

Summary
Corynebacterium glutamicum, an established industrial amino acid producer, has been genetically modified for efficient succinate production from the renewable carbon source glucose under fully aerobic conditions in minimal medium. The initial deletion of the succinate dehydrogenase genes (sdhCAB) led to an accumulation of 4.7 g l−1 (40 mM) succinate as well as high amounts of acetate (125 mM) as by‐product. By deleting genes for all known acetate‐producing pathways (pta‐ackA, pqo and cat) acetate production could be strongly reduced by 83% and succinate production increased up to 7.8 g l−1 (66 mM). Whereas overexpression of the glyoxylate shunt genes (aceA and aceB) or overproduction of the anaplerotic enzyme pyruvate carboxylase (PCx) had only minor effects on succinate production, simultaneous overproduction of pyruvate carboxylase and PEP carboxylase resulted in a strain that produced 9.7 g l−1 (82 mM) succinate with a specific productivity of 1.60 mmol g (cdw)−1 h−1. This value represents the highest productivity among currently described aerobic bacterial succinate producers. Optimization of the production conditions by decoupling succinate production from cell growth using the most advanced producer strain (C. glutamicumΔpqoΔpta‐ackAΔsdhCABΔcat/pAN6‐pycP458Sppc) led to an additional increase of the product yield to 0.45 mol succinate mol−1 glucose and a titre of 10.6 g l−1 (90 mM) succinate.
Introduction
The four‐carbon dicarboxylate succinate is used in the pharmaceutical, agricultural and food industry (Zeikus et al., 1999). The present market for succinate is rather small but as succinate chemistry is similar to that of maleic anhydride, it could serve as a replacement for the synthesis of bulk chemicals such as butanediol, tetrahydrofuran and γ‐butyrolactone, which are currently produced in a total annual amount of more than 500 000 t (McKinlay et al., 2007).
Presently, succinate is predominantly produced chemically on the basis of petroleum, but microbial production of succinate from renewable carbon sources is a promising environmentally friendly approach for obtaining this building block. The US Department of Energy identified biotechnologically derived succinate as one of the top 12 value‐added chemicals that could be produced in large quantities from biomass (Werpy and Peterson, 2004). Although the production of bio‐based succinate as bulk chemical is still cost‐intensive compared with petrochemically based alternatives, increasing oil and gas prices as well as the public's and the industry's growing interest for ‘green’ products raise the demand for the development of a competitive microbial production process (Hermann and Patel, 2007). These facts draw the attention to organisms which can produce succinate at high rates with little or no by‐products and to efficiently used substrates to achieve an inexpensive fermentation and purification process.
Succinate can be formed by a wide variety of bacteria in the course of anaerobic fermentative metabolism, such as, for example, Pasteurellaceae species, Escherichia coli and Corynebacterium glutamicum. Whereas Pasteurellaceae species such as Mannheimia succiniciproducens and Basfia succiniciproducens or Anaerobiospirillum succiniciproducens, a member of the Succinivibrionaceae, produce succinate as the major end‐product of fermentation, non‐modified E. coli strains accumulate succinate only as a minor product (Samuelov et al., 1991; Guettler et al., 1999; Lee et al., 2002; Inui et al., 2004b; Jantama et al., 2008; Scholten and Dagele, 2008). Under anaerobic conditions in the presence of bicarbonate, C. glutamicum does not grow, but efficiently converts glucose to succinate, lactate and acetate (Inui et al., 2004b). Based on the above mentioned bacterial species a number of processes with metabolically engineered strains and/or optimized production conditions have been developed (Samuelov et al., 1999; Okino et al., 2008; Oh et al., 2009; Scholten et al., 2009; Zhang et al., 2009). Although anaerobic succinate production allows high yields and titres, it also poses some problems: (i) the substrate spectrum which allows an optimal redox balance is limited. For example, glucose as substrate requires the formation of by‐products in order to provide sufficient reducing equivalents for the maximal possible reduction of oxaloacetate to succinate (San et al., 2002). In contrast, conversion of glycerol and carbon dioxide to succinate offers a closed redox balance and does not necessitate by‐product formation. (ii) Long production processes combined with slow anaerobic growth, which are typical for anaerobic fermentations, can result in overall low volumetric productivity (McKinlay et al., 2007). (iii) As most of the best anaerobic producers do not grow in minimal media (Lee et al., 2001; Oh et al., 2009; Scholten et al., 2009), complex media supplements are needed which do not only raise the costs for the educts, but potentially also complicate product purification (McKinlay et al., 2007). All these problems could be circumvented by using an aerobic succinate producer.
So far only a few aerobic succinate producers have been described. For example an extensively engineered E. coli strain has been developed that produces succinate from glucose under fully aerobic conditions and reaches the theoretical maximal yield of 1 mol mol−1, but with low specific productivity of 0.62 mmol g cell dry weight (cdw)−1 h−1. However, probably due to its genetic modifications, the growth‐coupled production process with this strain was performed in LB medium which contains trypton and yeast extract (Lin et al., 2005a). The yeast Yarrowia lipolytica uses glycerol as substrate and accumulates succinate in non‐buffered medium reaching a final pH of 3.2. By omitting pH neutralization, fermentation and purification costs could be decreased. However, the volumetric productivity is low (0.87 mM h−1) and like for the E. coli production process described above, complex media are necessary for growth and product formation with Y. lipolytica (Yuzbashev et al., 2010). A genetically engineered Saccharomyces cerevisiae strain for aerobic succinate production is also tolerant to low pH values and does not require pH neutralization, but accumulates only low succinate concentrations (30 mM) with a yield of 0.11 mol mol−1 and a volumetric productivity of 0.17 mM h−1 (Raab et al., 2010).
In view of these facts, we started to explore the potential of C. glutamicum for aerobic succinate production. Corynebacterium glutamicum is a facultatively anaerobic, Gram‐positive soil bacterium with GRAS status (generally regarded as safe), which is used for the large‐scale production of more than 2 million tons of l‐glutamate and 1.1 million tons of l‐lysine annually. In addition, C. glutamicum strains were developed for the production of several other industrially relevant products such as putrescine (Schneider and Wendisch, 2010), isobutanol (Blombach et al., 2011) or ethanol (Inui et al., 2004a). The genome of C. glutamicum is known (Ikeda and Nakagawa, 2003; Kalinowski et al., 2003; Yukawa et al., 2007) and numerous genetic tools are available allowing genetic engineering (Kirchner and Tauch, 2003). Moreover, extensive knowledge on the central metabolism of C. glutamicum is available due to 60 years of research on amino acid production (Eggeling and Bott, 2005; Burkovski, 2008). Novel results relevant for succinate production represent the identification of two genes coding for succinate importers, dccT (Youn et al., 2008) and dctA (Youn et al., 2009), and of a gene coding for a succinate exporter, sucE (Huhn et al., 2011). Based on the extensive knowledge available and the fact that anaerobic succinate production with this species has been demonstrated, a C. glutamicum strain for efficient aerobic succinate production from glucose was constructed in this work by metabolic engineering, using similar strategies as described for E. coli (Lin et al., 2005a,b; Wendisch et al., 2006). The key mutation required for succinate production was the deletion of the succinate dehydrogenase genes. Subsequent deletion of all known acetate‐producing pathways as well as overproduction of phosphoenolpyruvate carboxylase and pyruvate carboxylase led to significantly increased succinate production (Fig. 1). Finally, the production process was optimized by limiting biomass formation, thereby providing more carbon for succinate production. In summary, our results demonstrate for the first time the potential of C. glutamicum for aerobic succinate production in minimal medium.
Figure 1.
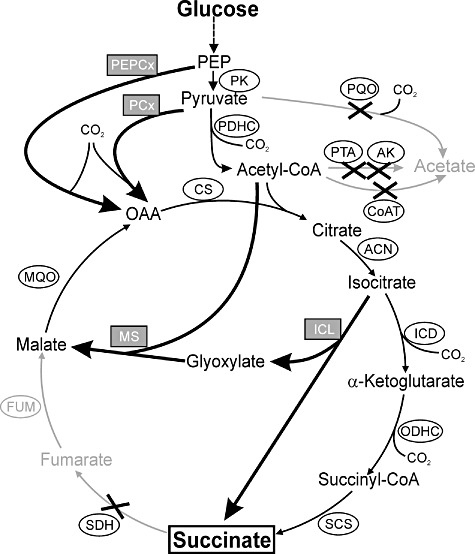
Scheme of the central metabolism of C. glutamicum showing the genetic modifications used in this work to construct a strain for aerobic succinate production. Enzymes whose genes were deleted are indicated by ‘X’. The reactions affected by these deletions and their products are displayed in grey. Enzymes whose genes were overexpressed are highlighted in grey boxes and the arrows for the corresponding reactions are thickened. Abbreviations: ACN, aconitase; AK, acetate kinase; CoAT, acetyl‐CoA:CoA transferase; CS, citrate synthase; FUM, fumarase; ICD, isocitrate dehydrogenase; ICL, isocitrate lyase; MQO, malate:menaquinone oxidoreductase; MS, malate synthase; OAA, oxaloacetate; ODHC, 2‐oxoglutarate dehydrogenase complex; PEP, phosphoenolpyruvate; PK, pyruvate kinase; PEPCx, PEP carboxylase; PCx, pyruvate carboxylase; PDHC, pyruvate dehydrogenase complex; PTA, phosphotransacetylase; PQO, pyruvate:menaquinone oxidoreductase; SCS, succinyl‐CoA synthetase; SDH, succinate dehydrogenase.
Results
Inactivation of the succinate dehydrogenase complex for aerobic succinate production
During aerobic metabolism C. glutamicum uses the TCA cycle for generation of reducing equivalents and precursors for amino acids (Bott, 2007). Under these conditions succinate is an intermediate of the oxidative TCA cycle and does not accumulate in the supernatant. In order to allow the accumulation of succinate as an end‐product of aerobic metabolism, the succinate dehydrogenase complex encoded by the sdhCAB genes must be inactivated. For this purpose, a C. glutamicumΔsdhCAB deletion mutant was constructed which is completely unable to oxidize succinate to fumarate (Fig. 1). The growth behaviour and product formation of this strain were compared with those of the wild type. For this purpose, independent batch cultivations in modified CGXII medium with 4% (w/v) glucose as carbon source were performed in the Multifors bioreactors. To prevent acidification of the medium caused by organic acid production the pH was kept at 7.0. Oxygen limitation was avoided by keeping pO2 at > 30% saturation. During cultivation growth, glucose consumption and organic acid production were measured (Fig. 2). Corynebacterium glutamicumΔsdhCAB exhibited a 9% reduced growth rate and 28% decreased biomass formation compared with the wild type (Table 1). The wild‐type cells consumed glucose with an uptake rate of 4.30 mmol g (cdw)−1 h−1 and did not produce measurable quantities of succinate or other organic acids in the culture broth. Corynebacterium glutamicumΔsdhCAB showed growth‐coupled accumulation of succinate from the beginning of the cultivation up to the end of the exponential growth phase with a specific production rate of 0.75 mmol g (cdw)−1 h−1. At the end of the cultivation after 22.5 h the succinate concentration in the supernatant reached 40 mM. The succinate yield (0.18 mol mol−1 glucose) represented 18% of the theoretically maximal yield (1 mol succinate mol−1 glucose). The major product formed during cultivation of C. glutamicumΔsdhCAB was acetate, which accumulated in parallel to succinate and reached a concentration of 125 mM (0.56 mol mol−1 glucose) after 22.5 h.
Figure 2.
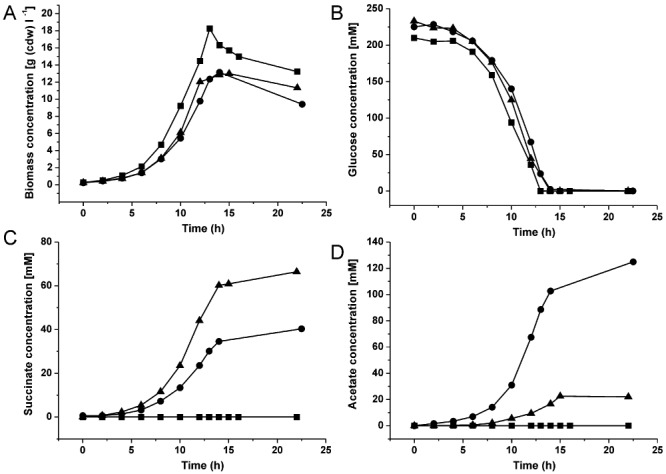
Comparison of biomass formation (A), glucose consumption (B), succinate production (C) and acetate production (D) of C. glutamicum wild type (squares, n = 3), C. glutamicumΔsdh (circles, n = 4) and C. glutamicum BL‐1 (triangles, n = 2) in aerobic batch cultivations in modified CGXII medium containing 4% (w/v) glucose under constantly controlled conditions of pH 7.0 and pO2 > 30% using a Multifors bioreactor system. The results displayed are mean values from n independent experiments.
Table 1.
Growth and organic acid production parameters of different C. glutamicum succinate producer strains during aerobic cultivation in modified CGXII medium containing 4% (w/v) glucose, 25 mg l−1 kanamycin (plasmid‐containing strains only) and 0.5 mM IPTG (plasmid‐containing strains only) at pH 7.0 and pO2 > 30% saturation.
| C. glutamicum strain | Growth rate (h−1) | Biomass [g (cdw) l−1] | Glucose consumed (mM) | Glucose uptake rate [mmol g (cdw)−1 h−1] | Final acetate titre (mM) | Final succinate titre (mM) | Succinate production rate [mmol g−1 (cdw) h−1] | Yield succinate/glucose (mol mol−1) |
|---|---|---|---|---|---|---|---|---|
| Wild type | 0.34 | 18.1 | 210 | 4.30 | n.d. | n.d. | n.d. | n.d. |
| Δsdh | 0.31 | 13.1 | 225 | 5.29 | 125 | 40 | 0.75 | 0.18 |
| BL‐1 | 0.33 | 13.0 | 233 | 5.54 | 22 | 66 | 1.39 | 0.28 |
| BL‐1 (pAN6‐pycP458S) | 0.30 | 12.2 | 242 | 5.49 | 20 | 68 | 1.44 | 0.28 |
| BL‐1 (pAN6‐pycP458Sppc) | 0.29 | 12.2 | 230 | 5.15 | 20 | 82 | 1.60 | 0.36 |
| BL‐1 (pAN6‐aceAaceB) | 0.27 | 10.5 | 235 | 5.19 | 22 | 72 | 1.38 | 0.30 |
All parameters describing rates were calculated for the exponential growth phase of the cultures. The data presented are mean values from at least two independent batch cultivations.
Reduction of acetate formation
To reduce acetate formation, the genes for the known acetate‐forming pathways in C. glutamicum, namely pqo, cat and pta‐ackA coding for pyruvate:menaquinone oxidoreductase (PQO), acetyl‐CoA:CoA transferase (CoAT), phosphotransacetylase (PTA) and acetate kinase (AK), respectively, were deleted in the ΔsdhCAB mutant. Because of organic acid production and the resulting growth defect of C. glutamicumΔsdhCAB, the deletion of pqo and pta‐ackA was performed first in C. glutamicum wild type and then the deletion of the sdhCAB operon was introduced. In the course of strain construction the identification of acetyl‐CoA:CoA transferase (CoAT) and its gene cat in C. glutamicum (Veit et al., 2009) revealed another target for reducing acetate production. The cat gene was deleted in the strain C. glutamicumΔpqoΔpta‐ackAΔsdhCAB resulting in strain C. glutamicum BL‐1 (ΔpqoΔpta‐ackAΔsdhCABΔcat) (Fig. 1). Subsequently, growth and organic acid production of strain BL‐1 was analysed under the same conditions as described above. As shown in Fig. 2, it exhibited comparable growth rates (µ = 0.33 h−1), biomass formation [13.0 g (cdw) l−1] and glucose uptake rates [5.54 mmol g (cdw)−1 h−1] as the strain C. glutamicumΔsdhCAB (Table 1). The succinate concentration in the supernatant increased exponentially from the beginning of the cultivation reaching a maximal concentration of 66 mM after 22.5 h. This value represented a 65% increase of the final succinate titre and a 55% increase in yield (0.28 mol mol−1 glucose) compared with C. glutamicumΔsdhCAB. The specific succinate production rate [1.39 mmol g (cdw)−1 h−1] was increased about 85% compared with that of the ΔsdhCAB strain. The deletion of pqo, pta‐ackA and cat led to an 82% reduction in acetate formation compared with the ΔsdhCAB strain; however, with 22 mM formed after 22.5 h acetate was still the major by‐product. The reasons for the residual acetate formation will be discussed below.
Role of anaplerotic reactions and of the glyoxylate shunt for aerobic succinate production by C. glutamicum BL‐1
The production of succinate from glucose under aerobic conditions is strongly dependent on anaplerotic reactions forming oxaloacetate as substrate for citrate synthase and thus for initiation of the oxidative TCA cycle. Therefore three strategies to improve the availability of oxaloacetate were tested (Fig. 1). (i) Overproduction of pyruvate carboxylase (PCx): previous work showed that the ATP‐dependent carboxylation of pyruvate to oxaloacetate catalysed by PCx is responsible for 90% of the oxaloacetate replenishment during aerobic growth of C. glutamicum on glucose (Petersen et al., 2000). The amino acid exchange P458S in PCx was reported to be beneficial for lysine production by C. glutamicum (Ikeda et al., 2006), although the molecular effect on PCx is not yet clear. To test the influence of a higher PCx activity on succinate production, the expression plasmid pAN6‐pycP458S was constructed. (ii) Simultaneous overproduction of pyruvate carboxylase (PCx) and phosphoenolpyruvate carboxylase (PEPCx): PEPCx, which catalyses the carboxylation of PEP to oxaloacetate (Mori and Shiio, 1985; Eikmanns et al., 1989), was described to be responsible for only 10% of the oxaloacetate replenishment in C. glutamicum during growth on glucose (Petersen et al., 2000). Nevertheless, the combined overproduction of PEPCx (encoded by the ppc gene) with PCxP458S was expected to further stimulate the synthesis from oxaloacetate and therefore the expression plasmid pAN6‐pycP458Sppc was constructed. (iii) Overproduction of the glyoxylate shunt enzymes isocitrate lyase (ICL) and malate synthase (MS): an enhanced carbon flux through the glyoxylate shunt by overproduction of ICL and MS could be a very promising option for anaplerotic oxaloacetate formation. By bypassing isocitrate dehydrogenase and the 2‐oxoglutarate dehydrogenase complex (ODHC) in the TCA cycle, less CO2 is produced and the glyoxylate shunt itself could be used as alternative succinate production pathway besides the oxidative part of the TCA cycle (Fig. 1). This strategy was also used for the design of the currently best aerobic succinate producer of E. coli (Lin et al., 2005b). For overproduction of ICL and MS the expression plasmid pAN6‐aceAaceB was constructed which contains the genes aceA and aceB coding for ICL and MS respectively (Reinscheid et al., 1994a,b).
For the succinate production experiments, strain BL‐1 was transformed with each of the three above mentioned plasmids resulting in the C. glutamicum strains BL‐1/pAN6‐pycP458S, BL‐1/pAN6‐pycP458Sppc and BL‐1/pAN6‐aceAaceB. These strains were analysed and compared in independent batch cultivations with modified CGXII medium containing 4% (w/v) glucose, 25 mg l−1 kanamycin and 0.5 mM IPTG at pH 7.0 and pO2 > 30% saturation (Fig. 3). Surprisingly, overproduction of PCx had no influence on the glucose uptake rate and on succinate production compared with the parental strain BL‐1, as no significant increase of the succinate titre (68 mM), the succinate production rate [1.44 mmol g (cdw)−1 h−1] or the succinate yield (0.28 mol mol−1 glucose) was detected (Table 1). Overproduction of PCx led to a decreased growth rate (0.30 h−1) and biomass formation [12.2 g (cdw) l−1]. Acetate accumulation was decelerated, but the final acetate concentration (20 mM) reached the level of the BL‐1 strain. The combined overexpression of pycP458S and ppc in the strain BL‐1/pAN6‐ pycP458Sppc led to an increased succinate production rate [1.60 mmol g (cdw)−1 h−1] and the final succinate titre was raised about 25% to 82 mM compared with the parental strain (Table 1), which is also reflected in a 28% higher succinate yield (0.36 mol mol−1 glucose). Interestingly, although the succinate production rate was increased by 15%, the glucose consumption rate was reduced by 7% to 5.15 mmol g (cdw)−1 h−1. Overexpression of pycP458S and ppc had no effect on acetate accumulation (Table 1). The functional overexpression of aceA and aceB in BL‐1/pAN6‐aceAaceB was confirmed by in vitro enzymatic activity measurements. The specific activity of ICL (encoded by aceA) was 0.79 U mg−1 protein, the specific activity of MS (aceB) was 0.51 U mg−1 protein. The overexpression of aceA and aceB in strain BL‐1/pAN6‐aceAaceB had a negative influence on the growth rate (0.27 h−1) and on biomass formation [10.5 g (cdw) l−1] compared with the ΔsdhCAB mutant (Table 1). The glucose uptake rate was reduced compared with the BL‐1 strain [5.19 versus 5.54 mmol g (cdw)−1 h−1], whereas the succinate production rate was the same as for the BL‐1 strain [1.38 versus 1.39 mmol g (cdw)−1 h−1]. The final succinate titre (72 versus 66 mM) as well as the succinate yield (0.30 versus 0.28 mol mol−1) were slightly increased compared with the BL‐1 strain. Overproduction of ICL and MS had no influence on the final acetate titre in the supernatant although its accumulation was delayed probably due to the slower growth.
Figure 3.
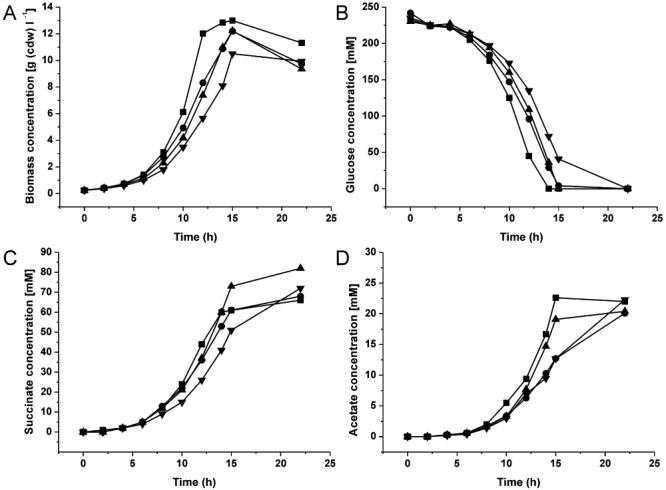
Influence of overexpression of genes for anaplerotic enzymes in C. glutamicum strain BL‐1 on biomass formation (A), glucose consumption (B), succinate production (C) and acetate production (D). The following strains were tested: BL‐1 (squares), BL‐1 with pAN6‐pycP458S (pyruvate carboxylase, circles), BL‐1 with pAN6‐pycP458Sppc (pyruvate carboxylase and PEP carboxylase, triangles pointing upwards) and BL‐1 with pAN6‐aceAaceB (isocitrate lyase and malate synthase, triangles pointing downwards). The strains were cultivated in modified CGXII medium containing 4% (w/v) glucose, 25 mg l−1 kanamycin and 0.5 mM IPTG under constantly control conditions of pH 7.0 and pO2 > 30% using a Multifors bioreactor system. Mean values from two independent cultivations are shown.
Carbon balance of selected succinate producers
The reduction of acetate formation combined with an improved oxaloacetate provision in a ΔsdhCAB deletion mutant resulted in the currently best aerobic succinate producer C. glutamicum BL‐1/pAN6‐pycP458Sppc. However, the succinate yield (0.36 mol mol−1 glucose) of this strain was still far below the theoretical maximum of 1 mol succinate mol−1 glucose. In order to identify which carbon pools (biomass, by‐products) could be diminished to increase succinate synthesis, the carbon balances of the wild type and the three succinate producer strains ΔsdhCAB, BL‐1 and BL‐1/pAN6‐pycP458Sppc were calculated and analysed (Fig. 4).
Figure 4.
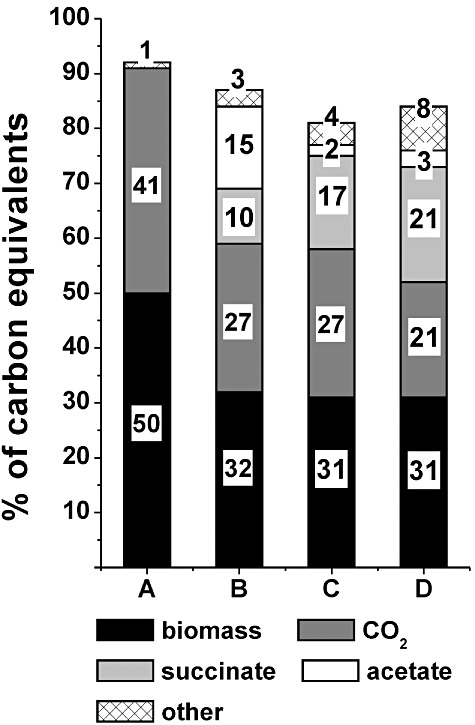
Comparison of the carbon balances of C. glutamicum aerobic succinate producer strains calculated as percentage of product (mmol carbon) of the consumed glucose (mmol carbon) at the end of glucose consumption phase presented as mean values from the performed experiments. For cultivation conditions see legend of Fig. 3. (A) Corynebacterium glutamicum wild type after 12 h; (B) C. glutamicumΔsdh after 14 h; (C) C. glutamicum BL‐1 after 14 h; (D) C. glutamicum BL‐1 with pAN6‐pycP458Sppc after 14 h.
In C. glutamicum wild type, 50% of the carbon consumed ended up in biomass, 41% in carbon dioxide and < 1% in lactate, completing the carbon balance to nearly 92%. As no additional products such as organic acids or amino acids were detected in the supernatant of the wild type by our analysis, the missing 8% were assigned as ‘unidentified products’ and ignored in the further comparison of the strains. Corynebacterium glutamicumΔsdhCAB as first‐generation aerobic succinate producer used 32% of the consumed glucose for biomass formation, 27% was released as CO2, 15% was converted to acetate and 10% to succinate. The sum of the minor products, i.e. lactate, pyruvate, α‐ketoglutarate, lysine, alanine and valine, was very low and represented only 3% of the carbon consumed. The carbon recovery of the analysis for the ΔsdhCAB strain reached 88%. In the improved succinate producer strain BL‐1, the percentage of carbon going into biomass (32%) and CO2 (27%) did not change compared with the ΔsdhCAB strain, but as a result of metabolic engineering the carbon equivalents converted to acetate decreased to 2%. The minor products (4%) showed a small increase compared with the parental strain ΔsdhCAB. The calculated carbon balance for the BL‐1 strain was completed to 82%. The carbon balance of the aerobic succinate producer strain BL‐1/pAN6‐pycP458Sppc was similar to that of BL‐1 with respect to carbon converted into biomass (31%), but only 21% of the carbon was released as CO2. The carbon incorporated into acetate (3%) remained similar to strain BL‐1, but the minor products rose to 8% of the total carbon. The most important feature of this strain was the increase in succinate by 4% compared with its parent BL‐1 due to the enhanced carboxylation, reaching a percentage of 21%. The carbon balance of strain BL‐1/pAN6‐pycP458Sppc was completed to 84%.
Growth‐decoupled succinate production
The carbon balance analysis described above demonstrated that the major portion of the utilized carbon source was converted into biomass and CO2. As the theoretically maximal yield of 1 mol succinate mol−1 glucose can only be obtained with non‐growing cells, the possibility was explored to redirect the carbon flux at a certain stage during cultivation from growth to product synthesis, thus increasing the yield. The strategy chosen to achieve this goal was to restrict growth of the culture after reaching half of the biomass formed in the previous experiments. For this purpose, the nitrogen supply was limited by omitting urea from the medium and reducing the (NH4)2SO4 concentration from 20 g l−1 to 2.71 g l−1. Nitrogen is essential for growth, but not necessary for succinate formation. The production experiments were performed with the best aerobic succinate producer strain BL‐1/pAN6‐pycP458Sppc in batch cultivations with modified CGXII medium at pH 7.0 and pO2 > 30% saturation. Growth, glucose utilization and product formation of the nitrogen‐limited (N‐limited) cultures as well as of control cultures containing 20 g l−1 (NH4)2SO4, which were run in parallel, were analysed and compared (Fig. 5).
Figure 5.
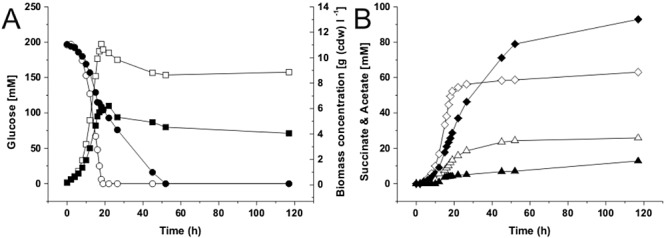
Growth (squares), glucose consumption (circles), succinate production (diamonds) and acetate production (triangles) of C. glutamicum BL‐1 with pAN6‐pycP458Sppc under conditions of nitrogen excess (open symbols) and nitrogen limitation (solid symbols) during batch cultivation in modified CGXII medium containing 4% (w/v) glucose, 25 mg l−1 kanamycin and 0.5 mM IPTG under constantly control conditions of pH 7.0 and pO2 > 30% in a Multifors bioreactor system. Under nitrogen excess, the medium contained 151 mM (NH4)2SO4 and no urea; under nitrogen limitation, the medium contained no urea and 20 mM (NH4)2SO4. Mean values from at least two independent cultivations are shown.
The cultivation process of the N‐limited cultures could be divided into two phases, a growth‐coupled and a growth‐decoupled production phase. The transition point was reached when the N‐limited culture had reached its maximal biomass concentration after 22 h. In contrast, the cultivation process under N‐excess conditions comprised only a growth‐coupled production phase (see above). The maximal biomass concentration reached under N limitation [6.05 g (cdw) l−1] was reduced by 45% compared with the control culture [11.06 g (cdw) l−1]. When reaching the maximal biomass concentration, the N‐limited cultures had consumed 52% of the initial glucose concentration and accumulated 40% (36 mM) of its final succinate titre. Thus, 48% (96 mM) of the initial glucose concentration was still available for product synthesis. In contrast, the non‐limited culture had consumed glucose almost completely when it reached its maximal biomass after 18 h and the accumulated succinate represented 78% (49 mM) of the final titre. At the end of the cultivation (117 h), the N‐limited cultures of strain BL‐1/pAN6‐pycP458Sppc had accumulated 90 mM succinate, corresponding to a 43% increase of the succinate titre compared with the non‐limited control cultures (63 mM). Further consequences of N limitation were the reduction of acetate formation by 50% (15 mM) and the formation of additional by‐products, pyruvate (25 mM) and α‐ketoglutarate (18 mM). The succinate yield of the N‐limited cultures reached 0.45 mol mol−1 glucose and thus was 41% higher than the yield of the N‐excess cultures. Interestingly, under N limitation CO2 production was lowered. At the time of glucose depletion, the N‐limited culture had formed 113 mM CO2, where the nitrogen excess culture had formed 219 mM CO2.
Discussion
In this work, C. glutamicum strains for aerobic succinate production from glucose were constructed and analysed. The initial deletion of the sdhCAB genes encoding succinate dehydrogenase led to a strain that produced 40 mM succinate, a titre which was quite high compared with the titre reached by strains of E. coli (< 2 mM) and S. cerevisiae (24 mM) with equivalent deletions (Lin et al., 2005b; Raab et al., 2010). This fact encouraged further metabolic engineering of C. glutamicum towards efficient succinate production. As the major by‐product of C. glutamicumΔsdhCAB was found to be acetate, the next step was to inactivate the known acetate‐forming pathways, i.e. the conversion of acetyl‐CoA to acetyl phosphate and then to acetate by phosphotransacetylase and acetate kinase (Reinscheid et al., 1999), the conversion of pyruvate to acetate and CO2 with concomitant reduction of menaquinone to menaquinol by pyruvate:menaquinone oxidoreductase (Schreiner et al., 2006) and the conversion of acetyl‐CoA to acetate by an acetyl‐CoA:CoA transferase, which requires a second substrate that is converted to its CoA derivative in this reaction (Yasuda et al., 2007; Veit et al., 2009). By deletion of the corresponding genes ackA, pta, pqo and cat (Fig. 1), acetate formation could be strongly reduced, but not completely abolished. The new strain BL‐1 formed 65% more succinate and its productivity was nearly doubled compared with the ΔsdhCAB strain. The gain of product was on the cost of acetate reduction, as seen by the comparison of the carbon balances (Fig. 4). It is yet unclear which reactions and enzymes are responsible for the remaining production of acetate in strain BL‐1. It is probably due to the hydrolysis and/or conversion of acetyl‐CoA to acetate. Several genes of C. glutamicum were annotated as putative acetyltransferases or hydrolases and might be involved in acetate formation. The genes cg0358 and cg2472 are annotated as putative ac(et)yltransferases/hydrolases, cg2860 as acyl‐CoA thioester hydrolase, and cg0592 as butyryl‐CoA:acetate CoA transferase (Kalinowski et al., 2003). Analysis of the corresponding protein sequences in the PFAM database for domain structures (PFAM version 24.0, http://pfam.sanger.ac.uk/) revealed that Cg0358, Cg2472 and Cg2860 contain domains of the thioesterase superfamily and in the case of Cg2472 even catalytic residues could be identified (Ollis et al., 1992; Benning et al., 1998). Therefore these enzymes might be able to catalyse acetyl‐CoA hydrolysis. For Cg0592 no significant domain match was found within PFAM. This protein is about one‐third of the length of cat gene product and has very high sequence similarity to the C‐terminal part of the Cat protein, but apparently lacks the N‐terminal catalytic domain. Deletion of one or more of these putative acetate‐producing enzymes will be a target for further strain development to reduce acetate formation and increase succinate production.
A second approach to improve succinate production was to enhance the anaplerotic reactions. Whereas overexpression of the pyc gene was without effect on succinate production, the combined overexpression of pyc and ppc improved succinate production. The succinate titre, yield and productivity were increased compared with the BL‐1 strain by 24%, 28% and 14% respectively. The enhanced succinate production could therefore be assigned to PEPCx. This was surprising since previous flux analysis had shown that during aerobic growth of C. glutamicum on glucose 90% of the anaplerotic C3 carboxylation to oxaloacetate was performed by PCx and only 10% by PEPCx (Petersen et al., 2000). In addition, PCx was reported to be the major bottleneck for the aerobic production of glutamate and lysine, both derivatives of TCA cycle intermediates (Peters‐Wendisch et al., 2001). Our data suggest that major (regulatory) differences exist with respect to the importance of PCx and PEPCx between succinate production strains and lysine or glutamate production strains. In the amino acid production strains, oxaloacetate and α‐ketoglutarate are removed from the TCA cycle, but the cycle is not interrupted and thus there is no accumulation of TCA cycle intermediates. In the aerobic succinate producers, the cycle is blocked at the succinate dehydrogenase reaction, causing the accumulation of succinate and possibly also other TCA cycle intermediates. Moreover, the replenishment of oxaloacetate for the citrate synthase reaction completely depends on anaplerotic reactions. If the rate of oxaloacetate formation is lower than the rate of acetyl‐CoA formation, the latter will accumulate, in particular in the absence of the most prominent acetate‐forming enzymes. The possibly elevated concentrations of succinate, other TCA cycle intermediates and acetyl‐CoA in C. glutamicum succinate producer strains might trigger regulatory effects on enzymes participating in the product synthesis pathway. In particular, it has been reported that PCx and PEPCx are regulated contrarily by acetyl‐CoA: 110 µM acetyl‐CoA inhibits PCx activity, whereas PEPCx is activated in the presence of 100 µM acetyl‐CoA (Mori and Shiio, 1985; Peters‐Wendisch et al., 1998). This opposed regulation of the PEP/pyruvate carboxylation reactions through acetyl‐CoA could explain the missing effect of pyc overexpression and the stimulating effect of ppc overexpression on succinate production. A beneficial effect of ppc overexpression in comparison with pyc overexpression was also reported for glutamate production by a ΔdtsR1 mutant in which fatty acid synthesis is disturbed, which might also result in higher acetyl‐CoA levels within the cell (Yao et al., 2009).
A highly active glyoxylate shunt could not only serve as OAA replenishment pathway, but also as an alternative succinate production route that avoids the loss of carbon as carbon dioxide in the isocitrate dehydrogenase and 2‐oxoglutarate dehydrogenase reactions. Therefore, overexpression of aceA and aceB was expected to improve aerobic succinate production. This approach was previously shown to be very efficient for construction of aerobic E. coli succinate producer strains (Lin et al., 2005a). Strain BL‐1 carrying pAN6‐aceAaceB showed a 9% increased succinate titre and a 7% increased yield compared with the reference strain BL‐1, but the growth rate was decreased by 18%. These results together with the enzyme activity measurements of the overproduced ICL and MS proved that the glyoxylate shunt was functional, but the improvements were rather small. This could be due to the inhibition of ICL from C. glutamicum by succinate (Ki of 1.48 mM) and also by intermediates of glycolysis (Reinscheid et al., 1994a). Although the BL‐1/pAN6‐aceAaceB strain has high in vitro ICL activity (0.79 U mg−1 protein), the in vivo activity is probably very low due to elevated intracellular levels of succinate and perhaps also glycolytic intermediates. The reason for the reduced growth rate remains unclear.
The carbon balance analysis and comparison of the three aerobic succinate producers ΔsdhCAB, BL‐1 and BL‐1/pAN6‐pycP458Sppc delivered three important conclusions. First, the increase in succinate production of the BL‐1 strain compared with the ΔsdhCAB mutant did not match with the reduction in acetate accumulation, as only about half of the carbon equivalents becoming available in the BL‐1 strain by the decreased acetate production (13%) were directed to succinate. This was probably due to an insufficient provision of oxaloacetate, as overexpression of ppc (together with pyc) in strain BL‐1 shifted another 4% of carbon equivalents towards succinate. Second, deletion of the sdhCAB genes caused the accumulation of unidentified products, as the total carbon recovery of the ΔsdhCAB mutant decreased by 4% compared with the wild type. This effect was further increased in strains BL‐1 and BL‐1/pAN6‐ pycP458Sppc, in which 10% and 8% of the consumed carbon equivalents from glucose could not be assigned to products. Third, despite the improvements made by metabolic engineering, the major proportion of the carbon source was still used for biomass and CO2 formation. By limiting biomass formation through a reduced nitrogen supply, CO2 production was strongly reduced and growth‐decoupled succinate production from glucose was obtained. Under these conditions, the succinate yield and the succinate titre of strain BL‐1/pAN6‐pycP458Sppc could be increased by more than 40%.
In summary, our results show for the first time the potential of C. glutamicum strains for aerobic succinate production in a minimal medium. The best strain obtained in this work, BL‐1/pAN6‐pycP458Sppc, is able to accumulate succinate with a yield of 0.45 mol mol−1 glucose and reaches the highest specific productivity [1.60 mmol g−1 (cdw) h−1] currently described for aerobic bacterial succinate producers. The use of minimal medium for succinate production with C. glutamicum offers an advantage to most alternative processes described for other microorganisms, which rely on complex medium additives (Lin et al., 2005a; Yuzbashev et al., 2010). Moreover, it can be envisaged that further improvements of our C. glutamicum strains and of the process, e.g. by using fed‐batch cultivation conditions, will allow significant further increases in the titre and the yield of succinate.
Experimental procedures
Bacterial strains, plasmids and culture conditions
All bacterial strains and plasmids used or constructed in the course of this work are listed in Table 2. Corynebacterium glutamicum strains were routinely cultivated at 30°C in Brain‐Heart‐Infusion medium (Difco Laboratories, Detroit, MI, USA) supplemented with 2% (w/v) glucose or in a modified CGXII medium containing (NH4)2SO4 (20 g l−1), urea (5 g l−1), KH2PO4 (1 g l−1), K2HPO4 (1 g l−1), MgSO4 × 7 H2O (0.25 g l−1), CaCl2 (10 mg l−1), FeSO4 × 7 H2O (10 mg l−1), MnSO4 × H2O (0.1 mg l−1), ZnSO4 × 7 H2O (1 mg l−1), CuSO4 × 5 H2O (0.2 mg l−1), NiCl2 × 6 H2O (20 µg l−1), biotin (0.2 mg l−1), 3‐morpholinopropanesulfonic acid (MOPS) (42 g l−1) and 4% (w/v) glucose as carbon and energy source. The pH of the medium was adjusted to pH 7 with KOH. For cultivations of the BL‐1 strain of C. glutamicum and its derivatives the biotin concentration was doubled to 0.4 mg l−1. Escherichia coli DH5α was routinely grown in LB medium at 37°C. If appropriate, kanamycin (25 µg ml−1 for C. glutamicum or 50 µg ml−1 for E. coli) and 0.5 mM isopropyl β‐d‐1‐thiogalactopyranoside (IPTG) were added.
Table 2.
Strains and plasmids used in this study.
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| Strains | ||
| C. glutamicum ATCC 13032 | Wild‐type strain, biotin auxothroph | Abe et al. (1967) |
| C. glutamicumΔsdhCAB | ATCC 13032 derivative with an in‐frame deletion of the sdhCAB operon | This work |
| C. glutamicum BL‐1 | ATCC 13032 derivative with in‐frame deletions of pqo, pta‐ackA, sdhCAB and cat | This work |
| C. glutamicum DM1727 | ATCC 13032 derivative with a Pro458Ser exchange in the pyruvate carboxylase gene (pycP458S) | B. Bathe, Evonik Degussa |
| E. coli DH5α | F‐φ80dlacΔ(lacZ)M15 Δ(lacZYA‐argF) U169 endA1 recA1 hsdR17 (rK‐, mK+) deoR thi‐1 phoA supE44λ‐gyrA96 relA1 | Invitrogen |
| Plasmids | ||
| pAN6 | KanR; C. glutamicum/E. coli shuttle vector for regulated gene expression (Ptac, lacIq, pBL1 oriVC.g., pUC18 oriVE.c.) derived from pEKEx2 | Frunzke et al. (2008) |
| pAN6‐pycP458S | KanR; pAN6 derivative containing the pycP458S gene from C. glutamicum DM1727 under control of the tac promotor | This work |
| pAN6‐ pycP458Sppc | KanR; pAN6 derivative containing the pycP458S gene from C. glutamicum DM1727 and the ppc gene from C. glutamicum ATCC13032 under control of the tac promotor | This work |
| pAN6‐aceAaceB | KanR; pAN6 derivative containing the aceA and aceB genes from C. glutamicum ATCC13032 under control of the tac promotor | This work |
| pK19mobsacB | KanR; vector for allelic exchange in C. glutamicum (pK18 oriVE.c. sacB lacZα) | Schäfer et al. (1994) |
| pk19mobsacB‐Δcat | KanR; pK19mobsacB derivative containing a 1.1 kb overlap‐extension PCR product (SalI/XbaI) which covers the flanking regions of the C. glutamicum cat gene | Veit et al. (2009) |
| pK19mobsacB‐Δpqo | KanR; pK19mobsacB derivative containing a 1 kb overlap‐extension PCR product (HindIII/EcoRI) which covers the flanking regions of the C. glutamicum pqo gene | This work |
| pk19mobsacB‐Δpta‐ΔackA | KanR; pK19mobsacB derivative containing a 1.1 kb overlap‐extension PCR product (HindIII/XbaI) which covers the flanking regions of the C. glutamicum pta‐ackA genes | This work |
| pK19mobsacB‐ΔsdhCAB | KanR; pK19mobsacB derivative containing a 1.1 kb overlap‐extension PCR product (HindIII/XbaI) which covers the flanking regions of the C. glutamicum sdhCAB genes | This work |
Recombinant DNA work
The enzymes for recombinant DNA work were obtained from Roche Diagnostics (Mannheim, Germany) or New England Biolabs (Frankfurt am Main, Germany). All oligonucleotides were synthesized by Eurofins MWG Operon (Table S1). Routine methods like PCR, restriction or ligation were carried out according to standard protocols (Sambrook et al., 2001). The generation of all PCR products was performed with KOD Hot Start polymerase (Novagen, Darmstadt, Germany). Plasmids were isolated from E. coli with the QIAprepspin miniprep kit (Qiagen, Hilden, Germany). Escherichia coli was transformed by the RbCl method (Hanahan, 1985). Transformation of C. glutamicum was performed as described previously (van der Rest et al., 1999). All plasmid constructs described below were controlled by DNA sequencing (LGC genomics, Berlin, Germany).
Construction of deletion mutants and plasmids
Corynebacterium glutamicum mutants with in‐frame deletions of sdhCAB (ΔsdhCAB), pta‐ackA (Δpta‐ackA), pqo (Δpqo) and cat (Δcat) were constructed via a two‐step homologous recombination procedure as described previously (Niebisch and Bott, 2001). The regions up‐ and downstream (approximately 500 bp each) of the gene/operon to be deleted were amplified using pairs of oligonucleotides designated Δ‐gene/operon‐1 and Δ‐gene/operon‐2 and Δ‐gene/operon‐3 and Δ‐gene/operon‐4 (Table S1), respectively, and the two PCR products served as templates for an overlap‐extension PCR using the oligonucleotides Δ‐gene/operon‐1 and Δ‐gene/operon‐4. The resulting PCR product of about 1 kb was digested with the restriction enzymes indicated in Table S1 and cloned into pK19mobsacB cut with the same enzymes. The plasmid pK19mobsacB‐Δcat was kindly provided by Volker F. Wendisch (University of Bielefeld). The transfer of the resulting plasmids, designated pK19mobsacB‐Δgene/operon, into C. glutamicum and selection for the first and second recombination events were performed as described previously (Niebisch and Bott, 2001). Kanamycin‐sensitive and saccharose‐resistant clones were tested by colony PCR with an oligonucleotide pair designated Δ‐gene/operon‐out‐fw and Δ‐gene/operon‐out‐rv (Table S1). Clones which had the desired in‐frame deletion of the gene/operon revealed a 1 kb fragment in which all nucleotides except for the first six codons (in case of Δcat the first seven codons) and the last 12 codons were replaced by a 21 bp tag.
For the construction of the expression plasmid pAN6‐pycP458S, the pycP458S gene was amplified using the oligonucleotides NdeI‐pyc‐for and NheI‐SbfI‐pyc‐rev and chromosomal DNA of C. glutamicum DM1727 (exchanging the native start codon GTG to ATG). The PCR product was digested with NdeI and NheI and cloned into the vector pAN6. For the construction of the expression plasmid pAN6‐pycP458Sppc, the coding region of ppc together with a 9 bp linker and an artificial ribosome binding site (AAGGA) was amplified using chromosomal DNA of C. glutamicum wild type and the oligonucleotide pair SbfI‐ppc‐for/NheI‐ppc‐rev. The PCR product was digested with SbfI and NheI and cloned into pAN6‐pycP458S, resulting in pAN6‐pycP458Sppc. For construction of pAN6‐aceAaceB, the aceA gene was amplified using the oligonucleotides NdeI‐aceA‐for and NdeI‐aceA‐rev and chromosomal DNA of C. glutamicum wild type. The PCR product was digested with NdeI and cloned into the pAN6 vector. The correct orientation of the insert was confirmed by restriction analysis and DNA sequencing. Subsequently the aceB gene was amplified from chromosomal DNA of C. glutamicum wild type using the oligonucleotides NheI‐RBS‐aceB‐for and NheI‐aceB‐rev, which introduce an NheI restriction site and a ribosomal binding site (AAGGA) together with a 9 bp linker or only an NheI restriction site respectively. The PCR product was digested with NheI and cloned into plasmid pAN6‐aceA. The correct orientation of the aceB gene was again confirmed by restriction analysis and DNA sequencing.
Cultivation conditions for aerobic succinate production
For aerobic succinate production, 5 ml of BHI medium supplemented with 2% (w/v) glucose were inoculated with one single colony of the desired C. glutamicum strain from a fresh BHI agar plate and incubated on a rotary shaker for 8 h at 30°C. Subsequently the cells were used to inoculate a 500 ml baffled shake flask filled with 50 ml of modified CGXII medium. After approximately 16 h incubation at 120 r.p.m. and 30°C the cells were used to inoculate a 1.4 l bioreactor (Multifors Multi‐Fermenter System with six independently controllable bioreactors, Infors, Einsbach, Germany) to an optical density at 600 nm (OD600) of approximately 1. The bioreactor contained 600 ml modified CGXII medium with 4% (w/v) glucose, but without the buffer substance 3‐morpholinopropanesulfonic acid. When required, 25 µg ml−1 kanamycin was used in the pre‐ and main cultivations. For induction of the target genes in the pAN6‐based expression plasmids, 0.5 mM IPTG was added. The bioreactors were sparged with 0.9 l min−1 air. Oxygen saturation was measured online with a polarimetric oxygen electrode (Mettler Toledo, Giessen, Germany) and was held permanently over 30% by gradually increasing stirrer speed from 600 r.p.m. up to 1000 r.p.m. The pH was determined online using a standard pH electrode (Mettler Toledo) and adjusted to pH 7 with 3 M potassium hydroxide and 3 M hydrochloric acid. Foam formation was suppressed automatically by titration of 25% (v/v) Antifoam 204/water suspension (Sigma‐Aldrich, Steinheim, Germany). Carbon dioxide in the exit gas flow was measured continuously with an Exit Gas Analyser (Infors, Einsbach, Germany). To explore growth‐decoupled succinate production, the maximal biomass formation of C. glutamicum was reduced to approximately 50% by decreasing the nitrogen content of the medium. In these experiments, urea was omitted and the (NH4)2SO4 concentration reduced to 2.71 g l−1. In the control cultures, urea was also omitted, but the (NH4)2SO4 concentration remained at 20 g l−1.
Enzyme assay of ICL and MS in cell extracts
Cell cultivation, crude extract preparation and enzyme measurements were performed as described elsewhere (Gerstmeir et al., 2004).
Growth parameter determination
Growth was followed by measuring the OD600 with an Ultrospec 500‐Pro spectrophotometer (Amersham Biosciences, Freiburg, Germany). The biomass concentration was calculated from OD600 values using an experimentally determined correlation factor of 0.25 g (cdw) l−1 for an OD600 of 1 (Kabus et al., 2007).
Quantification of organic acids and glucose in the culture supernatant
The quantification of glucose and organic acids in the supernatant of the cultures was performed using an Agilent 1100 LC system (Agilent Technologies, Waldbronn, Germany) equipped with a 300 × 8 mm organic acid column (polystyrol‐divinylbenzol resin, CS Chromatographie Service GmbH, Langerwehe, Germany) and a guard cartridge (40 × 8 mm) filled with the same material. Isocratic elution was performed for 38 min at 40°C with 100 mM sulphuric acid at a flow rate of 0.4 ml min−1. Glucose was detected via an Agilent 1100 Refractive Index Detector and the organic acids were detected via an Agilent 1100 Diode Array Detector at 215 nm. All substances were quantified with the help of an experimentally determined linear correlation factor between the area measured in the chromatogram for a peak with a substance‐specific retention time and the concentration of the pure substance in water solution. Quantification was performed using calibration curves obtained with external standards.
Acknowledgments
We thank the Federal Ministry of Food, Agriculture, and Consumer Protection (BMELV) for financial support within the ERA‐IB project ‘BioProChemBB’, Volker F. Wendisch (University of Bielefeld) for providing the plasmid pK19mobsacB‐Δcat and Brita Weil for technical assistance.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Oligonucleotides used in this study.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abe S., Takayama K., Kinoshita S. Taxonomical studies on glutamic acid producing bacteria. J Gen Appl Microbiol. 1967;13:279–301. [Google Scholar]
- Benning M.M., Wesenberg G., Liu R., Taylor K.L., Dunaway‐Mariano D., Holden H.M. The three‐dimensional structure of 4‐hydroxybenzoyl‐CoA thioesterase from Pseudomonas sp. strain CBS‐3. J Biol Chem. 1998;273:33572–33579. doi: 10.1074/jbc.273.50.33572. [DOI] [PubMed] [Google Scholar]
- Blombach B., Riester T., Wieschalka S., Ziert C., Youn J.W., Wendisch V.F., Eikmanns B.J. Corynebacterium glutamicum tailored for efficient isobutanol production. Appl Environ Microbiol. 2011;77:3300–3310. doi: 10.1128/AEM.02972-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott M. Offering surprises: TCA cycle regulation in Corynebacterium glutamicum. Trends Microbiol. 2007;15:417–425. doi: 10.1016/j.tim.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Burkovski A. Caister Academic Press; 2008. [Google Scholar]
- Eggeling L., Bott M. CRC Press, Taylor & Francis Group; 2005. [Google Scholar]
- Eikmanns B.J., Follettie M.T., Griot M.U., Sinskey A.J. The phosphoenolpyruvate carboxylase gene of Corynebacterium glutamicum: molecular cloning, nucleotide sequence, and expression. Mol Gen Genet. 1989;218:330–339. doi: 10.1007/BF00331286. [DOI] [PubMed] [Google Scholar]
- Frunzke J., Engels V., Hasenbein S., Gätgens C., Bott M. Co‐ordinated regulation of gluconate catabolism and glucose uptake in Corynebacterium glutamicum by two functionally equivalent transcriptional regulators, GntR1 and GntR2. Mol Microbiol. 2008;67:305–322. doi: 10.1111/j.1365-2958.2007.06020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstmeir R., Cramer A., Dangel P., Schaffer S., Eikmanns B. RamB, a novel transcriptional regulator of genes involved in acetate metabolism of Corynebacterium glutamicum. J Bacteriol. 2004;186:2798–2809. doi: 10.1128/JB.186.9.2798-2809.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guettler M.V., Rumler D., Jain M.K. Actinobacillus succinogenes sp. nov., a novel succinic‐acid‐producing strain from the bovine rumen. Int J Syst Bacteriol. 1999;49:207–216. doi: 10.1099/00207713-49-1-207. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Techniques for transformation of E. coli. In: Glover D.M., editor. IRL Press; 1985. pp. 109–135. [Google Scholar]
- Hermann B.G., Patel M. Today's and tomorrow's bio‐based bulk chemicals from white biotechnology: a techno‐economic analysis. Appl Biochem Biotechnol. 2007;136:361–388. doi: 10.1007/s12010-007-9031-9. [DOI] [PubMed] [Google Scholar]
- Huhn S., Jolkver E., Kramer R., Marin K. Identification of the membrane protein SucE and its role in succinate transport in Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2011;89:327–335. doi: 10.1007/s00253-010-2855-1. [DOI] [PubMed] [Google Scholar]
- Ikeda M., Nakagawa S. The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl Microbiol Biotechnol. 2003;62:99–109. doi: 10.1007/s00253-003-1328-1. [DOI] [PubMed] [Google Scholar]
- Ikeda M., Ohnishi J., Hayashi M., Mitsuhashi S. A genome‐based approach to create a minimally mutated Corynebacterium glutamicum strain for efficient l‐lysine production. J Ind Microbiol Biotechnol. 2006;33:610–615. doi: 10.1007/s10295-006-0104-5. [DOI] [PubMed] [Google Scholar]
- Inui M., Kawaguchi H., Murakami S., Vertes A.A., Yukawa H. Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen‐deprivation conditions. J Mol Microbiol Biotechnol. 2004a;8:243–254. doi: 10.1159/000086705. [DOI] [PubMed] [Google Scholar]
- Inui M., Murakami S., Okino S., Kawaguchi H., Vertès A., Yukawa H. Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions. J Mol Microbiol Biotechnol. 2004b;7:182–196. doi: 10.1159/000079827. [DOI] [PubMed] [Google Scholar]
- Jantama K., Haupt M.J., Svoronos S.A., Zhang X., Moore J.C., Shanmugam K.T., Ingram L.O. Combining metabolic engineering and metabolic evolution to develop nonrecombinant strains of Escherichia coli C that produce succinate and malate. Biotechnol Bioeng. 2008;99:1140–1153. doi: 10.1002/bit.21694. [DOI] [PubMed] [Google Scholar]
- Kabus A., Niebisch A., Bott M. Role of cytochrome bd oxidase from Corynebacterium glutamicum for growth and lysine production. Appl Environ Microbiol. 2007;73:861–868. doi: 10.1128/AEM.01818-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowski J., Bathe B., Bartels D., Bischoff N., Bott M., Burkovski A. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of l‐aspartate‐derived amino acids and vitamins. J Biotechnol. 2003;104:5–25. doi: 10.1016/s0168-1656(03)00154-8. et al. [DOI] [PubMed] [Google Scholar]
- Kirchner O., Tauch A. Tools for genetic engineering in the amino acid‐producing bacterium Corynebacterium glutamicum. J Biotechnol. 2003;104:287–299. doi: 10.1016/s0168-1656(03)00148-2. [DOI] [PubMed] [Google Scholar]
- Lee P.C., Lee W.G., Lee S.Y., Chang H.N. Succinic acid production with reduced by‐product formation in the fermentation of Anaerobiospirillum succiniciproducens using glycerol as a carbon source. Biotechnol Bioeng. 2001;72:41–48. [PubMed] [Google Scholar]
- Lee P.C., Lee S.Y., Hong S.H., Chang H.N. Isolation and characterization of a new succinic acid‐producing bacterium, Mannheimia succiniciproducens MBEL55E, from bovine rumen. Appl Microbiol Biotechnol. 2002;58:663–668. doi: 10.1007/s00253-002-0935-6. [DOI] [PubMed] [Google Scholar]
- Lin H., Bennett G.N., San K.Y. Metabolic engineering of aerobic succinate production systems in Escherichia coli to improve process productivity and achieve the maximum theoretical succinate yield. Metab Eng. 2005a;7:116–127. doi: 10.1016/j.ymben.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Lin H., Bennett G., San K. Genetic reconstruction of the aerobic central metabolism in Escherichia coli for the absolute aerobic production of succinate. Biotechnol Bioeng. 2005b;89:148–156. doi: 10.1002/bit.20298. [DOI] [PubMed] [Google Scholar]
- McKinlay J.B., Vieille C., Zeikus J.G. Prospects for a bio‐based succinate industry. Appl Microbiol Biotechnol. 2007;76:727–740. doi: 10.1007/s00253-007-1057-y. [DOI] [PubMed] [Google Scholar]
- Mori M., Shiio I. Purification and some properties of phosphoenolpyruvate carboxylase from Brevibacterium flavum and its aspartate‐overproducing mutant. J Biochem. 1985;97:1119–1128. doi: 10.1093/oxfordjournals.jbchem.a135156. [DOI] [PubMed] [Google Scholar]
- Niebisch A., Bott M. Molecular analysis of the cytochrome bc1‐aa3 branch of the Corynebacterium glutamicum respiratory chain containing an unusual diheme cytochrome c1. Arch Microbiol. 2001;175:282–294. doi: 10.1007/s002030100262. [DOI] [PubMed] [Google Scholar]
- Oh I.J., Kim D.H., Oh E.K., Lee S.Y., Lee J. Optimization and scale‐up of succinic acid production by Mannheimia succiniciproducens LPK7. J Microbiol Biotechnol. 2009;19:167–171. doi: 10.4014/jmb.0807.447. [DOI] [PubMed] [Google Scholar]
- Okino S., Noburyu R., Suda M., Jojima T., Inui M., Yukawa H. An efficient succinic acid production process in a metabolically engineered Corynebacterium glutamicum strain. Appl Microbiol Biotechnol. 2008;81:459–464. doi: 10.1007/s00253-008-1668-y. [DOI] [PubMed] [Google Scholar]
- Ollis D.L., Cheah E., Cygler M., Dijkstra B., Frolow F., Franken S.M. The alpha/beta hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. et al. [DOI] [PubMed] [Google Scholar]
- Petersen S., de Graaf A.A., Eggeling L., Mollney M., Wiechert W., Sahm H. In vivo quantification of parallel and bidirectional fluxes in the anaplerosis of Corynebacterium glutamicum. J Biol Chem. 2000;275:35932–35941. doi: 10.1074/jbc.M908728199. [DOI] [PubMed] [Google Scholar]
- Peters‐Wendisch P.G., Kreutzer C., Kalinowski J., Patek M., Sahm H., Eikmanns B.J. Pyruvate carboxylase from Corynebacterium glutamicum: characterization, expression and inactivation of the pyc gene. Microbiology. 1998;144:915–927. doi: 10.1099/00221287-144-4-915. [DOI] [PubMed] [Google Scholar]
- Peters‐Wendisch P.G., Schiel B., Wendisch V.F., Katsoulidis E., Möckel B., Sahm H., Eikmanns B.J. Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microbiol Biotechnol. 2001;3:295–300. [PubMed] [Google Scholar]
- Raab A.M., Gebhardt G., Bolotina N., Weuster‐Botz D., Lang C. Metabolic engineering of Saccharomyces cerevisiae for the biotechnological production of succinic acid. Metab Eng. 2010;12:518–525. doi: 10.1016/j.ymben.2010.08.005. [DOI] [PubMed] [Google Scholar]
- Reinscheid D.J., Eikmanns B.J., Sahm H. Characterization of the isocitrate lyase gene from Corynebacterium glutamicum and biochemical analysis of the enzyme. J Bacteriol. 1994a;176:3474–3483. doi: 10.1128/jb.176.12.3474-3483.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinscheid D.J., Eikmanns B.J., Sahm H. Malate synthase from Corynebacterium glutamicum: sequence analysis of the gene and biochemical characterization of the enzyme. Microbiology. 1994b;140:3099–3108. doi: 10.1099/13500872-140-11-3099. [DOI] [PubMed] [Google Scholar]
- Reinscheid D.J., Schnicke S., Rittmann D., Zahnow U., Sahm H., Eikmanns B.J. Cloning, sequence analysis, expression and inactivation of the Corynebacterium glutamicum pta‐ack operon encoding phosphotransacetylase and acetate kinase. Microbiology. 1999;145:503–513. doi: 10.1099/13500872-145-2-503. [DOI] [PubMed] [Google Scholar]
- van der Rest M.E., Lange C., Molenaar D. A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA. Appl Microbiol Biotechnol. 1999;52:541–545. doi: 10.1007/s002530051557. [DOI] [PubMed] [Google Scholar]
- Sambrook J., MacCallum P., Russell D. Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Samuelov N.S., Lamed R., Lowe S., Zeikus J.G. Influence of CO2‐HCO3‐ levels and pH on growth, succinate production, and enzyme activities of Anaerobiospirillum succiniciproducens. Appl Environ Microbiol. 1991;57:3013–3019. doi: 10.1128/aem.57.10.3013-3019.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelov N.S., Datta R., Jain M.K., Zeikus J.G. Whey fermentation by Anaerobiospirillum succiniciproducens for production of a succinate‐based animal feed additive. Appl Environ Microbiol. 1999;65:2260–2263. doi: 10.1128/aem.65.5.2260-2263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San K.Y., Bennett G.N., Berrios‐Rivera S.J., Vadali R.V., Yang Y.T., Horton E. Metabolic engineering through cofactor manipulation and its effects on metabolic flux redistribution in Escherichia coli. Metab Eng. 2002;4:182–192. doi: 10.1006/mben.2001.0220. et al. [DOI] [PubMed] [Google Scholar]
- Schäfer A., Tauch A., Jäger W., Kalinowski J., Thierbach G., Pühler A. Small mobilizable multi‐purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994;145:69–73. doi: 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]
- Schneider J., Wendisch V.F. Putrescine production by engineered Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2010;88:859–868. doi: 10.1007/s00253-010-2778-x. [DOI] [PubMed] [Google Scholar]
- Scholten E., Dagele D. Succinic acid production by a newly isolated bacterium. Biotechnol Lett. 2008;30:2143–2146. doi: 10.1007/s10529-008-9806-2. [DOI] [PubMed] [Google Scholar]
- Scholten E., Renz T., Thomas J. Continuous cultivation approach for fermentative succinic acid production from crude glycerol by Basfia succiniciproducens DD1. Biotechnol Lett. 2009;31:1947–1951. doi: 10.1007/s10529-009-0104-4. [DOI] [PubMed] [Google Scholar]
- Schreiner M.E., Riedel C., Holatko J., Patek M., Eikmanns B.J. Pyruvate:quinone oxidoreductase in Corynebacterium glutamicum: molecular analysis of the pqo gene, significance of the enzyme, and phylogenetic aspects. J Bacteriol. 2006;188:1341–1350. doi: 10.1128/JB.188.4.1341-1350.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit A., Rittmann D., Georgi T., Youn J.W., Eikmanns B.J., Wendisch V.F. Pathway identification combining metabolic flux and functional genomics analyses: acetate and propionate activation by Corynebacterium glutamicum. J Biotechnol. 2009;140:75–83. doi: 10.1016/j.jbiotec.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Wendisch V.F., Bott M., Eikmanns B.J. Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for biotechnological production of organic acids and amino acids. Curr Opin Microbiol. 2006;9:268–274. doi: 10.1016/j.mib.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Werpy T., Peterson G. US Department of Energy; 2004. pp. 1–76. [Google Scholar]
- Yao W., Deng X., Zhong H., Liu M., Zheng P., Sun Z., Zhang Y. Double deletion of dtsR1 and pyc induce efficient l‐glutamate overproduction in Corynebacterium glutamicum. J Ind Microbiol Biotechnol. 2009;36:911–921. doi: 10.1007/s10295-009-0569-0. [DOI] [PubMed] [Google Scholar]
- Yasuda K., Jojima T., Suda M., Okino S., Inui M., Yukawa H. Analyses of the acetate‐producing pathways in Corynebacterium glutamicum under oxygen‐deprived conditions. Appl Microbiol Biotechnol. 2007;77:853–860. doi: 10.1007/s00253-007-1199-y. [DOI] [PubMed] [Google Scholar]
- Youn J.W., Jolkver E., Kramer R., Marin K., Wendisch V.F. Identification and characterization of the dicarboxylate uptake system DccT in Corynebacterium glutamicum. J Bacteriol. 2008;190:6458–6466. doi: 10.1128/JB.00780-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn J.W., Jolkver E., Kramer R., Marin K., Wendisch V.F. Characterization of the dicarboxylate transporter DctA in Corynebacterium glutamicum. J Bacteriol. 2009;191:5480–5488. doi: 10.1128/JB.00640-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukawa H., Omumasaba C.A., Nonaka H., Kos P., Okai N., Suzuki N. Comparative analysis of the Corynebacterium glutamicum group and complete genome sequence of strain R. Microbiology. 2007;153:1042–1058. doi: 10.1099/mic.0.2006/003657-0. et al. [DOI] [PubMed] [Google Scholar]
- Yuzbashev T.V., Yuzbasheva E.Y., Sobolevskaya T.I., Laptev I.A., Vybornaya T.V., Larina A.S. Production of succinic acid at low pH by a recombinant strain of the aerobic yeast Yarrowia lipolytica. Biotechnol Bioeng. 2010;107:673–682. doi: 10.1002/bit.22859. et al. [DOI] [PubMed] [Google Scholar]
- Zeikus J.G., Jain M.K., Elankovan P. Biotechnology of succinic acid production and markets for derived industrial products. Appl Environ Microbiol. 1999;51:545–552. [Google Scholar]
- Zhang X., Jantama K., Shanmugam K.T., Ingram L.O. Reengineering Escherichia coli for succinate production in mineral salts medium. Appl Environ Microbiol. 2009;75:7807–7813. doi: 10.1128/AEM.01758-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oligonucleotides used in this study.