

Summary
Developments in biocatalysis have been largely fuelled by consumer demands for new products, industrial attempts to improving existing process and minimizing waste, coupled with governmental measures to regulate consumer safety along with scientific advancements. One of the major hurdles to application of biocatalysis to chemical synthesis is unavailability of the desired enzyme to catalyse the reaction to allow for a viable process development. Even when the desired enzyme is available it often forces the process engineers to alter process parameters due to inadequacies of the enzyme, such as instability, inhibition, low yield or selectivity, etc. Developments in the field of enzyme or reaction engineering have allowed access to means to achieve the ends, such as directed evolution, de novo protein design, use of non‐conventional media, using new substrates for old enzymes, active‐site imprinting, altering temperature, etc. Utilization of enzyme discovery and improvement tools therefore provides a feasible means to overcome this problem. Judicious employment of these tools has resulted in significant advancements that have leveraged the research from laboratory to market thus impacting economic growth; however, there are further opportunities that have not yet been explored. The present review attempts to highlight some of these achievements and potential opportunities.
Introduction
Chemical process development has made a huge impact on economic growth; however, it has also raised several concerns on environmental safety. Market demands for new therapeutics and socio‐political demands for environmental sustainability coupled with diminishing fossil reserves have led to increased utilization of ‘bio’‐based technologies for industrial use. A report by McKinsey and Co. predicted that industrial biotechnology would account for 10–20% of sales in the chemical industry by the year 2010 (McKinsey Report, 2006). In response to such estimates, the leading industrialized nations such as the USA (Advanced Technology Platform, National Institute of Standards and Technology US, 2003), Japan (Japan Bioindustry Association, Biotechnology Strategy Council, 2002) and the EU (European Technology Platform for Sustainable Chemistry, 2003) have recognized industrial biotechnology as one of the key drivers of sustainable growth. Accordingly, industrial corporations worldwide are attempting to leverage industrial biotechnology into their process development efforts to fuel their innovation pipeline. However, a biological option is often considered only when the chemical arsenal has failed to achieve synthesis of the target molecule. This is primarily because the desired biocatalyst is either unavailable or unable to execute the required transformation in an efficient manner. In order to ensure sustained progress, search and discovery tools may be utilized to ideally generate a library of enzyme preparations ready for use in organic synthesis. Hence, continued success of the chemical industry significantly depends on effective utilization of search tools for quickly identifying an enzyme for a particular reaction. Accordingly, need for systematic methods to (i) screen for novel enzyme sources with improved characteristics as a favourable starting point for process development, (ii) fine tune the existing enzyme using protein engineering approaches, (iii) identify and manipulate metabolic pathways for natural product biosynthesis, and finally (iv) engineer the reaction components as an alternative to enzyme manipulation to overcome catalyst limitation has stimulated significant developments in the field, allowing greater integration of biotechnology into process development and drug discovery programmes (Fig. 1). Nature provides an immensely vast bounty of resources produced by complex metabolic pathways that are controlled by enzymes. The interplay of these pathways provides a rich source of valuable natural products and metabolites with a wide range of applications. Our ability to tap into this bio‐‘treasure’ depends on the tools available to access the nature's pool. This largely limits our ability to pick pathways and/or enzymes from the nature's toolbox and add to a chemist's toolbox for practical utility. Recent success of genome sequencing programmes has resulted in an explosion of information available from sequence databases, thus creating an opportunity to explore the possibility of finding new enzymes by database mining. Genome annotations are typically based on general relation to the closest homologous sequence, rather than precise functional analysis. It remains the task of laboratory research to assign function to these putative protein sequences (by determining their natural substrates), and provide the answer to ‘what to apply where’ question. In order to reach a stage to execute such molecular approaches, one must normally practice the conventional screening strategies; however, requirement for purifying the enzyme for its detailed characterization makes the approach highly labour‐intensive. The advantage of genome mining approach also referred as reverse genetics approach is that it bypasses these time‐consuming steps for enzymes with too low activity (Stewart, 2006). Some of the noteworthy success stories have allowed retrieval of important biocatalysts, such as ketoreductases (Kaluzna et al., 2004), epoxide hydrolases (Loo et al., 2006), esterases (Henke and Bornscheuer, 2002; Dherbécourt et al., 2008), Baeyer‐Villiger monooxygenases (Fraaije et al., 2002; 2004), nitrilases (Seffernick et al., 2009), glycosyltransferases (Henrissat et al., 2001), Cytochromes P450 (Agematu et al., 2006), to mention a few.
Figure 1.
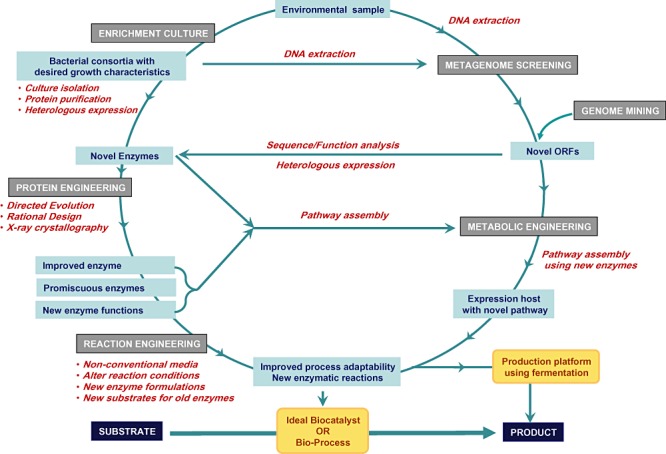
Biocatalyst discovery and implementation cycle.
The present review attempts to encapsulate the developments in the field of discovery and implementation of microbes (as a source of natural products) and enzymes (as biocatalysts for synthetic applications) for use in industrial biotechnology. However, to ensure commercial success it is important to perform an overall cost analysis very early during the process development (Tufvesson et al., 2010).
Ease of executing stereoselective transformations without the need of employing protection and deprotection strategies has led to increased acceptance of enzymes as enantioselective catalysts, since these are relatively difficult to achieve using the chemical counterparts. Chirality has become a central theme in drug discovery and development programmes (Ariens, 1984), which has led to increased regulatory pressure on the manufacturers to market only the homochiral form of the drug wherever applicable (Strong, 1999). Hence, the pharmaceutical companies are now also using the strategy of ‘chiral switch’ (development of a single enantiomer from a previously marketed racemate) to promote life cycle extension of their products (Agranat et al., 2002). This further strengthens the justification for employing enzymes in addition to their already illustrated benefits (Koeller and Wong, 2001). Experimental objective often governs the adoption of a screening strategy and one may sift through natural habitats or databases for desired microorganisms or enzymes. Consequently, the present review will also attempt to highlight some noteworthy developments in biocatalytic process development achieved using tools for enzyme discovery and improvement, successfully applied on industrial scale and also indicate the potential opportunities to be explored.
Screening for enzyme activity
In order to obtain microorganisms with special growth characteristics, use of enrichment culture technique is employed. It uses the principle of natural selection, wherein a mixed microbial population is inoculated in a medium of defined (but limited) chemical composition and allowed to grow under controlled conditions (temperature, air supply, light, pH, etc.) in such a way that it only suits growth of a particular type of microorganisms with specific characteristics. After successive transfers (over a period of several months), some bacterial species become more dominant than others and are therefore said to have been ‘enriched’. Perhaps, the best example of use of this approach has been for obtaining strains capable of hydrolysing acrylonitrile for acrylamide production for which extensive screening was conducted. Thus, Rhodococcus sp. N‐774 (Watanabe et al., 1987), Rhodococcus rhodochrous J‐1 (Asano et al., 1980) and Pseudomonas chlororaphis B23 (Asano et al., 1982) were isolated and led to establishment of a commercial plant. Acrylamide biosynthesis is now the largest enzyme‐catalysed process worldwide, functioning at industrial scale of more than 10 000 tonnes per year by Nitto Chemical Industry (Tokyo, Japan). Nitrile hydratase has, therefore, become one of the most important industrial biocatalysts used for production of acrylamide (Nagasawa, Shimizu, Yamada, 1993), nicotinamide (Nagasawa et al., 1988), 5‐cyanovaleramide (Hann et al., 1999), etc.
The physiological role of nitrile‐degrading and synthesizing enzymes in plants and microbes is not clearly understood. Plants are believed to utilize cyanogenic glucosides for protection against herbivore attack by producing hydrogen cyanide which is highly toxic. As a result of lack of information on these enzyme systems, there were no literature reports describing biosynthesis of nitriles and it remains uncertain whether these compounds have a biological role or are just petrochemicals. The picture became clear when aldoxime dehydratase was discovered in microorganisms and found to be linked physiologically and genetically with nitrile hydratase (Kato et al., 2000a). Acclimation of soil samples resulted in isolation of Bacillus sp. strain OxB‐1 (Asano and Kato, 1998), the first report on enzymatic synthesis of nitriles from aldoximes by using the new enzyme aldoxime dehydratase. These studies illuminated for the first time, coexistence of aldoxime dehydratase and nitrilase gene cluster in the genome of Bacillus sp. strain OxB‐1 and various other strains (Kato et al., 1998; 1999a; 2000a,b). In general, it was found that most microorganisms exhibiting nitrile‐degrading activity also expressed the aldoxime dehydratase activity now termed as aldoxime‐nitrile pathway (Fig. 2) (Asano, 2002).
Figure 2.
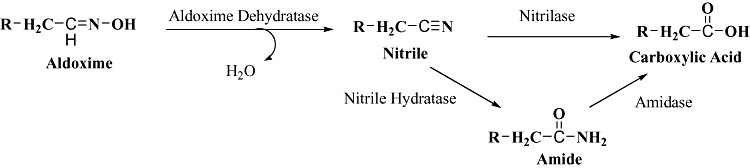
Aldoxime‐nitrile pathway involving nitrile hydrolases and aldoxime dehydratase as a route to optically pure nitriles, amides and carboxylic acids.
Not even best of the screening strategies can guarantee obtaining a viable hit. In such cases, one may access known databases of culture collection centres and screen from the library of microorganisms. One of the prime example of this strategy resulted in identification of Alcaligenes faecalis ATCC 8750 as a very potent expresser of a highly enantioselective nitrilase acting on racemic mandelonitrile and produced (R)‐(‐)‐mandelic acid, a versatile chiral building block (Yamamoto et al., 1991a). The cyanohydrin undergoes in situ racemization, making it a dynamic kinetic resolution. An advantage of the nitrilase process is that no organic solvents are required and it avoids the synthesis of a nitrile, as the cyanohydrin is formed from benzaldehyde and hydrogen cyanide under the reaction conditions. The recombinant nitrilase expressed in Escherichia coli is presently being used by BASF (Ludwigshafen, Germany) (Ress‐Loschke et al., 2000), Nitto Chemical Industry (Tokyo, Japan) (Endo and Tamura, 1991; 1994; Endo et al., 1994) and Asahi Kasei Kogyo (Osaka, Japan) (Yamamoto et al., 1991b) on a multiton scale to produce optically pure mandelic acid along with its substituted derivatives. A. faecalis nitrilase has also been employed for other commercial processes. An immobilized form of the recombinant host (E. coli) expressing genetically modified nitrilase was also utilized for production of hydroxy methionine derivatives that find use as nutritional additive in cattle feed (Favre‐Bulle et al., 2001; Pierrard et al., 2001; Rey et al., 2004) (Fig. 3).
Figure 3.
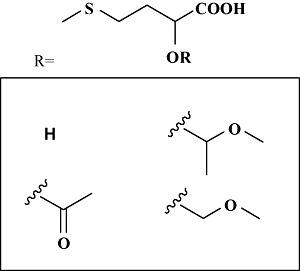
Hydroxy‐methionine analogues produced using nitrilase.
By envisaging an enzymatic route for an existing chemical process, one may devise a screening strategy to acquire enzymes catalysing the desired transformation. Success in such endeavours depends solely on the approach adopted for microorganism isolation. Such an investigation led to development of the first route for production of 12‐aminolauric acid from ω‐laurolactam by enzymatic trans‐crystallization (Asano et al., 2008; Fukuta et al., 2009) (Fig. 4) and hydrolase activity was found in various microorganisms such as Cupriavidus sp. T7, Acidovorax sp. T31, Cupriavidus U124, Rhodococcus sp. U224 and Sphingomonas sp. U238. The product finds applications in the synthesis of nylon (Kojima et al., 2000), adhesives and hardening agents (Chen et al., 2000). Ideally, enzymatic trans‐crystallization is well suited for substrate candidates that have low aqueous solubility by virtue of their crystalline solid state. It typically involves addition of high substrate loadings (forming a heterogeneous mixture) which gradually dissolves in aqueous phase, where the enzyme resides. Simultaneously, the substrate (in aqueous phase) is acted upon by the enzyme, thus forming an equilibrium to make up for the depleted substrate (in the aqueous phase from the solid phase), resulting in product formation until the point of its solubility, beyond which the product precipitates as a crystalline form. This strategy therefore also allows overcoming both substrate and product inhibition.
Figure 4.

Enzymatic hydrolysis of ω‐laurolactam to 12‐aminolauric acid.
Sometimes novel enzymatic reactions may also be accessed by using new substrates for known enzymes. l‐lysine has been prepared on an industrial scale by Toray Industries (Tokyo, Japan) by hydrolysis of dl‐α‐amino‐ε‐caprolactam (ACL) using l‐specific hydrolase and concomitant racemization of the d‐substrate enantiomer using ACL racemase (Fukumura, 1977a,b). Screening for ACL racemase activity led to identification of Achromobacter obae (Fukumura, 1977a) and the enzyme was found to accept only ACL, α‐amino‐δ‐valerolactam, α‐amino‐β‐thio‐ε‐caprolactam (Ahmed et al., 1986). However, by altering the reported substrate of ACL racemase with amino acid amides, it was possible to achieve racemization of these molecules for the first time (Asano and Yamaguchi, 2005a). This resulted in the first report on preparation of optically pure amino acids from corresponding dl‐amino acid amides using ACL racemase coupled with a stereospecific amidase (Asano and Yamaguchi, 2005b).
Metagenome screening
Traditional microbiological studies to access the natural diversity rely on the use of culturable microorganisms. Nature represents an enormous diversity, specially considering what we have been able to access so far accounts for less that 1% of the total biodiversity (in terms of prokaryotic genomes) since the majority of 99% are unculturable (Kamagata and Tamaki, 2005; Sekiguchi, 2006). Tools of metagenome analysis obviate this need by directly extracting DNA from environmental samples (collected from diverse geographical location), preparing a genomic library and systematically screening (based on sequence or functional analysis) the library for the open reading frames potentially encoding putative novel enzymes (Streit et al., 2004; Cowan et al., 2005; Green and Keller, 2006; Schmeisser et al., 2007; Singh and Pelaez, 2008; Steele et al., 2009). It was more than a decade after the first report on genomic DNA extraction from soil samples was published (Torsvik and Goksoyr, 1980; Pace et al., 1986), that systematic approaches were undertaken which allowed earliest and decisive insight into the precise dimensions of natural biodiversity (Torsvik et al., 2002) by finding more than a million previously unknown genes sampled from marine plankton in Sargossa Sea (Venter et al., 2004). Accordingly, numerous industrial houses have realized the promise that metagenomics holds and attempted to harness the biotechnological potential of unculturable microorganisms, notably among are them Diversa (San Diego, California, USA, now Verenium Corporation, Cambridge, Massachusetts, USA) (DeSantis et al., 2002; Robertson et al., 2004), TerraGen Discovery (Vancouver, Canada, now taken over by Cubist Pharmaceuticals, Cambridge, Massachusetts, USA) (Radomski et al., 1998), Genencor International (Palo Alto, California, USA) (Knietsch et al., 2003a), and BRAIN AG (Zwingenberg, Germany) (Breves et al., 2003; Liebeton and Eck, 2004) who have partnered with other mainstream pharmaceutical and chemical companies and various academic institutions.
By accessing diverse geographical locations such as deep ocean beds, volcanic vents, arctic tundra, etc. exploratory efforts to screen for novel enzyme sources resulted in recovery of more than 100 new nitrilase genes (Robertson et al., 2004) allowing the novel library to serve as catalytic toolbox for production of high value carboxylic acid derivatives from the corresponding nitriles (DeSantis et al., 2002). These novel nitrilases were also used for enantioselective hydrolysis of 3‐hydroxyglutaronitrile to (R)‐cyano‐3‐hydroxybutyric acid, an intermediate for the production of Atorvastatin, a blockbuster (a drug with annual sales of more than US$ 1 billion) lipid‐lowering drug (sold under the trade name of Lipitor) from Pfizer (New York, USA) (Fig. 5). Interestingly, the work also lead to identification of novel (S)‐selective nitrilases (albeit low enantioselectivity, 30% enantiomeric excess) for mandelonitrile, for which primarily only (R)‐selective nitrilases are reported. Numerous studies to access the metagenome have yielded enzymes with potential for biocatalytic applications, such as lipase (Jeon et al., 2009; Fernández‐Álvaro et al., 2010), oxidoreductase (Knietsch et al., 2003b), amidase (Gabor et al., 2004), amylase (Rondon et al., 2000), nitrilase (Bayer et al., 2011), β‐glucosidase (Wang et al., 2010; Jiang et al., 2011), decarboxylase (Jiang et al., 2009), epoxide hydrolase (Kotik et al., 2010), among many others.
Figure 5.
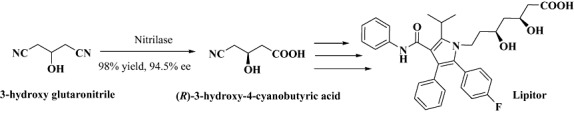
Nitrilase‐mediated desymmetrization of prochiral dinitrile to Lipitor intermediate.
In addition to searching for new enzymes, the metagenome approach has also allowed identification of novel natural products with medicinal properties. One of the notable achievements led to identification of genes encoding for the enzymes polyketide synthase type I and II, which form part of a large gene cluster involved in biosynthesis of polyketide antibiotics (Piel, 2002; Courtois et al., 2003; Piel et al., 2004). Using a phylogenetic approach, the authors were also able to clone genes putatively involved in the biosynthesis of antitumour compounds from the metagenomic DNA of beetles and sponges and their bacteria (Piel et al., 2004). TerraGen Discovery provided the first discovery of polyketide synthase type II genes from microbial soil metagenome (Seow et al., 1997) and subsequently cloned these genes for expression in Streptomyces lividans as a host for production of novel bioactive metabolites (Wang et al., 2000). Successful implementation of a screening effort, in particular metagenome screening, also requires careful considerations of a number of associated parameters, such as site for sample collection (Wexler et al., 2005; Ferrer et al., 2007; Brennerova et al., 2009; de Vasconcellos et al., 2010), choosing the appropriate expression vector and host (Rondon et al., 2000; Wang et al., 2000; Wild et al., 2002; Martinez et al., 2004; Li et al., 2005; Taupp et al., 2009), and development of effective screening system (Gloux et al., 2007; Litthauer et al., 2010).
In general, metagenome approaches allow access to biodiversity based either on screening for function (using enzyme activity measurements) or nucleotide sequence analysis (using PCR primers or hybridization probes). The inherent limitations of the above approaches, such as requirement for high‐throughput screening methods and expression of the gene cluster in the host (for former) and bias towards known homologous sequences (for latter), prompted development of a new method based on substrate induced gene expression (SIGEX) (Uchiyama et al., 2005). Since the genes and regulatory elements for expression of catabolic enzymes (induced by substrates or metabolites) are located in close proximity to one another, the method allows screening of these catabolic genes by inducing their expression in response to a stimulus (such as presence of a chemical). The approach was used to screen and isolate genes induced by aromatic hydrocarbons from a groundwater metagenome library (Uchiyama et al., 2005). In order to increase the throughput of screening, an operon‐trap vector was constructed, making it appropriate for use in shotgun cloning and used in conjunction with fluorescence activated cell sorter (FACS) for selection of positive clones in liquid culture that harbour stimulus responsive metagenome fragments upstream of the gfp gene. By using SIGEX it was possible to isolate 33 benzoate‐inducible and two naphthalene‐inducible clones from a library of 152 000 clones, in addition to discovering a novel enzyme Bzo71‐8 P450.
The method offers numerous advantages in the form of amenability to high‐throughput screening and accessibility to catabolic genes for which no assay method has been established. However, it also suffers from disadvantages like; it misses constitutively expressed genes, is sensitive to orientation of genes and cannot be used for substrates that do not migrate to the cytoplasm. Hence choice of an appropriate method for the desired screening approach must involve careful consideration of various influencing parameters. In accordance with this approach, another interesting report describes an attempt to maximize the potential of metagenome. Designated as METREX (metabolite‐regulated expression), the method utilizes induction of a quorum sensing biosensor by an inducer produced by a metagenome clone to yield fluorescence triggered by GFP production (conveniently screened using fluorescence microscopy of FACS) (Williamson et al., 2005).
Use of such molecular approaches often results in a large library size which must be screened to identify potential hits. Developments in associated fields have promoted substantial advancement in our ability to access the biodiversity pool by using automated high‐throughput screening platforms (Betton, 2004; Bradbury, 2004; Ho et al., 2004; Lafferty and Dycaico, 2004; Lorenz, 2004; Pajak et al., 2004). This significantly improves the ability to not only access a number of novel enzymes but also impacts the speed with which they are screened. This has led to some important findings, such as novel xylanases obtained from bacteria residing in termite gut (Brennan et al., 2004), thus providing proof of concept that it is possible to access novel sequence space that are unique to their origin.
Protein engineering
Enzymes obtained by screening approaches (traditional or metagenome) do not necessarily fulfil all process requirements and need further fine tuning for adaptability to industrial scale production. Often inadequacies such as low stability, substrate and/or product inhibition, narrow substrate spectrum, enantioselectivity, etc. limit the application of the enzyme at hand for practical purposes. Using the tools of protein engineering, researchers are now able to reprogram the enzyme characteristics thus providing opportunities for tailoring enzymes for specific reactions. A practical approach for protein design is to use the three‐dimensional enzyme structure and rational design to systematically alter specific amino acid residues in the active site. This approach although more pragmatic, requires detailed knowledge of the structure and function of the protein to make desired changes. One of the important landmark studies using this approach was reported recently for production of the antidiabetic drug Sitagliptin (Savile et al., 2010). Chemical methods (rhodium‐catalysed hydrogenation) used for the production of the drug are highly undesirable and offer problems such as product contamination, inadequate stereoselectivity, etc. This prompted search for a transaminase mediated alternative using prositagliptin ketone (precursor for the product) as a substrate (Fig. 6). However, initial screening suggested that no enzyme activity could be obtained for the bulky substrate. By using the approach of structure‐based rational protein design and directed evolution, it was possible to alter the substrate specificity and stability of transaminase from Arthrobacter sp. (Koszelewski et al., 2008) to obtain variants that could not only accept the bulky ketone as substrate at high concentrations, but also tolerate elevated temperatures and high solvent concentrations (to enhance solubility of the insoluble bulky ketone). Using the transaminase variants it has been possible to produce various trifluoromethyl amines and phenylethylamines with electron rich substituents with high enantioselectivity, a feat that was impossible to achieve using the existing chemical methods. The process advancement efforts (developed jointly by Merck and Codexis) were also recognized by awarding the prestigious Presidential Green Chemistry Award from US Environmental Protection Agency (EPA) in June 2010.
Figure 6.
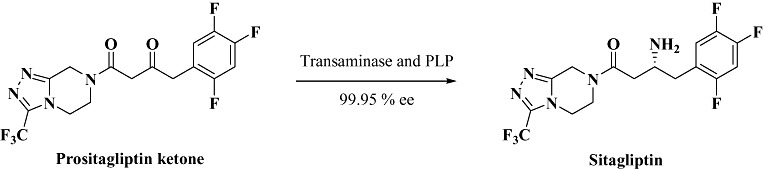
Transaminase‐mediated route to synthesis of the antidiabetic drug Sitagliptin.
However, use of such structure‐guided approaches has a major drawback since it requires detailed structural knowledge of a protein, which is unavailable for a vast majority of proteins. Even when such details are available, it can be extremely difficult to predict the effects of various mutations. In the absence of any detailed information available about enzyme structure, one may use directed evolution strategies to design de novo protein templates exhibiting improvements in desired characteristics or novel traits. The technique of in vitro directed evolution mimics natural evolution without having the knowledge of enzyme structure and function by using tools such as random mutagenesis and sexual recombination to obtain gradual improvement in enzyme characteristics. With an enzyme at hand to catalyse the desired reaction, one may also execute the approach of directed evolution to evolve desired characteristics such as improved enantioselectivity (van Loo et al., 2004), inverting enantioselectivity (May et al., 2000), altered substrate specificity (Asano et al., 2005; Hibbert et al., 2008), enhanced thermostability (Uchiyama et al., 2000), improving catalytic activity (Komeda et al., 2003), enhancing stability to organic solvents (Wong et al., 2004; Kawata and Ogino, 2009), overcoming product or substrate inhibition (Kim et al., 2004), enhancing enzyme activity (Crameri et al., 1998; Castle et al., 2004) to improve process adaptability of the biocatalytic scheme.
One may also screen sequence space using computational approaches comparable to screening effort for libraries obtained during directed evolution. Ability to design protein structure de novo also opens a fascinating opportunity to evolve new enzyme functions. Recently, a synthetic Diels‐Alderase was created to execute intramolecular cycloaddition using the computational transition state model for well‐studied Diels‐Alder reaction and accordingly designing the associated enzyme active site (Siegel et al., 2010). Such bimolecular, bond‐forming reactions represent a significant challenge, since the two substrates must align in a precise orientation to allow for reaction rate acceleration and control of stereoselectivity. There have only been two reports on naturally occurring Diels‐Alderases; however, they are restricted only to conducting intramolecular conjugation (Oikawa et al., 1995; Auclair et al., 2000). This prompted the efforts to design from scratch an artificial enzyme capable of executing such conjugation with high selectivity. Transitions state kinetics of the Diels‐Alder reaction suggests that the highest occupied molecular orbital (HOMO) of the diene and the lowest occupied molecular orbital (LOMO) of the dienophile interact, and reaction rate acceleration is highest when this energy gap is lowest (Oikawa, 2010). By defining the geometrical constraints of the substrate molecules in the transition state, it was possible to define the active site of the enzyme by placing hydrogen bond donating and accepting residues near the two substrates. Using quantum mechanical predictions and the core active‐site design, it was possible to generate 109 three‐dimensional models of minimal active sites, referred to as theozymes. This ensemble of theozymes was next matched to known protein scaffolds to shortlist potential candidates that were subsequently subjected to design using scientific instinct guided by Rosetta design software (for optimization of active‐site geometry), followed by site‐directed mutagenesis (for fine tuning the novel templates) to obtain enzyme variants that performed intermolecular conjugation with high enantio‐ and diastereoselectivity. Although, reaction rates of these novel Diels‐Alderases is lower as compared with other enzyme‐catalysed reactions, but the option of de novo protein design represents a landmark achievement in defining our ability to create tailor made enzymes. Directed evolution approaches (Turner, 2003; 2009; Tracewell and Arnold, 2009) along with the screening methods (Aharoni et al., 2005) have been reviewed extensively and offer a powerful tool to evolve promiscuous enzyme activity (Bornscheuer and Kazlauskas, 2004; Hult and Berglund, 2007). Enzyme promiscuity refers to the ability of an enzyme to catalyse a reaction distinct from its natural reaction with mechanistic connections (Khersonsky and Tawfik, 2010). Nature possibly employs this strategy for providing jump‐off points to evolve new enzymes. Protein databases typically contain sequence information for which no function is known. This limits our understanding of Nature's enzymatic diversity and lack of knowledge about its function means that industrial application will be difficult to implement. Accordingly, there have been efforts to use computational approaches (like in silico ligand docking) (Song et al., 2007), mechanistic enzymology (Palmer et al., 1999; Taylor Ringia et al., 2004; Sakai et al., 2009) and structural biology (Gulick et al., 2001; Thoden et al., 2004) for guiding laboratory exploration to assign function to unknown and uncharacterized enzymes (Gerlt, 2007).
Using computational design and in vitro evolution, retro‐aldolases were designed (Röthlisberger et al., 2008) to catalyse Kemp elimination reactions for which naturally occurring biocatalysts are not available (Casey et al., 1973; Kemp and Casey, 1973). The evolved retro‐aldolases exhibited catalytic efficiencies comparable to most of the Kemp elimination catalysts such as 5‐nitro‐benzisoxazole and catalysed cleavage of C–C bonds in unnatural substrates. Additionally, the X‐ray structure of the evolved variants exhibited exact superimposition to the designed models (Röthlisberger et al., 2008). Computational approaches have also been applied to improving thermostability (Korkegian et al., 2005) and altering catalytic activity of enzymes (Lassila et al., 2005) and further developments should enhance our ability to cope with more complex and multistep reaction mechanisms. A combination of computational design (to construct active‐site framework) and directed evolution (to fine tune) represent powerful tools to evolve new enzyme functions which are not known to exist in nature.
Enzymatic process engineering
As an alternative to labour‐intensive approach of protein engineering or screening for new enzyme activities, one may also employ the strategy of systematic alteration of reaction components and/or conditions, such as solvent, additive, temperature, enzyme formulation, substrate, etc. to access desired activity and explore possibilities of new reactions using known enzymes.
Use of enzymes in organic solvents has now become a major thrust area on biocatalysis since it allows solubilization of insoluble substrates and modification of enzyme properties (Klibanov, 2001). However, the choice of an organic solvent requires considerations of enzyme behaviour, partitioning of substrate and product from the enzyme, reaction equilibrium, hydrolytic side reactions, etc. (Martineka et al., 1981; Halling, 1994; Heinemann et al., 2003). At present there exist no precise rules for making choice of an appropriate solvent; however, certain studies have correlated enzyme behaviour to various solvent properties (Kaul and Banerjee, 2008), particularly logP (Laane et al., 1987). The scope and breadth of enzyme‐catalysed reactions in organic media has been extensively reviewed (Zaks and Russel, 1988; Gupta, 1992; Carrea and Riva, 2008; Doukyua and Ogino, 2010) and the techniques have been applied particularly to lipases. Interestingly, lipases catalyse esterification in organic medium (Fig. 7A) and hydrolysis in aqueous medium (Fig. 7B), both reactions yielding opposite enantiomers of the product under different conditions (Breitgoff et al., 1986; Terao et al., 1988; Wang and Wong, 1988). Lipase‐catalysed transesterification reactions have largely been executed in hydrophobic solvents; however, a study describing asymmetric transesterification of glycerol with acyl donors (such as vinyl benzoate) utilized hydrophilic solvents (Kato et al., 1999b; 2000c) (Fig. 8). About 40 commercially available lipase sources were screened for the asymmetric transesterification reaction and finally CHIRAZYME L‐2 (Candida antarctica) provided access to (R)‐α‐monobenzoyl glycerol in 1,4‐dioxane. The study therefore highlighted a novel yet simple single step route to optically active chiral building block from a prochiral substrate. Since one of the major impediments to use non‐aqueous enzymology is maintaining high catalytic activity of enzymes, one may use approaches such as enzyme modification, inclusion of additives and molecular imprinting to enhance enzyme properties. Dissolving the enzyme in a solution of ligand (usually a substrate or its analogue) and subsequent lyophilization results in an enzyme preparation with embossed active site capable of retaining memory upon placement in anhydrous solvent (due to more rigid active‐site conformation). The memory is however lost if the enzyme is placed in aqueous medium since the proteins become more flexible (due to high dielectric constant of water) and tend to retain their original active‐site conformation. This provides a means to fine tune not only enzyme activity and specificity (Rich and Dordick, 1997) but also its enantioselectivity (Okahata et al., 1995). For successful implementation of this approach, the ligand must be completely soluble in aqueous medium (from which the enzyme is lyophilized). However, most of the compounds of commercial interest are practically insoluble in water which significantly limits the applicability of molecular imprinting. Overcoming this limitation requires one to modify the ligand or the aqueous medium in order to enhance its solubility to achieve improved enzyme performance in organic solvents (Rich et al., 2002). Ionic liquids have been used as a replacement of organic solvents and offer the advantage of allowing solubilization of both hydrophobic and hydrophilic molecules (Kragl et al., 2002; Park and Kazlauskas, 2003; van Rantwijk et al., 2003; Song, 2004). These liquids are generally known to be mild and significantly enzyme‐friendly and have allowed enzymes to exhibit improved yield and selectivity, e.g. lipase‐B from C. antarctica (CAL‐B) when used in ionic liquids resulted in significantly higher yield and regioselectivity for acylation of glucose (99% yield and 93% selectivity) compared with the reaction performed in organic solvents (73% yield and 76% selectivity) (Park and Kazlauskas, 2001).
Figure 7.

Lipase‐mediated route to both enantiomers of monoacetate of benzoyl glycerol by engineering the reaction medium. Panel A: Transesterification on organic solvent. Panel B: Hydrolysis in aqueous media.
Figure 8.
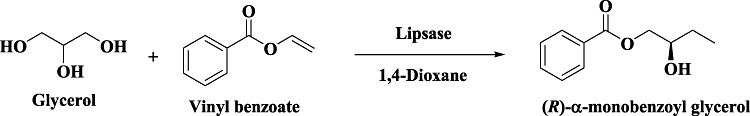
Lipase‐catalysed asymmetric transesterification to produce α‐monobenzoyl glycerol.
Reaction temperature has been known to have a significant effect on enzyme enantioselectivity (Phillips, 1992; 1996) by influencing activation energies of the substrate enantiomers and thereby providing a means to regulate the enantiomer ‘traffic’ at the active site. Typically, the activation‐free energy may be separated into enthalpic and entropic components, which are normally known to counteract one another (referred to as enthalpy‐entropy compensation phenomenon) and a number of studies have reported enhancement of enantioselectivity by decreasing temperature (result of greater contribution from activation enthalpy) (Persson et al., 2002; Kaul et al., 2007) and also enhanced enantioselectivity with increasing temperature (result of greater contribution from activation entropy) (Ottosson et al., 2001). However, this also sets a limitation on practical utility of the approach due to too low reaction rates at low temperatures and rapid enzyme inactivation at elevated temperatures. Modulating the reaction medium with use of organic solvents has been shown to be an effective way to overcome the problem of biocatalysis at subzero temperatures (Sakai et al., 1998; 2004).
This section represents only a tiny fraction of the large volume of literature available on this subject, to provide the reader with an overview of the possibilities achievable with reaction engineering.
Prospects and opportunities
For practical application an enzyme must ideally demonstrate process adaptability (in terms of stability, substrate spectrum, inhibition profile, etc.) without forcing the process engineer to alter production parameters to suit the enzyme (resulting in a compromised process). In spite of greater amalgamation of biocatalysis into chemical synthesis, industrial applications have been modest mainly due to lack of the desired enzyme. However, application of these search tools can help create opportunities for adopting a biostrategy by not only providing access to such enzyme systems but also lead to process improvements from a commercial view point. Some of these potential opportunities will be discussed below.
Sulfation chemistry and sulfated scaffolds generated using these techniques find numerous applications in various industrial sectors ranging from analytical (Stalcup and Gahm, 1996) to pharmaceutical (Dorfman et al., 2006) and consumer products (Salka and Bator, 1990). However, majority of these scaffolds are prepared using chemical methods that suffer from numerous disadvantages. Although sulfation appears to be a single‐step reaction, its execution by chemical methods presents significant challenges (Al‐Horania and Desai, 2010). Lability of sulfate groups to harsh conditions (acidic pH and high temperature) coupled by lack of manoeuvrability following introduction of sulfate group complicates the process conditions. Introduction of a sulfate group allows only a few functional group transformations to be successfully executed forcing the design of the synthetic scheme to include sulfation as the final step. The above complications generally increase geometrically for poly‐sulfated scaffolds. Additionally, the chemical sulfation process is highly energy intensive which requires excessive cooling (exothermic reaction). A still larger segment of the consumer product industry uses sulfated detergents in most finished products such as shampoos, soaps, cleaners, etc. Most of these products (using sulfated esters and detergents) are contaminated by 1,4‐dioxane, a potential carcinogen that is introduced by a process of ethoxylation (Black et al., 2001). Federal policies governing consumer safety allow carcinogenic contaminants to be present in small amounts and do not compel the manufacturers to remove them completely, which has led to many companies being sued for negligence in the past (NewsInferno, 2001; PRLog Press Release, 2008). These problems can be circumvented by introducing an enzymatic step of sulfation, which would allow precise control over reaction selectivity and achieve sulfation of complex chemical scaffolds in a single step, without the need for employing protection and deprotection strategies, under mild reaction conditions. Sulfotransferases catalyse transfer of a sulfate group from a donor molecule to an acceptor (usually and alcohol or amine) (Negishi et al., 2001). Bacterial sources of the enzyme are known to utilize p‐nitropheynl sulfate (PNPS) as a donor which has relatively a low cost and favours process economics for industrial application and also allows execution of high‐throughput screening approach, since enzymatic sulfation can be easily monitored by measuring absorbance at 400 nm for formation of p‐nitrophenol (PNP) (Fig. 9). Screening of human faeces samples leads to identification of Eubacterium A‐44 as a producer of arylsulfotransferase (Kobashi et al., 1986) which has been used for sulfation to produce molecules of therapeutic interest, such as cholecystokinin (Hagiwara et al., 1990a), angiotensin‐II (Hagiwara et al., 1990b), tannins (Koizumi et al., 1991), among many others. Application of sulfotransferases to a broader range of molecules has been restricted mainly due to narrow substrate spectrum of these enzymes. This provides a favourable starting point for using discovery and engineering tools to this enzyme for executing a broad range of sulfation reactions of different templates with applications in chemical and pharmaceutical industry.
Figure 9.
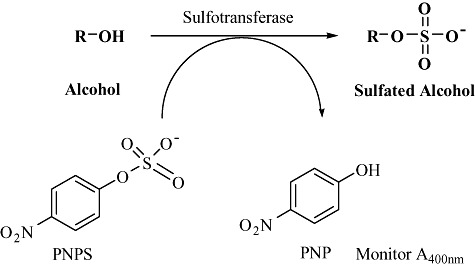
Enzymatic synthesis of sulfated scaffolds using bacterial sulfotransferases that utilize PNPS as cofactor and allow activity determination in high‐throughput format.
Methylation reactions involve the use of electrophilic methyl sources, such as methyl halides, sulfates, carbonates, etc.; however, executing it in a regioselective manner is very difficult chemically and requires approaches such as solvent and/or solute manipulation to alter nucleophilicity of competing nucleophiles (Duran et al., 2004) or using selective protection strategies (Bruneta et al., 2007). Enzymatically this can be achieved by using methyltransferases; however, factors such as low reactivity rate and use of costly cofactors (S‐adenosyl methionine or SAM) have significantly hindered their application on a commercial scale. These enzymes are known to be involved in the terminal step of antibiotic synthesis such as erythromycin, saunomycin, rapamycin, etc. (Seno and Baltz, 1982; Weber et al., 1989). Application of the discovery tools to engineer the cofactor specificity, regioselectivity of the enzyme along with its stability and reaction rate represent attractive opportunities for industrial sectors. Additionally, a methyltransferase that could execute methylation of 6‐hydroxy group of erythromycin A could provide a convenient single‐step route to clarithromycin which presently requires multiple synthetic steps (Watanabe et al., 1993).
Some of the most recent advancements achieved in the field of biocatalytic approaches on industrial scale have been covered in this review and potential opportunities discussed. It remains uncertain as to which technique specifically offers a significant edge in terms of identifying a potential hit; however, devising a discovery strategy by using some or all of these tools renders opportunities to be explored further. There is no doubt that the uncultivable resource of microorganisms (> 99%) symbolizes an immense opportunity for discovering novel enzyme systems with unique properties and we have hardly scratched the surface. Information available from sequence databases should strengthen our abilities to predict enzyme function, specially by using more insightful tools such as the 3DM database to execute protein engineering (Kourist et al., 2010). Prudent mining of genome databases may actually be a smart way to discover new enzymes and results in findings where nature has optimized a biocatalyst beyond human wisdom (Grosse et al., 2010). Increasing body of knowledge and information gathered through utilization of the tools (described herein) will facilitate the pursuit for the development of an ideal biocatalytic process. Future applications will witness greater integration of traditional and molecular approaches to achieve tailor‐made enzymes and integrated metabolic pathways for use in industrial sectors.
References
- Advanced Technology Platform, National Institute of Standards and Technology US. 1998. ) Catalysis and biocatalysis technologies: leveraging resources and targeting performance [WWW document]. URL http://www.atp.nist.gov/atp/97wp‐cat.htm.
- Agematu H., Matsumoto N., Fujii Y., Kabumoto H., Doi S., Machida K. Hydroxylation of testosterone by bacterial cytochromes P450 using the Escherichia coli expression system. Biosci Biotechnol Biochem. 2006;70:307–311. doi: 10.1271/bbb.70.307. et al. [DOI] [PubMed] [Google Scholar]
- Agranat I., Caner H., Caldwell J. Putting chirality to work: the strategy of chiral switches. Nat Rev Drug Discov. 2002;1:753–768. doi: 10.1038/nrd915. [DOI] [PubMed] [Google Scholar]
- Aharoni A., Griffiths A.D., Tawfik D.S. High‐throughput screens and selections of enzyme‐encoding genes. Curr Opin Chem Biol. 2005;9:210–216. doi: 10.1016/j.cbpa.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Ahmed S.A., Esaki N., Tanaka H., Soda K. Mechanism of α‐amino‐ε‐caprolactam racemase. Biochemistry. 1986;25:385–388. doi: 10.1021/bi00350a017. [DOI] [PubMed] [Google Scholar]
- Al‐Horania R.A., Desai U.R. Chemical sulfation of small molecules – advances and challenges. Tetrahedron. 2010;66:2907–2918. doi: 10.1016/j.tet.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariens E.J. Stereochemistry: a basis for sophisticated nonsense in pharmacokinetics and clinical pharmacology. Eur J Clin Pharmacol. 1984;26:663–668. doi: 10.1007/BF00541922. [DOI] [PubMed] [Google Scholar]
- Asano Y. Overview of screening for new microbial catalysts and their uses in organic synthesis‐selection and optimization of biocatalysts. J Biotechnol. 2002;94:65–72. doi: 10.1016/s0168-1656(01)00419-9. [DOI] [PubMed] [Google Scholar]
- Asano Y., Kato Y. Z‐Phenylacetaldoxime degradation by a novel aldoxime dehydratase from Bacillus sp. strain OxB‐1. FEMS Microbiol Lett. 1998;158:185–190. [Google Scholar]
- Asano Y., Yamaguchi S. Dynamic kinetic resolution of amino acid amide racemizing catalysed by d‐aminopeptidase and α‐amino‐ε‐caprolactam racemase. J Am Chem Soc. 2005a;127:7696–7697. doi: 10.1021/ja050300m. [DOI] [PubMed] [Google Scholar]
- Asano Y., Yamaguchi S. Discovery of new substrates amino acid amides for α‐amino‐ε‐caprolactam racemase from Achromobacter obae. J Mol Catal B Enzym. 2005b;36:22–29. [Google Scholar]
- Asano Y., Tani Y., Yamada H. A new enzyme ‘nitrile hydratase’ which degrades acetonitrile in combination with amidase. Agric Biol Chem. 1980;44:2251–2252. [Google Scholar]
- Asano Y., Yasuda T., Tani Y., Yamada H. A new enzymatic method of acrylamide production. Agric Biol Chem. 1982;46:1183–1189. [Google Scholar]
- Asano Y., Kira I., Yokozeki K. Alteration of substrate specificity of aspartase by directed evolution. Biomol Eng. 2005;22:95–101. doi: 10.1016/j.bioeng.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Asano Y., Fukuta Y., Yoshida Y., Komeda H. The screening, characterization, and use of omega‐laurolactam hydrolase: a new enzymatic synthesis of 12‐aminolauric acid. Biosci Biotechnol Biochem. 2008;72:2141–2150. doi: 10.1271/bbb.80210. [DOI] [PubMed] [Google Scholar]
- Auclair K., Sutherland A., Kennedy K., Witter D.J., Van den Heever J.P., Hutchinson C.R., Vederas J.C. Lovastatin nonaketide synthase catalyzes an intramolecular Diels‐Alder reaction of a substrate analogue. J Am Chem Soc. 2000;122:11519–11520. [Google Scholar]
- Bayer S., Birkemeyer C., Ballschmiter M. A nitrilase from a metagenomic library acts regioselectively on aliphatic dinitriles. Appl Microbiol Biotechnol. 2011;89:91–98. doi: 10.1007/s00253-010-2831-9. [DOI] [PubMed] [Google Scholar]
- Betton J.M. High throughput cloning and expression strategies for protein production. Biochimie. 2004;86:601–605. doi: 10.1016/j.biochi.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Black R.E., Hurley F.J., Havery D.C. Occurrence of 1,4‐dioxane in cosmetic raw materials and finished cosmetic products. J AOAC Int. 2001;84:666–670. [PubMed] [Google Scholar]
- Bornscheuer U.T., Kazlauskas R.J. Reaction specificity of enzymes: catalytic promiscuity in biocatalysis: using old enzymes to form new bonds and follow new pathways. Angew Chem Int Ed. 2004;43:6032–6040. doi: 10.1002/anie.200460416. [DOI] [PubMed] [Google Scholar]
- Bradbury B. Automated colony picker for high throughput genomics/proteomics. Proteomics. 2004;4:893. [Google Scholar]
- Breitgoff D., Laumen K., Schneider M.P. Enzymatic differentiation of the enantiotopic hydroxymethyl groups of glycerol; synthesis of chiral building blocks. J Chem Soc Chem Commun. 1986;20:1523–1524. [Google Scholar]
- Brennan Y., Callen W.N., Christoffersen L., Dupree P., Goubet F., Healey S. Unusual microbial xylanases from insect guts. Appl Environ Microbiol. 2004;70:3609–3617. doi: 10.1128/AEM.70.6.3609-3617.2004. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennerova M.V., Josefiova J., Brenner V., Pieper D.H., Junca H. Metagenomics reveals diversity and abundance of meta‐cleavage pathways in microbial communities from soil highly contaminated with jet fuel under air‐sparging bioremediation. Environ Microbiol. 2009;11:2216–2227. doi: 10.1111/j.1462-2920.2009.01943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breves R., Maurer K.H., Eck J., Lorenz P., Zinke H. 2003.
- Bruneta E., Muñoza D.M., Parraa F., Mantecóna S., Juanesa O., Rodríguez‐Ubisa J.C. Preparation of clarithromycin. Selective 6‐O‐methylation of the novel erythromycin A 9‐O‐(2‐pyrimidyl)oxime. Tetrahedron Lett. 2007;48:1321–1324. [Google Scholar]
- Carrea G., Riva S. Wiley‐VCH; 2008. [Google Scholar]
- Casey M.L., Kemp D.S., Paul K.G., Cox D.D. The physical organic chemistry of benzisoxazoles I. The mechanism of the base‐catalyzed decomposition of benzisoxazoles. J Org Chem. 1973;38:2294–2301. [Google Scholar]
- Castle L.A., Siehl D.L., Gorton R., Patten P.A., Chen Y.H., Bertain S. Discovery and directed evolution of a glyphosate tolerance gene. Science. 2004;304:1151–1154. doi: 10.1126/science.1096770. et al. [DOI] [PubMed] [Google Scholar]
- Chen T.K., Tien Y.I., Wei K.H. Synthesis and characterization of novel segmented polyurethane/clay nanocomposites. Polymer. 2000;41:1345–1353. [Google Scholar]
- Courtois S., Cappellano C.M., Ball M., Francou F.X., Normand P., Helynck G. Recombinant environmental libraries provide access to microbial diversity for drug discovery from natural products. Appl Environ Microbiol. 2003;69:49–55. doi: 10.1128/AEM.69.1.49-55.2003. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan D., Meyer Q., Stafford W., Muyanga S., Cameron R., Wittwer P. Metagenomic gene discovery: past, present and future. Trends Biotechnol. 2005;23:321–329. doi: 10.1016/j.tibtech.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Crameri A., Raillard S.A., Bermudez E., Stemmer W.P. DNA shuffling of a family of genes from diverse species accelerates directed evolution. Nature. 1998;391:288–291. doi: 10.1038/34663. [DOI] [PubMed] [Google Scholar]
- DeSantis G., Zhu Z., Greenberg W.A., Wong K., Chaplin J., Hanson S.R. An enzyme library approach to biocatalysis: development of nitrilases for enantioselective production of carboxylic acid derivatives. J Am Chem Soc. 2002;124:9024–9025. doi: 10.1021/ja0259842. et al. [DOI] [PubMed] [Google Scholar]
- Dherbécourt J., Falentin H., Canaan S., Thierry A. A genomic search approach to identify esterases in Propionibacterium freudenreichii involved in the formation of flavour in Emmental cheese. Microb Cell Fact. 2008;7:16. doi: 10.1186/1475-2859-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfman T., Moore M.J., Guth A.C., Choe H., Farzan M. A tyrosine‐sulfated peptide derived from the heavy‐chain CDR3 region of an HIV‐1‐neutralizing antibody binds gp120 and inhibits HIV‐1 infection. J Biol Chem. 2006;281:28529–28535. doi: 10.1074/jbc.M602732200. [DOI] [PubMed] [Google Scholar]
- Doukyua N., Ogino H. Organic solvent‐tolerant enzymes. Biochem Eng J. 2010;48:270–282. [Google Scholar]
- Duran D., Aviyente V., Baysal C. Solvent effect on the synthesis of clarithromycin: a molecular dynamics study. J Comput Aided Mol Des. 2004;18:145–154. doi: 10.1023/b:jcam.0000030037.67742.cb. [DOI] [PubMed] [Google Scholar]
- Endo T., Tamura K. 1991.
- Endo T., Tamura K. 1994. , and ) Process for producing R(‐)‐mandelic acid or a derivative thereof from a mandelonitrile using Rhodococcus. US Patent 5296373.
- Endo T., Yamagami T., Tamura K. 1994.
- European Technology Platform for Sustainable Chemistry. 2003. ) White biotechnology: gateway to a more sustainable future [WWW document]. URL http://www.europabio.org/documents/100403/Innenseiten_final_screen.pdf.
- Favre‐Bulle O., Pierrard J., David C., Morel P., Horbez D. 2001.
- Fernández‐Álvaro E., Kourist R., Winter J., Böttcher D., Liebeton K., Naumer C. Enantioselective kinetic resolution of phenylalkyl carboxylic acids using metagenome‐derived esterases. Microb Biotechnol. 2010;3:59–64. doi: 10.1111/j.1751-7915.2009.00141.x. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer M., Beloqui A., Golyshina O.V., Plou F.J., Neef A., Chernikova T.N. Biochemical and structural features of a novel cyclodextrinase from cow rumen metagenome. Biotechnol J. 2007;2:207–213. doi: 10.1002/biot.200600183. et al. [DOI] [PubMed] [Google Scholar]
- Fraaije M.W., Kamerbeeka N.M., van Berkelb W.J.H., Janssen D.B. Identification of a Baeyer‐Villiger monooxygenase sequence motif. FEBS Lett. 2002;518:43–47. doi: 10.1016/s0014-5793(02)02623-6. [DOI] [PubMed] [Google Scholar]
- Fraaije M.W., Wu J., Heuts D.P.H.M., van Hellemond E.W., Spelberg J.H.L., Janssen D.B. Discovery of a thermostable Baeyer‐Villiger monooxygenase by genome mining. Appl Microbiol Biotechnol. 2004;66:393–400. doi: 10.1007/s00253-004-1749-5. [DOI] [PubMed] [Google Scholar]
- Fukumura T. Conversion of d‐ and dl‐α‐amino‐ε‐caprolactam into l‐lysine using both yeast cells and bacterial cells. Agric Biol Chem. 1977a;41:1327–1330. [Google Scholar]
- Fukumura T. Bacterial racemisation of α‐amino‐ε‐caprolactam. Agric Biol Chem. 1977b;41:1321–1325. [Google Scholar]
- Fukuta Y., Komeda H., Yoshida Y., Asano Y. High yield synthesis of 12‐aminolauric acid by ‘enzymatic transcrystallization’ of omega‐laurolactam using omega‐laurolactam hydrolase from Acidovorax sp. T31. Biosci Biotechnol Biochem. 2009;73:980–986. doi: 10.1271/bbb.80551. [DOI] [PubMed] [Google Scholar]
- Gabor E.M., de Vries E.J., Janssen D.B. Construction, characterization, and use of small‐insert gene banks of DNA isolated from soil and enrichment cultures for the recovery of novel amidases. Environ Microbiol. 2004;6:948–958. doi: 10.1111/j.1462-2920.2004.00643.x. [DOI] [PubMed] [Google Scholar]
- Gerlt J.A. A protein structure (or function ?) initiative. Structure. 2007;15:1353–1356. doi: 10.1016/j.str.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Gloux K., Leclerc M., Iliozer H., L'Haridon R., Manichanh C., Corthier G. Development of high‐throughput phenotyping of metagenomic clones from the human gut microbiome for modulation of eukaryotic cell growth. Appl Environ Microbiol. 2007;73:3734–3737. doi: 10.1128/AEM.02204-06. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green B.D., Keller M. Capturing the uncultivated majority. Curr Opin Biotechnol. 2006;17:236–240. doi: 10.1016/j.copbio.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Grosse S., Bergeron H., Imura A., Boyd J., Wang S., Kubota K. Nature versus nurture in two highly enantioselective esterases from Bacillus cereus and Thermoanaerobacter tengcongensis. Microb Biotechnol. 2010;3:65–73. doi: 10.1111/j.1751-7915.2009.00142.x. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick A.M., Schmidt D.M., Gerlt J.A., Rayment I. Evolution of enzymatic activities in the enolase superfamily: crystal structures of the l‐Ala‐d/l‐Glu epimerases from Escherichia coli and Bacillus subtilis. Biochemistry. 2001;40:15716–15724. doi: 10.1021/bi011641p. [DOI] [PubMed] [Google Scholar]
- Gupta M.N. Enzyme function in organic solvents. Eur J Biochem. 1992;203:25–32. doi: 10.1111/j.1432-1033.1992.tb19823.x. [DOI] [PubMed] [Google Scholar]
- Hagiwara M., Ohuchi E., Hongo K., Oki M., Nakano M., Amemiya M. Pharmacological activities of synthetic human cholecystokinin‐33 of which tyrosine was sulfated by arylsulfotransferase. Chem Pharm Bull. 1990a;38:1369–1372. doi: 10.1248/cpb.38.1369. et al. [DOI] [PubMed] [Google Scholar]
- Hagiwara M., Ohuchi E., Hongo K., Oki M., Wada K., Morikawa T., Kobashi K. Pharmacological activity of angiotensin‐II modified by tyrosine sulfation. Jpn J Pharmacol. 1990b;52:493–495. doi: 10.1254/jjp.52.493. [DOI] [PubMed] [Google Scholar]
- Halling P.J. Thermodynamic predictions for biocatalysis in nonconventional media: theory, tests, and recommendations for experimental design and analysis. Enzyme Microb Technol. 1994;16:178–206. doi: 10.1016/0141-0229(94)90043-4. [DOI] [PubMed] [Google Scholar]
- Hann E.C., Eisenberg A., Fager S.K., Perkins N.E., Gallagher F.G., Cooper S.M. 5‐Cyanovaleramide production using immobilized Pseudomonas chlororaphis B23. Bioorg Med Chem. 1999;7:2239–2245. doi: 10.1016/s0968-0896(99)00157-1. et al. [DOI] [PubMed] [Google Scholar]
- Heinemann M., Kümmel A., Giesen R., Ansorge‐Schumacher M.B., Buchs J. Experimental and theoretical analysis of phase equilibria in a two‐phase system used for biocatalytic esterifications. Biocatal Biotransform. 2003;21:115–121. [Google Scholar]
- Henke E., Bornscheuer U. Esterases from Bacillus subtilis and B. stearothermophilus share high sequence homology but differ substantially in their properties. Appl Microbiol Biotechnol. 2002;60:320–326. doi: 10.1007/s00253-002-1126-1. [DOI] [PubMed] [Google Scholar]
- Henrissat B., Coutinho P.M., Davies G.J. A census of carbohydrate‐active enzymes in the genome of Arabidopsis thaliana. Plant Mol Biol. 2001;47:55–72. [PubMed] [Google Scholar]
- Hibbert E.G., Senussi T., Smith M.E.B., Costelloe S.J., Ward J.M., Hailes H.J., Dalby P.A. Directed evolution of transketolase substrate specificity towards an aliphatic aldehyde. J Biotechnol. 2008;134:240–245. doi: 10.1016/j.jbiotec.2008.01.018. [DOI] [PubMed] [Google Scholar]
- Ho K., Xiao Q., Fach E.M., Hulmes J.D., Bethea D., Opiteck G.J. Semi‐automated sample preparation for plasma proteomics. J Assoc Lab Autom. 2004;9:238–249. et al. [Google Scholar]
- Hult K., Berglund P. Enzyme promiscuity: mechanism and applications. Trends Biotechnol. 2007;25:231–238. doi: 10.1016/j.tibtech.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Japan Bioindustry Association, Biotechnology Strategy Council. 2002. ) Biotechnology strategy guidelines [WWW document]. URL http://www.jba.or.jp/jabex/pdf/BT%20Strategy%20Guideline%20(translated%20by%20JETRO).pdf.
- Jeon J.H., Kim J.T., Kim Y.J., Kim H.K., Lee H.S., Kang S.G. Cloning and characterization of a new cold‐active lipase from a deep‐sea sediment metagenome. Appl Microbiol Biotechnol. 2009;81:865–874. doi: 10.1007/s00253-008-1656-2. [DOI] [PubMed] [Google Scholar]
- Jiang C., Shen P., Yan B., Wua B. Biochemical characterization of a metagenome‐derived decarboxylase. Enzyme Microb Technol. 2009;45:58–63. [Google Scholar]
- Jiang C., Li S.X., Luo F.F., Jin K., Wang Q., Hao Z.Y. Biochemical characterization of two novel β‐glucosidase genes by metagenome expression cloning. Bioresour Technol. 2011;102:3272–3278. doi: 10.1016/j.biortech.2010.09.114. et al. [DOI] [PubMed] [Google Scholar]
- Kaluzna I.A., Matsuda T., Sewell A.K., Stewart J.D. Systematic investigation of Saccharomyces cerevisiae enzymes catalyzing carbonyl reductions. J Am Chem Soc. 2004;126:12827–12832. doi: 10.1021/ja0469479. [DOI] [PubMed] [Google Scholar]
- Kamagata Y., Tamaki H. Cultivation of uncultured fastidious microbes. Microbes Environ. 2005;20:85–91. [Google Scholar]
- Kato Y., Ooi R., Asano Y. Isolation and characterization of a bacterium possessing a novel aldoxime‐dehydration activity and nitrile‐degrading enzymes. Arch Microbiol. 1998;170:85–90. doi: 10.1007/s002030050618. [DOI] [PubMed] [Google Scholar]
- Kato Y., Ooi R., Asano Y. A new enzymatic method of nitrile synthesis by Rhodococcus sp. strain YH3‐3. J Mol Catal B Enzym. 1999a;6:249–256. [Google Scholar]
- Kato Y., Fujiwara I., Asano Y. A novel method for preparation of optically active α‐monobenzoyl glycerol via lipase catalyzed asymmetric transesterification of glycerol. Bioorg Med Chem Lett. 1999b;9:3207–3210. doi: 10.1016/s0960-894x(99)00556-9. [DOI] [PubMed] [Google Scholar]
- Kato Y., Ooi R., Asano Y. Distribution of aldoxime dehydratase in microorganisms. Appl Environ Microbiol. 2000a;66:2290–2296. doi: 10.1128/aem.66.6.2290-2296.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y., Nakamura K., Sakiyama H., Mayhew S.G., Asano Y. A novel heme‐containing lyase, phenylacetaldoxime dehydratase from Bacillus sp. strain OxB‐1: purification, characterization, and molecular cloning of the gene. Biochemistry. 2000b;39:800–809. doi: 10.1021/bi991598u. [DOI] [PubMed] [Google Scholar]
- Kato Y., Fujiwara I., Asano Y. Synthesis of optically active α‐monobenzoyl glycerol by asymmetric transesterification of glycerol. J Mol Catal B Enzym. 2000c;9:193–200. [Google Scholar]
- Kaul P., Banerjee U.C. Predicting enzyme behavior in nonconventional media: correlating nitrilase function with solvent properties. J Ind Microbiol Biotechnol. 2008;35:713–720. doi: 10.1007/s10295-008-0332-y. [DOI] [PubMed] [Google Scholar]
- Kaul P., Stolz A., Banerjee U.C. Cross‐linked amorphous nitrilase aggregates for enantioselective nitrile hydrolysis. Adv Synth Catal. 2007;349:2167–2176. [Google Scholar]
- Kawata T., Ogino H. Enhancement of the organic solvent‐stability of the LST‐03 lipase by directed evolution. Biotechnol Prog. 2009;25:1605–1611. doi: 10.1002/btpr.264. [DOI] [PubMed] [Google Scholar]
- Kemp D.S., Casey M.L. Physical organic chemistry of benzisoxazoles II. Linearity of the bronsted free energy relationship for the base‐catalyzed decomposition of benzisoxazoles. J Am Chem Soc. 1973;95:6670–6680. [Google Scholar]
- Khersonsky O., Tawfik D.S. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu Rev Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- Kim J.H., Choi G.S., Kim S.B., Kim W.H., Lee J.Y., Ryu Y.W., Kim G.J. Enhanced thermostability and tolerance of high substrate concentration of an esterase by directed evolution. J Mol Catal B Enzym. 2004;27:169–175. [Google Scholar]
- Klibanov A.M. Improving enzymes by using them in organic solvents. Nature. 2001;409:241–246. doi: 10.1038/35051719. [DOI] [PubMed] [Google Scholar]
- Knietsch A., Bowien S., Whited G., Gottschalk G., Daniel R. Identification and characterization of coenzyme B12‐dependent glycerol dehydratase‐ and diol dehydratase‐encoding genes from metagenomic DNA libraries derived from enrichment cultures. Appl Environ Microbiol. 2003a;69:3048–3060. doi: 10.1128/AEM.69.6.3048-3060.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knietsch A., Waschkowitz T., Bowien S., Henne A., Daniel R. Construction and screening of metagenomic libraries derived from enrichment cultures: generation of a gene bank for genes conferring alcohol oxidoreductase activity on Escherichia coli. Appl Environ Microbiol. 2003b;69:1408–1416. doi: 10.1128/AEM.69.3.1408-1416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobashi K., Fukaya Y., Kim D.H., Akao T., Takebe S. A novel type of aryl sulfotransferase obtained from an anaerobic bacterium of human intestine. Arch Biochem Biophys. 1986;245:537–539. doi: 10.1016/0003-9861(86)90247-x. [DOI] [PubMed] [Google Scholar]
- Koeller K.M., Wong C.H. Enzymes for chemical synthesis. Nature. 2001;409:232–240. doi: 10.1038/35051706. [DOI] [PubMed] [Google Scholar]
- Koizumi M., Akao T., Kibuchi T., Okuda T., Kobashi K. Enzymatic sulfation of polyphenols related to tannins by arylsulfotransferase. Chem Pharm Bull. 1991;39:2638–2643. doi: 10.1248/cpb.39.2638. [DOI] [PubMed] [Google Scholar]
- Kojima Y., Usuki A., Kawasumi M., Okada A., Kurauchi T., Kamigaito O. One‐pot synthesis of nylon 6–clay hybrid. J Polym Sci [A1] 2000;31:1755–1758. [Google Scholar]
- Komeda H., Ishikawa N., Asano Y. Enhancement of the thermostability and catalytic activity of stereospecific amino‐acid amidase from Ochrobactrum anthropi SV3 by directed evolution. J Mol Catal B Enzym. 2003;21:283–290. [Google Scholar]
- Korkegian A., Black M.E., Baker D., Stoddard B.L. Computational thermostabilization of an enzyme. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koszelewski D., Lavandera I., Clay D., Rozzell D., Kroutil W. Asymmetric synthesis of optically pure pharmacologically relevant amines employing ω‐transaminases. Adv Synth Catal. 2008;350:2761–2766. [Google Scholar]
- Kotik M., Štěpánek V., Grulich M., Kyslík P., Archelas A. Access to enantiopure aromatic epoxides and diols using epoxide hydrolases derived from total biofilter DNA. J Mol Catal B Enzym. 2010;65:41–48. [Google Scholar]
- Kourist R., Jochens H., Bartsch S., Kuipers R., Padhi S.K., Gall M. The α/β‐hydrolase fold 3DM database (ABHDB) as a tool for protein engineering. Chembiochem. 2010;11:1635–1643. doi: 10.1002/cbic.201000213. et al. [DOI] [PubMed] [Google Scholar]
- Kragl U., Eckstein M., Kaftzik N. Enzyme catalysis in ionic liquids. Curr Opin Biotechnol. 2002;13:565–571. doi: 10.1016/s0958-1669(02)00353-1. [DOI] [PubMed] [Google Scholar]
- Laane C., Tramper J., Lilly M.D. Elsevier; 1987. [Google Scholar]
- Lafferty M., Dycaico M.J. GigaMatrix™: an ultra high‐throughput tool for accessing biodiversity. J Assoc Lab Autom. 2004;9:200–208. [Google Scholar]
- Lassila J.K., Keeffe J.R., Oelschlaeger P., Mayo S.L. Computationally designed variants of Escherichia coli chorismate mutase show altered catalytic activity. Protein Eng Des Sel. 2005;18:161–163. doi: 10.1093/protein/gzi015. [DOI] [PubMed] [Google Scholar]
- Li Y., Wexler M., Richardson D.J., Bond P.L., Johnston A.W.B. Screening a wide host‐range, waste‐water metagenomic library in tryptophan auxotrophs of Rhizobium leguminosarum and of Escherichia coli reveals different classes of cloned trp genes. Environ Microbiol. 2005;7:1927–1936. doi: 10.1111/j.1462-2920.2005.00853.x. [DOI] [PubMed] [Google Scholar]
- Liebeton K., Eck J. Identification and expression in E. coli of novel nitrile hydratases from the metagenome. Eng Life Sci. 2004;4:554–562. [Google Scholar]
- Litthauer D., Abbai N.S., Piater L.A., van Heerden E. Pitfalls using tributyrin agar screening to detect lipolytic activity in metagenomic studies. Afr J Biotechnol. 2010;9:4282–4285. [Google Scholar]
- Loo B., Kingma J., Arand M., Wubbolts M.G., Janssen D.B. Diversity and biocatalytic potential of epoxide hydrolases identified by genome analysis. Appl Environ Microbiol. 2006;72:2905–2917. doi: 10.1128/AEM.72.4.2905-2917.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loo B., Spelberg J.H.L., Kingma J., Sonke T., Wubbolts M.G., Janssen D.B. Directed evolution of epoxide hydrolase from A. radiobacter toward higher enantioselectivity by error‐prone PCR and DNA shuffling. Chem Biol. 2004;11:982–990. doi: 10.1016/j.chembiol.2004.04.019. [DOI] [PubMed] [Google Scholar]
- Lorenz M.G.O. Liquid‐handling robotic workstations for functional genomics. J Assoc Lab Autom. 2004;9:262–267. [Google Scholar]
- McKinsey Report. 2006. ) By 2010 industrial biotechnology will account for 10 percent of sales within the chemical industry [WWW document]. URL http://www.chemeurope.com/en/news/56388/mckinsey‐by‐2010‐industrial‐biotechnology‐will‐account‐for‐10‐percent‐of‐sales‐within‐the‐chemical‐industry.html?pw=a&defop=and&wild=yes&sdate=01%2F01%2F1995&edate=07%2F18%2F2006.
- Martineka K., Semenova A.N., Berezina I.Y. Enzymatic synthesis in biphasic aqueous‐organic systems. I. Chemical equilibrium shift. Biochim Biophys Acta Enzymol. 1981;658:76–89. doi: 10.1016/0005-2744(81)90251-5. [DOI] [PubMed] [Google Scholar]
- Martinez A., Kolvek S.J., Yip C.L.T., Hopke J., Brown K.A., MacNeil I.A., Osburne M.S. Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl Environ Microbiol. 2004;70:2452–2463. doi: 10.1128/AEM.70.4.2452-2463.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May O., Nguyen P.T., Arnold F.H. Inverting enantioselectivity by directed evolution of hydantoinase for improved production of l‐methionine. Nat Biotechnol. 2000;18:317–320. doi: 10.1038/73773. [DOI] [PubMed] [Google Scholar]
- Nagasawa T., Mathew C.D., Mauger J., Yamada Y. Nitrile hydratase‐catalyzed production of nicotinamide from 3‐cyanopyridine in Rhodococcus rhodochrous J‐1. Appl Environ Microbiol. 1988;54:1766–1769. doi: 10.1128/aem.54.7.1766-1769.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa T., Shimizu H., Yamada H. The superiority of the third generation catalyst, Rhodococcus rhodochrous J 1 nitrile hydratase, for the industrial production of acrylamide. Appl Microbiol Biotechnol. 1993;40:189–195. [Google Scholar]
- Negishi M., Pedersen L.G., Petrotchenko E., Shevtsov S., Gorokhov A., Kakuta Y., Pedersen L.C. Structure and function of sulfotransferases. Arch Biochem Biophys. 2001;390:149–157. doi: 10.1006/abbi.2001.2368. [DOI] [PubMed] [Google Scholar]
- NewsInferno. 2001. ) Whole foods, other firms sued for carcinogen in ‘natural’ soap [WWW document]. URL http://www.newsinferno.com/archive/whole‐foods‐other‐firms‐sued‐for‐carcinogen‐in‐natural‐soap/
- Oikawa H. Diels‐Alderases. In: Mander L., Lui H.‐W., editors. Vol. 8. Elsevier; 2010. pp. 277–314. [Google Scholar]
- Oikawa H., Katayama K., Suzuki Y., Ichihara A. Enzymatic activity catalysing exo‐selective Diels‐Alder reaction in solanapyrone biosynthesis. J Chem Soc Chem Commun. 1995;13:1321–1322. [Google Scholar]
- Okahata Y., Hatano A., Ijiro K. Enhancing enantioselectivity if a lipid coated lipase via imprinting methods for esterification in organic solvents. Tetrahedron Asymmetry. 1995;6:1311–1322. [Google Scholar]
- Ottosson J., Rotticci‐Mulder J.‐C., Rotticci D., Hult K. Rational design of enantioselective enzymes requires considerations of entropy. Protein Sci. 2001;10:1769–1774. doi: 10.1110/ps.13501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace N.R., Stahl D.A., Lane D.J., Olsen G.J. The analysis of natural microbial populations by ribosomal RNA sequences. Adv Microb Ecol. 1986;9:1–55. [Google Scholar]
- Pajak L., Zhang R., Pittman C., Roby K., Boyer S. Automated genomic and proteomic applications on the Biomek® NX laboratory automation workstation. J Assoc Lab Autom. 2004;9:177–184. [Google Scholar]
- Palmer D.R., Garrett J.B., Sharma V., Meganathan R., Babbitt P.C., Gerlt J.A. Unexpected divergence of enzyme function and sequence: ‘N‐acylamino acid racemase’ is o‐succinylbenzoate synthase. Biochemistry. 1999;38:4252–4258. doi: 10.1021/bi990140p. [DOI] [PubMed] [Google Scholar]
- Park S., Kazlauskas R.J. Improved preparation and use of room‐temperature ionic liquids in lipase‐catalyzed enantio‐ and regioselective acylations. J Org Chem. 2001;66:8395–8401. doi: 10.1021/jo015761e. [DOI] [PubMed] [Google Scholar]
- Park S., Kazlauskas R.J. Biocatalysis in ionic liquids – advantages beyond green technology. Curr Opin Biotechnol. 2003;14:432–437. doi: 10.1016/s0958-1669(03)00100-9. [DOI] [PubMed] [Google Scholar]
- Persson M., Costes D., Wehtje E., Adlercreutz P. Effects of solvent, water activity and temperature on lipase and hydroxynitrile lyase enantioselectivity. Enzyme Microb Technol. 2002;30:916–923. [Google Scholar]
- Phillips R.S. Temperature effects on stereochemistry of enzymatic reactions. Enzyme Microb Technol. 1992;14:417–419. [Google Scholar]
- Phillips R.S. Temperature modulation of the stereochemistry of enzymatic catalysis: prospects for exploitation. Trends Biotechnol. 1996;14:13–16. [Google Scholar]
- Piel J. A polyketide synthase‐peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles. Proc Natl Acad Sci USA. 2002;99:14002–14007. doi: 10.1073/pnas.222481399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel J., Hui D., Fusetani N., Matsunaga S. Targeting modular polyketide synthases with iteratively acting acyltransferases from metagenomes of uncultured bacterial consortia. Environ Microbiol. 2004;6:921–927. doi: 10.1111/j.1462-2920.2004.00531.x. [DOI] [PubMed] [Google Scholar]
- Pierrard J., Favre‐Bulle O., Jourdat C. 2001.
- PRLog Press Release. 2008. ) California lawsuit; whole foods, avalon and others with products containing carcinogenic 1,4‐dioxane [WWW document]. URL http://www.prlog.org/10079593‐california‐lawsuit‐whole‐foods‐avalon‐and‐others‐with‐products‐containing‐carcinogenic‐1‐4‐dioxane.html.
- Radomski C.C.A., Seow K.T., Warren R.A.J., Yap W.H. 1998.
- van Rantwijk F., Madeira L.R., Sheldon R.A. Biocatalytic transformations in ionic liquids. Trends Biotechnol. 2003;21:131–138. doi: 10.1016/S0167-7799(03)00008-8. [DOI] [PubMed] [Google Scholar]
- Ress‐Loschke M., Friedrich T., Hauer B., Mattes R., Engels D. 2000.
- Rey P., Rossi J., Taillades J., Gros G., Nore O. Hydrolysis of nitriles using an immobilized nitrilase: applications to the synthesis of methionine hydroxy analogue derivatives. J Agric Food Chem. 2004;52:8155–8162. doi: 10.1021/jf048827q. [DOI] [PubMed] [Google Scholar]
- Rich J.O., Dordick J.S. Controlling subtilisin activity and selectivity in organic media by imprinting with nucleophilic substrates. J Am Chem Soc. 1997;119:3245–3252. [Google Scholar]
- Rich J.O., Mozhaev V.V., Dordick J.S., Clark D.S., Khmelnitsky Y.L. Molecular imprinting of enzymes with water‐insoluble ligands for nonaqueous biocatalysis. J Am Chem Soc. 2002;124:5254–5255. doi: 10.1021/ja012219z. [DOI] [PubMed] [Google Scholar]
- Robertson D.E., Chaplin J.A., DeSantis G., Podar M., Madden M., Chi E. Exploring nitrilase sequence space for enantioselective catalysis. Appl Environ Microbiol. 2004;70:2429–2436. doi: 10.1128/AEM.70.4.2429-2436.2004. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondon M.R., August P.R., Bettermann A.D., Brady S.F., Grossman T.H., Liles M.R. Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol. 2000;66:2541–2547. doi: 10.1128/aem.66.6.2541-2547.2000. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röthlisberger D., Khersonsky O., Wollacott1 A.M., Jiang L., DeChancie J., Betker J. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. et al. [DOI] [PubMed] [Google Scholar]
- Sakai A., Fedorov A.A., Fedorov E.V., Schnoes A.M., Glasner M.E., Brown S. Evolution of enzymatic activities in the enolase superfamily: stereochemically distinct mechanisms in two families of cis,cis‐muconate lactonizing enzymes. Biochemistry. 2009;48:1445–1453. doi: 10.1021/bi802277h. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T. Low‐temperature method for a dramatic improvement in enantioselectivity in lipase‐catalyzed reactions. Tetrahedron Asymmetry. 2004;15:2749–2756. [Google Scholar]
- Sakai T., Kishimoto T., Tanaka Y., Ema T., Utaka M. Low‐temperature method for enhancement of enantioselectivity in the lipase‐catalyzed kinetic resolutions of solketal and some chiral alcohols. Tetrahedron Lett. 1998;39:7881–7884. [Google Scholar]
- Salka B.A., Bator P.E. 1990.
- Savile C.K., Janey J.M., Mundorff E.C., Moore J.C., Tam S., Jarvis W.R. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to Sitagliptin manufacture. Science. 2010;329:305–309. doi: 10.1126/science.1188934. et al. [DOI] [PubMed] [Google Scholar]
- Schmeisser C., Steele H., Streit W.R. Metagenomics, biotechnology with non‐culturable microbes. Appl Microbiol Biotechnol. 2007;75:955–962. doi: 10.1007/s00253-007-0945-5. [DOI] [PubMed] [Google Scholar]
- Seffernick J.L., Samanta S.K., Louie T.M., Wackett L.P., Subramanian M. Investigative mining of sequence data for novel enzymes: a case study with nitrilases. J Biotechnol. 2009;143:17–26. doi: 10.1016/j.jbiotec.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Sekiguchi Y. Yet to be cultured microorganisms relevant to methane production fermentation processes. Microbes Environ. 2006;21:1–15. [Google Scholar]
- Seno E.T., Baltz R.H. S‐Adenosyl‐l‐methionine: Macrocin O‐methyltransferase activities in a series of Streptomyces fradiae mutants that produce different levels of the macrolide antibiotic tylosin. Antimicrob Agents Chemother. 1982;21:758–763. doi: 10.1128/aac.21.5.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seow K.T., Meurer G., Gerlitz M., Wendt‐Pienkowski E., Hutchinson C.R., Davies J. A study of iterative type II polyketide synthases, using bacterial genes cloned from soil DNA: a means to access and use genes from uncultured microorganisms. J Bacteriol. 1997;179:7360–7368. doi: 10.1128/jb.179.23.7360-7368.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel J.B., Zanghellini A., Lovick H.M., Kiss G., Lambert A.R., St.Clair J.L. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels‐Alder reaction. Science. 2010;329:309–313. doi: 10.1126/science.1190239. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.B., Pelaez F. Biodiversity, chemical diversity and drug discovery. Prog Drug Res. 2008;65:143–174. doi: 10.1007/978-3-7643-8117-2_4. [DOI] [PubMed] [Google Scholar]
- Song C.E. Enantioselective chemo‐ and bio‐catalysis in ionic liquids. Chem Commun. 2004;7:1033–1043. doi: 10.1039/b309027b. [DOI] [PubMed] [Google Scholar]
- Song L., Kalyanaraman C., Fedorov A.A., Fedorov E.V., Glasner M.E., Brown S. Prediction and assignment of function for a divergent N‐succinyl amino acid racemase. Nat Chem Biol. 2007;3:486–491. doi: 10.1038/nchembio.2007.11. et al. [DOI] [PubMed] [Google Scholar]
- Stalcup A.M., Gahm K.H. A sulfated cyclodextrin chiral stationary phase for high‐performance liquid chromatography. Anal Chem. 1996;68:1369–1374. doi: 10.1021/ac951199e. [DOI] [PubMed] [Google Scholar]
- Steele H.L., Jaeger K.E., Daniel R., Streit W.R. Advances in recovery of novel biocatalysts from metagenomes. J Mol Microbiol Biotechnol. 2009;16:25–37. doi: 10.1159/000142892. [DOI] [PubMed] [Google Scholar]
- Stewart J.D. Genomes as resources for biocatalysis. Adv Appl Microbiol. 2006;59:31–52. doi: 10.1016/S0065-2164(06)59002-1. [DOI] [PubMed] [Google Scholar]
- Streit W.R., Daniel R., Jaeger K.‐E. Prospecting for biocatalysts and drugs in the genomes of non‐cultured microorganisms. Curr Opin Biotechnol. 2004;15:285–290. doi: 10.1016/j.copbio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Strong M. FDA policy and regulation of stereoisomers: paradigm shift and the future of safer more effective drugs. Food Drug Law J. 1999;54:463–487. [PubMed] [Google Scholar]
- Taupp M., Lee S., Hawley A., Yang J., Hallam S.J. Large insert environmental genomic library production. J Vis Exp. 2009;31 doi: 10.3791/1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor Ringia E.A., Garrett J.B., Thoden J.B., Holden H.M., Rayment I., Gerlt J.A. Evolution of enzymatic activity in the enolase superfamily: functional studies of the promiscuous o‐succinylbenzoate synthase from Amycolatopsis. Biochemistry. 2004;43:224–229. doi: 10.1021/bi035815+. [DOI] [PubMed] [Google Scholar]
- Terao Y., Murata M., Achiwa K., Nishio T., Akamtsu M., Kamimura M. Highly efficient lipase‐catalyzed asymmetric synthesis of chiral glycerol derivatives leading to practical synthesis of s‐propranolol. Tetrahedron Lett. 1988;29:5173–5176. [Google Scholar]
- Thoden J.B., Taylor Ringia E.A., Garrett J.B., Gerlt J.A., Holden H.M., Rayment I. Evolution of enzymatic activity in the enolase superfamily: structural studies of the promiscuous o‐succinylbenzoate synthase from Amycolatopsis. Biochemistry. 2004;43:5716–5727. doi: 10.1021/bi0497897. [DOI] [PubMed] [Google Scholar]
- Torsvik V.L., Goksoyr J. Determination of bacterial DNA in soil. Soil Biol Biochem. 1980;10:7–12. [Google Scholar]
- Torsvik V., Ovreas L., Thingstad T.F. Prokaryotic diversity – magnitude, dynamics, and controlling factors. Science. 2002;296:1064–1066. doi: 10.1126/science.1071698. [DOI] [PubMed] [Google Scholar]
- Tracewell C.A., Arnold F.H. Directed enzyme evolution: climbing fitness peaks one amino acid at a time. Curr Opin Chem Biol. 2009;13:3–9. doi: 10.1016/j.cbpa.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufvesson P., Ramos J.L., Nordblad M., Woodley J.M. Guidelines and cost analysis for catalyst production in biocatalytic processes. Org Process Res Dev. 2010;15:266–274. [Google Scholar]
- Turner N.J. Directed evolution of enzymes for applied biocatalysis. Trends Biotechnol. 2003;21:474–478. doi: 10.1016/j.tibtech.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Turner N.J. Directed evolution drives the next generation of biocatalysts. Nat Chem Biol. 2009;5:567–573. doi: 10.1038/nchembio.203. [DOI] [PubMed] [Google Scholar]
- Uchiyama H., Inaoka T., Ohkuma‐Soyejima T., Togame H., Shibanaka Y., Yoshimoto T., Kokubo T. Directed evolution to improve the thermostability of prolyl endopeptidase. J Biochem. 2000;128:441–447. doi: 10.1093/oxfordjournals.jbchem.a022772. [DOI] [PubMed] [Google Scholar]
- Uchiyama T., Abe T., Ikemura T., Watanabe K. Substrate‐induced gene‐expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat Biotechnol. 2005;23:88–93. doi: 10.1038/nbt1048. [DOI] [PubMed] [Google Scholar]
- de Vasconcellos S.P., Angolini C.F.F., Garcíaa I.N.S., Dellagnezzea B.M., da Silvaa C.C., Marsaioli A.J. Screening for hydrocarbon biodegraders in a metagenomic clone library derived from Brazilian petroleum reservoirs. Org Geochem. 2010;41:675–681. [Google Scholar]
- Venter J.C., Remington K., Heidelberg J.F., Halpern A.L., Rusch D., Eisen J.A. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. et al. [DOI] [PubMed] [Google Scholar]
- Wang G.Y., Graziani E., Waters B., Pan W., Li X., McDermott J. Novel natural products from soil DNA libraries in a streptomycete host. Org Lett. 2000;2:2401–2404. doi: 10.1021/ol005860z. et al. [DOI] [PubMed] [Google Scholar]
- Wang K., Li G., Yu S.Q., Zhang C.T., Liu Y.H. A novel metagenome‐derived β‐galactosidase: gene cloning, overexpression, purification and characterization. Appl Microbiol Biotechnol. 2010;88:155–165. doi: 10.1007/s00253-010-2744-7. [DOI] [PubMed] [Google Scholar]
- Wang Y.F., Wong C.H. Lipase‐catalyzed irreversible transesterification for preparative synthesis of chiral glycerol derivatives. J Org Chem. 1988;53:3127–3129. [Google Scholar]
- Watanabe I., Satoh Y., Enomoto K. Screening, isolation and taxonomical properties of microorganisms having acrylonitrile‐hydrating activity. Agric Biol Chem. 1987;51:3193–3199. [Google Scholar]
- Watanabe Y., Kashimura M., Asaka T., Adachi T., Morimoto S. Chemical modification of erythromycins. XI. Synthesis of clarithromycin (6‐O‐methylerythromycin A) via erythromycin A quaternary ammonium salt derivative. Heterocycles. 1993;36:243–247. [Google Scholar]
- Weber J.M., Schoner B., Losick R. Identification of a gene required for the terminal step in erythromycin A biosynthesis in Saccharopolyspora erythraeaStreptomyces erythreus. Gene. 1989;75:235–241. doi: 10.1016/0378-1119(89)90269-2. [DOI] [PubMed] [Google Scholar]
- Wexler M., Bond P.L., Richardson D.J., Johnston A.W. A wide host‐range metagenomic library from a waste water treatment plant yields a novel alcohol/aldehy dedehydrogenase. Environ Microbiol. 2005;7:1917–1926. doi: 10.1111/j.1462-2920.2005.00854.x. [DOI] [PubMed] [Google Scholar]
- Wild J., Hradecna Z., Szybalski W. Conditionally amplifiable BACs: switching from single‐copy to high‐copy vectors and genomic clones. Genome Res. 2002;12:1434–1444. doi: 10.1101/gr.130502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson L.L., Borlee B.R., Schloss P.D., Guan C., Allen H.K., Handelsman J. Intracellular screen to identify metagenomic clones that induce or inhibit a quorum‐sensing biosensor. Appl Environ Microbiol. 2005;71:6335–6344. doi: 10.1128/AEM.71.10.6335-6344.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong T.S., Arnold F.H., Schwaneberg U. Laboratory evolution of cytochrome P450 BM‐3 monooxygenase for organic co‐solvents. Biotechnol Bioeng. 2004;85:351–358. doi: 10.1002/bit.10896. [DOI] [PubMed] [Google Scholar]
- Yamamoto K., Oishi K., Fujimatsu I., Komatsu K. Production of R‐(‐)‐mandelic acid from mandelonitrile by Alcaligenes faecalis ATCC 8750. Appl Environ Microbiol. 1991a;57:3028–3032. doi: 10.1128/aem.57.10.3028-3032.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K., Otsubo K., Oishi K. 1991b.
- Zaks A., Russel A.J. Enzymes in organic solvents: properties and applications. J Biotechnol. 1988;8:259–262. [Google Scholar]