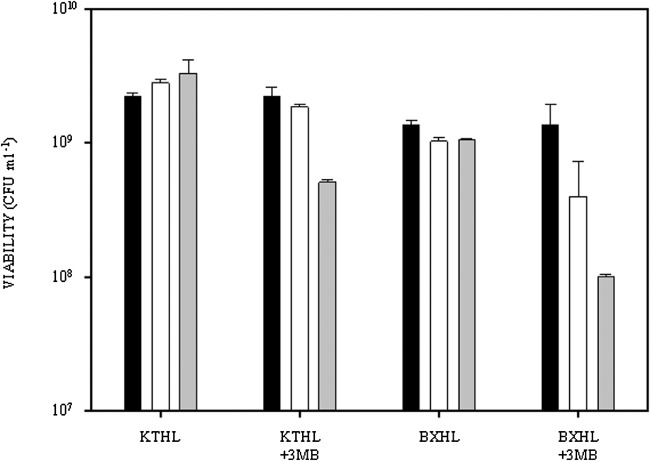
Number of viable cells of P. putida KTHL and P. putida BXHL grown under two steps PHA production conditions. Strains were grown with and without 3MB (5 mM), added at the beginning of the second step (see Experimental procedures). Black bars, values at 0 time; white bars, values at 6 h; gray bars, values at 24 h.
After fermentation, cultures were subjected to sucrose density step‐gradient ultracentrifugation to check the presence of PHA granules in the culture supernatant because of the cell disruption induced in the P. putida KTHL strain (Fig. 4A). This method allows the separation of the PHA granules from the cell fraction (non‐disrupted cells and cell debris) as PHA granules have a specific gravity lower than cell fraction, which sediments at the bottom of the tube (Moldes et al., 2004). In the absence of inducer, PHA granules were not released into the medium and sedimented together with the cell fraction (Fig. 4A, tube 2). However, in the presence of the 3MB inducer, a PHA white band was visible at the sucrose step‐gradient interface, indicating the release of the granules to the extracellular medium (Fig. 4A, tube 3). In the case of the wild‐type strain, extracellular PHA granules were not detected even in the presence of 3MB (Fig. 4A, tube 1). Thus, it was clearly demonstrated that PHA granules were effectively released by the self‐disruption system integrated into the recombinant P. putida KTHL strain, proving the potentiality of this system.
Figure 4.
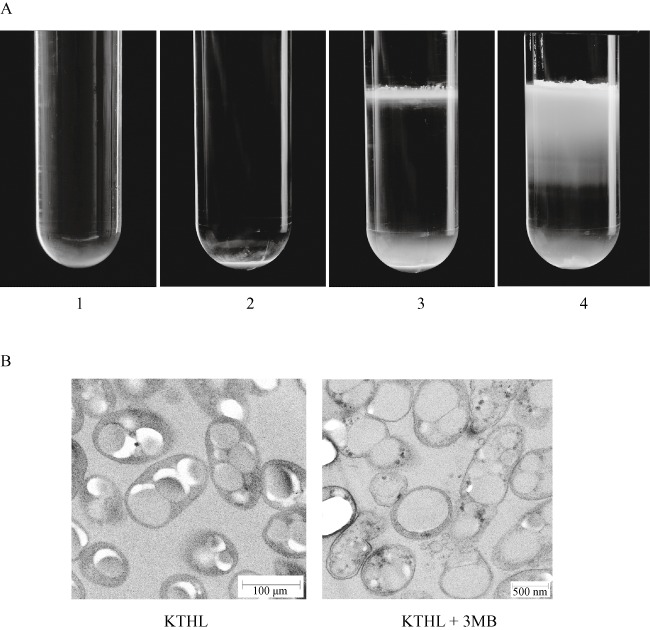
Sucrose density step‐gradient ultracentrifugation of P. putida strains. Cells were grown under two steps PHA production conditions, with or without 3MB (5 mM) as inducer (see Experimental procedures). A. Tube 1, P. putida KT2440 wild type grown with 3MB; tube 2, P. putida KTHL grown without 3MB; tube 3, P. putida KTHL grown with 3MB; tube 4, P. putida KTHL grown with 3MB broken by a fourfold passage through a French press. The white band located at the interfaces of tubes 3 and 4 corresponds to the free PHA granules released to the extracellular medium. B. Transmition electronic microscopy views of mcl‐PHA accumulating P. putida KTHL cells. Samples were taken after 24 h growing under the two phases fermentation system in the presence (right) or absence (left) of 3MB inducer.
To study the efficiency of the lytic system in P. putida KTHL, the amount of self‐released PHA was compared with the total PHA obtained after breaking the cells through the French press (Fig. 4A, tube 4). The PHA content detected extracellularly by the French press standard procedure was 84.9% of CDW, while only 7.6% of CDW was detected in the case of KTHL strain grown in the presence of 3MB. The wide white band clearly visible at the sucrose step‐gradient interface after the French press cell disruption procedure demonstrated that the lytic system was indeed active in P. putida KTHL strain, but the efficiency was still low. The incomplete autolytic effect of the xylS/Pm::ejh‐ejl cassette induction, when 3MB was added to the medium, was monitored by transmition electronic microscopy (Fig. 4B), demonstrating that, although cell envelopes were considerably disturbed, most granules maintained their intracellular location.
Sensitivity of P. putida KTHL strain to detergents and chelating agents
Cell viability and biomass production data derived from the results described above were in agreement with those obtained in P. putida KTHL cells growing in rich medium (Luria–Bertani, LB) in the presence of 3MB, which showed, under the phase contrast microscope, a majority of ‘ghost cells’ (Fig. 5). Morphology of these ‘ghosts’ is a typical consequence of holin action, where the cells are still keeping their shape but lack their cytoplasmic content and, consequently, there is a significant drop in viability when 3MB is added to the medium. This was demonstrated by the viability analyses as function of membrane integrity by staining with the LIVE/DEAD BacLight bacterial viability kit (Fig. 6). In this assay, cells with a compromised membrane, which are considered to be dead or dying, stained red (Fig. 6B and D), whereas cells with an intact membrane stained green (Fig. 6A and C). Figure 6 showed a drastic effect on the green/red cell ratio after the addition of the lytic inducer. The cell viability was also affected in the presence of 3MB when cells were grown under PHA production conditions (Tables 2 and 3). These findings confirmed that when the ejh‐ejl cassette was expressed, it caused the death of most of the cells but they were not totally disrupted, probably as a result of an intact outer membrane or a reduced efficiency of the phage endolysin. Thus, to improve the yield of the extraction procedure we tested the sensitivity of the strains to detergents or chelating agents in rich medium. To this aim, P. putida KTHL and wild‐type cells were incubated for 18 h at 30°C in LB medium and in LB medium supplemented with 0.075% (w/v) deoxycholate (DOC) (Fig. 7), 0.01% SDS or 0.2 mM EDTA (data not shown). At these concentrations, the 3MB induced P. putida KTHL cells were sensitive to the chemical agents, while no effect was observed on the growth of the wild‐type strain or the non‐induced recombinant strain. In addition, we have determined that DOC, SDS and EDTA did not alter the growth of the 3MB induced P. putida KTHL cells when they were added to LB medium at final concentrations lower than 0.01%, 0.005% and 0.1 mM respectively. Besides, we also determined that the addition of concentrations higher than 0.2% DOC and 0.4 mM EDTA changed the growth of both, the wild type and the non‐induced KTHL strains. All the above results clearly indicated that when the cells grow in a rich medium the cell envelope of the P. putida KTHL mutant strain is significantly altered as a result of the expression of the Ejh and Ejl proteins.
Figure 5.
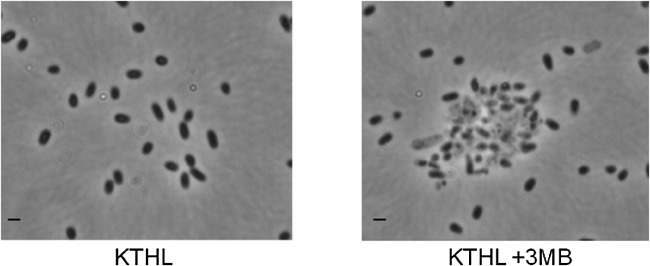
Phase contrast microscopy of P. putida KTHL ghost cells as consequence of the expression of the lytic cassette. Cells were grown in rich medium (LB) with (right) and without (left) 3MB for 24 h. Black bars correspond to 2 µm.
Figure 6.
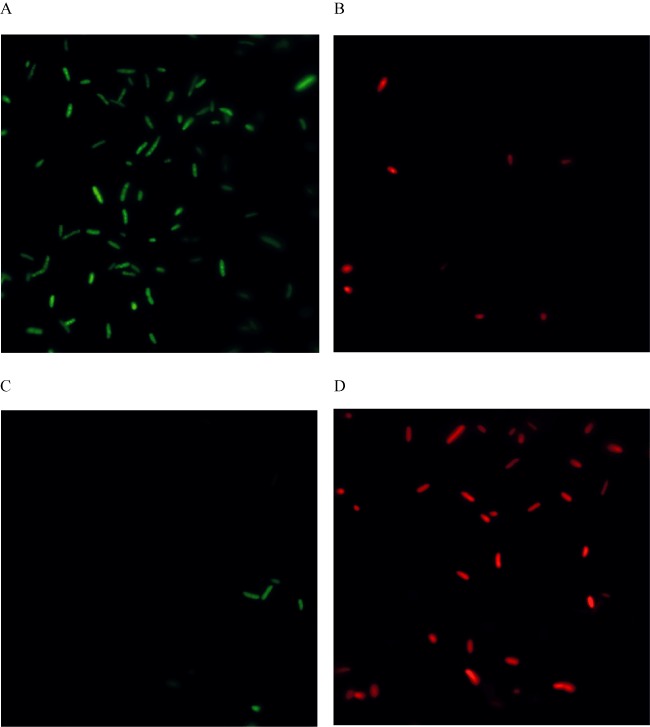
Monitoring the viability of P. putida KTHL population as function of the membrane integrity. LIVE/DEAD bacterial viability assay of KTHL cells grown in LB in the presence (panels C and D) and absence (panels A and B) of 3MB.
Figure 7.
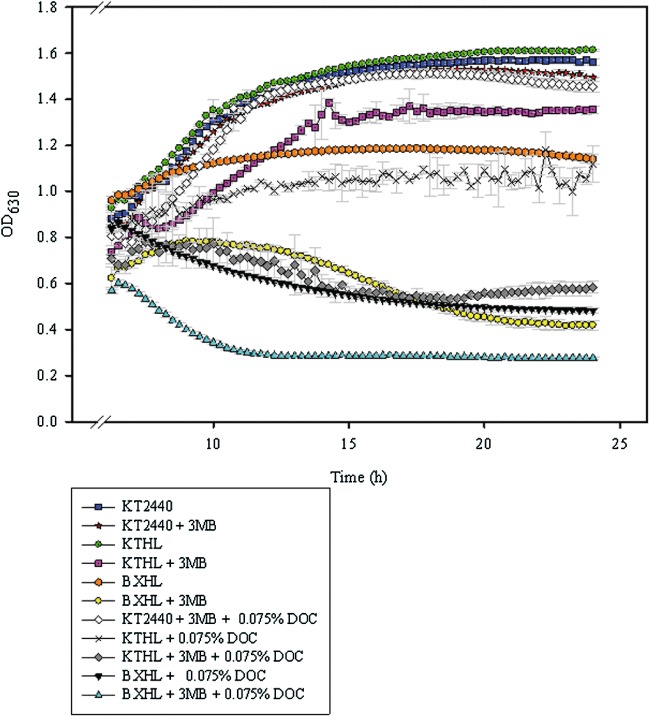
Growth curves in LB medium of P. putida strains in the presence of DOC. P. putida KT2440, KTHL and BXHL strains were grown in LB medium with and without 3MB (see Experimental procedures). After 6 h of incubation at 30°C, aliquots of each culture were supplemented with 0.075% (w/v) DOC. The ability for growing in the presence of the detergent was determined by measuring the OD630 of the cultures during 18 h in 96‐microwell plates (see Experimental procedures for details). As control, aliquots of the cultures with no added agent were also analysed. Error bars represent standard deviation found in three different experiments.
Conversely, when cells were cultured under two steps PHA production conditions with 3MB and in the presence of these chemical agents for 18 h, the release of the granules into the extracellular medium did not increase in P. putida KTHL strain compared with similar experiments performed in the absence of chemical agents (7.6% of CDW). Even at high concentrations of EDTA (10 mM) and SDS (0.1%) simultaneously added to the medium, the detected PHA at the extracellular medium was 9.2% of CDW. This result suggested that the intracellular presence of PHA granules renders the cells more resistant to cell disruption.
Expression of the lytic cassette in P. putida tol‐pal mutant strains
Our previous results indicated that cells expressing the ejh‐ejl cassette were more sensitive to outer membrane disturbing agents, such as chelating agents and detergents, when growing in rich medium than under optimal PHA production conditions in nitrogen limited minimal medium. Therefore, to improve the cell disruption for recovering PHA granules we tested a different strategy to alter the stability of the cell envelope. With this aim, we used tol‐pal mutants of P. putida KT2440 as recipients of the lytic system instead of wild‐type strain, because these mutants exhibit severe alterations in outer membrane integrity (Table 1). A comprehensive scheme of the Tol/Pal membrane proteins is depicted in Fig. 8.
Figure 8.
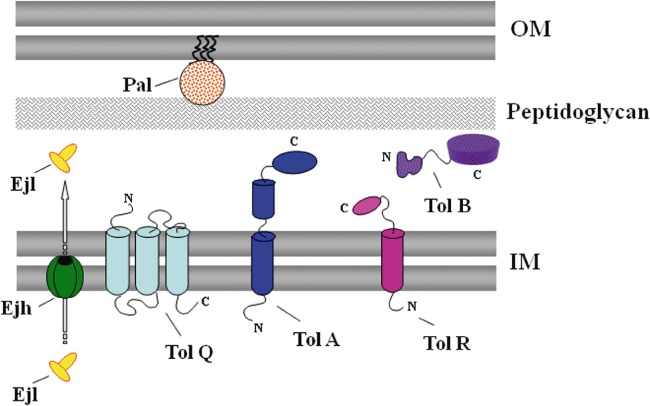
Schematic localization of the Ejh holin, the Ejl endolysin and the Tol–Pal proteins in the cell envelope of P. putida strains. TolQ, TolR and TolA are cytoplasmic membrane proteins that form a complex in the cytoplasmic (inner) membrane (IM). TolQ is an integral inner membrane protein containing three transmembrane domains with two cytoplasmic regions. TolR and TolA are anchored to the cytoplasmic membrane leaving most of the protein exposed to the periplasm. Pal is an outer membrane (OM) peptidoglycan‐associated lipoprotein and forms a complex with TolB, which is a periplasmic protein. Ejh holin is a small membrane protein that oligomerizes in the IM to form ‘holes’ that allow the translocation of the Ejl endolysin to the peptidoglycan, where it degrades the cell wall and lyses the cell.
First, we tested the growth and PHA production capacities in the two steps culture fermentation system of the four tol‐pal mutant strains AX, BX, QX and RX, mutated in the tolA, tolB, tolQ and tolR genes respectively. Gas chromatography‐mass spectrometry (GC‐MS) analyses confirmed that the four mutants were able to grow and produce PHA under these fermentation conditions and that the quantity of PHA accumulated by the four strains was similar to that of the wild‐type strain (Table 3). Afterwards, the lytic expression system was transferred, through a transposition‐mediated mechanism, to the chromosome of mutant strains rendering the recombinant AXHL, BXHL, QXHL and RXHL strains (Table 1). When we tested the ability of these four new strains for growing and producing PHA in the two steps culture fermentation system, in the presence and absence of 3MB, we observed that P. putida BXHL was the only strain that reached similar OD600 values than those of the wild type, while the other mutants grew very poorly under these conditions (Table 3). In addition, the intracellular PHA contents of AXHL, RXHL and QXHL strains were lower than 0.4 g l−1, while the PHA content of BXHL was 0.86 g l−1. Interestingly, cell viability of BXHL was reduced by one order of magnitude when the cells were cultured in the presence of the 3MB lytic inducer (Table 3, Fig. 3). Taking into account the above results, P. putida BXHL was selected as the best candidate strain to apply the autolytic disruption strategy.
Pseudomonas putida BXHL strain was monitored by transmition electronic microscopy (Fig. 9) showing that, similarly to KTHL strain, the addition of 3MB generated deep alterations in the cell envelopes but most of the granules remained inside the cells. Remarkably, when we tested the susceptibility of P. putida BXHL strain grown in rich medium to the effect of membrane disrupting agents such as DOC, SDS and EDTA, we observed that this strain was even more sensitive than P. putida KTHL mutant, suggesting that tolB mutation contributes to alter the stability of cell envelope. As an example, Fig. 7 shows the growth curves of P. putida strains in LB medium with and without inducer and in the presence or absence of 0.075% DOC. It can be observed that induced P. putida BXHL is up to three times more sensitive to the detergent than induced P. putida KTHL. In addition, we compared the PHA granule release in P. putida wild type and BXHL mutant strains after simultaneous incubation with 10 mM EDTA and 0.1% SDS. In this experiment, both strains were cultured using the two steps culture fermentation system with 3MB. After 24 h of incubation in the PHA production conditions media (this is, the end of second step), the cultures were centrifuged and resulting sediments were suspended in distilled water supplemented with 10 mM EDTA and 0.1% SDS. After 8 h of incubation at room temperature, cultures were subjected to sucrose density step‐gradient ultracentrifugation for analysing the presence of PHA granules in the culture supernatant (Fig. 10). It is worth to mention that none viable cell was detected at this condition. P. putida BXHL induced to self‐disruption and incubated with the chemical agents (Fig. 10A, tube 2) showed a much thicker white band when compared with that of the wild‐type strain (Fig. 10A, tube 1). These findings indicated the release of almost all granules to the extracellular medium. To confirm these results, the intracellular PHA content of the cell fraction present in the sediments after the ultracentrifugation step of the 3MB induced P. putida BXHL strain incubated with and without the chemical agents was quantified by GC‐MS (see Experimental procedures). When no chemical agents were added, the PHA content present in the culture sediment was 0.7 g l−1, similar to that found in the total culture (0.86 g l−1). However, PHA was not detected at all in the sediment of cells incubated with 10 mM EDTA and 0.1% SDS, suggesting that this sediment is only composed of cell debris and that most of PHA (93% of CDW) was released to the extracellular medium, as shown in Fig. 10B.
Figure 9.
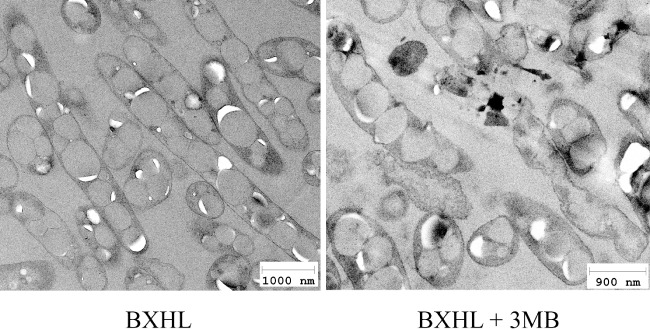
Transmition electronic microscopy views of mcl‐PHA accumulating P. putida BXHL cells. Cell samples were taken after 24 h growing under the two phases fermentation system in the presence or absence of 3MB inducer. P. putida BXHL grown in the presence of 3MB showed disturbed membranes.
Figure 10.
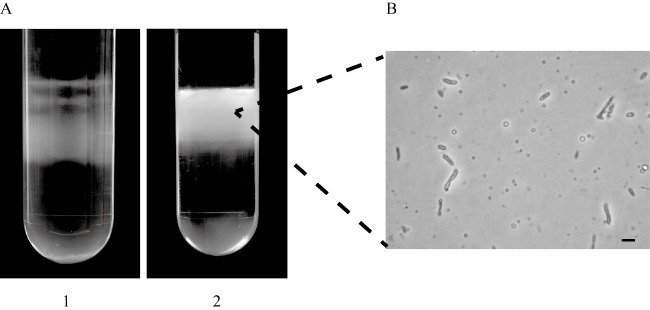
PHA granule release analysis by sucrose density step‐gradient ultracentrifugation of P. putida BXHL strain. Cells were grown under two steps PHA production conditions in the presence of the inducer 3MB for 24 h (see Experimental procedures for details). A. Tube 1, cells of P. putida KT2440 wild type; tube 2, cells of P. putida BXHL. The white band in the interface of tube 2 corresponds to the free PHA granules released to the extracellular medium. B. Phase contrast microscopy of the white band obtained in tube 2, where free PHA granules secreted to the extracellular medium and some cells can be observed. Black bar corresponds to 2 µm.
Direct cell extraction of PHA by organic solvents
In addition to the advantage that the lytic systems could simplify the self‐release of PHA from the cells to the medium, the alterations of the cell envelope might also be useful to facilitate the direct cell extraction of PHA by organic solvents. To analyse this assumption, we compared the PHA recovery in the wild‐type, KTHL and BXHL strains, cultured in the presence or absence of the lytic inducer. PHA was extracted from the cells with organic solvents just after centrifugation or filtration (wet biomass) without additional treatments such as lyophilization (dried biomass) or physical cell disrupting methods plus separation, which are time‐ and energy‐consuming processes. Direct solvent extraction of PHA from wet biomass could be beneficial, but has been demonstrated inoperative for wild‐type cells (Table 3).
Cultures of 24 h of P. putida strains grown in the two steps culture fermentation system with and without 3MB were directly suspended after centrifugation in several organic solvents such as methylene chloride, ethyl acetate or acetone (see Experimental procedures for details). Then, dissolved PHA was precipitated by adding methanol. Remarkably, only ethyl acetate gave positive results when wet biomass was used for direct PHA extraction (Table 3). As expected, wild‐type strain grown with and without 3MB inducer did not render polymer after precipitation whereas 0.13 g l−1 of PHA was obtained from the 3MB induced KTHL cells. BXHL cells induced with 3MB yielded higher recovery rates (0.28 g l−1 of PHA) with a purity of 97.65% ± 2.16, indicating that this strain has been improved to facilitate the direct solvent extraction of PHA from wet biomass. Although the PHA direct recovery in organic solvents is still far from being useful at industrial scale, these experiments demonstrated the possibility of improving the wet biomass direct solvent extraction of PHA by modifying the characteristics of the bacterial cell envelope and the properties of the solvents.
Discussion
Polyhydroxyalkanoate is the paradigmatic example of a bacterial biopolymer that accumulates only in the cytoplasm (de Smet et al., 1983; Madison and Huisman, 1999). Depending on the organism, PHA production can reach levels as high as 90% of the cell dry weight (Olivera et al., 2001a). The cytoplasm space limits the amount of polymer that can be produced by a microbial cell, and the yield per volume is limited by the number of cells and the biopolymer fraction in the biomass. This limitation increases the complexity of the production and downstream processes for preparing purified PHA, implying the use of cell disruption procedures as well as processes for extracting PHA from crude lysates (Zinn et al., 2001). Metabolic engineering approaches have succeeded in improving microorganisms for PHA production and in fact, a considerable variety of strategies have been designed to increase the yield of mcl‐PHA production in homologous and heterologous microorganisms (Qi et al., 1998; Prieto et al., 1999; Ren et al., 2000; Olivera et al., 2001b). However, only one example has been reported to date for engineering Alcanivorax borkumensis in order to overproduce mcl‐PHA and facilitate its extraction as the polymer was deposited extracellularly by an unknown mechanism (Sabirova et al., 2006). Several attempts have been carried out to mimic such process by using phage lysis genes to disrupt recombinant cells that produce PHB (Fidler and Dennis, 1992; Resch et al., 1998; Yu et al., 2000; Hori et al., 2002), but a similar approach in mcl‐PHA producing strains remained to be explored.
In this study, we report the construction of a programmed self‐disruptive P. putida BXHL strain that facilitates the release of the mcl‐PHAs granules to the extracellular medium. The engineered cell disruption system is based on two proteins from the pneumococcal bacteriophage EJ‐1, Ejh holin and Ejl endolysin (amidase), which have been transferred to the chromosome of a tolB mutant of P. putida KT2440 to increase the strain stability by reducing the gene copy number. The coupled expression of phage genes involved in host lysis is a transcriptional late event, in the phage physiological cycle, which has been shown to have only a reduced specificity for the bacterial host, i.e. the phage lytic systems are effective when transferred to other bacteria different than their specific phage hosts (Young, 2005). In this sense, phage holin proteins are able to oligomerize in different heterologous bacteria to form holes in the inner membrane (Wang et al., 2000). Moreover, some phage murein hydrolases can degrade the peptidoglycan of non‐host bacteria, although its optimal enzymatic activity might depend on specific structural requirements (López and García, 2004). In our case, combined results of growth curves, viability data and microscopic observations strongly suggest that Ejh holin is fully functional in P. putida, although the Ejl amidase appears to be less efficient than in pneumococcus because of its specific requirement for choline‐containing cell walls (López and García, 2004). Remarkably, the low activity of Ejl is certainly an advantage for our engineered system as it allows us to diminish the toxicity of the lytic system and facilitates its control. In fact, in spite of the low hydrolytic activity we have observed a high toxicity of the lytic system when it was expressed in multicopy plasmids. Therefore, it seems extremely important to tightly control the lytic systems and partially reduce their activities when used for biotechnological purposes. In this context, it would appear surprising that not all the cell population of the same culture could be lysed at the same time and with the same efficiency, but because of need of balancing the efficiency and toxicity of the lytic system it is assumable that not all the cells reach the instability threshold required to become lysed.
It is worth to mention the unexpected lytic resistance phenotype of P. putida KTHL observed even in the presence of chemical agents when the cells were cultured in minimal medium under PHA production conditions, suggesting that accumulation of PHA granules renders the cells more resistant to cell lysis. Although the mechanism supporting this resistance is not yet well understood, we can speculate on several possible reasons: one could be a reduced production of the lytic proteins in the nitrogen limited minimal medium; another could be related to the presence of large amounts of lipid monolayers in the PHA granules that could trap holin monomers and thus, disturb the oligomerization of the holin in the membrane lipid bilayer. We cannot discard also that the own synthesis of PHA granules could interfere with hole formation and endolysin secretion or that the presence of granules could result in a more rigid cell structure. Whether the structure of the cell envelope of PHA producing cells is reinforced to support the pressure generated by the presence of the PHA granules in the cytoplasm rendering these cells more resistant to membrane perturbations could merit to be investigated.
The advantage of the self‐disruptive P. putida BXHL strain to fulfil the proposed task is based on the introduction of a mutation in the TolB protein, which forms part of the Tol–Pal protein system (Llamas et al., 2000; Godlewska et al., 2009) (Fig. 8). In P. putida, like in the majority of Gram‐negative bacteria, the Tol–Pal complex forms a membrane‐spanning multiprotein system composed of five core proteins: TolQ, TolR, TolA, TolB and Pal (Llamas et al., 2003) (Fig. 8). As documented in E. coli, the Tol‐Pal system is organized into two protein complexes: a cytoplasmic membrane complex composed of the TolQ, TolR and TolA proteins, which interact with each other via their transmembrane domains (Derouiche et al., 1995; Koebnik, 1995; Germon et al., 1998; 2001; Journet et al., 1999) and an outer membrane complex made up of TolB and Pal, which also interact with Lpp, OmpA, and the peptidoglycan layer (Bouveret et al., 1995; Koebnik, 1995; Clavel et al., 1998) (Fig. 8). The Tol–Pal complexes appear to confer stability to the cell envelope because the Tol mutants present some envelope defects that render the cells more sensitive to lysis (Llamas et al., 2000). Therefore, the combination of Tol mutations with the engineered lytic system provided a novel approach to investigate the possibility to induce a controlled cell lysis under PHA producing conditions or, at least, to produce PHA containing cells that were more susceptible to lytic treatments at the end of the fermentation process in order to facilitate PHA extraction. In a few words, we have demonstrated that PHA extraction by detergents or organic solvents can be significantly facilitated in the BXHL strain (Fig. 10 and Table 3). In fact, we have demonstrated that a significant fraction of highly pure PHA generated in lytic induced cells can be extracted, without additional treatments, with ethyl acetate directly from wet biomass. This work opens new prospects for using lytic facilitators not necessarily to release PHA to the medium but for altering the stability of cell envelope to simplify the extraction of pure polymers without using other expensive and low environmentally friendly procedures. The different behaviour of the tol‐pal mutants revealed the importance of finding the critical equilibrium between cell viability/stability, PHA production and facilitation of PHA extraction. Although other solvents should be tested, ethyl acetate is a friendly chemical as a result ot its low cost, low toxicity and nice odour, avoiding the halogenated solvents that are frequently used for PHA extraction (Thakor et al., 2005). The combination of different lytic facilitator tools appears to be very useful to fine tune the cell suitability for optimizing the PHA extraction process providing a wide range of different conditions that probably cannot be reached using a single lytic system.
Although additional experiments should be performed to optimize the use of self‐disruptive systems for scaling up conditions, this proof of concept settles the bases to demonstrate that engineering cells for facilitating PHA extraction is feasible, showing new perspectives to satisfy the needs for an environmentally friendly and economical production process of bioplastics.
Experimental procedures
Bacterial strains, plasmids and growth conditions
The bacterial strains, plasmids and oligonucleotides used are listed in Table 1. Unless otherwise stated, E. coli and P. putida strains were grown in LB medium (Sambrook and Russell, 2001) with shaking at 37°C and 30°C respectively. The appropriate selection antibiotics, kanamycin (50 µg ml−1) or ampicillin (100 µg ml−1) were added when needed. Growth was monitored with a Shimadzu UV‐260 spectrophotometer at 600 nm (OD600). Solid media were supplemented with 1.5% (w/v) agar. Depending on the experiments, cells could be cultured in liquid media in one or two phases fermentation systems: (i) one step culture system, where the strains are grown in one step under PHA production conditions in order to reach as much cell biomass/PHA yield as possible, and (ii) two steps culture system, where cells are initially cultured in an undefined rich medium (LB) and afterwards transferred to the PHA production medium to fill up of storage biopolymer. For mcl‐PHA accumulation in one phase fermentation process, P. putida strains were cultured in minimal medium 0.1 N M63 (de Eugenio et al., 2010) using 15 mM octanoic acid as carbon source in a 500 ml flask. In the second strategy by using two steps fermentation procedure, 100 ml of LB were inoculated with 0.5 ml of each overnight preculture of P. putida strains. After 14 h of incubation at 30°C with shaking (250 rpm), the cultures were centrifuged and resuspended in 200 ml of 0.1 N M63 mineral medium with 15 mM octanoic acid as the carbon source, in 500 ml flask. The strains were cultivated at 30°C with shaking for 24 h. Unless otherwise stated, the 3MB inducer was added at 5 mM to the PHA production medium. Biomass in different cultures was determined as previously reported (de Eugenio et al., 2010).
DNA manipulations and plasmid constructions
DNA manipulations and other molecular biology techniques were essentially performed as described previously (Sambrook and Russell, 2001). Plasmid pEDF12 is a pNM185 derivative (Mermod et al., 1986) that contains a SacI‐SacII restriction fragment encoding holin (Ejh) and lysin (Ejl) proteins from the genome of the pneumococcal bacteriophage EJ‐1. The DNA fragment containing ejh and ejl genes was isolated by PCR using as template the pEDF12 vector and the oligonucleotides V08 and V09 (Table 1). For PCR amplifications, we used 2 U of AmpliTaq DNA polymerase (Perkin‐Elmer Applied Biosystems, Norwalk, CT, USA), 0.1 µg of DNA template, 0.5 µg of each deoxynucleoside triphosphate and 2.5 mM of MgCl2 in the buffer recommended by the manufacturer. Conditions for amplification were chosen according to the G + C content of the corresponding oligonucleotides. The PCR product was digested with appropriate endonucleases and cloned in pUC18Not, producing the plasmid pUCNotHL. The ejh‐ejl cassette was transferred as NotI fragment to pCNB1 plasmid (pUT/mini‐Tn5 xylS/Pm) (Fig. 2), for stable insertion in the chromosome of P. putida KT2440 under transcriptional control of the Pm promoter. The resulting strain is called P. putida KTHL (Fig. 2). All these constructions were confirmed by sequencing using an ABI Prism 3730 DNA Sequencer. Nucleotide sequences were determined directly with the same oligonucleotides used for cloning.
Granule isolation and quantification
Granule fraction was essentially isolated as described (Moldes et al., 2004). Briefly, cultures were centrifuged for 1 h at 31 000 g to separate culture supernatants from cells and the resulting pellets were resuspended in phosphate buffer, pH 8.0. Fifty percent of each sample was lyophilized and analysed by GC‐MS for total PHA quantification of the culture, as previously reported (de Eugenio et al., 2007). The other 50% of each sample was placed on sucrose gradient layers preformed by overloading 11 ml of 20% and 15% sucrose solutions (Moldes et al., 2004). After centrifugation at 120 000 g for 15 h, the resulting bands were collected and observed under an optical microscope for granule visualization. The intracellular PHA content was calculated by quantifying the PHA content of the sediment after ultracentrifugation. The extracellular PHA content was determined indirectly as the difference between the total PHA content of the culture and the intracellular PHA content.
Transmission electron microscopy
Cells were harvested, washed twice in PBS, and fixed in 5% (w/v) glutaraldehyde in the same solution. Afterwards, cells were incubated with 2.5% (w/v) OsO4 for 1 h, gradually dehydrated in ethanol solutions [30, 50, 70, 90 and 100% (v/v); 30 min each] and propylene oxide (1 h). Finally, they were embedded in Epon 812 resin. Ultrathin sections (thickness 70 nm) were cut with a microtome using a Diatome diamond knife. The sections were picked up with 400 mesh cupper grids coated with a layer of carbon and subsequently observed in a Jeol‐1230 electron microscope (JeolLtd. Akishima, Japan).
Cell viability calculation
To calculate cell viability, serial dilutions from 10−1 to 10−7 were made in saline solution (0.9% NaCl). 10 µl of each dilution from 10−2 to 10−7 was plated on LB solid medium and colony‐forming units were counted. For each strain, three different experiments were carried out. A LIVE/DEAD BacLight bacterial viability kit l‐13152 (Invitrogen‐Molecular Probes) was also used for monitoring the viability of bacterial populations as function of the membrane integrity of the cell according to the instruction of the manufacturers.
Cell sensitivity to detergents and chelating compounds
To determine the bacterial sensitivity to SDS, DOC and EDTA, overnight cultures of each strain were diluted in fresh LB medium with and without 3MB at an OD600 of 0.5. After 6 h of incubation at 30°C in an orbital shaker at 250 rpm, a 10 ml aliquot of each culture was taken and supplemented with 0.075, 0.01 or 0.2% (w/v) DOC; 0.01 or 0.005 (w/v) SDS; 0.1, 0.2 or 0.4 mM EDTA. The ability for growing in the presence of each agent was determined by measuring the OD630 of the cultures during 18 h in 96‐microwell plates with 200 µl of the cultures in each well. Plates were incubated at 30°C during 24 h, with 2 min of orbital shaking every 15 min (Multiskan Ascent, Thermo). The growth rates shown are the mean values from three replicates. As controls, cultures with no added agent were also analysed.
Facilitated PHA extraction analysis
Thirty five ml of each P. putida strain grown for 24 h under two steps PHA producing conditions with and without 3MB were centrifuged and the resulting sediments were directly resuspended in 0.5 ml of different organic solvents (methylene chloride, ethyl acetate or acetone) and incubated for 24 h at room temperature under mild stirring. Solvent phases were collected, evaporated at room temperature to reach a final volume of 0.1 ml and precipitated at −20°C by adding 10 times the volume of methanol. Finally, the polymer was dried under vacuum for 10 min. The resulting polymers were analysed by GC‐MS for determination of PHA content and purity.
Acknowledgments
We thank Dr E. Díaz for helpful discussions. We are indebted to E. Duque and J. L. Ramos for sending the tol mutants of P. putida KT2440. The technical works of A. Valencia and V. Morales are greatly appreciated. This work was supported by grants from the Comunidad Autónoma de Madrid (P‐AMB‐259‐0505), the Ministry of Science and Innovation (BIO2007‐67304, BIO2010‐21049, CSD2007‐00005) and by European Union Grants (GEN 2006‐27750‐C5‐3‐E and NMP2‐CT‐2007‐026515).
References
- Bouveret E., Derouiche R., Rigal A., Lloubès R., Lazdunski C., Bénédetti H. Peptidoglycan‐associated lipoprotein‐TolB interaction. J Biol Chem. 1995;270:11071–11077. doi: 10.1074/jbc.270.19.11071. [DOI] [PubMed] [Google Scholar]
- Clavel T., Germon P., Vianney A., Portalier R., Lazzaroni J.C. TolB protein of Escherichia coli K‐12 interacts with the outer membrane peptidoglycan‐associated proteins Pal, Lpp and OmpA. Mol Microbiol. 1998;29:359–367. doi: 10.1046/j.1365-2958.1998.00945.x. [DOI] [PubMed] [Google Scholar]
- Derouiche R., Bénédetti H., Lazzaroni J.C., Lazdunski C., Lloubès R. Protein complex within Escherichia coli inner membrane. J Biol Chem. 1995;270:11078–11084. doi: 10.1074/jbc.270.19.11078. [DOI] [PubMed] [Google Scholar]
- Díaz E., Munthali M., Lunsdorf H., Holtje J.V., Timmis K.N. The two‐step lysis system of pneumococcal bacteriophage EJ‐1 is functional in gram‐negative bacteria: triggering of the major pneumococcal autolysin in Escherichia coli. Mol Microbiol. 1996;19:667–681. doi: 10.1046/j.1365-2958.1996.399929.x. [DOI] [PubMed] [Google Scholar]
- Elbahloul Y., Steinbüchel A. Large‐scale production of poly(3‐hydroxyoctanoic acid) by Pseudomonas putida GPo1 and a simplified downstream process. Appl Environ Microbiol. 2009;75:643–651. doi: 10.1128/AEM.01869-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Eugenio L.I., García P., Luengo J.M., Sanz J., San Román J., García J.L., Prieto M.A. Biochemical evidence that phaZ gene encodes a specific intracellular medium chain length polyhydroxyalkanoate depolymerase in Pseudomonas putida KT2442: characterization of a paradigmatic enzyme. J Biol Chem. 2007;282:4951–4962. doi: 10.1074/jbc.M608119200. [DOI] [PubMed] [Google Scholar]
- de Eugenio L.I., Escapa I.F., Morales V., Dinjaski N., Galán B., García J.L., Prieto M.A. The turnover of medium‐chain‐length polyhydroxyalkanoates in Pseudomonas putida KT2442 and the fundamental role of PhaZ depolymerase for the metabolic balance. Environ Microbiol. 2010;12:207–221. doi: 10.1111/j.1462-2920.2009.02061.x. [DOI] [PubMed] [Google Scholar]
- Fidler S., Dennis D. Polyhydroxyalkanoate production in recombinant Escherichia coli. FEMS Microbiol Rev. 1992;103:231–236. doi: 10.1016/0378-1097(92)90314-e. [DOI] [PubMed] [Google Scholar]
- Furrer P., Panke S., Zinn M. Efficient recovery of low endotoxin medium‐chain‐length poly([R]‐3‐hydroxyalkanoate) from bacterial biomass. J Microbiol Methods. 2007;69:206–213. doi: 10.1016/j.mimet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Gagnon K.D., Lenz R.W., Farris R.J., Fuller R.C. Crystallization behaviour and its influence on the mechanical properties of a thermoplastic elastomer produced by Pseudomonas oleovorans. Macromolecules. 1992;25:3723–3728. [Google Scholar]
- Gavrilescu M., Chisti Y. Biotechnology‐ a sustainable alternative for chemical industry. Biotechnol Adv. 2005;23:471–499. doi: 10.1016/j.biotechadv.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Germon P., Clavel T., Vianney A., Portalier R., Lazzaroni J.C. Mutational analysis of the Escherichia coli K‐12 TolA N‐terminal region and characterization of its TolQ‐interacting domain by genetic suppression. J Bacteriol. 1998;180:6433–6439. doi: 10.1128/jb.180.24.6433-6439.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germon P., Ray M.C., Vianney A., Lazzaroni J.C. Energy dependent conformational change in the TolA protein of Escherichia coli involves its N‐terminal domain, TolQ, and TolR. J Bacteriol. 2001;183:4110–4114. doi: 10.1128/JB.183.14.4110-4114.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godlewska R., Wisniewska K., Piedras Z., Jagusztyn‐Krynicka E.K. Peptidoglycan‐associated lipoprotein (Pal) of Gram‐negative bacteria: function, structure, role in pathogenesis and potential application in immunoprophylaxis. FEMS Microbiol Lett. 2009;298:1–11. doi: 10.1111/j.1574-6968.2009.01659.x. [DOI] [PubMed] [Google Scholar]
- Gorenflo V., Schmack G., Vogel R., Steinbüchel A. Development of a process for the biotechnological large‐scale production of 4‐hydroxyvalerate‐containing polyesters and characterization of their physical and mechanical properties. Biomacromolecules. 2001;2:45–57. doi: 10.1021/bm0000992. [DOI] [PubMed] [Google Scholar]
- van Hee P., Elumbaring A.C., van der Lans R.G., van der Wielen L.A. Selective recovery of polyhydroxyalkanoate inclusion bodies from fermentation broth by dissolved‐air flotation. J Colloid Interface Sci. 2006;297:595–606. doi: 10.1016/j.jcis.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Herrero M., de Lorenzo V., Timmis K.N. Transposon vectors containing non‐antibiotic resistance selection markers for cloning and stable chromosomal insertions of foreign genes in Gram negative bacteria. J Bacteriol. 1990;172:6557–6567. doi: 10.1128/jb.172.11.6557-6567.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori K., Kaneko M., Tanji Y., Xing X.H., Unno H. Construction of self‐disruptive Bacillus megaterium in response to substrate exhaustion for polyhydroxybutyrate production. Appl Microbiol Biotechnol. 2002;59:211–216. doi: 10.1007/s00253-002-0986-8. [DOI] [PubMed] [Google Scholar]
- Huijberts G.N., Eggink G., de Waard P., Huisman G.W., Witholt B. Pseudomonas putida KT2442 cultivated on glucose accumulates poly(3‐hydroxyalkanoates) consisting of saturated and unsaturated monomers. Appl Environ Microbiol. 1992;58:536–544. doi: 10.1128/aem.58.2.536-544.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Journet L., Rigal A., Lazdunski C., Bénédett H. Role of TolR N‐terminal, central, and C‐terminal domains in dimerization and interaction with TolA and TolQ. J Bacteriol. 1999;181:4476–4484. doi: 10.1128/jb.181.15.4476-4484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellerhals M.B., Kessler B., Witholt B. Closed‐loop control of bacterial high‐cell‐density fed‐batch cultures: production of mcl‐PHAs by Pseudomonas putida KT2442 under single‐substrate and cofeeding conditions. Biotechnol Bioeng. 1999;56:306–315. [PubMed] [Google Scholar]
- Koebnik R. Proposal for a peptidoglycan‐associating alpha‐helical motif in the C‐terminal regions of some bacterial cell‐surface proteins. Mol Microbiol. 1995;16:1269–1270. doi: 10.1111/j.1365-2958.1995.tb02348.x. [DOI] [PubMed] [Google Scholar]
- de Koning G.J.M., Kellerhals M., van Meurs C., Witholt B. A process for the recovery of poly(hydroxyalkanoates) from Pseudomonads. Part 2: process development and economic evaluation. Bioprocess Biosyst Eng. 1997;17:15–21. [Google Scholar]
- Llamas M.A., Ramos J.L., Rodríguez‐Herva J.J. Mutations in each of the tol genes of Pseudomonas putida reveal that they are critical for maintenance of outer membrane stability. J Bacteriol. 2000;182:4764–4772. doi: 10.1128/jb.182.17.4764-4772.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llamas M.A., Ramos J.L., Rodríguez‐Herva J.J. Transcriptional organization of the Pseudomonas putida tol‐oprL genes. J Bacteriol. 2003;185:184–195. doi: 10.1128/JB.185.1.184-195.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López R., García E. Recent trends on the molecular biology of pneumococcal capsules, lytic enzymes, and bacteriophage. FEMS Microbiol Rev. 2004;28:553–580. doi: 10.1016/j.femsre.2004.05.002. [DOI] [PubMed] [Google Scholar]
- de Lorenzo V., Fernández S., Herrero M., Jakubzik U., Timmis K.N. Engineering of alkyl‐ and haloaromatic‐responsive gene expression with mini‐transposons containing regulated promoters of biodegradative pathways of Pseudomonas. Gene. 1993;130:41–46. doi: 10.1016/0378-1119(93)90344-3. [DOI] [PubMed] [Google Scholar]
- Madison L.L., Huisman G.W. Metabolic engineering of poly (3‐hydroxyalkanoates): from DNA to plastic. Microbiol Mol Biol Rev. 1999;63:21–53. doi: 10.1128/mmbr.63.1.21-53.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermod N., Ramos J.L., Lehrbach P.R., Timmis K.N. Vector for regulated expression of cloned genes in a wide range of Gram‐negative bacteria. J Bacteriol. 1986;167:447–454. doi: 10.1128/jb.167.2.447-454.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldes C., García P., García J.L., Prieto M.A. In vivo immobilization of fusion proteins on bioplastics by the novel tag BioF. Appl Environ Microbiol. 2004;70:3205–3212. doi: 10.1128/AEM.70.6.3205-3212.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T. Travels of a Pseudomonas, from Japan around the world. Environ Microbiol. 2002;4:782–786. doi: 10.1046/j.1462-2920.2002.00310.x. [DOI] [PubMed] [Google Scholar]
- Nelson K.E., Weinel C., Paulsen I.T., Dodson R.J., Hilbert H., Martins dos Santos V.A. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ Microbiol. 2002;4:799–808. doi: 10.1046/j.1462-2920.2002.00366.x. et al. [DOI] [PubMed] [Google Scholar]
- Olivera E.R., Carnicero D., Jodra R., Minambres B., García B., Abraham G.A. Genetically engineered Pseudomonas: a factory of new bioplastics with broad applications. Environ Microbiol. 2001a;3:612–618. doi: 10.1046/j.1462-2920.2001.00224.x. et al. [DOI] [PubMed] [Google Scholar]
- Olivera E.R., Carnicero D., García B., Miñambres B., Moreno M.A., Cañedo L. Two different pathways are involved in the beta‐oxidation of n‐alkanoic and n‐phenylalkanoic acids in Pseudomonas putida U: genetic studies and biotechnological applications. Mol Microbiol. 2001b;39:863–874. doi: 10.1046/j.1365-2958.2001.02296.x. et al. [DOI] [PubMed] [Google Scholar]
- Prieto M.A. From oil to bioplastics, a dream come true? J Bacteriol. 2007;189:289–290. doi: 10.1128/JB.01576-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto M.A., Kellerhals M.B., Bozzato G.B., Radnovic D., Witholt B., Kessler B. Engineering of stable recombinant bacteria for production of chiral medium‐chain‐length poly‐3 hydroxyalkanoates. Appl Environ Microbiol. 1999;65:3265–3271. doi: 10.1128/aem.65.8.3265-3271.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto M.A., de Eugenio L.I., Galán B., Luengo J.M., Witholt B. Synthesis and degradation of polyhydroxyalkanoates. In: Ramos J.L., Filloux A., editors. Springer; 2007. pp. 397–428. [Google Scholar]
- Qi Q., Steinbüchel A., Rehm B.H. Metabolic routing towards polyhydroxyalkanoic acid synthesis in recombinant Escherichia colifadR): inhibition of fatty acid beta‐oxidation by acrylic acid. FEMS Microbiol Lett. 1998;167:89–94. doi: 10.1111/j.1574-6968.1998.tb13212.x. [DOI] [PubMed] [Google Scholar]
- Ramsay J.A., Berger E., Voyer R., Chavarie C., Ramsay B.A. Extraction of poly‐3‐hydroxybutyrate using chlorinated solvents. Biotechnol Tech. 1994;8:589–594. [Google Scholar]
- Ren Q., Sierro N., Kellerhals M., Kessler B., Witholt B. Properties of engineered poly‐3‐hydroxyalkanoates produced in recombinant Escherichia coli strains. Appl Environ Microbiol. 2000;66:1311–1320. doi: 10.1128/aem.66.4.1311-1320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resch S., Gruber K., Wanner G., Slater S., Dennis D., Lubitz W. Aqueous release and purification of poly(β‐hydroxybutyrate) from Escherichia coli. J Biotechnol. 1998;65:173–182. doi: 10.1016/s0168-1656(98)00127-8. [DOI] [PubMed] [Google Scholar]
- Sabirova J.S., Ferrer M., Lunsdorf H., Wray V., Kalscheuer R., Steinbüchel A. Mutation in a ‘tesB‐like’ hydroxyacyl‐coenzyme A‐specific thioesterase gene causes hyperproduction of extracellular polyhydroxyalkanoates by Alcanivorax borkumensis SK2. J Bacteriol. 2006;188:8452–8459. doi: 10.1128/JB.01321-06. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Russell D.W. 3rd. Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- de Smet M., Eggink G., Witholt B., Kingma J., Wynberg H. Characterization of intracellular inclusions formed by Pseudomonas oleovorans during growth on octane. J Bacteriol. 1983;154:870–878. doi: 10.1128/jb.154.2.870-878.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z., Ramsay J.A., Guay M., Ramsay B.A. Fermentation process development for the production of medium‐chain‐length poly‐3‐hyroxyalkanoates. Appl Microbiol Biotechnol. 2007;75:475–485. doi: 10.1007/s00253-007-0857-4. [DOI] [PubMed] [Google Scholar]
- Thakor N., Lütke‐Eversloh T., Steinbüchel A. Application of the BPEC pathway for large‐scale biotechnological production of poly(3‐mercaptopropionate) by recombinant Escherichia coli, including a novel in situ isolation method. Appl Environ Microbiol. 2005;71:835–841. doi: 10.1128/AEM.71.2.835-841.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang I.N., Smith D.L., Young R. Holins: the protein clocks of bacteriophage infections. Annu Rev Microbiol. 2000;54:799–825. doi: 10.1146/annurev.micro.54.1.799. [DOI] [PubMed] [Google Scholar]
- Williams S.F., Martin D.P., Horowitz D.M., Peoples O.P. PHA applications: addressing the price performance issue: I. Tissue engineering. Int J Biol Macromol. 1999;25:111–121. doi: 10.1016/s0141-8130(99)00022-7. [DOI] [PubMed] [Google Scholar]
- Young R. Phage lysis. In: Waldor M., Friedman D.I., Adhya S.L., editors. ASM Press; 2005. pp. 92–127. [Google Scholar]
- Yu H., Yin J., Li H., Yang S., Shen Z. Construction and selection of the novel recombinant Escherichia coli strain for poly(β‐hydroxybutyrate) production. J Biosci Bioeng. 2000;89:307–311. doi: 10.1016/s1389-1723(00)88950-1. [DOI] [PubMed] [Google Scholar]
- Zinn M., Witholt B., Egli T. Occurrence, synthesis and medical application of bacterial polyhydroxyalkanoate. Adv Drug Deliv Rev. 2001;53:5–21. doi: 10.1016/s0169-409x(01)00218-6. [DOI] [PubMed] [Google Scholar]