

Summary
Using directed evolution and site‐directed mutagenesis, we have isolated a highly thermostable variant of Aspergillus niger glucoamylase (GA), designated CR2‐1. CR2‐1 includes the previously described mutations Asn20Cys and Ala27Cys (forming a new disulfide bond), Ser30Pro, Thr62Ala, Ser119Pro, Gly137Ala, Thr290Ala, His391Tyr and Ser436Pro. In addition, CR2‐1 includes several new putative thermostable mutations, Val59Ala, Val88Ile, Ser211Pro, Asp293Ala, Thr390Ser, Tyr402Phe and Glu408Lys, identified by directed evolution. CR2‐1 GA has a catalytic efficiency (kcat/Km) at 35°C and a specific activity at 50°C similar to that of wild‐type GA. Irreversible inactivation tests indicated that CR2‐1 increases the free energy of thermoinactivation at 80°C by 10 kJ mol−1 compared with that of wild‐type GA. Thus, CR2‐1 is more thermostable (by 5 kJ mol−1 at 80°C) than the most thermostable A. niger GA variant previously described, THS8. In addition, Val59Ala and Glu408Lys were shown to individually increase the thermostability in GA variants by 1 and 2 kJ mol−1, respectively, at 80°C.
Introduction
Glucoamylase (GA) from Aspergillus niger (EC3.2.1.3) is used as an industrial enzyme to hydrolyse maize starch into glucose. The first step of this process involves the liquefaction of starch into starch dextrin by α‐amylase. This step is carried out at 105°C for 5 min and then at 95°C for 1 h. Glucoamylase is used in the second step (saccharification) to convert the dextrin into glucose monomers. This step involves cooling the dextrin to 60°C, the operational temperature of GA. At this temperature, the reaction can take several days to complete. The resulting glucose may be used directly as feedstock for ethanol or other fermentation, or converted enzymatically to fructose in the production of high‐fructose syrup (Crabb and Shetty, 1999; Ford, 1999; Reilly, 1999). Increasing GA thermostability, and thus its operating temperature and catalytic rate, is important from an industrial standpoint because it is costly both to cool dextrin from 95°C to 60°C and to hold it for several days during the saccharification process.
Glucoamylase from A. niger has been characterized both biochemically and genetically (Aleshin et al., 1992; 1994; 1996). The enzyme has two domains and a linker: an N‐terminal catalytic domain of 440 amino acids with an (α,α)6‐barrel structure that has been structurally characterized by X‐ray crystallography (Fig. 1); a C‐terminal starch‐binding domain of 108 amino acids, necessary for hydrolysis of insoluble starch granules (but not necessary for hydrolysis of soluble substrates); and a heavily O‐glycosylated linking region of 68 amino acids. The GA gene from A. niger has been cloned and can be expressed conveniently in the yeast Saccharomyces cerevisiae (Cole et al., 1988). Numerous approaches have successfully been taken to improve the thermostability of A. niger GA by site‐directed mutagenesis (Ford, 1999; Reilly, 1999; Liu and Wang, 2003). Recently, Wang and colleagues (2006) have used directed evolution in conjunction with site‐directed mutagenesis to construct the most thermostable A. niger GA previously described (THS8). THS8 contains eight mutations (Asp20Cys, Ala27Cys, Ser30Pro, Thr62Ala, Ser119Pro, Gly137Ala, Thr290Ala and His391Tyr) with a cumulative increase in thermostability (ΔG) over wild‐type GA of 5 kJ mol−1 at 80°C without a reduction in catalytic activity.
Figure 1.
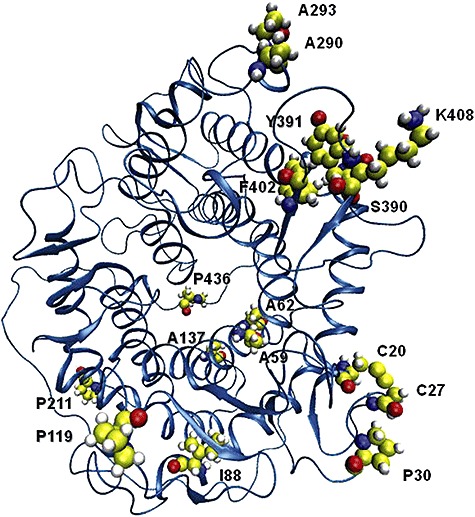
Three‐dimensional structure of the catalytic domain of GA showing the location of the mutations in CR2‐1.
We have continued the directed‐evolution isolation of new thermostable mutations, and we have now constructed a multiple mutant including several new mutations designated CR2‐1, which doubles the thermostability of THS8. CR2‐1 is more thermostable than THS8 at 80°C by 5 kJ mol−1, and is more thermostable than wild‐type GA by 10 kJ mol−1, making it the most thermostable A. niger GA variant described to date.
Results
Mutant isolation
A diagram of the isolation of the multiply‐mutant variants RE5, RE15, SRE15, CR1‐1 and CR2‐1 is shown in Fig. 2. Twenty‐four multiply‐mutated GA variants previously isolated by random and site‐directed mutagenesis were subjected to random in vitro recombination. Eleven thousand colonies resulting from in vitro recombination of these mutations were then screened for GA thermostability at 78°C using the plate thermostability screen. One hundred and five colonies with enhanced GA thermostability were chosen for a second round of screening at 79°C. Two of these, RE5 and RE15, were chosen for further characterization based on their thermostability. DNA sequencing showed that both RE5 and RE15 included the following previously described thermostability mutations in the catalytic domain: Asn20Cys and Ala27Cys (forming a new disulfide bond), Ser30Pro, Ser119Pro, Gly137Ala and Thr290Ala (Table 1). In addition, RE5 included the previously described catalytic‐domain mutation His391Tyr, whereas RE15 included two new amino acid replacement mutations in the catalytic domain, Ser211Pro and Thr390Ser, and a mutation, Tyr527Cys, in the starch‐binding domain (subsequently removed).
Figure 2.
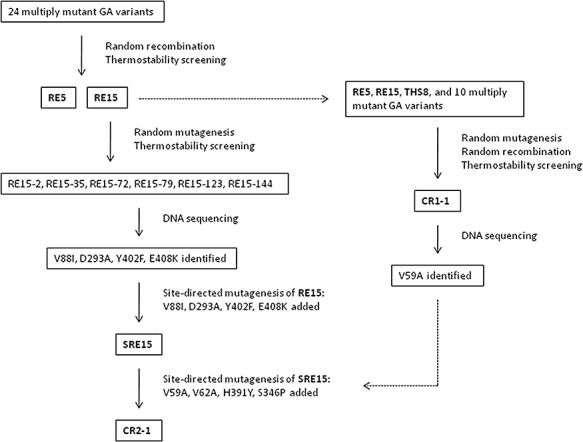
Mutant isolation diagram.
Table 1.
Amino acid substitutions of thermostable GA mutants.
| Mutant | Amino acid substitutions | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N20C | A27C | S30P | V59A | T62A | V88I | S119P | G137A | S211P | T290A | D293A | T390S | H391Y | Y402F | E408K | S436P | |
| RE5 | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||||||
| RE15 | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||||
| SRE15 | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| CR1‐1 | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||
| CR2‐1 | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
The GA gene of RE15 was subjected to random PCR mutagenesis, and 40 200 resulting colonies were screened for GA thermostability at 80°C. Of these, six new mutants that were more thermostable than RE15, designated RE15‐2, RE15‐35, RE15‐72, RE15‐79, RE15‐123 and RE15‐144, were subjected to DNA sequencing. From the sequences of these six new mutants, a total of 11 new point mutations in the catalytic domain were identified. Seven of the new mutations were silent, whereas four resulted in amino acid replacement: Val88Ile, Asp293Ala, Tyr402Phe and Glu408Lys. Asp293Ala and Tyr402Phe were each found in only one of the six mutants (RE15‐2 and RE15‐123 respectively), whereas Val88Ile was found in RE15‐35 and RE15‐79, and Glu408Lys was found in RE15‐35, RE15‐72 and RE15‐144. SRE15 was constructed by site‐directed mutagenesis to include all of the mutations of RE15 plus the four new mutations: Val88Ile, Asp293Ala, Tyr402Phe and Glu408Lys. To focus on mutations of the catalytic domain, the starch‐binding domain sequences of both RE15 and SRE15 were replaced with the wild‐type sequence for all subsequent work.
Site‐directed mutagenesis was used to recreate Val88Ile, Ser211Pro, Asp293Ala, Tyr402Phe and Glu408Lys singly in an otherwise wild‐type GA background for further thermostability tests. Thr390Ser was not isolated in a wild‐type GA background for further thermostability testing due to the similarity of the replacement amino acid side‐group, and its likely neutrality with respect to thermostability. A thermostability plate assay was used to test the individual phenotypic effect of the single mutations Val88Ile, Ser211Pro, Asp293Ala, Tyr402Phe and Glu408Lys compared with wild‐type GA, Ser30Pro (Allen et al., 1998), RE15 and SRE15 (Fig. 3). Of the new single mutations, only Glu408Lys showed a slightly increased resistance to thermoinactivation compared with wild‐type GA. In contrast, both RE15 and SRE15 showed substantially increased thermostability.
Figure 3.
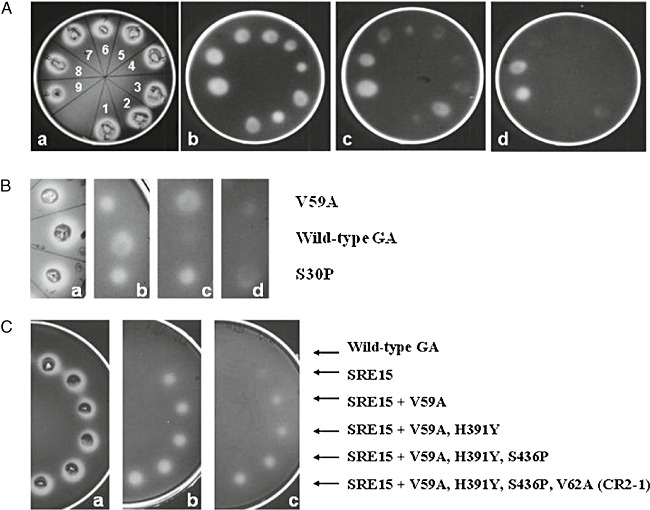
Thermostability plate assays. A. Plate a shows yeast colonies growing at 30°C on media containing 1% soluble starch and stained with iodine to reveal halos of secreted GA activity. Plates b, c and d show the residual GA activity at 50°C on 1% starch plates after transfer of GA to a nitrocellulose membrane, and inactivation at 76°C for 1, 3 and 4 min respectively. (1) Wild‐type GA; (2) Ser30Pro; (3) Val881le; (4) Ser211Pro; (5) Asp293Ala; (6) Tyr402Phe; (7) Glu408Lys; (8) RE15; (9) SRE15. B. Plate a shows yeast colonies growing at 30°C on media containing 1% soluble starch and stained with iodine to reveal halos of secreted GA activity. Plates b, c and d show the residual GA activity at 50°C on 1% starch plates after transfer of GA to a nitrocellulose membrane and inactivated at 78°C for 1, 2 and 3 min respectively. C. Plate a shows yeast colonies growing at 30°C on media containing 1% soluble starch and stained with iodine to reveal halos of secreted GA activity. Plates b and c show the residual GA activity at 50°C on 1% starch plates after transfer of GA to a nitrocellulose membrane, and inactivation at 84°C for 2 min and 5 min respectively.
In a separate directed evolution experiment, RE5 and RE15 plus 10 other multiply‐mutated GA thermostable variants including THS8 were subjected to random mutagenesis and random recombination, resulting in the isolation of CR1‐1. DNA sequencing of CR1‐1 showed that this GA variant contained a new mutation, Val59Ala, and the previously characterized mutations Asn20Cys/Ala27Cys, Ser30Pro, Ser119Pro, Gly137Ala, Ser211Pro, Thr290Ala, and His391Tyr, and Ser436Pro (Table 1) (Chen et al., 1996; Li et al., 1997; 1998; Allen et al., 1998; Wang et al., 2006). Ser436Pro was isolated independently as a random thermostable mutation in CR1‐1, but it had previously been constructed by site‐directed mutagenesis and had its thermostability characterized (Li et al., 1998). Val59Ala was recreated by site‐directed mutagenesis in a wild‐type GA background to test its individual phenotype. A thermostability plate assay with Ser30Pro as a control showed that Val59Ala was more thermostable than wild‐type GA, with a thermostability similar to that of Ser30Pro (Fig. 3).
CR2‐1 was constructed by site‐directed mutagenesis of SRE15 to contain all of the thermostable mutations previously isolated by directed evolution in the Ford lab, including those of CR1‐1. In addition to the mutations of SRE15, CR2‐1 contains Val59Ala, Thr62Ala (Wang and colleagues 2006), His391Tyr and Ser436Pro (Table 1). Thr62Ala was one of the mutations of THS8, and has been shown to increase thermostability by 2 kJ mol−1 at 65°C (Wang et al., 2006).
Figure 3 shows a plate thermostability assay of wild‐type GA, SRE15, and a stepwise addition to SRE15 by site‐directed mutagenesis of each of the mutations that differ between SRE15 and CR2‐1. The results indicate the cumulative thermostability obtained by addition of these thermostability mutations. CR2‐1 still has strong residual activity after heat inactivation at 84°C for 5 min compared with an almost complete loss of activity by SRE15.
Val59Ala and Glu408Lys were each recreated in a multiply‐mutated thermostable genetic background by site‐directed mutagenesis to test for their individual contribution to thermostability. Val59Ala was added to SRE15 (designated SRE15+V59A) so that its thermostability could be compared with that of SRE15, while Glu408Lys was added to RE5 (designated RE5+E408K) for comparison with RE5.
Kinetic analysis
RE5, RE15, SRE15, CR1‐1, CR2‐1, RE5+E408K, SRE15+V59A and wild‐type GA enzymes were purified by affinity chromatography on an acarbose‐Sepharose column from S. cerevisiae shake‐flask cultures and characterized kinetically. The specific activity of the enzymes using maltose as a substrate was measured at 50°C (Table 2). The specific activities of the mutant enzymes were similar to or greater than that of wild‐type GA. Enzyme kinetics were measured at 35°C for comparison with previous data (Allen et al., 1998; Li et al., 1998; Wang et al., 2006) (Table 2). All of the mutant enzymes showed catalytic efficiencies (kcat/Km) in the same range as that of wild‐type GA, with values similar to that previously reported for other active thermostable mutants (Allen et al., 1998; Li et al., 1998; Wang et al., 2006).
Table 2.
Catalytic properties of wild‐type and mutant GAs at pH 4.5, using maltose as substrate.
| GA form | Specific activitya (IU mg−1) | kcatc (s−1) | Km (mM) | kcat/Km (s−1 mM−1) |
|---|---|---|---|---|
| Wild type | 13.9 ± 0.2b | 7.3 ± 0.1d | 0.7 ± 0.0 | 10 |
| RE5 | 14.7 ± 1.1 | 7.7 ± 0.3 | 1.4 ± 0.1 | 5.5 |
| RE5+E408K | 15.4 ± 0.6 | 8.1 ± 0.2 | 1.4 ± 0.1 | 5.8 |
| CR1‐1 | 13.8 ± 0.4 | 6.4 ± 0.1 | 0.8 ± 0.0 | 7.7 |
| RE15 | 21.3 ± 0.7 | 11.7 ± 0.4 | 1.0 ± 0.1 | 12 |
| SRE15 | 23.1 ± 1.9 | 12.9 ± 0.6 | 1.8 ± 0.2 | 8.0 |
| SRE15+V59A | 12.2 ± 0.7 | 6.5 ± 0.2 | 1.5 ± 0.0 | 4.3 |
| CR2‐1 | 13.9 ± 0.3 | 7.5 ± 0.1 | 1.1 ± 0.0 | 6.8 |
Measured at 50°C.
Standard deviation from six assays.
Measured at 35°C.
Standard error from duplicate assays.
Kinetic parameters for irreversible thermoinactivation of the mutants and wild‐type GA are shown in Table 3. The free energy of thermoinactivation (ΔG) for all of the mutant GAs was higher than that of wild‐type GA at both 72.5°C and 80°C. CR2‐1 showed the most thermostability of the GA mutants, and is the most thermostable A. niger GA variant reported to date, with a ΔG value 10 kJ mol−1 higher than that of wild‐type GA at both 72.5°C and 80°C, and with a calculated melting temperature 10.0°C higher than that of wild‐type GA. At 80°C, SRE15 has a ΔG 8 kJ mol−1 higher than that of wild‐type GA, and is more thermostable than CR1‐1, RE5 and RE15. At 72.5°C, however, both CR1‐1 and RE5 are more thermostable than SRE15. RE5+E408K is more thermostable than RE5 by 1 kJ mol−1 at 72.5°C and 2 kJ mol−1 at 80°C, with an increased calculated melting temperature of 1.1°C, indicating that Glu408Lys is a thermostabilizing mutation in this genetic background. Similarly, SRE15+V59A is more thermostable than SRE15 by 4 kJ mol−1 at 72.5°C and 1 kJ mol−1 at 80°C, with an increased calculated melting temperature of 3.5°C, indicating that V59A is also a thermostabilizing mutation.
Table 3.
Parameters for irreversible thermoinactivation of wild‐type and mutant GAs.
| GA form | ΔG72.5°C (kJ mol−1) | ΔG‡80°C (kJ mol−1) | Tm (°C) |
|---|---|---|---|
| Wild type | 101 | 96 | 69.2a |
| RE5 | 107 | 101 | 74.6 |
| RE5+E408K | 108 | 103 | 75.7 |
| CR1‐1 | 107 | 103 | 75.4 |
| RE15 | 105 | 101 | 72.8 |
| SRE15 | 106 | 104 | 75.0 |
| SRE15+V59A | 110 | 105 | 78.5 |
| CR2‐1 | 111 | 106 | 79.2 |
Temperature at which enzymes are 50% inactivated after 10 min.
Figure 4 is an irreversible thermoinactivation plot showing the effect of temperatures between 65°C and 87.5°C on the thermoinactivation constant (kd) of RE15, CR1‐1, CR2‐1 and wild‐type GAs. The results show that at all temperatures, each of the mutant GAs is substantially more thermostable than wild‐type GA, with CR2‐1 being the most thermostable. Figure 5 shows the effect of temperature on the kd values of SRE15+V59A versus SRE15 as a control, and RE5+E408K versus RE5 as a control. The results show that at every temperature tested, SRE15+V59A and RE5+E408K are more thermostable than SRE15 and RE5 respectively.
Figure 4.
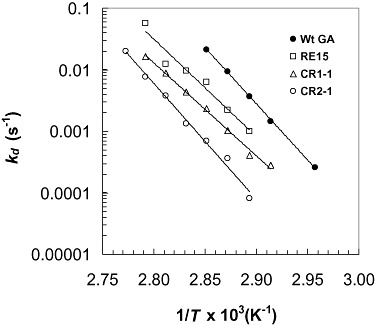
Irreversible thermoinactivation plots of RE15, CR1‐1 and CR2‐1, and wild‐type (Wt) GAs.
Figure 5.
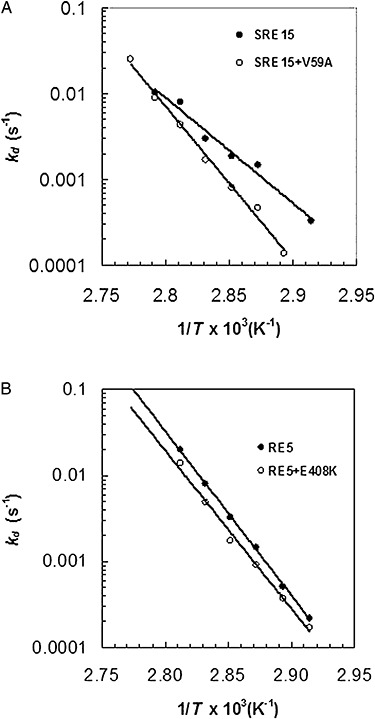
Irreversible thermoinactivation. A. SRE15+V59A is compared with SRE15. B. RE5+E408K is compared with RE5.
Discussion
We have used the techniques of directed evolution (phenotype screening following random mutagenesis and random recombination) and site‐directed mutagenesis to isolate several multiply‐mutated GA variants (RE5, RE15, SRE15, CR1‐1, CR2‐1), with enhanced thermostability compared with wild‐type GA (Fig. 2). CR2‐1 combines all of the mutations isolated by directed evolution to produce the most thermostable A. niger GA variant described to date, with a significant increase (10 kJ mol−1) in the free energy of inactivation over that of wild‐type GA, and an increased calculated melting temperature of 10.0°C.
RE5 and RE15 were isolated following random recombination and screening of previously isolated (but not necessarily characterized) thermostable GA variants. The seven catalytic‐domain mutations identified in RE5 by DNA sequencing were all previously characterized and individually produce small incremental increases in thermostability at 65°C compared with wild‐type GA: Asn20Cys/Ala27Cys (forming a new disulfide bond) (ΔΔG = 2 kJ mol−1 at 65°C) (Li et al., 1998); Ser30Pro (ΔΔG = 2 kJ mol−1 at 65°C) (Allen et al., 1998); Ser119Pro (ΔΔG = 1 kJ mol−1 at 65°C) (Wang et al., 2006); Gly137Ala (ΔΔG = 1 kJ mol−1 at 65°C) (Chen et al., 1996); Thr290Ala (ΔΔG = 1 kJ mol−1 at 65°C); and His391Tyr (ΔΔG = 1 kJ mol−1 at 65°C) (Wang et al., 2006).
RE15 has all of the same mutations as RE5 except for His391Tyr, and has two additional active‐site mutations, Ser211Pro and Thr390Ser. Although Ser211Pro and Thr390Ser were included in the subsequently constructed variants SRE15 and CR2‐1, there is no evidence that they enhance A. niger GA thermostability. No difference in thermostability has been detected between Ser211Pro and wild‐type GA either in plate assays (Fig. 1) or in thermostability assays (data not shown). Ser211Pro is located at the N1 position of GA helix 7. Proline is one of the preferred residues at this position of α‐helices (Richardson and Richardson, 1988). Thr390Ser was considered to be a neutral or non‐enhancing substitution with respect to thermostability, and was not studied in isolation due to the similarity of the side‐group substitution and its location outside of the highly conserved GA domains. Serine residues are present at a low frequency in thermophilic proteins compared with mesophilic proteins (Kumar et al., 2000).
SRE15 was constructed by adding four new mutations identified by directed evolution to the mutations of RE15: Val88Ile, Asp293Ala, Tyr402Phe and Glu408Lys. The cumulative effect of the addition of these four mutations was an increase in thermostability (ΔG) of SRE15 over that of RE15 by 3 kJ mol−1 at 80°C, with little effect on either catalytic efficiency or specific activity. Each of the individual mutations was isolated in a wild‐type GA genetic background to test for thermostability compared with wild‐type GA in plate assays (Fig. 3). Of these four mutations, only Glu408Lys showed a slight increase in thermostability compared with wild‐type GA in plate thermostability tests. A rationale for the putative contribution to the thermostability of SRE15 by each of the new mutations can be made nonetheless, because in combination they increase the thermostability of RE15 by 3 kJ mol−1 at 80°C. The location of each of these amino acid substitutions in the GA catalytic‐domain three‐dimensional structure can be seen in Fig. 1.
Val88 is located in a buried interhelical loop near α‐helix H3 in a hydrophobic microdomain. Substitution with the similar but more hydrophobic residue isoleucine may contribute to the stability of the hydrophobic microdomain, resulting in a small individual contribution by Val88Ile to GA thermostability.
Asp293Ala may make an individual contribution to GA thermostability by removing the thermolabile sequence Asp–Gly at this position. Chen and colleagues (1995) replaced Ala293 with two other amino acids, Glu and Gln, and found that these substitutions slightly increased GA thermostability at 70°C, presumably due to the removal of the Asp–Gly sequence at this position.
Tyr402Phe replaces a polar aromatic amino acid with a non‐polar aromatic residue in an internal semi‐hydrophobic environment in the protein. The phenyalanine ring at residue 402 would be in close proximity to the imadazole ring of His391, with the two rings facing each other about 3.5–4.0 Å apart. At lower pH values, the nitrogen atom on the imadazole ring of His391 is protonated, making it a powerful proton donor. His391 may thus form a stabilizing cation–π interaction with the π electrons of the benzyl ring of Phe402, contributing to the cumulative thermostability of the enzyme (Gallivan and Dougherty, 1999; Scheiner et al., 2002).
The thermostability of Glu408Lys was further tested by adding it to the thermostable genetic background of RE5. RE5+E408K had an increased ΔG of 1 and 2 kJ mol−1 at 72.5°C and 80°C, respectively, compared with RE5 alone (Table 3). These results confirm that Glu408Lys is a thermostabilizing mutation. Lys408 is located near residues Asp403 and Asp406 on the enzyme surface in contact with the surrounding solvent. A molecular dynamics simulation modelling the potentially stabilizing interaction of Lys408 with nearby residues showed that the NH3+ group on Lys408 is close enough (1.81 Å) to form a stabilizing salt bridge with the COO‐ group on Asp406, with potential to contribute to enzyme thermostability (Fig. 6). Salt bridges can be very stabilizing at higher temperatures, and are more prevalent in thermophilic than mesophilic proteins (Kumar et al., 2000).
Figure 6.
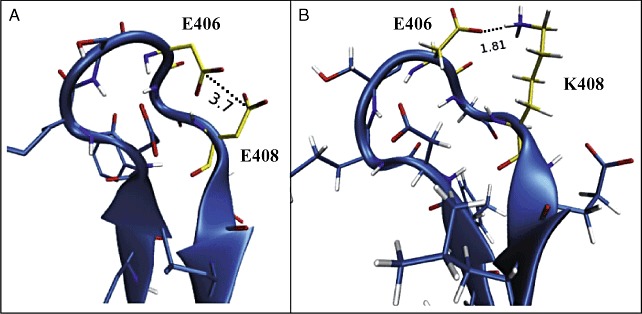
Molecular dynamics simulation of mutant E408K interactions. A. Wild type with glutamate at residue 408 (E408). The E408 and E406 side‐chains are separated by 3.7 Å. B. Mutant E408K with lysine substituting for glutamate at residue 408. The surface loop is stabilized by the formation of a 1.8 Å salt bridge between K408 and E406.
CR1‐1 was isolated in a second set of directed evolution experiments starting with RE5, RE15, THS8 (Wang et al., 2006), and other thermostable GA variants (Fig. 2). CR1‐1 was shown by DNA sequencing to contain several previously described mutations, and one new mutation, Val59Ala. CR1‐1 is more thermostable at 72.5°C and 80°C than either RE5 or RE15. Although CR1‐1 is slightly more thermostable than SRE15 at 72.5°C, it is less thermostable than SRE15 at 80°C.
To determine if Val59Ala is a thermostabilizing mutation, it was reconstructed both in a wild‐type GA background and in a thermostable SRE15 genetic background, and compared with wild‐type GA and SRE15 respectively. The plate assay results (Fig. 3) are consistent with those of the thermoinactivation test shown in Table 3 and Fig. 5, which indicate that Val59Ala is a strongly thermostabilizing mutation at 72.5°C (4 kJ mol−1) and a more weakly thermostabilizing mutation at 80°C (1 kJ mol−1). Ala59 is an unusual thermostabilizing substitution, as it is located in an interior, hydrophobic region of the enzyme but takes up less space and has fewer potential interactions with other residues than the larger and more hydrophobic Val59. For this reason, a molecular dynamics simulation of Val59 was not performed. Valine is more typically found as a substitution for the smaller alanine side‐group in other thermostable proteins. Perhaps the less bulky Ala residue enhances protein folding at residue 59 of A. niger GA.
Our results show that incremental increases in GA thermostability may be made by the accumulation of mutations in directed evolution, even though mutations identified by thermostability screening may not be detectable as thermostable when reconstructed in an otherwise wild‐type genetic background. Although a rationale for increasing thermostability could be made for all of the mutations (except Thr390Ser), the only individual mutations recreated in a wild‐type GA genetic background that demonstrated increased thermostability compared with wild‐type GA were Val59Ala and Glu408Lys. These two mutations also demonstrated thermostability when recreated in thermostable backgrounds.
The relatively weak thermostability phenotype of Glu408Lys in a wild‐type GA background (seen in the plate thermostability tests, Fig. 3) may be due to thermolability at sites distant from Glu408Lys. Perhaps only when the enzyme is more stable at these distant sites is the stability conferred by the Glu408Lys mutation effective. Thus the phenotypic ‘window’ of effectiveness of the mutation may depend on the stability of the entire enzyme. This may also be true for the other new mutations isolated in this study which cumulatively contributed to the thermostability of SRE15 over RE15 by 3 kJ mol−1 at 80°C. These mutations were initially detected and isolated in a genetic background containing multiple thermostability mutations, in enzymes that were already more thermostable than wild‐type GA. It is possible that these mutations would show a stronger individual phenotypic effect if tested in a more thermostable genetic background.
Experimental procedures
Yeast culture
The A. niger GA gene was expressed in S. cerevisiae using the yeast episomal vector YEpPM18, and the S. cerevisiae host strain C468 (α leu 2‐3 leu 2‐112 his 3‐11 his 3‐15 mal‐), both generous gifts from Cetus Corporation (Cole et al., 1988). Yeast cultures were grown following the methods described in Flory and colleagues (1994).
Plate thermostability assay
Plate thermostability assays were performed as described in Wang and colleagues (2006), adapted for GA from the methods of Okkels and colleagues (1997). Yeast were grown on SD‐His plates containing 1% soluble starch (Flory et al., 1994) at 30°C, overlaid with a nitrocellulose membrane. During yeast growth, soluble GA secreted from the yeast colonies adsorbed onto the membrane where it contacted the colony. Membranes were removed from the plates, rinsed, and submerged in 0.05 M NaOAc buffer, pH 4.5 at high temperatures for various lengths of time to partially or completely inactivate the adsorbed enzymes. After thermal treatment, membranes were overlaid onto fresh plates containing 1% soluble starch, and incubated at 50°C for 48 h. Residual GA activity from the membrane created ‘halos’ of starch degradation where the adsorbed enzyme contacted starch in the plate. Halos were detected by staining plates with iodine vapours.
DNA manipulation
The Altered Sites II (Promega) and QuickChange (Stratagene) in vitro mutagenesis kits were used for site‐directed mutagenesis. DNA oligonucleotide synthesis and sequencing were performed at the Iowa State University DNA Facility. Other DNA manipulations were performed using standard methods (Sambrook and Russell, 2001).
Random in vitro mutagenesis of the GA gene and random in vitro recombination of mutations were performed as described in Wang and colleagues (2006). Polymerase chain reaction mutagenesis of the GA gene from YEpPM18 was performed following the method of Leung and colleagues (1989). In vivo recombination of the mutagenized GA gene products and gapped YEpPM18 vectors (without the GA gene) was used to introduce the mutated GA genes into yeast, following the methods of Muhlrad and colleagues (1992) and Staples and Dieckmann (1993).
Random recombination of mutations was performed by in vivo recombination in yeast of pooled mutated and overlapping restricted GA gene products and gapped YEpPM18 vectors (without the GA gene) as described in Wang and colleagues (2006).
Enzyme analysis
Yeast expressing wild‐type and mutant GAs were grown in shake‐flask cultures as described in Wang and colleagues (2006). Enzymes were collected by centrifugation, and concentrated by diafiltration in an Amicon S1 spiral membrane ultrafiltration cartridge using 0.5 M NaCl/0.1 M NaOAc buffer, pH 4.5. Glucoamylase was purified by affinity chromatography on an acarbose‐Sepharose column using the method of Chen and colleagues (1994). Protein concentrations were determined against bovine serum albumen as a standard, using a bicinchoninic acid protein assay (Pierce).
Specific activity using maltose (3.3%) as substrate was measured using the method of Chen and colleagues (1994). One unit (IU) of GA was defined as the amount required to produce 1 µmol min−1 glucose at 50°C.
Steady‐state kinetic analysis was performed as described by Wang and colleagues (2006). Maltose concentrations from 0.2 Km to 4 Km were used to determine activity at 35°C using 0.05 M NaOAc buffer, pH 4.5. Reactions contained 4 µg of enzyme in 1.2 ml. EnziFitter software (Elsevier–Biosoft) was used to calculate the kinetic constants Km and kcat using the Michaelis–Menten equation.
Irreversible thermoinactivation was measured as described in Wang and colleagues (2006). Enzymes in 0.05 M NaOAc buffer at pH 4.5 were inactivated at temperatures between 65°C and 87.5°C for various amounts of time. After inactivation, enzymes were chilled on ice, and incubated at 4°C for 24 h. Residual activity was determined at 35°C in 0.05 M NaOAc buffer, pH 4.5 using 4.0% maltose as substrate. Coefficients of irreversible thermoinactivation, kd, were determined by calculating the linear regression of the ln (residual activity) versus time of thermoinactivation.
Irreversible thermoinactivation plots of kd/T versus 1/T were made to indicate thermostability of the enzymes at different temperatures (Figs 4 and 5). ΔG values at 72.5°C and 80°C were calculated from linear regressions of the slopes of these plots using transition state theory (Chen et al., 1994). Melting temperature (Tm) was determined by extrapolation from the irreversible thermoinactivation plots at the kdvalue where half the residual activity is lost after 10 min (Allen et al., 1998).
Computational methods
The three‐dimensional structure of GA showing the mutations of CR2‐1 (Fig. 1) was generated by substituting the mutations into the wild‐type GA crystal structure (Aleshin et al., 1996) using VMD 1.8.5 (Visual Molecular Dynamics) graphics (Humphrey et al., 1996).
The interaction of Lys408 with surrounding residues was modelled by force‐field molecular dynamics simulation of the GA crystal structure (Aleshin et al., 1996) using NAMD 2.5 (NAnoscale Molecular Dynamics) (Phillips et al., 2005). The enzyme was isolated from substrate and glycosylation moieties, and was constrained during the simulation, except for the loop containing the mutation (amino acids 401–411). All the water molecules were allowed to move. Hydrogen atoms were added to the crystal structure using VMD 1.8.5 (Visual Molecular Dynamics) (Humphrey et al., 1996). The enzyme was virtually mutated using the PyMol molecular graphics system (DeLano, 2002). All the water molecules observed in the crystal structure were retained for the simulation, and additional water molecules were added with the VMD package SOLVATE (Humphrey et al., 1996) to form a water box with a 5 Å cushion around the surface of the protein. The water molecules were modelled using the TIP3P force‐field (Jorgensen et al., 1983). The CHARMM 22 (Chemistry at HARvard Molecular Mechanics) force‐field (MacKerell et al., 1998) was used to model the protein. The entire simulation consisted of 20 000 steps of energy minimization, followed by 1 × 106 steps of actual molecular dynamics run. The integration time was 1 fs. The protein in the water box was subjected to periodic boundary conditions. The temperature and pressure of the system were maintained to 37°C and 101 kPa respectively. These conditions were applied using the Nosé–Hoover Langevin piston model (Nosé, 1984; Hoover, 1985). The simulation was carried out in an IBM Blue Gene computer with 1024 nodes, containing dual‐core PPC‐440 700 MHz processors. The processors have 512 MB of RAM each.
Acknowledgments
The authors wish to thank James Meade for the wild‐type A. niger GA gene, and J.K. Shetty for the gift of Acarbose. We are grateful to Melissa Abreu, Houdy Khosravi, Roni D. Mukerjea, Bill Rockey, April Ross and Vera Williams for technical assistance in the laboratory; to Chandrika Mulakala for assistance analysing enzyme structure and function; to Luis Petersen for computational analysis of GA and for assistance analysing enzyme structure and function, and to Peter J. Reilly for many helpful discussions and assistance in manuscript preparation.
References
- Aleshin A.E., Golubev A., Firsov L.M., Honzatko R.B. Crystal structure of glucoamylase from Aspergillus awamori var X100 to 2.2‐Å resolution. J Biol Chem. 1992;267:19291–19298. doi: 10.2210/pdb1gly/pdb. [DOI] [PubMed] [Google Scholar]
- Aleshin A.E., Hoffman C., Firsov L.M., Honzatko R.B. Refined crystal structures of glucoamylase from Aspergillus awamori var X100. J Mol Biol. 1994;238:575–591. doi: 10.1006/jmbi.1994.1316. [DOI] [PubMed] [Google Scholar]
- Aleshin A.E., Stoffer B., Firsov L.M., Svenssopn B., Honzatko R.B. Crystallographic complexes of glucoamylase with maltooligosaccharide analogs: relationship of stereochemical distortions at the nonreducing end to the catalytic mechanism. Biochemistry. 1996;35:8319–8328. doi: 10.1021/bi960321g. [DOI] [PubMed] [Google Scholar]
- Allen M.J., Coutinho P.M., Ford C.F. Stabilization of Aspergillus awamori glucoamylase by proline substitution and combining stabilizing mutations. Protein Eng. 1998;11:783–788. doi: 10.1093/protein/11.9.783. [DOI] [PubMed] [Google Scholar]
- Chen H.‐M., Bakir U., Reilly P.J., Ford C. Increased thermostability of Asn182Ala mutant glucoamylase. Biotechnol Bioeng. 1994;43:101–105. doi: 10.1002/bit.260430113. [DOI] [PubMed] [Google Scholar]
- Chen H.‐M., Ford C.F., Reilly P.J. Identification and elimination by site‐directed mutagenesis of thermolabile aspartyl bonds in Aspergillus awamori glucoamylase. Protein Eng. 1995;8:575–582. doi: 10.1093/protein/8.6.575. [DOI] [PubMed] [Google Scholar]
- Chen H.‐M., Li Y., Panda T., Buehler F.U., Ford C., Reilly P.J. Effect of replacing helical glycine residues with alanines on reversible and irreversible stability and production of Aspergillus awamori glucoamylase. Protein Eng. 1996;9:499–505. doi: 10.1093/protein/9.6.499. [DOI] [PubMed] [Google Scholar]
- Cole G.E., McCabe P.C., Inlow D., Geland D.H., Ben‐Bassat A., Innis M.A. Stable expression of Aspergillus awamori glucoamylase in distiller's yeast. Bio/Technology. 1988;6:417–421. [Google Scholar]
- Crabb W.D., Shetty J.K. Commodity scale production of sugars from starches. Curr Opin Microbiol. 1999;2:252–256. doi: 10.1016/S1369-5274(99)80044-7. [DOI] [PubMed] [Google Scholar]
- DeLano W. 2002.
- Flory N., Gorman M., Coutinho P.M., Ford C., Reilly P.J. Thermosensitive mutants of Aspergillus awamori glucoamylase by random mutagenesis: inactivation kinetics and structural interpretation. Protein Eng. 1994;7:1005–1012. doi: 10.1093/protein/7.8.1005. [DOI] [PubMed] [Google Scholar]
- Ford C. Improving operating performance of glucoamylase by mutagenesis. Curr Opin Biotechnol. 1999;10:353–357. doi: 10.1016/S0958-1669(99)80064-0. [DOI] [PubMed] [Google Scholar]
- Gallivan J.P., Dougherty D.A. Cation–π interactions in structural biology. Proc Natl Acad Sci USA. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover W.G. Canonical dynamics: equilibrium phase‐space distributions. Phys Rev A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jorgensen W., Chandrasekhar J., Madura J., Impey R., Klein M. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- Kumar S., Tsai C.J., Nussinov K. Factors enhancing protein thermostability. Protein Eng. 2000;13:179–191. doi: 10.1093/protein/13.3.179. [DOI] [PubMed] [Google Scholar]
- Leung D.W., Chen E., Goeddel D.V. A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique. 1989;1:11–15. [Google Scholar]
- Li Y., Reilly P.J., Ford C.F. Effect of introducing disulfide bonds into Aspergillus awamori glucoamylase. Protein Eng. 1997;10:1199–1204. [Google Scholar]
- Li Y., Coutinho P.M., Ford C. Effect on thermostability and catalytic activity of introducing disulfide bonds into Aspergillus awamori glucoamylase. Protein Eng. 1998;11:661–667. doi: 10.1093/protein/11.8.661. [DOI] [PubMed] [Google Scholar]
- Liu H.‐M., Wang W.‐C. Protein engineering to improve the thermostability of glucoamylase from Aspergillus awamori based on molecular dynamics simulations. Protein Eng. 2003;16:19–25. doi: 10.1093/proeng/gzg007. [DOI] [PubMed] [Google Scholar]
- MacKerell J.A., Bashford D., Bellot M., Dunbrack J.R., Evanseck J., Field M. All‐Atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. et al. [DOI] [PubMed] [Google Scholar]
- Muhlrad D., Hunter R., Parker R. A rapid method for localized mutagenesis of yeast genes. Yeast. 1992;8:79–82. doi: 10.1002/yea.320080202. [DOI] [PubMed] [Google Scholar]
- Nosé S. A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys. 1984;81:511–519. [Google Scholar]
- Okkels J.S., Svendsen A., Borch K., Thellersen M., Patkar S.A., Petersen D.A. 1997. et al) Novel lypolytic Enzymes. WO Patent 97/07202.
- Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly P.J. Protein engineering of glucoamylase to improve industrial performance – a review. Starch/Stärke. 1999;51:269–274. [Google Scholar]
- Richardson J.S., Richardson D.C. Amino acid preferences for specific locations at the ends of α‐helices. Science. 1988;240:1648–1652. doi: 10.1126/science.3381086. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Russell D.W. 3rd. Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Scheiner S., Karr T., Pattanayak J. Comparisons of various types of hydrogen bonds involving aromatic amino acids. J Am Chem Soc. 2002;124:13257–13264. doi: 10.1021/ja027200q. [DOI] [PubMed] [Google Scholar]
- Staples R.R., Dieckmann C.L. Generation of temperature‐sensitive cbp1 strains of Saccharomyces cerevisiae by PCR mutagenesis and in vivo recombination: characteristics of the mutant strains imply that CBP1 is involved in stabilization and processing of cytochrome b Pre‐mRNA. Genetics. 1993;135:981–991. doi: 10.1093/genetics/135.4.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Fuchs E., Silva R., McDaniel A., Seibel J., Ford C. Improvement of Aspergillus niger glucoamylase thermostability by directed evolution. Starch/Stärke. 2006;58:501–508. [Google Scholar]