

Summary
Significant achievements in polyketide gene expression have made Escherichia coli one of the most promising hosts for the heterologous production of pharmacologically important polyketides. However, attempts to produce glycosylated polyketides, by the expression of heterologous sugar pathways, have been hampered until now by the low levels of glycosylated compounds produced by the recombinant hosts. By carrying out metabolic engineering of three endogenous pathways that lead to the synthesis of TDP sugars in E. coli, we have greatly improved the intracellular levels of the common deoxysugar intermediate TDP‐4‐keto‐6‐deoxyglucose resulting in increased production of the heterologous sugars TDP‐L‐mycarose and TDP‐d‐desosamine, both components of medically important polyketides. Bioconversion experiments carried out by feeding 6‐deoxyerythronolide B (6‐dEB) or 3‐α‐mycarosylerythronolide B (MEB) demonstrated that the genetically modified E. coli B strain was able to produce 60‐ and 25‐fold more erythromycin D (EryD) than the original strain K207‐3, respectively. Moreover, the additional knockout of the multidrug efflux pump AcrAB further improved the ability of the engineered strain to produce these glycosylated compounds. These results open the possibility of using E. coli as a generic host for the industrial scale production of glycosylated polyketides, and to combine the polyketide and deoxysugar combinatorial approaches with suitable glycosyltransferases to yield massive libraries of novel compounds with variations in both the aglycone and the tailoring sugars.
Introduction
Polyketides are one of the most important groups of bioactive compounds, because of their great diversity of chemical structures and biological activities (Hopwood, 1997). The most medically and agriculturally important compounds are produced mainly by members of the actinomycete order of bacteria, and include important antibiotics (erythromycin), insecticides (spinosyn), antiparasitic (avermectins), immunosuppressive (rapamycin) and anticancer drugs (doxorubicin) (Mendez and Salas, 2001). Most polyketide‐producing organisms (e.g. marine microorganisms) present limited growth characteristics and are often difficult to manipulate genetically. Thus, the use of a more genetically and physiologically tractable heterologous host for polyketide production became an attractive alternative to overcome these limitations. Heterologous polyketide synthase (PKS) expression, utilizing Escherichia coli as a host, has been largely employed to provide proteins for mechanistic and structural analyses. Further, since the successful de novo production of the 6‐dEB aglycone in E. coli (Pfeifer et al., 2001), there has been a considerable effort to use this bacterium as a host for the biosynthesis of polyketides (Mutka et al., 2006). Moreover, the use of E. coli as a host to explore combinatorial biosynthesis represents a significant advance to speed‐up combinatorial experiments in the continuous effort to obtain compounds with novel or improved pharmaceutical properties (Menzella and Reeves, 2007).
Recently, we have demonstrated the feasibility of reconstituting heterologous glycosylation pathways in E. coli, a critical step in the biosynthesis of many polyketides that contain deoxysugars attached to the aglycone core. These sugar components generally participate in the molecular recognition of the cellular target by the bioactive compound, and their presence is often essential to impart or enhance their specific biological activity.
In proof‐of‐concept experiments, we have shown the production of the potent antibiotic erythromycin C in E. coli, constituting the first glycosylated macrolide synthesized by expression of a heterologous PKS and tailoring enzymes in this microorganism (Peiru et al., 2005). In that work, the complete biosynthetic pathways for two deoxysugars, TDP‐l‐mycarose and TDP‐d‐desosamine, and two P450 hydroxylases were successfully coexpressed with the three DEBS subunits in the E. coli strain K207‐3. However, the glycosylation levels obtained with this recombinant E. coli strain were low. One possible reason could be a limited availability of substrates for the synthesis of the heterologous TDP‐sugars in this host. Alternatively, the presence of macrolide efflux pumps operating in E. coli could have also been detrimental to the glycosylation process by depleting the intracellular polyketide substrates (Lee and Khosla, 2007).
Most of the deoxysugars in natural bioactive compounds belong to the 6‐deoxyhexoses (6DOH) family (Salas and Mendez, 2007). Many of them, like TDP‐l‐mycarose and TDP‐d‐desosamine from the erythromycin and megalomicin biosynthetic pathways, are synthesized through the common precursor TDP‐4‐keto‐6‐deoxyglucose (TKDG). This metabolite is generated from glucose‐1‐phosphate activated to TDP‐d‐glucose by a TDP‐d‐glucose synthase (e.g. RmlA), and further dehydratated to TDP‐4‐keto‐6‐deoxy‐d‐glucose by a TDP‐d‐glucose‐4,6‐dehydratase (e.g. RmlB) (Fig. 1A). In E. coli, TKDG is a precursor of TDP‐4‐acetamido‐4,6‐dideoxy‐d‐galactose (TDP‐d‐Fuc4NAc) (Fig. 1A), a nucleotide‐activated sugar involved in the biosynthesis of the enterobacterial common antigen (ECA), a glycolipid located on the cell surface of all Gram‐negative enteric bacteria (Kajimura et al., 2006). TKDG is also an intermediate in the synthesis of sugars that form the O‐specific polysaccharide chain of the lipopolysaccharide (LPS) described in several E. coli serotypes, like TDP‐l‐rhamnose and TDP‐4‐acetamido‐4,6‐dideoxyglucose (TDP‐d‐Qui4NAc) of the O7 LPS in E. coli VW187 (Marolda et al., 1999; Fig. 1A). The presence of endogenous TKDG‐consuming pathways in E. coli suggests the possibility of a limited availability of this compound for the heterologous biosynthesis of TDP‐l‐mycarose and TDP‐d‐desosamine which, in turn, might result in low levels of glycosylated polyketides in this host.
Figure 1.
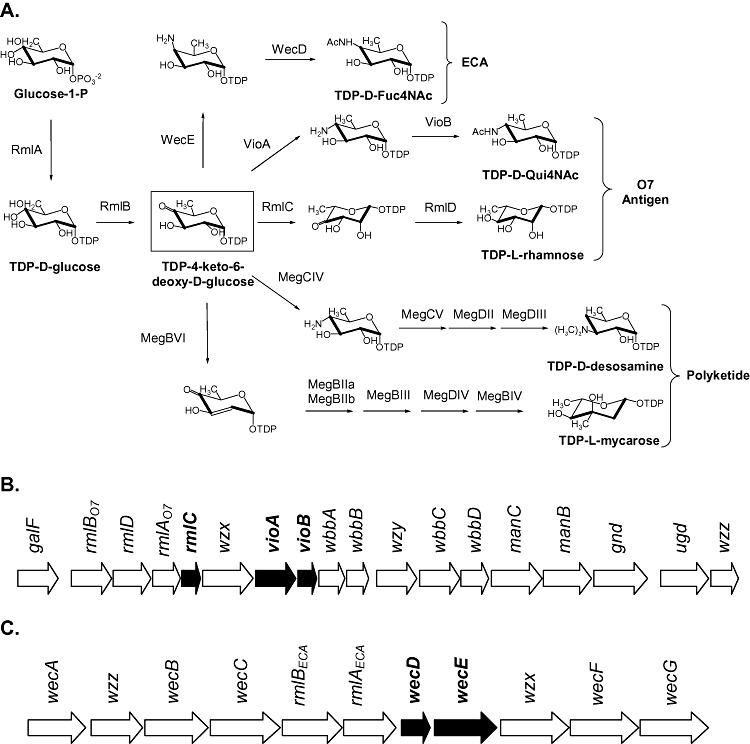
(A) Schematic representation of sugar biosynthetic pathways involving TKDG as an intermediate. Endogenous TDP sugars from E. coli VW187 strain are shown, which are further incorporated as carbohydrate components of the ECA (TDP‐d‐Fuc4NAc) or the O7 polysaccharide (TDP‐l‐rhamnose and TDP‐d‐Qui4NAc). TDP‐l‐mycarose and TDP‐d‐desosamine biosynthetic routes are also described, which are incorporated as sugar components of several polyketides. Genetic organization of the biosynthesis clusters of (B) O7 antigen and (C) ECA. Genes involved in the biosynthetic steps described in this work are highlighted in dark.
On the other hand, it is known that several efflux systems in E. coli contribute to its intrinsic resistance to toxic compounds such as antibiotics, antiseptics, detergents and dyes (Sulavik et al., 2001). Some of these efflux pumps were shown to be involved in macrolides resistance through active efflux of these drugs out of the cell (Nishino and Yamaguchi, 2001; Sulavik et al., 2001). In this regard, it was recently proposed that MEB could be prematurely secreted by macrolide efflux pumps in E. coli during the diglycosylation process of 6‐dEB, thereby lowering the efficiency of desosamine attachment to this mycarosylated precursor (Lee and Khosla, 2007).
To overcome these putative limitations on the synthesis of glycosylated polyketides we have performed metabolic engineering in E. coli in order to improve the glycosylation process. Here we report the identification and knock‐out of several endogenous TKDG‐consuming pathways in the polyketides producer E. coli strain K207‐3, and their effects on the accumulation of the common TDP‐sugars intermediate TKDG and the production of heterologous TDP‐sugars. Additionally, the effect of a knockout of a macrolide efflux system was explored as a means to further increase the production of glycosylated polyketides.
Results
Identification of TKDG‐consuming pathways
K207‐3 is an E. coli B‐derived strain originally developed for the heterologous expression of polyketide biosynthetic genes, and further tested for the production of fully decorated polyketides (Murli et al., 2003; Peiru et al., 2005). However, the polyketide glycosylation efficiency of this strain is very low. To optimize this strain for increased production of novel TKDG‐derived TDP‐sugars, we first decided to increase the intracellular levels of TKDG by identifying and then disrupting the competing endogenous pathways. Although the genes involved in the biosynthesis of TDP‐l‐rhamnose and TDP‐d‐Qui4NAc have not been identified, previous works revealed the presence of these TDP‐sugars in E. coli B, indicating that their biosynthetic pathways would remain functional (Fig. 2A, see below; Okazaki et al., 1960). Moreover, even though E. coli strain B does not display O‐specific side‐chain LPS, genetic evidence suggests that this strain could be a former O7 E. coli, in which its O antigen synthesis was inactivated by an insertion sequence (Marolda et al., 1999; Schneider et al., 2002). Therefore, based on the available sequencing data from the O7‐specific LPS biosynthetic gene cluster of E. coli VW187 (Fig. 1B), we designed primers for the isolation of the rmlC and vioA genes, involved in the synthesis of TDP‐l‐rhamnose and TDP‐d‐Qui4NAc respectively. rmlC encodes the enzyme that catalyses 3′,5′‐epimerization of TKDG in the TDP‐l‐rhamnose pathway, while vioA encodes the TKDG aminotransferase involved in the synthesis of TDP‐d‐Qui4NAc (Fig. 1A) (Marolda et al., 1999; Wang et al., 2007). PCR products were obtained using K207‐3 chromosomal DNA and their nucleotide sequences were 100% identical to the previously reported rmlC and vioA gene sequences from the O7‐specific LPS biosynthesis gene cluster of E. coli VW187.
Figure 2.
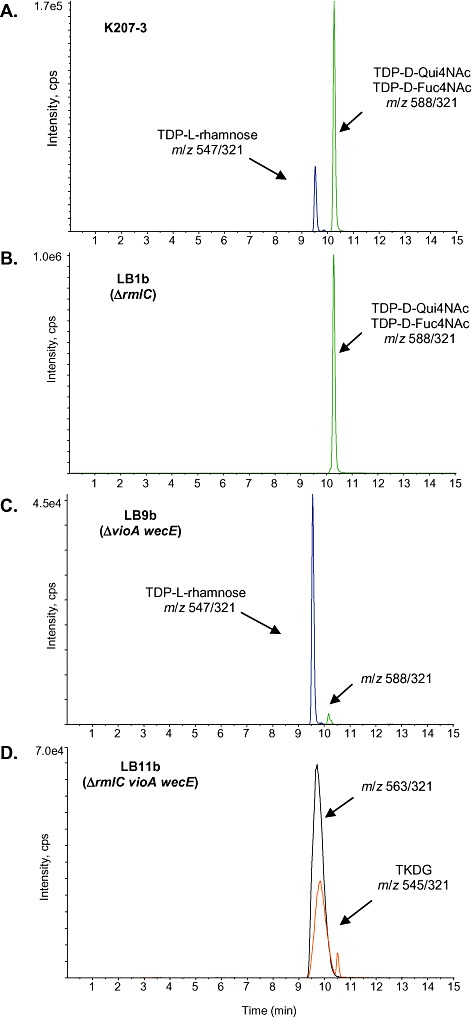
LC/MS/MS analysis of TDP‐sugars in cell‐free extracts of different E. coli strains: (A) K207‐3; (B) LB1b; (C) LB9b; (D) LB11b. m/z parent/daughter pairs for each compound are indicated.
As mentioned above, enteric bacteria synthesize TDP‐d‐Fuc4NAc as one of the sugar components of the polysaccharide portion of ECA. TKDG is an intermediate in the TDP‐d‐Fuc4NAc biosynthetic pathway, which is the substrate for an aminotransferase encoded by wecE (Fig. 1A). Based on the wec gene cluster sequence from E. coli K12 (Fig. 1C), oligonucleotides were designed to amplify wecE using K207‐3 chromosomal DNA as a template. The PCR product was cloned, and its nucleotide sequence was 100% identical to the wecE gene from E. coli K12.
Inactivation of TKDG‐consuming pathways
Once the possible TKDG‐consuming enzymes were identified, we proceeded to inactivate the corresponding genes by the method of Datsenko and Wanner (2000), and to analyse the endogenous TDP‐sugars accumulated in the wild‐type and the mutant strains by a specific LC/MS/MS method (Rodriguez et al., 2006). The LC/MS/MS analysis of cell‐free extracts from K207‐3 showed two main peaks with parent/daughter pairs of m/z 547/321, which correspond to TDP‐l‐rhamnose, and 588/321, which correspond to either TDP‐d‐Qui4NAc or TDP‐d‐Fuc4NAc (Fig. 2A). Deletion of rmlC in K207‐3 resulted in LB1b strain (Table 1), and its TDP‐sugars analysis showed that the TDP‐rhamnose peak became almost negligible compared with the prominent TDP‐d‐Qui4NAc/TDP‐d‐Fuc4NAc peak (Fig. 2B). This result confirmed that rmlC is a functional gene in strain K207‐3, and is consistent with its proposed role in the synthesis of TDP‐l‐rhamnose.
Table 1.
Strains and plasmids used in this study.
| Strain | Relevant genotype | Source or reference |
|---|---|---|
| DH5α | lacZΔM15, recA1 | Promega |
| K207‐3 | F‐ompT hsdSB(r‐m‐), gal dcm (DE3), panD::panDS25A,ΔprpRBCD::T7prom‐sfp, T7prom‐prpE, ygfG::T7prom‐accA1‐T7prom‐pccB | Murli et al. (2003) |
| LB1b | K207‐3 ΔrmlC | This work |
| LB7b | K207‐3 ΔwecE | This work |
| LB8b | K207‐3 ΔvioA | This work |
| LB9b | K207‐3 ΔwecE vioA | This work |
| LB11b | K207‐3 ΔrmlC wecE vioA wzx | This work |
| LB12 | K207‐3 ΔwecDE::kan | This work |
| LB13b | K207‐3 ΔrmlC vioAB wzx | This work |
| LB14b | K207‐3 ΔrmlC wecDE vioAB wzx | This work |
| LB15b | K207‐3 ΔrmlC wecE vioAB wzx | This work |
| LB17b | K207‐3 ΔacrAB::kan | This work |
| LB19b | K207‐3 ΔrmlC wecDE vioAB wzx acrAB | This work |
| Plasmid | Description | Source or reference |
| pCR‐Blunt‐TOPO | General Blunt‐end cloning vector | Invitrogen |
| pET28a | E. coli expression vector, ColE1 ori, kan | Novagen |
| pKOS431‐39.1 | E. coli expression vector, RSF1030 ori, kan | Peiru et al. (2005) |
| pKOS506‐72B | megCIV, megCV, megDII, megDIII, megCII, megCIII, ermE; CloDF13 ori, str. | Rodriguez et al. (2006) |
| pLB353 | megBVI, megBIV, megBV, megBIII, megDIV, megBIIa, megBIIb, megF; RSF1030 ori, kan | This work |
| pGro7 | PBADgroES ‐groEL; P15A ori, cat | Takara |
| pKD4 | Template plasmid, contains kan flanked by FRT, bla kan | Datsenko and Wanner (2000) |
| pKD46 | PBADλβ oxo, pSC101 oriTSbla | Datsenko and Wanner (2000) |
| pCP20 | λ cI857+, PRflp, pSC101 oriTS, bla cat | Datsenko and Wanner (2000) |
str, streptomycin resistance gene; kan, kanamycin resistance gene; cat, chloramphenicol acetyltransferase gene; bla, ampicillin resistance gene.
Inactivation of wecE (LB7b) or vioA (LB8b), instead, did not result in significant changes on the TDP‐sugar profile of these strains compared with K207‐3 (data not shown), which is consistent with the notion that the isomeric compounds TDP‐d‐Qui4NAc and TDP‐d‐Fuc4NAc run as overlapped peaks. Thus, the functionality of WecE and its proposed role in the biosynthesis of TDP‐d‐Fuc4NAc were demonstrated indirectly by analysing the effect of the wecE mutation on the synthesis of ECA. Western blot studies demonstrated that the LB7b mutant lacked ECA on its cell wall, confirming that WecE is involved in the biosynthesis of this sugar nucleotide precursor of the trisaccharide repeated unit of the glycolipid (Fig. S1).
Based on the previous results and to confirm the role of VioA in the biosynthesis of TDP‐d‐Qui4NAc we constructed a double mutant wecE vioA and named it LB9b. LC/MS/MS analysis of this strain showed a prominent TDP‐l‐rhamnose peak and only traces of the m/z 588/321 peak, in agreement with the proposed functions of WecE and VioA (Fig. 2C).
Once the functionality of these three TKDG‐consuming pathways was confirmed in K207‐3, we pursued the construction of the rmlC, wecE and vioA triple mutant. This involved a one‐step inactivation of both rmlC and vioA, followed by a deletion of wecE, resulting in the mutant strain LB11b. LC/MS/MS analysis of this mutant strain revealed two major peaks with parent/daughter pairs of m/z 545/321 and m/z 563/321, consistent with the mass of TKDG and its hydrated form, and exhibiting their typical broad shape (Fig. 2D). These peaks could not be observed in any of the strains previously analysed confirming our initial hypothesis that the inactivation of the three TDP‐sugar pathways under study should result in the accumulation of the common intermediate TKDG.
Expression of heterologous TDP‐sugar operons in E. coli
In order to test if the accumulation of the TKDG intermediate in LB11b could be translated into an improved production of heterologous TDP‐sugars, we thought to express the complete TDP‐d‐desosamine pathway from Micromonospora megalomicea in this engineered strain. However, the possibility that the deleted genes from the endogenous TKDG‐consuming pathways could be complemented by orthologous genes present in the heterologous TDP‐sugar pathways (e.g. megCIV from the TDP‐d‐desosamine route and vioA) led us to introduce two new mutations affecting the enzymatic steps found downstream VioA and WecE. The target genes were vioB and wecD, proposed to encode the acetyltransferases involved in the last step of TDP‐d‐Qui4NAc and TDP‐d‐Fuc4NAc biosynthesis, respectively (Fig. 1A; Hung et al., 2006; Wang et al., 2007). Both genes were amplified by PCR using K207‐3 chromosomal DNA as a template, exhibiting 100% identity to vioB and wecD from E. coli VW187 and E. coli K12 respectively. The construction of this new strain, named LB14b, involved a one‐step inactivation of rmlC, vioA and vioB, followed by a second inactivation step, where the wecE and wecD were both deleted.
Expression of the TDP‐d‐desosamine pathway was tested in K207‐3 and LB14b transformed with pKOS506‐72B. This plasmid harbours all the genes required for the synthesis of TDP‐d‐desosamine from TKDG, the glycosyltransferases megCII/megCIII and the erythromycin resistance gene ermE (Table 1). The resulting strains were next transformed with plasmid pGro7, which overproduces GroES/EL chaperones, shown to improve the solubility of heterologously expressed proteins (Peiru et al., 2005). TDP‐sugar analysis of the cell‐free extracts obtained of both strains showed a peak with a parent/daughter pair of m/z 558/321, which corresponds to TDP‐d‐desosamine (Fig. 3A and B). The relative peak height corresponding to the intracellular accumulation of TDP‐d‐desosamine observed in LB14b extracts exhibited a 50‐fold increase over the K207‐3 first‐generation strain, confirming that the increase of TKDG pools is essential to achieve larger amounts of heterologous TDP‐sugars in E. coli. This was further confirmed through the production of a second TDP‐sugar from plasmid pLB353, which encodes the complete TDP‐l‐mycarose biosynthetic pathway (Table 1). The expression of this pathway in LB14b resulted in a similar increase of TDP‐sugar production when compared with the parental strain, as observed with TDP‐d‐desosamine (data not shown), validating this new host for the improved production of heterologous TDP‐sugars.
Figure 3.
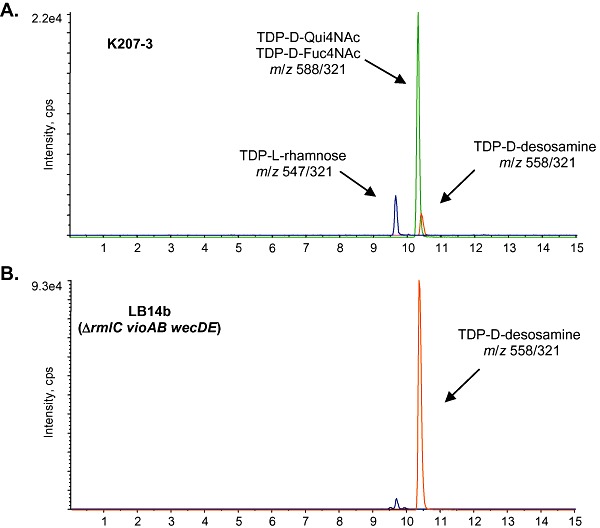
LC/MS/MS analysis of TDP‐sugars in cell‐free extracts of different E. coli mutants expressing the complete TDP‐d‐desosamine biosynthetic operon from plasmid pKOS506‐72B. (A) K207‐3/pKOS506‐72B; (B) LB14b/pKOS506‐72B. m/z parent/daughter pairs for each compound are indicated.
Production of EryD in E. coli
The main goal in the construction of these metabolically engineered strains was to obtain an improved system for polyketide glycosylation. For this reason, once we demonstrated that LB14b was able to accumulate high levels of intracellular TDP‐d‐desosamine and TDP‐l‐mycarose, we tested the capacity of this strain to yield high levels of glycosylated polyketides through bioconversion experiments (Fig. 4A). These experiments were performed in flask cultures of strains K207‐3 and LB14b transformed with pGro7 and pKOS506‐72B. After induction with IPTG, MEB was added to the cultures to a final concentration of 100 mg l−1 and incubated for 24 h at 22°C. LC/MS analysis of extracts obtained from these cultures showed a dramatic increase in the conversion of MEB to EryD by LB14b compared with the parental K207‐3 strain (Fig. 4B and C). The concentration of EryD was estimated through a Micrococcus luteus inhibition assay using an EryA standard as reference, and considering the activity of EryD against this microorganism as four times lower than that of EryA (Kibwage et al., 1985). The estimated production of EryD was 3 mg l−1 in K207‐3 and 80 mg l−1 in LB14b cultures (about 2% and 60% conversion of the total MEB fed respectively; Fig. 5). This represents a > 25‐fold increase in the production of EryD in LB14b compared with its parental strain K207‐3, validating the effectiveness of our metabolic engineering approach to enhance the heterologous glycosylation of polyketides in E. coli.
Figure 4.

(A) Schematic representation of the post‐PKS modification steps involved in bioconversion of the aglycone 6‐dEB to the bioactive compound EryD. LC‐MS analysis of the bioconversion of MEB by E. coli strains expressing the TDP‐d‐desosamine pathway: (B) K207‐3; (C) LB14b. MEB and EryD are indicated.
Figure 5.
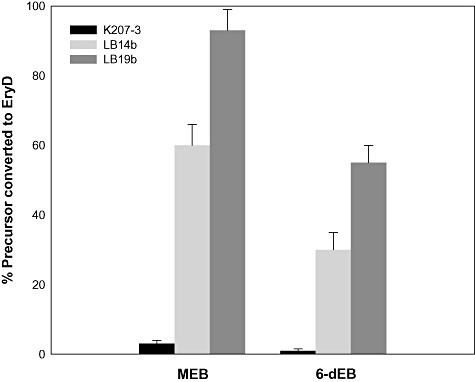
Production yields of EryD from bioconversion experiments in cultures of K207‐3, LB14b and LB19b expressing TDP‐sugar operons. Strains harbouring pKOS506‐72B were fed with 100 mg l−1 MEB, and strains harbouring both pKOS506‐72B and pLB353 were fed with 100 mg l−1 6‐dEB. EryD values were obtained from three independent experiments.
The biosynthesis of EryD from the 6‐dEB aglycone requires the attachment of two deoxysugars, TDP‐l‐mycarose and TDP‐d‐desosamine, to the macrolactone ring. This biosynthetic route also involves a hydroxylation step prior to the addition of the deoxysugars, catalysed by MegF, which converts 6‐dEB to erythronolide B (EB) (Fig. 4A). In order to evaluate the glycosylation system developed, we analysed this diglycosylation process through bioconversion studies performed with K207‐3 and LB14b. Both strains were co‐transformed with plasmids pGro7, pKOS506‐72B and pLB353, and cultures were fed with 100 gl l−1 of 6‐dEB. After 48 h of incubation at 22°C, production of EryD was quantified in cell‐free extracts by inhibition assays and analysed by LC/MS. EryD concentrations were 1 mg l−1 in K207‐3 cultures, and 60 mg l−1 in LB14b cultures (about 0.5% and 30% conversion respectively; Fig. 5). This impressive 60‐fold enhancement of 6‐dEB diglycosylation by LB14b over its parental strain was also evidenced by LC/MS analysis (Fig. 6).
Figure 6.
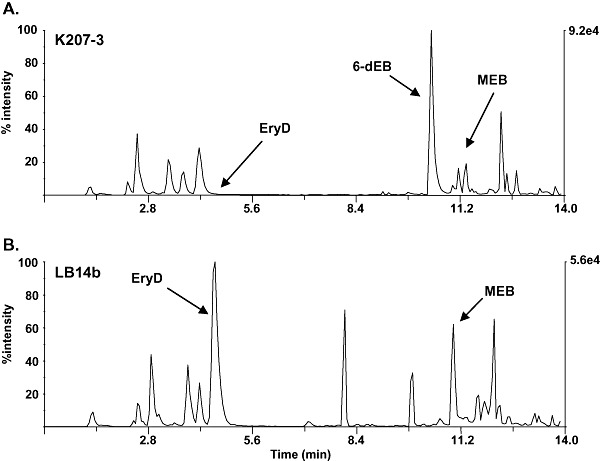
LC‐MS analysis of the bioconversion of 6‐dEB by E. coli strains expressing the TDP‐l‐mycarose and TDP‐desosamine pathways: (A) K207‐3; (B) LB14b. 6‐dEB, MEB and EryD are indicated.
Macrolide efflux pumps and glycosylation efficiency improvement
Considering that the AcrAB‐TolC pump is the main transporter involved in erythromycin resistance in E. coli (Sulavik et al., 2001), we decided to analyse whether inactivation of this pathway could improve further the efficiency of our macrolide glycosylation system. For this, we proceeded to disrupt the acrAB genes in LB14b as described in Experimental procedures to obtain strain LB19b. This new strain was tested for production of EryD in bioconversion experiments from both MEB and 6‐dEB, and compared with K207‐3 and LB14b. Bioconversion of MEB was performed in cultures of LB19b transformed with plasmids pGro7 and pKOS506‐72B, in the conditions described above. The antibiotic activity observed in culture supernatants obtained after 24 h of induction corresponded to an estimated production of 120 mg l−1 of EryD, which represents a 95% bioconversion of the MEB added to the culture (Fig. 5). This represents a 30% higher conversion of MEB to EryD than with LB14b. Cultures of LB19b transformed with plasmids pGro7, pKOS506‐72B and pLB353 were tested for diglycosylation by feeding with 100 mg l−1 of 6‐dEB. After 48 h of incubation at 22°C, production of EryD was estimated to be 100 mg l−1, about 40% higher than by LB14b (Fig. 5). This EryD production represents a 100‐fold improvement for the diglycosylation of 6‐dEB in the engineered strain LB19b compared with the parental K207‐3 strain.
It is worth mentioning that in a K207‐3 ΔacrAB mutant (LB17b) no significant differences were observed in EryD production levels compared with K207‐3 (data not shown). This suggests that the AcrAB‐TolC efflux system influences EryD production only at higher glycosylation levels, indicating that the main bottleneck in this process is TKDG availability.
Discussion
In the present work we report a metabolic engineering approach in E. coli K207‐3 to improve the glycosylation process of 6‐dEB for the production of EryD. A systematic analysis by PCR and LC/MS/MS enabled us to identify in K207‐3 three metabolic pathways that result in the consumption of TKDG, a key intermediate in the biosynthesis of many polyketide‐related 6‐DOHs, like TDP‐l‐mycarose and TDP‐d‐desosamine. Two of these pathways were previously described for the biosynthesis of the O7 antigen in E. coli VW187 (Marolda et al., 1999). Here we present data confirming the presence of the sugars TDP‐l‐rhamnose and TDP‐d‐Qui4NAc in K207‐3, and the partial characterization of their biosynthetic gene cluster that suggests a horizontal transfer from VW187. The third pathway described corresponds to TDP‐d‐Fuc4NAc biosynthesis, which is highly conserved among enteric bacteria and has been analysed in detail in E. coli K12 (Meier‐Dieter et al., 1992; Hwang et al., 2004; Hung et al., 2006).
By means of a series of genetic deletions, we were able to inactivate the three metabolic routes mentioned at the level of their TKDG‐consuming steps, and the resulting strain, LB14b, exhibited increased intracellular amounts of this metabolite (Fig. 2D). This strain was tested for its capacity to glycosylate MEB and 6‐dEB to obtain EryD through the heterologous expression of TDP‐d‐desosamine and TDP‐l‐mycarose biosynthetic pathways. LB14b exhibited a significantly enhanced glycosylation capacity, producing 80 mg l−1 of EryD from MEB in only 24 h of incubation in a batch process with a final OD600 of ~3 (Fig. 5). Thus, production of gram quantities of this compound seems achievable in high cell‐density cultures of E. coli where OD600 greater than 100 can be maintained at high productivity for more than a week (Lau et al., 2004). A further improvement in the production of glycosylated polyketides was obtained by the additional disruption of the genes encoding the AcrAB‐TolC pump, confirming the detrimental role of this efflux system in the macrolide glycosylation process (Fig. 5). The final strain, LB19b, produced 100‐fold higher amounts of EryD from 6‐dEB than K207‐3 (Fig. 5).
The current paper reports the first ‘rational’ strain improvement for the production of glycosylated polyketides in E. coli, consolidating this host as a robust platform for the heterologous production of these compounds. We believe this system constitutes not only a promising tool for the heterologous production of therapeutically important glycosylated polyketides synthesized by unculturable organisms, but also for the generation of novel bioactive compounds through sugars and/or polyketides combinatorial biosynthesis approaches (Salas and Mendez, 2007). Although natural products glycorandomization can also be accomplished by synthesis/semi‐synthesis, the enormous structural complexity of many glycosylated natural products renders this strategy less advantageous than in vivo methods, which allow to access new compounds via cost‐effective fermentation processes (Yang et al., 2004). For instance, Salas and co‐workers have developed a series of sugar biosynthesis plasmids that permitted to generate several new derivatives of the anti‐tumour compound elloramycin in a Streptomyces host (Lombo et al., 2004). These constructs enable genes to be easily incorporated, removed or replaced by others through a ‘plug and play’ cassette system, to perform combinatorial engineering of deoxysugar biosynthesis pathways. The high glycosylation yields obtained with LB19b would allow implementing a similar strategy in this strain, with the additional advantages related to the easier genetic manipulations, faster culturing processes and many more genetic tools available for E. coli. Finally, these sugar pathways could be combined, together with a suitable glycosyltransferase, with polyketide combinatorial approaches developed for E. coli (Menzella et al., 2005; 2007), to yield massive libraries of novel compounds with variations in both the aglycone core and the tailoring sugars.
Experimental procedures
Bacterial strains, plasmids and growth media
The bacterial strains and plasmids used in this study are listed in Table 1. Luria–Bertani (LB), LB agar and Antibiotic Medium 11 were obtained from Difco. Antibiotics were obtained from Sigma, and were used at the following concentrations in LB or LB agar when necessary: kanamycin (50 µg ml−1), streptomycin (50 µg ml−1), ampicillin (100 µg ml−1) and chloramphenicol (20 µg ml−1).
DNA manipulation
DNA restriction enzymes were used as recommended by the manufacturer (New England Biolabs). DNA manipulations were performed in E. coli Dh5α using standard protocols (Sambrook et al., 1989). DNA fragments were purified from agarose gels with the GFX PCR DNA and Gel Band Purification Kit (GE Helthcare). Plasmids were prepared using a QIAprep Spin Miniprep Kit (Qiagen). Deep Vent DNA polymerase was used in all PCR reactions according to the supplier's instructions (New England Biolabs).
Plasmid constructions
Each gene of the TDP‐l‐mycarose biosynthetic pathway was amplified by PCR from M. megalomicea genomic DNA and sequenced (Peiru et al., 2005; 2007). The 5′ primers used were designed to have an NdeI site overlapping the translational initiation codon, changing GTG start codons to ATG when necessary. The 3′ primers contained EcoRI and adjacent SpeI sites downstream from the stop codon. Primer sequences and amplification conditions were previously described (Peiru et al., 2005; 2007). Each gene was inserted as NdeI/EcoRI fragments into the pET28a vector (Novagen).
The general strategy for the construction of the mycarose operon was as previously described (Peiru et al., 2005). The mycarose operon contained in a pET28a vector was finally removed as an XbaI/EcoRI fragment and cloned into pKOS431‐39.1 to give the plasmid pLB353.
Construction of deletion strains
Deletion mutant strains were constructed according to the method described by Datsenko and Wanner (2000), using the kanamycin resistance gene kan. The specific primers used to construct the deletions and to confirm the allelic exchange are listed in Table S1. Gene replacement by the kan cassette was first verified by PCR using primers xxx‐up and xxx‐do (where xxx refers to any given targeted gene), corresponding to sequences flanking the target gene. A second control PCR was carried out to confirm the correct integration of the kan gene using primers xxx‐up and k2, resulting in a 0.5 kb amplification product. The kan insert was further removed as described by Datsenko and Wanner, 2000, and its deletion verified by PCR analysis using primers flanking the deleted region.
Strains LB1b, LB7b and LB8b were constructed using the pairs of hybrid primers rmlC‐del1/rmlC‐del2, wecE‐del1/wecE‐del2 and vioA‐del1/vioA‐del2 respectively. Strain LB9b was constructed by P1‐mediated transduction of ΔwecE::kan from the LB7b strain prior to removal of the kanamycin resistance cassette into the LB8b strain. The kan insert was then removed through the FLP recombinase as mentioned before. The LB10b strain was constructed using the pair of hybrid primers rmlC‐del1/vioA‐del2. This involves the replacement of a 3.1 kb fragment comprising rmlC, wzx and vioA by the kanamycin resistance cassette, which was further removed. Strain LB11b was constructed by P1‐mediated transduction of ΔwecE::kan from LB7b into LB10b, as described for LB8b construction. Strain LB12 was constructed using the pair of hybrid primers wecD‐del1/wecE‐del2 for a single‐step replacement of wecD and wecE by the kanamycin resistance cassette. The LB13b strain was constructed using the pair of hybrid primers rmlC‐del1/vioB‐del2, resulting in the replacement of a 3.6 kb fragment comprising rmlC, wzx, vioA and vioB by the kanamycin resistance cassette, which was finally removed. Strain LB14b was constructed by P1‐mediated transduction of ΔwecDE::kan from LB12 strain into LB13b, and the kan insert was further removed. Strain LB15b was constructed by P1‐mediated transduction of ΔwecE::kan from LB7b into LB13b, as described for LB8b construction. Strain LB17 was constructed using the pair of hybrid primers acrA‐del1/acrB‐del2 for a single step replacement of acrA and acrB by the kanamycin resistance cassette. Strain LB19b was constructed by P1‐mediated transduction of ΔacrAB::kan from LB17 strain into LB14b, and the kan insert was finally removed.
ECA analysis
Exopolysaccharide preparation, electrophoresis and detection were performed as previously described (Marolda et al., 2006). Briefly, membrane fractions were prepared as described previously (Barr et al., 1999), boiled for 10 min and then incubated overnight at 60°C with 1.6 µg µl−1 proteinase K. Membranes were boiled again for 10 min, and sample buffer was added. Samples were separated on a 14% tricine SDS‐PAGE gel and transferred to nitrocellulose membranes. Blots were reacted with anti‐O14 polyclonal antiserum to detect ECA.
TDP‐sugars analysis on cell‐free extracts
Escherichia coli strains were grown at 37°C in shake flasks in LB medium and in the presence of the corresponding antibiotics for plasmid maintenance when required. Overnight cultures were diluted 1:100 in fresh medium and grown to an OD600 of 0.6 before the addition of 2 mg ml−1l‐arabinose and 0.5 mM IPTG to a final concentration of 0.5 mM when needed. Induction was allowed to proceed for 24 h at 22°C. The cells were harvested, re‐suspended in 20 mM Tris buffer pH 7.6 and disrupted by sonication. After centrifugation at 15 000 g for 20 min, the supernatants were analysed by LC/MS/MS for the detection of TDP‐sugars, as described previously (Rodriguez et al., 2006).
Bioconversion experiments and polyketide analysis
Escherichia coli strains harbouring pGro7 and the different expression plasmids were cultured overnight at 37°C in LB with appropriate antibiotics, then subcultured by 1:100 dilution in the same medium and grown to an OD600 of 0.6. Chaperones and sugar gene expression were induced by addition of 2 mg ml−1l‐arabinose and 0.5 mM IPTG, respectively, and cultures were supplemented with 100 µg ml−1 of 6‐dEB or MEB. Cultures were grown at 22°C for 24–48 h, centrifuged at 15 000 g for 5 min, and culture broths were analysed by LC/MS as described previously (Peiru et al., 2005).
Micrococcus luteus inhibition assays
The EryD produced in bioconversion experiments was quantified through both an agar diffusion assay and a serial dilution test using M. luteus ATCC 9341 as test strain and EryA as standard. Samples were prepared as mentioned above but without addition of antibiotics, and supernatants were clarified with 0.2 µm filters. Agar diffusion plates were prepared with Antibiotic Medium 11 (Difco) seeded with a 24 h LB culture of M. luteus (0.2% v/v), and 6 mM wells were cut and removed to be filled with 25 µl of samples dilutions. The antibiotic concentrations of the test samples were determined by measuring the diameters of inhibition zones around the wells after incubation overnight at 30°C, using a standard of EryA as reference. The serial dilution test was performed by inoculating 2 ml samples of serial twofold dilutions in LB of the bioconversion samples with 2 × 106M. luteus cells. Cultures were grown with shaking at 30°C for 48 h, and bacterial growth inhibition was used to estimate the antibiotic concentration, using EryA as standard.
Acknowledgments
We would like to thank M.E. Castelli for her helpful assistance on ECA analysis, and D. A. Hopwood for critical comments on the manuscript. This work was supported by ANPCyT grants 01‐13705 and 15‐31969 and PIP 6436 from CONICET to H. Gramajo.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Exopolysaccharide preparations of K207-3 (lane 1) and LB7b (lane 2) strains were analysed by immunodetection with anti-O14 antiserum to detect ECA.
Table S1. Primers used in this study.
This material is available as part of the online article from http://www.blackwell-synergy.com
Please note: Blackwell Publishing is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Barr K., Klena J., Rick P.D. The modality of enterobacterial common antigen polysaccharide chain lengths is regulated by o349 of the wec gene cluster of Escherichia coli k‐12. J Bacteriol. 1999;181:6564–6568. doi: 10.1128/jb.181.20.6564-6568.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko K.A., Wanner B.L. One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopwood D.A. Genetic contributions to understanding polyketide synthases. Chem Rev. 1997;97:2465–2498. doi: 10.1021/cr960034i. [DOI] [PubMed] [Google Scholar]
- Hung M.N., Rangarajan E., Munger C., Nadeau G., Sulea T., Matte A. Crystal structure of TDP‐fucosamine acetyltransferase (WecD) from Escherichia coli, an enzyme required for enterobacterial common antigen synthesis. J Bacteriol. 2006;188:5606–5617. doi: 10.1128/JB.00306-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang B.Y., Lee H.J., Yang Y.H., Joo H.S., Kim B.G. Characterization and investigation of substrate specificity of the sugar aminotransferase WecE from E. coli K12. Chem Biol. 2004;11:915–925. doi: 10.1016/j.chembiol.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Kajimura J., Rahman A., Hsu J., Evans M.R., Gardner K.H., Rick P.D. O acetylation of the enterobacterial common antigen polysaccharide is catalyzed by the product of the yiaH gene of Escherichia coli K‐12. J Bacteriol. 2006;188:7542–7550. doi: 10.1128/JB.00783-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibwage I.O., Hoogmartens J., Roets E., Vanderhaeghe H., Verbist L., Dubost M. Antibacterial activities of erythromycins A, B, C, and D and some of their derivatives. Antimicrob Agents Chemother. 1985;28:630–633. doi: 10.1128/aac.28.5.630. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau J., Tran C., Licari P., Galazzo J. Development of a high cell‐density fed‐batch bioprocess for the heterologous production of 6‐deoxyerythronolide B in Escherichia coli. J Biotechnol. 2004;110:95–103. doi: 10.1016/j.jbiotec.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Lee H.Y., Khosla C. Bioassay‐guided evolution of glycosylated macrolide antibiotics in Escherichia coli. PLoS Biol. 2007;5:e45. doi: 10.1371/journal.pbio.0050045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombo F., Gibson M., Greenwell L., Brana A.F., Rohr J., Salas J.A., Mendez C. Engineering biosynthetic pathways for deoxysugars: branched‐chain sugar pathways and derivatives from the antitumor tetracenomycin. Chem Biol. 2004;11:1709–1718. doi: 10.1016/j.chembiol.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Marolda C.L., Feldman M.F., Valvano M.A. Genetic organization of the O7‐specific lipopolysaccharide biosynthesis cluster of Escherichia coli VW187 (O7:K1) Microbiology. 1999;145:2485–2495. doi: 10.1099/00221287-145-9-2485. [DOI] [PubMed] [Google Scholar]
- Marolda C.L., Tatar L.D., Alaimo C., Aebi M., Valvano M.A. Interplay of the Wzx translocase and the corresponding polymerase and chain length regulator proteins in the translocation and periplasmic assembly of lipopolysaccharide o antigen. J Bacteriol. 2006;188:5124–5135. doi: 10.1128/JB.00461-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier‐Dieter U., Barr K., Starman R., Hatch L., Rick P.D. Nucleotide sequence of the Escherichia coli rfe gene involved in the synthesis of enterobacterial common antigen. Molecular cloning of the rfe‐rff gene cluster. J Biol Chem. 1992;267:746–753. [PubMed] [Google Scholar]
- Mendez C., Salas J.A. Altering the glycosylation pattern of bioactive compounds. Trends Biotechnol. 2001;19:449–456. doi: 10.1016/s0167-7799(01)01765-6. [DOI] [PubMed] [Google Scholar]
- Menzella H.G., Reeves C.D. Combinatorial biosynthesis for drug development. Curr Opin Microbiol. 2007;10:238–245. doi: 10.1016/j.mib.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Menzella H.G., Reid R., Carney J.R., Chandran S.S., Reisinger S.J., Patel K.G. Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat Biotechnol. 2005;23:1171–1176. doi: 10.1038/nbt1128. et al. [DOI] [PubMed] [Google Scholar]
- Menzella H.G., Carney J.R., Santi D.V. Rational design and assembly of synthetic trimodular polyketide synthases. Chem Biol. 2007;14:143–151. doi: 10.1016/j.chembiol.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Murli S., Kennedy J., Dayem L.C., Carney J.R., Kealey J.T. Metabolic engineering of Escherichia coli for improved 6‐deoxyerythronolide B production. J Ind Microbiol Biotechnol. 2003;30:500–509. doi: 10.1007/s10295-003-0073-x. [DOI] [PubMed] [Google Scholar]
- Mutka S.C., Carney J.R., Liu Y., Kennedy J. Heterologous production of epothilone C and D in Escherichia coli. Biochemistry. 2006;45:1321–1330. doi: 10.1021/bi052075r. [DOI] [PubMed] [Google Scholar]
- Nishino K., Yamaguchi A. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J Bacteriol. 2001;183:5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki R., Okazaki T., Kuriki Y. Isolation of thymidine diphosphate rhamnose and a novel thymidine diphosphate sugar compound from Escherichia coli strain B. Biochim Biophys Acta. 1960;38:384–386. doi: 10.1016/0006-3002(60)91271-3. [DOI] [PubMed] [Google Scholar]
- Peiru S., Menzella H.G., Rodriguez E., Carney J., Gramajo H. Production of the potent antibacterial polyketide erythromycin C in Escherichia coli. Appl Environ Microbiol. 2005;71:2539–2547. doi: 10.1128/AEM.71.5.2539-2547.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiru S., Rodriguez E., Tran C.Q., Carney J.R., Gramajo H. Characterization of the heterodimeric MegBIIa:MegBIIb aldo‐keto reductase involved in the biosynthesis of L‐mycarose from Micromonospora megalomicea. Biochemistry. 2007;46:8100–8109. doi: 10.1021/bi700396n. [DOI] [PubMed] [Google Scholar]
- Pfeifer B.A., Admiraal S.J., Gramajo H., Cane D.E., Khosla C. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- Rodriguez E., Peiru S., Carney J.R., Gramajo H. In vivo characterization of the dTDP‐D‐desosamine pathway of the megalomicin gene cluster from Micromonospora megalomicea. Microbiology. 2006;152:667–673. doi: 10.1099/mic.0.28680-0. [DOI] [PubMed] [Google Scholar]
- Salas J.A., Mendez C. Engineering the glycosylation of natural products in actinomycetes. Trends Microbiol. 2007;15:219–232. doi: 10.1016/j.tim.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Sambrook J. Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Schneider D., Duperchy E., Depeyrot J., Coursange E., Lenski R., Blot M. Genomic comparisons among Escherichia coli strains B, K‐12, and O157:H7 using IS elements as molecular markers. BMC Microbiol. 2002;2:18. doi: 10.1186/1471-2180-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulavik M.C., Houseweart C., Cramer C., Jiwani N., Murgolo N., Greene J. Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes. Antimicrob Agents Chemother. 2001;45:1126–1136. doi: 10.1128/AAC.45.4.1126-1136.2001. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Xu Y., Perepelov A.V., Qi Y., Knirel Y.A., Wang L., Feng L. Biochemical characterization of dTDP‐D‐Qui4N and dTDP‐D‐Qui4NAc biosynthetic pathways in Shigella dysenteriae type 7 and Escherichia coli O7. J Bacteriol. 2007;189:8626–8635. doi: 10.1128/JB.00777-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Hoffmeister D., Liu L., Fu X., Thorson J.S. Natural product glycorandomization. Bioorg Med Chem. 2004;12:1577–1584. doi: 10.1016/j.bmc.2003.12.046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Exopolysaccharide preparations of K207-3 (lane 1) and LB7b (lane 2) strains were analysed by immunodetection with anti-O14 antiserum to detect ECA.
Table S1. Primers used in this study.