

Summary
The bioleaching of metal sulfide has developed into a very important industrial process and understanding the microbial dynamic is key to advancing commercial bioleaching operations. Here we report the first quantitative description of the dynamic of active communities in an industrial bioleaching heap. Acidithiobacillus ferrooxidans was the most abundant during the first part of the leaching cycle, while the abundance of Leptospirillum ferriphilum and Ferroplasma acidiphilum increased with age of the heap. Acidithiobacillus thiooxidans kept constant throughout the leaching cycle, and Firmicutes group showed a low and a patchy distribution in the heap. The Acidiphilium‐like bacteria reached their highest abundance corresponding to the amount of autotrophs. The active microorganisms in the leaching system were determined using two RNA‐based sensitive techniques. In most cases, the 16S rRNA copy numbers of At. ferrooxidans, L. ferriphilum, At. thiooxidans and F. acidiphilum, was concomitant with the DNA copy numbers, whereas Acidiphilium‐like bacteria and some Firmicutes members did not show a clear correlation between 16S rRNA accumulation and DNA copy numbers. However, the prokaryotic acidophile microarray (PAM) analysis showed active members of Alphaproteobacteria in all samples and of Sulfobacillus genus in older ones. Also, new active groups such as Actinobacteria and Acidobacterium genus were detected by PAM. The results suggest that changes during the leaching cycle in chemical and physical conditions, such as pH and Fe3+/Fe2+ ion rate, are primary factors shaping the microbial dynamic in the heap.
Introduction
Heap bioleaching is currently the most successful technology for the extraction of base metals from low‐grade sulfide ores (Watling, 2006), and during the last few decades this technology has become increasingly important due to the depletion of high‐grade copper ores and the existence of huge natural reserves of copper in the form of secondary copper sulfides (Galleguillos et al., 2008). In recent years scientific and commercial interest has emerged to study the microbial ecology of industrial bioleaching processes (Demergasso et al., 2005; Hawkes et al., 2006; Xie et al., 2007; Wakeman et al., 2008), because the understanding of the microbiological aspects would facilitate the design and operation of industrial heaps to improve this technology (Brierley, 2001; Rawlings, 2002; Watling, 2006; Yin et al., 2007). In case of metal sulfide bioleaching, metal sulfides are oxidized to metal ions and sulfate by aerobic, acidophilic Fe(II) and/or sulfur‐compound oxidizing Bacteria or Archaea (Schippers, 2007). These microorganisms are responsible for producing the ferric iron and sulfuric acid for the bioleaching reactions (Rawlings, 2007; Johnson and Hallberg, 2007). In addition to those acidophiles that have direct roles in accelerating mineral dissolution, other acidophiles, most of them heterotrophic, could have a positive impact on the overall process (Johnson and Hallberg, 2007).
Some recent studies have corroborated that Acidithiobacillus ferrooxidans and Leptospirillum species seem to be the most abundant microorganisms in heap leaching and mine waste environments (Diaby et al., 2007; Remonsellez et al., 2007; Xie et al., 2007; Galleguillos et al., 2008; He et al., 2008; Kock and Schippers, 2008). Several mesophilic and moderately thermophilic species of phylum Firmicutes have been identified and isolated from sulfide ore heaps, stirred tanks and mine waste, but many of them are not validly described yet (Diaby et al., 2007; Schippers, 2007; Xie et al., 2007; He et al., 2008; Johnson et al., 2008). Some acidophilic heterotrophic bacteria have been found in bioleaching operations and environments (Diaby et al., 2007; Rowe et al., 2007; Xie et al., 2007; Kock and Schippers, 2008). Ferroplasma and Thermoplasma lineages have been regularly found in bioleaching operations as well (Hawkes et al., 2006; Xie et al., 2007; Galleguillos et al., 2008; He et al., 2008), and other Archaea microorganisms like Sulfurisphaera and Sulfolobus genera have been identified in a copper test‐heap (Demergasso et al., 2005).
A variety of molecular techniques have been widely used for the analyses of mineral‐leaching populations (Johnson and Hallberg, 2007). After that, the use of PCR associated with fluorescence emission (q‐PCR) has emerged as a new approach to quantitatively describe the community composition, and several works have reported the quantification of Bacteria and Archaea in water, soil and sediment samples, and even in mine waste tailing and leaching solutions (Fey et al., 2004; Kock and Schippers, 2006; 2008; Liu et al., 2006; Schippers and Neretin, 2006; Remonsellez et al., 2007). Despite the available techniques, the studies of bioleaching heaps have described the microbial diversity by relative abundance of communities (Demergasso et al., 2005; Wakeman et al., 2008) and most of them in some specific samples from the processes (Rawlings and Johnson, 2007; Xie et al., 2007; He et al., 2008). Therefore, the predominance of certain members of the population at specific stages of the bioleaching heap process and the reasons of that dynamic is just being described (Demergasso et al., 2005; Galleguillos et al., 2008; Wakeman et al., 2008).
The DNA approaches used in the analysis of acidophiles are based in the detection and amplification of one specific gene (mainly the 16S rRNA gene), indicating the presence of the microorganism containing that gene (Johnson and Hallberg, 2007). However, a large part of the microorganisms could be dormant or ever dead and yet retain stable DNA. Instead, experiences with pure cultures have shown that cells with significant ribosome content are living and metabolically active (Schippers et al., 2005). To get information on the active microorganisms in a process, RNA‐based analyses should be performed. The last years have brought rapid advances especially in tools used for molecular microbial ecology that merit to be explode (Johnson and Hallberg, 2007). One important advance is the use of oligonucleotide arrays to detect and to analyse microbial communities (Zhou, 2003). Oligonucleotide arrays have been used successfully in environmental studies in the last years (Wu et al., 2001; Valinsky et al., 2002; Brodie et al., 2006; Gentry et al., 2006). Recently, two oligonucleotide microarrays have been developed to monitor the diversity in extremely acidic environments (Yin et al., 2007; Garrido et al., 2008). One of them has been used to detect the most metabolically active microorganisms using labelled environmental RNA (Garrido et al., 2008).
In order to get new insights and understanding the role that microorganisms play in mineral processing operations, we analysed the composition and the dynamic of the microbial communities in an industrial bioleaching heap in Chile (Fig. 1). To achieve this objective, we determined the active microorganisms during the leaching cycle in the industrial heap by means of a combination of two sensitive molecular techniques, such as quantitative real‐time PCR and a microarray for prokaryotic acidophiles.
Figure 1.
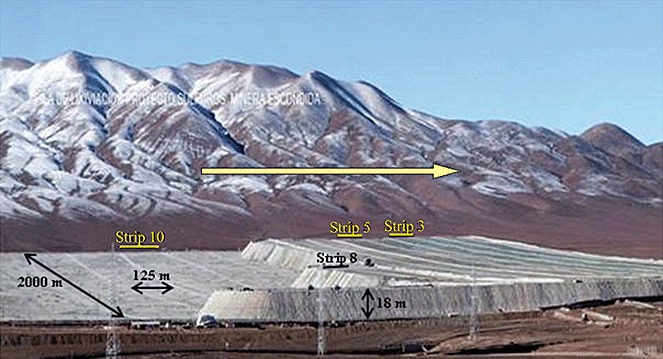
Image of the industrial bioleaching heap in Escondida Mine in January 2007. Arrows indicate the dimension of strips, and lines indicate the main strips (S3, S5, S8 and S10) analysed in this study. The direction of the yellow arrow indicates the increase in days of operation of the strips.
Results
Composition of the microbial community in the industrial bioleaching system
Since the beginning of the bioleaching cycle in the heap, DGGE and 16S rRNA clone libraries were used to identify the microbial diversity in early strips in operation and in the feed solution (year 2006). Comparative sequence analysis of DGGE bands showed the presence of organisms related to At. ferrooxidans, At. thiooxidans, Leptospirillum ferriphilum and the archaeon Ferroplasma acidiphilum (Table 1) with percentages of similarity between 97% and 99% with‐type strains. The rest of the bands showed sequence similarities between 80% and 85% to Gram‐positive Sulfobacillus spp. (called Sulfobacillus‐like bacteria), Alicyclobacillus disulfidooxidans and an uncultured Alicyclobacillus. The results showed in the Table 1 were comparable with DGGE bands analysed from other new strips in different times of the leaching cycle (data not shown).
Table 1.
Summary of the microbial community composition of the industrial heap using DGGE and 16S rRNA gene clone libraries during year 2006.
| Microorganism | DGGE | Clone libraries | ||||||
|---|---|---|---|---|---|---|---|---|
| August | November | September | August | |||||
| S1 | S3 | S1 | S3 | Feed | S1 | S3 | S4 | |
| Bacteria | ||||||||
| At. ferrooxidans | 2 | 2 | 2 | 2 | 57 | 18 | 87 | 96 |
| At. thiooxidans | 1 | 1 | 1 | 1 | 9 | 5 | 11 | – |
| L. ferriphilum | 1 | 1 | 1 | 1 | 8 | 13 | – | 2 |
| Ab. disulfidooxidans | 1 | 1 | – | – | – | – | 2 | – |
| Uncultured Alicyclobacillus | 1 | – | 1 | 1 | – | – | – | 1 |
| Sulfobacillus spp. | – | – | 1 | – | – | – | – | – |
| Acidiphilum‐like | – | – | – | – | – | – | – | 1 |
| Archaea | ||||||||
| F. acidiphilum | 2 | 2 | 2 | 2 | 5 | 6 | ND | ND |
DGGE, number of sequenced bands. Clone libraries, number of 16S rRNA clones analysed.
ND, not done. –, not found.
S1, strip 1; S2, strip 2; S3, strip 3; Feed, irrigation solution.
Moreover, we constructed bacterial and archaeal 16S rRNA clone libraries to complement the data obtained by DGGE. The analysis of all different clones showed that the community is mainly composed by sequences related to Acidithiobacillus genus (Table 1). We found two different phylotypes of At. ferrooxidans (called D2 and DM) and around of 75% of analysed clones were At. ferrooxidans D2. At. ferrooxidans D2 is similar to the sequence of At. ferrooxidans strain D2 (AJ278723) and these sequences have just 99% of identity with At. ferrooxidans DM (Fig. S1). Moreover, we found two different phylotypes of L. ferriphilum with 98% of identity by using blast algorithm (Altschul et al., 1997), and one of them is similar to L. ferriphilum type Warwick (Fig. S2). Sequences ≥ 98% related with At. thiooxidans, ≥ 93% with Ab. disulfidooxidans, 94% with uncultured Alicyclobacillus, and one sequence 94% similar to Acidiphilium spp. (called Acidiphilium‐like bacteria), were also identified. Sequences with high similarity (≥ 99%) related to F. acidiphilum from the Archaea domain were also detected (Table 1).
These results showed that the identified microorganisms correspond to phylogenetic groups such as Alphaproteobacteria, Gammaproteobacteria, Nitrospira, Firmicutes and Thermoplasmata, normally found in heap‐ and tank‐bioleaching processes (Espejo and Romero, 1997; Demergasso et al., 2005; Rawlings and Johnson, 2007) and acid mine waste (Johnson and Hallberg, 2003; Diaby et al., 2007).
Dynamic of the microbial communities in the heap
Sequences obtained by DGGE and clone libraries were used to design specific primers (Table 2) and, following standardization, real‐time PCR was used to quantify the dynamic of microorganisms inhabiting the industrial heap.
Table 2.
Oligonucleotides used in quantitative real‐time PCR analysis.
| Targeted group | Primer | Sequence (5′→3′) | Reference |
|---|---|---|---|
| Universala,b | 907R | CCGTCAATTCMTTTGAGTTT | Casamayor et al. (2002) |
| Bacteriab | UBactF | TCCTACGGGAGGCAGCAGT | Nadkarni et al. (2002) |
| Bacteriab | UBactR | GGACTACCAGGGTATCTAATCCTGTT | Nadkarni et al. (2002) |
| Archaeab | ARCH349F | GYGCASCAGKCGMGAAW | Takai and Horikoshi (2000) |
| Archaeab | ARCH806R | GGACTACVSGGGTATCTAAT | Takai and Horikoshi (2000) |
| Archaeaa,b | ARCH‐R | TGCTCCCCCGCCAATTCC | This study |
| At. ferrooxidans D2b | ATFD2‐F | CGGGTCCTAATACGATCTGCT | This study |
| At. ferrooxidans DMb | ATFDM‐F | TGGTTCCTAATACGAGCTACTG | This study |
| At. thiooxidansb | ATT‐F | GGGTGCTAATANCGCCTGCT | This study |
| L. ferriphilumb | Lferri‐F | CGTCAGAAIACGGCGCTTC | This study |
| L. ferriphilum Warwickb | LferriW‐F | GATGTCAGAACACGGCATTT | This study |
| Sulfobacillus‐likeb | Sesc‐F | GGAGACCGTGCCGTCG | This study |
| Ab. disulfidooxidansb | SG1‐F | AGTGGCGAAGGCGCCTTGCTGG | This study |
| Uncultured Alicyclobacillusa | Adunc‐F | CCTCTCCGACCCTCAAGTCT | This study |
| Uncultured Alicyclobacillusb | Adunc‐R | AGGAGAGGGAATGCTTTTGG | This study |
| Acidiphilium‐likeb | Acesc‐F | AGGCGGCTTRTACAGTCAGGC | This study |
| F. acidiphilumb | Fer‐F | GAAGCTTAACTCCANAAAGTCTG | This study |
cDNA synthesis for quantitative real‐time PCR and real‐time PCR amplification analysis.
Real‐time PCR amplification analysis.
The microorganisms inhabiting the heap throughout the leaching cycle are mainly bacterial species and reached total 16S rRNA gene copy numbers greater than 107 copies ml−1 and, in general, this value decreased around one order of magnitude in most of the strips when these aged more than 200 days (Fig. 2A and B; all other strips: data not shown). At. ferrooxidans phylotypes were the most abundant microorganisms during the first part of the leaching cycle (around 200 days of operation) and specifically At. ferrooxidans D2 reached around of 107 copies ml−1 during this period (Fig. 2A and B). These 16S rRNA gene copy numbers have been observed in all strips during the first month of operation (Remonsellez et al., 2007; Galleguillos et al., 2008; data not shown). At. ferrooxidans DM reached between 105 and 106 copies ml−1 in most of the strips (Fig. 2A and B and Table 3; all other strips: data not shown). In marked contrast, L. ferriphilum phylotypes were present with lower abundance (between 102 and 103 copies ml−1) during the first month of operation in all strips, but copy numbers of both Leptospirillum phylotypes, increased with age reaching around of 105 copies ml−1 (Fig. 2A and B and Table 3; all other strips: data not shown). The sulfur‐oxidizer At. thiooxidans maintained a constant copy numbers (105 copies ml−1) throughout the leaching cycle in all strips (Fig. 2A and B and Table 3; most of other strips: data not shown). The last microorganism has been identified from other heap operations (Goebel and Stackebrandt, 1994; Espejo and Romero, 1997), but its dynamic had not been reported until this study.
Figure 2.
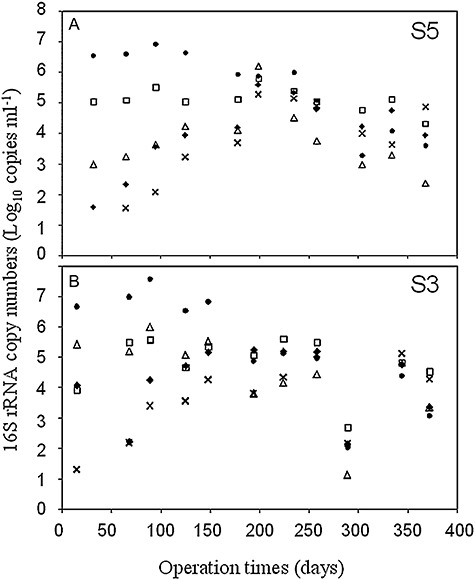
Dynamic of main microorganisms present in the bioleaching industrial process. DNA copy numbers of 16S rRNA genes of main bacterial species during the leach cycle of strip S3 (A) and strip S5 (B) are shown. Strips S3 and S5 began its operations in May and August 2006 respectively.  , At. ferrooxidans D2;
, At. ferrooxidans D2;  , At. ferrooxidans DM;
, At. ferrooxidans DM;  , At. thiooxidans;
, At. thiooxidans;  , L. ferriphilum; and
, L. ferriphilum; and  , L. ferriphilum Warwick.
, L. ferriphilum Warwick.
Table 3.
Copy numbers and accumulation levels of 16S rRNA of microorganisms present in the industrial heap (samples of August 2007) by real‐time PCR analysis.
| Microorganism | Copy numbers (copies ml−1) | Transcripts levels (copies ml−1) | ||||
|---|---|---|---|---|---|---|
| Strips | Strips | |||||
| S10 | S8 | S5 | S10 | S8 | S5 | |
| Bacteria | 6.7 × 106 | 9.0 × 105 | 3.2 × 105 | ND | ND | ND |
| At. ferrooxidans D2 | 3.4 × 106 | 3.1 × 105 | 1.2 × 103 | 1.1 × 107 | 5.9 × 104 | 2.1 × 104 |
| At. ferrooxidans DM | 2.9 × 106 | 8.1 × 104 | 2.3 × 103 | 1.0 × 107 | 4.1 × 104 | 4.7 × 104 |
| At. thiooxidans | 1.3 × 105 | 8.9 × 104 | 3.4 × 104 | 6.9 × 105 | 7.5 × 104 | 2.7 × 105 |
| L. ferriphilum | 3.8 × 103 | 1.9 × 104 | 1.4 × 105 | 5.3 × 102 | 3.8 × 103 | 2.2 × 105 |
| L. ferriphilum Warwick | 2.2 × 103 | 4.2 × 105 | 5.6 × 104 | 1.5 × 103 | 4.0 × 104 | 2.8 × 104 |
| Ab. disulfidooxidans | 7.4 × 103 | 2.1 × 104 | 2.7 × 103 | 3.3 × 103 | 1.3 × 102 | 3.3 × 103 |
| Sulfobacillus‐like | 8.9 × 102 | 6.0 × 102 | BDL | BDL | 1.9 × 102 | 1.3 × 103 |
| Uncultured Alicyclobacillus | 2.2 × 102 | 1.1 × 102 | BDL | BDL | BDL | BDL |
| Acidiphilum‐like | 9.6 × 103 | 9.2 × 103 | BDL | 5.1 × 104 | 2.9 × 104 | 1.4 × 103 |
| Archaea | BDL | 2.0 × 102 | 2.0 × 103 | ND | ND | ND |
| F. acidiphilum | BDL | 1.4 × 102 | 2.1 × 103 | 4.1 × 102 | 2.2 × 103 | 1.3 × 104 |
BDL, below detection limit. ND, not done.
The Firmicutes group, such as Ab. disulfidooxidans, uncultured Alicyclobacillus and Sulfobacillus‐like bacteria, generally were detected in low copy numbers in comparison with the other groups of Bacteria, but all of them reached number between 103 and 104 copies ml−1 and showed a patchy distribution in all strips (data not shown). The Acidiphilium‐like bacteria presented copy numbers between 102 and 104 cells ml−1 in all strips, but reached its highest abundance when autotrophs microorganisms presented high copy numbers, generally between 150 and 200 days of operation in all strips (data not shown). The archaeon F. acidiphilum showed the same behaviour than Leptospirillum species but reached a low copy numbers around of 103 copies ml−1 (Table 3; all strips: data not shown). In all cases, the data from quantitative real‐time PCR indicated that 16S rRNA gene copy numbers of Bacteria showed similar values to the sum of all species analysed, and 16S rRNA gene copy numbers of Archaea were almost identical to F. acidiphilum copy numbers (Table 3; data not shown).
The younger parts of the heap (strips) running since May 2008 (S7–S14) also showed remarkable differences in the microbial communities depending on the strip age. At. ferrooxidans phylotypes were found between 80% and 98% of abundance in the youngest strips (S13 and S14) with approximately 30 days of operation (Fig. 3). We observed one gap between the dominance of At. ferrooxidans and L. ferriphilum phylotypes in the strips S11 and S12 (150 and 120 days of operation respectively, Fig. 3), while L. ferriphilum phylotypes were found between 89% and 96% of relative abundance in the older strips (S7, S8, S9 and S10, over 250 days of operation, Fig. 3). The archaeon F. acidiphilum showed the same behaviour than L. ferriphilum but with lower abundance (4–9%). The rest of the bacteria such as At. thiooxidans, uncultured Alicyclobacillus and Sulfobacillus‐like bacteria were the less abundant (between 4.5% and 0.4%), with the exception of the strip S11 where Sulfobacillus‐like bacteria accounted for the 15% (Fig. 3). Ab. disulfidooxidans and Acidiphilium‐like bacteria species were not represented in the Fig. 3 because they showed relative abundances below 0.5%.
Figure 3.
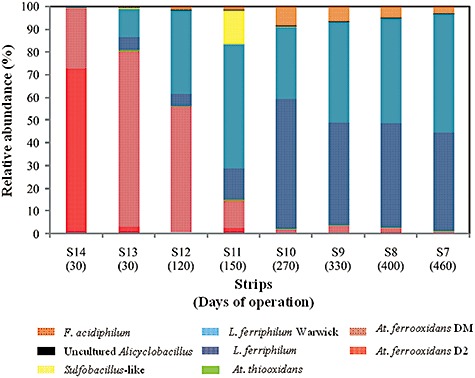
Community structures based on 16S rRNA genes of Bacteria and Archaea species from different strips of the industrial heap. Relative abundances of different microorganisms were evaluated by quantitative real‐time PCR of total DNA from different strips in operation until May 2008.
16S rRNA accumulation levels analysis
The 16S rRNA accumulation levels have been determined in different strips, as indicative of physiological activity (Parro et al., 2007). In this work we show the results of the main phylogenetic groups that were detected throughout the leaching cycle from three strips (S5, S8 and S10) with different days of operations (Figs 2 and 3, Table 3). The ranges of the physicochemical parameters of the strips (S5, S8 and S10) are shown in Table 4. In correlation with the time of operation, the pH decreased with age strip; total iron, Fe3+ and redox potential showed higher values in strips over 150 days of operation (S5 and S8), while the copper concentration showed a higher value in the strip S10 with less than 50 days of operation (Table 4).
Table 4.
Physicochemical parameters governing three strips that represent the bioleaching cycle (samples of August 2007).
| Strips (days of operation) | |||
|---|---|---|---|
| Parameters | S10 (30) | S8 (150) | S5 (330) |
| pH | 2.3 ± 0.14 | 1.83 ± 0.12 | 1.67 ± 0.1 |
| Total Fe (g l−1) | 1.18 ± 0.19 | 1.9 ± 0.2 | ND |
| Fe3+ (g l−1) | 0.87 ± 0.19 | 1.86 ± 0.19 | ND |
| Eh (mV) | 682 ± 30.5 | 849 ± 11.5 | 837 ± 43.9 |
| Cu (g l−1) | 4.1 ± 0.25 | 2.55 ± 0.6 | 1.62 ± 0.49 |
ND, not done. Total Fe and Fe3+ of other strips (S8, S9, S10 and S11) with 300 days of operations reached average values of 1.9 ± 0.185 and 1.88 ± 0.183 g l−1 respectively.
The 16S rRNA accumulation levels indicate that At. ferrooxidans phylotypes were the most active microorganism in the strip S10 with a shorter operation time (Table 3). The same behaviour has been observed in other strips in the first months of operation (Galleguillos et al., 2008). On the contrary, the results suggested that L. ferriphilum phylotypes were more actives in strips S5 and S8 with more than 150 days of operation (Table 3). The sulfur‐oxidizer At. thiooxidans was active in all analysed strips (Table 3), indicating that this microorganism could have an important role throughout the leaching cycle. The archaeon F. acidiphilum also showed higher 16S rRNA accumulation in strips with a longer operation time (Table 3), and the same behaviour has been observed in other strips with more than 200 days of operation (Galleguillos et al., 2008).
However, the rest of acidophilic microorganisms analysed did not show a clear correlation between DNA copy numbers and 16S rRNA accumulation. The Acidiphilium‐like bacteria was active in all strips analysed, but was not detected in the strip S5 by DNA copy numbers analysis (Table 3). Ab. disulfidooxidans and Sulfobacillus‐like bacteria showed a lower activity in all strips analysed, and a patchy activity of these Firmicutes members was observed. Finally, although the uncultured Alicyclobacillus showed low DNA copy numbers in strips S8 and S10, it was not active in the strips analysed (Table 3).
Time‐course strip monitoring with total RNA and prokaryotic acidophile microarray
We used the prokaryotic acidophile microarray (PAM) developed and validated by Garrido and colleagues (2008), for a fast monitoring of the most active groups of microorganisms at the industrial heap (Figs S3 and 4). The same samples used for the 16S rRNA accumulation studies were subjected to PAM analysis. Total RNA of these samples was amplified following the method described by Moreno‐Paz and Parro (2006), which was used for the amplification of total RNA from an acidophilic environment in transcriptome analysis (Parro et al., 2007). The amplified RNAs from strips S5, S8 and S10 were labelled with Cy5 or Cy3, and hybridized with the PAM microarray in a classical simultaneous dual hybridization (S5 and S10; S8 and S10; S5 and S8) as shown in the Fig. 4. The relative proportion of each phylogenetic group in the sample was estimated by calculating the ratio between the fluorescent signals (Cy5/Cy3) for each probe on the microarray as described (Garrido et al., 2008). The relative signal intensity for the analysed strips showed a dramatic change in the prokaryotic profile, from Acidithiobacillus spp. to Nitrospira group, Thermoplasmata class and Actinobacteria, from younger to older strips (Fig. 4A). Interestingly, strips closer in time of operation (strips S5 and S8, with more than 150 days of operation) showed similar preferential rRNA accumulation pattern (Fig. 4A) with some bias to Actinobacteria, Archaea and At. thiooxidans to the older one (strip S5). In agreement with this result, the patterns observed in older strips (strips S5 and S8) were compared with the youngest one (strip S10). The PAM results showed a sharp preferential distribution of some phylogenetic groups, with Acidithiobacillus spp., and Acidobacterium spp. dominating the first moments of the operation (strip S10, green colour in Fig. 4B), and L. ferriphilum, some Actinobacteria, Sulfobacillus spp. and members of the Thermoplasmata class dominating the older strips (strips S5 and S8, red colour in Fig. 4B). The total Bacteria showed a preferential expression in the strip S10, which correlated with data obtained from DNA copy numbers and 16 rRNA accumulation (Fig. 4A and Table 3). The Alphaproteobacteria and At. thiooxidans do not showed a preferential expression between the old and new strips (Fig. 4). Although members of the Alphaproteobacteria should be present in all the analysed samples, none of the specific probes for Acidiphilium spp. showed positive results, neither that for At. ferrooxidans group D, nor those for the L. ferrooxidans phylotypes (Fig. 4).
Figure 4.
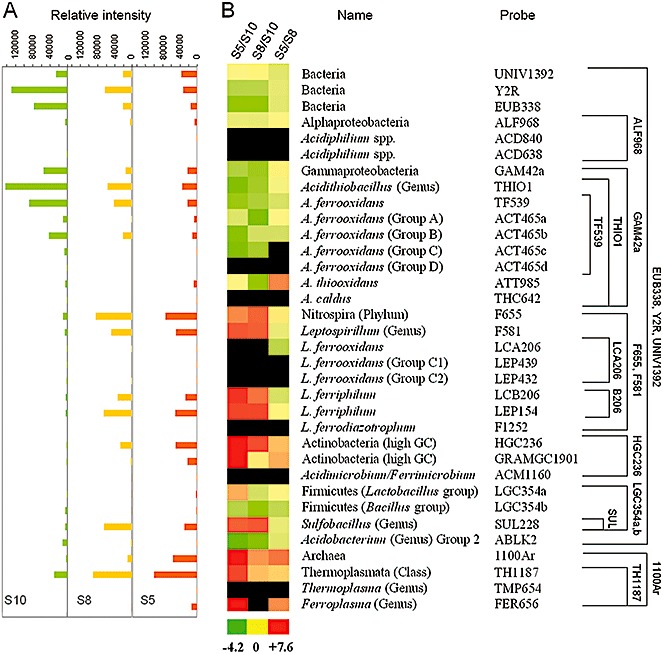
A barcode for industrial biolixiviation process by assaying total industrial heap RNA with a prokaryotic acidophile microarray (PAM). A. Histograms showing the average relative signal intensity of two different hybridizations for the different analysed strips, from younger to older ones (S10‐S8‐S5). Each bar of the histogram aligns horizontally with the corresponding strain name or phylogenetic group and probe (in B). B. Relative proportion of the different metabolically active phylogenetic groups by using prokaryotic acidophile microarray (PAM). The PAM results were obtained after two‐colour simultaneous hybridization with total fluorescent labelled environmental RNA from three samples of the industrial heap of August 2007 (S5, Cy5 labelled; S8, Cy5 and Cy3 labelled; S10, Cy3 labelled). After two‐colour simultaneous hybridizations (S5/S10, S8/S10 and S5/S8), the Log2 ratio between the signal intensity from each sample was calculated and clustered for comparison along sampling times (coloured figure). The log2 ratio values ranged from −4.2 (the most green), through 0 (yellow), to +7.6 (the most red). Black colour indicates no signal detected with the corresponding probe. Representative phyla and species are indicated, as well as a phylogenetic tree showing the probe range (right scheme).
Discussion
Acidophilic microorganisms in the heap leaching solutions
The most prominent microorganisms making up these communities are Gammaproteobacteria (At. ferrooxidans and At. thiooxidans) and some members of phylum Nitrospira (L. ferriphilum). These microorganisms have been found in most studies in bioleaching heaps (Table S1). The community consists to a lesser extent of Alphaproteobacteria, Firmicutes and Thermoplasmata groups, which have also been identified in bioleaching heaps (Table S1). Our results also confirm previous observations that identified new groups that could participate in such process, like Acidimicrobium genus or some Actinobacteria (Rawlings and Johnson, 2007).
In this work we used quantitative real‐time PCR to monitor the microbial diversity and speculate about the dynamic and physiological state of the microbial communities with respect to leaching cycle in industrial heaps. At. ferrooxidans phylotypes have always reached their highest abundance when pH values are over 2, and the Fe3+ ion and total iron concentrations are less than 1 and 1.2 g l−1 respectively (Tables 3 and 4). Moreover, we observed that the redox potential was around 680 mV when At. ferrooxidans reached higher abundance, and these results are consistent with the report of Boon and colleagues (1999) and Meruane and colleagues (2002), which showed that At. ferrooxidans are not capable of oxidation at higher redox potentials. Possibly in our case, the metabolism of At. ferrooxidans should change as the heap ages (Galleguillos et al., 2008) to sulfur oxidation coupled to oxygen like At. thiooxidans (Schippers and Sand, 1999) and/or to sulfur oxidation coupled to Fe3+ ion reduction (Sand, 1989). Leptospirillum species reached their highest abundance when the pH was below 2, high Fe3+ ion concentrations prevailed, and the concentration of copper was under 3 g l−1 (Tables 3 and 4). Their physiological particularities, such as tolerance to high redox potential and Fe3+ ion concentration (Rawlings et al., 1999; Bond et al., 2000), could be the reason for their dominance in these conditions as also described previously (Demergasso et al., 2005). The archaeon F. acidiphilum, as Lepstospirillum species, showed its highest abundance in old strips (Table 3 and Fig. 4), and possibly these microorganisms contribute to the mineral dissolution in environments with extremely low pH values (Bond et al., 2000). A recent study about the archaeal diversity in two bioleaching systems revealed that the presence of Thermoplasma and Ferroplasma was related to highest amounts of total iron and low pH values (Xiao et al., 2008).
One important finding in our work was the high abundance of the sulfur‐oxidizer At. thiooxidans throughout the leaching cycle in all strips. In our case, the presence of acidic solution in the system could decompose the copper sulfide to elemental sulfur as previously described Schippers and Sand (1999), and we suggest that At. thiooxidans microorganism have high abundance in the process due to the constant production of sulfur and intermediate reduced sulfur compounds by Fe3+ ion‐mediated chemical sulfide oxidation.
The species belonging to Firmicutes occurred in low copy number and generally showed a patchy distribution in all strips. Sulfobacillus spp. has been described as chemolithotroph and show mixotrophic and autotrophic growth (Bond et al., 2000). But, despite that several species of Firmicutes group have been isolated from bioleaching systems or thermal springs, many of them have not been described and characterized yet (Schippers, 2007). Recently, some species of Firmicutes have been identified also from other bioleaching sites and sulfide columns‐test (Xie et al., 2007; He et al., 2008; Wakeman et al., 2008). Some acidophilic heterotrophic Bacteria have been found usually in bioleaching operations (Hallberg and Johnson, 2001; Xie et al., 2007), but whether their capabilities contribute to the bioleaching efficiency of a microbial consortium in practice is still unclear (Johnson, 1998, Hallberg and Johnson, 2001). But whit respect to Acidiphilum‐like bacteria, our data suggest that their abundance may vary significantly in the bioleaching cycle possibly depending of the autotrophic oxidizer dynamic.
Active microorganisms by using RNA from an industrial heap
Despite the results reviewed above, the question remains whether the 16S rRNA gene copy numbers in this study originate from livings cells or from cell debris. Therefore, using RNA one expects to detect the most active microorganisms in the system (Schippers et al., 2005). The 16S rRNA accumulation of At. ferrooxidans, L. ferriphilum, At. thiooxidans and F. acidiphilum were in agreement with the PAM analysis. Therefore, we suggest that At. ferrooxidans and L. ferriphilum would produce Fe3+ at different stages of the bioleaching cycle, At. thiooxidans would generate sulfuric acid to supply protons throughout the bioleaching cycle and, possibly under low Fe2+ concentrations (over 200 days of operation), At. ferrooxidans acts like At. thiooxidans by acid production.
Acidiphilium‐like bacteria presented a high activity in all analysed strips (Table 3) and the PAM analysis showed active microorganisms belonging to Alphaproteobacteria with no preferential dominance between old and new strips (Fig. 4). We propose the hypothesis that the possible activity of heterotrophic bacteria could depend on the autotrophic oxidizer dynamics. Similarly, Schippers and colleagues (1995) described a positive correlation between cells count of acidophilic chemoorganotrophics microorganisms and those of At. ferrooxidans in two different uranium mine waste heaps.
With respect to the Firmicutes group, we did not find a clear activity depending on age strip (Table 3), possibly due to the fact that they could use sulfur and iron compounds as energy source (Bond et al., 2000). On the contrary, the Sulfobacillus genus was also detected by using PAM and was preferentially active in older strips (Fig. 4), as described previously (Demergasso et al., 2005) in one sulfide test‐heap in Chile. Recently, some sequences related to Firmicutes group have been isolated from solutions with high iron concentration from Tong Shankou copper Mine in China (Xie et al., 2007).
Interestingly, the PAM detected new active groups of microorganisms such as Actinobacteria and Acidobacterium genus. Therefore, the use of RNA as target is expected to be more sensitive than DNA analysis (Galleguillos et al., 2008; Garrido et al., 2008). Members of Actinobacteria group have been identified in another industrial operation (Bruhn et al., 1999). Some microorganisms related with Acidobacterium species have been isolated from metal‐rich mine waters with pH values between 2.4 and 2.7 (Hallberg et al., 2006; Rowe et al., 2007). Despite these studies, few microorganisms belonging to this phylogenetic group have been cultivated and detected in industrial bioleaching processes. The slight discrepancies between transcript levels by real‐time PCR and PAM can be explained by the methodological differences, mainly in the RNA amplification step and the specificity of the probes used (Garrido et al., 2008). Finally, PAM microarray can proportionate a sort of ‘barcode’ or ‘fingerprint’ for characterizing certain industrial bioleaching processes.
Using pregnant leaching solution as an indicator of the microbial communities within an industrial bioheap
Based on the necessity of understanding the microbiology of heaps (Brierley, 2001), we monitored the composition and dynamic of the microbial communities from an industrial bioleaching sulfide heap.
Notwithstanding that the bioleaching reactions are carried out mainly by bacteria attached to the sulfide mineral inside the heap in order to provide Fe3+ ion and protons (Schippers, 2007), we decided to analyse the leach solutions for several reasons:
-
(i)
The implications of taking ore samples during the bioleaching process at industrial level are enormous (Galleguillos et al., 2008), regarding the cost, the complexity of this process and the amount of samples needed because of the high heterogeneity inside the heap. In some previous studies, At. ferrooxidans, L. ferrooxidans, L. ferriphilum and Sulfobacillus spp. have been detected in sulfidic mine waste dumps (Diaby et al., 2007); in addition At. thiooxidans, Acidithiobacillus spp. and Thiomonas spp. have been detected in metal sulfide mine wastes (tailings) (Wielinga et al., 1999). A work published recently by Kock and Schippers (2008), in which depth profiles have shown that the composition of the microbial communities varied between zones of oxidized and unoxidized tailings, and maximum cell numbers have been determined in the pyrite oxidation tailings zones, shows just an example of the factors that affect the microbial community associated to specific ore samples (oxygen levels, oxidation rates, sulfide–sulfur content). This is much more relevant because the system analysed in this work is composed of non‐agglomerated run‐of‐mine ore, where heterogeneous niches must be common (Kock and Schippers, 2008; Wakeman et al., 2008).
-
(ii)
The comparison between the microbial communities in associated mineral and solutions performed in industrial and laboratory samples have shown enough similarity to be considered as indicator of the community inside the heap. We have confirmed that the most abundant communities in ore samples of this heap correspond to those commonly found as predominant in leaching solutions. In the oldest strip S1 with more than 400 days of operation At. ferrooxidans, At. thiooxidans and L. ferriphillum are the most abundant microorganisms with 6 × 103 copies g−1, 5 × 104 copies g−1 and 1 × 104 copies g−1 respectively. Also, we determined that At. ferrooxidans was the most abundant microorganism in one ore sample from strip S9, with 60 days of operation, reached 107 copies g−1 (F. Remonsellez, F. Galleguillos, A. Echeverria and C. Demergasso, unpublished data).
-
(iii)
It is accepted for bioleaching that bacterial cells can affect the sulfide dissolution by ‘contact’ and ‘non‐contact’ mechanisms (Rohwerder et al., 2003). The contact mechanism requires attachment of bacteria to the sulfide surface. Then, Fe2+ ions, elemental sulfur and other key intermediate sulfur compounds in oxidative sulfide degradation (Schippers et al., 1996; Boon et al., 1998; Sand et al., 2001), are biologically oxidized. This mechanism does not require the attachment of cells to the sulfide mineral (Sand et al., 2001).
Taking into account those arguments, we argue that the microorganisms detected in the leaching solutions represent an overview of what is happening in each strip. Therefore, we support the hypothesis that in large bioleaching operations the analysis of leaching solution could be a good alternative for monitoring these systems. Finally, we proposed that one periodic analysis of ore samples at different depths and locations to complement the data obtained from leach solutions could help to control the industrial bioleaching process from a microbial and a productive point of view.
Conclusion
The selection, control and monitoring of the microbial communities in biomining had been neither well studied nor fully understood. The question arise, therefore, which species are more abundant and effective during industrial bioleaching operations, and to answer this, more studies of microbial dynamics during different leaching stages containing different sulfide minerals are required. This is the first work to describe the dynamics of an active microbial community in an industrial heap using powerful RNA‐based tools and, these techniques can rapidly evaluate the levels of acidophilic microorganisms present in these systems. Finally, we are proposing that the chemical and physical conditions (like pH and the Fe3+/Fe2+ ratio) determine which bacteria are likely to dominate commercial bioleaching processes.
Experimental procedures
Industrial heap and samples
Escondida Mine is located 170 km South‐East from Antofagasta, Chile. The heap was built 2 years ago with low‐grade run‐of‐mine sulfide copper ore and air was supplied at the base of the heap through blowers. The ore was characterized as low‐grade sulfide material averaging 0.60% total Cu consisting of chalcocite (40%), covellite (10%) and chalcopyrite (50%). The heap was designed to have one raffinate irrigation solution directed from the solvent‐extraction plant that feeds 14 ore strips (S1–S14) at steady state. Each strip (125 m wide by 2000 m long) generates its own pregnant leaching solution (PLS) and the start of irrigation was approximately 1 month apart with the strip 1 being the oldest (Fig. 1). We defined that around of 400 days of operation correspond to the leaching cycle of each strip. Samples of Feed and PLS from each ore strip were collected every month during 2 years (from June 2006 to May 2008).
DNA and RNA extraction
DNA was extracted from cells collected by filtering 1 l of PLS through a 0.2 µm pore size membrane (Whatman) as described previously (Demergasso et al., 2005).
RNA was extracted from cells collected by filtering 4 l of PLS through a 0.2 µm pore size membrane (Whatman). To preserve the RNA the cells were recovered by scrapping with a sterile spatula in 2 ml of RNAlater solution (Ambion). RNA was purified using RNeasy kit (QIAGEN). The yield of RNA was determined spectrophotometrically using a Nanodrop ND‐1000 (NanoDrop technologies).
DGGE and 16S rRNA clone libraries analysis
DNA from industrial samples was directly used as template for PCR amplification of the 16S rRNA gene for DGGE and clone libraries. For DGGE analysis, PCR products were generated using the universal Bacteria and Archaea primer set as previously described (Muyzer et al., 1993), and the DGGE analysis was performed as previously described (Casamayor et al., 2002; Demergasso et al., 2005). The excision of bands and reamplification was performed according to Casamayor and colleagues (2002). The bacterial clone libraries were constructed using the primer set EUB27F (5′‐AGAGTTTGATCCTGGCTCAG‐3′) and 1492R (5′‐GGTTACCTTGTTACGACTT‐3′) and the archaeal clone libraries were constructed using the set ARQ21F (5′‐TCCGGTTGATCCYGCCGG‐3′) and 1492R. PCR products were cloned into the pGEM®T Easy Vector (Promega) and transformed into JM109 High Efficiency Competent Cells (Promega) according to the manufacturer's instructions. Sequences from DGGE and clone libraries were analysed using the blast algorithm (Altschul et al., 1997) at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov).
Quantitative real‐time PCR analysis
Real‐time PCR was performed with a Rotor‐GeneTM 6200 (Corbett Research Pty Ltd) using SYBR Green PCR Master Mix (Biotools, Spain) to determine the copy numbers of the 16S rRNA gene with specific primers target to Bacteria, Archaea, two phylotypes of At. ferrooxidans, two phylotypes of L. ferriphilum, At. thiooxidans, Sulfobacillus‐like bacteria, Ab. disulfidooxidans, uncultured Alicyclobacillus, Acidiphilium‐like bacteria and F. acidiphilum. The real‐time PCR analysis was used also to determine the transcription levels of 16S rRNA genes of the same analysed microorganisms. Table 2 shows the primer sequences used in this study.
Standard curves were generated extracting plasmid DNA from clones of 16S rRNA gene libraries containing sequences of bioleaching microorganisms inhabiting the industrial heap using a QIAprep Spin Miniprep Kit (QIAGEN). The DNA concentration in nanograms per µl was transformed to 16S rRNA gene copy number per µl as described previously (Remonsellez et al., 2007). Calibrations curves were generated by the RotorGene software 1.7 (Corbett Research), and for each standard the concentration was plotted against the cycle number, and the value at which the fluorescence signal increased above the threshold value was the Cycle threshold (Ct value).
To determine the transcript levels of 16S rRNA genes, the synthesis of cDNA from RNA samples was performed using the Sensiscript RT kit (QIAGEN), according to the manufacturer's instructions using 10 pmol of specific reverse primers (Table 2) and the reaction mixtures were incubated at 37°C for 30 min (Galleguillos et al., 2008).
The reaction mixture contained 10 µl of SYBR® Green PCR Master Mix (Biotools, Spain), 1 µl of DNA or cDNA, 1 µl of the corresponding oligonucleotide primers (Table 2), and nuclease‐free H2O added to a total of 20 µl. The amplification program was developed as described previously (Galleguillos et al., 2008). The melting curves were measured with a ramp raising the temperature from 50°C to 95°C by 1°C each 5 s.
RNA amplification, cDNA labelling and PAM
The RNA quality analysis, total RNA amplification and cDNA labelling were done as described elsewhere (Moreno‐Paz and Parro, 2006). The oligonucleotide PAM construction, hybridization and analysis were done as described previously (Garrido et al., 2008).
Acknowledgments
This work was supported in part by FONDEF Proyect D04I1169 from CONICYT and a technological stay in the Centro de Astrobiología (CSIC‐INTA) from CORFO. F. Remonsellez is supported in part by the Technology and Science Bicentennial Program from CONICYT. V. Parro had a ‘Ramón y Cajal’ contract from the Spanish Ministerio de Ciencia e Innovación. We thank Alex Echeverria for his help in the phylogenetic tree analysis.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Phylogenetic tree based on comparative analysis of 16S rRNA gene sequences representatives of At. ferrooxidans type strains. Sequences were aligned using the alignment tool of ARB program (Strunk and Ludwing, 1995). Phylogenetic tree was generated using the maximum-parsinomy algorithm in the ARB program. The bar indicates a 1% estimated sequence divergence. The sequences obtained in this study are indicated in bold.
Fig. S2. Phylogenetic tree based on comparative analysis of 16S rRNA gene sequences representatives of Leptospirillum strains. Sequences were aligned using the alignment tool of ARB program (Strunk and Ludwing, 1995). Phylogenetic tree was generated using the maximum-parsinomy algorithm in the ARB program. The bar indicates a 1% estimated sequence divergence. The sequences obtained in this study are indicated in bold.
Fig. S3. Assaying total industrial heap RNA with a Prokaryotic acidophile microarray (PAM).
A. Examples of two PAM images corresponding to hybridization with total RNA from strips S10 and S5. The yellow rectangles point out the universal probes UNI1392, Y2R and EU338 for a better orientation.
B. Names and the position of each probe which were described by Garrido et al. (2008). Different coloured rectangles indicate some relevant probes: universal (yellow), those showing higher intensity in earlier (S10, in green) or older (S5, red) stages of the industrial process.
C. Histograms showing the average relative signal intensity of two different hybridizations for the different analyzed strips, from younger to older ones (S10-S8-S5). BACT, Bacteria; Alpha, Alphaproteobacteria; HGC, probes for High GC containing bacteria; LGC, probes for Low GC containing bacteria, Firmicutes; Beta, Betaproteobacteria.
Table S1. Acidophilic microorganisms identified in heap processes.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;268:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond P.L., Druschel G.K., Banfield J.F. Comparison of acid mine drainage microbial communities in physically and geochemically distinct ecosystem. Appl Environ Microbiol. 2000;66:4962–4971. doi: 10.1128/aem.66.11.4962-4971.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon M., Heijnen J.J., Hansford G.S. The mechanism and kinetics of bioleaching sulphide minerals. Min Pro Ext Met Rev. 1998;19:107–115. [Google Scholar]
- Boon M., Brasser H.J., Hansford G.S., Heijnen J.J. Comparison of the oxidation kinetics of different pyrites in the presence of Thiobacillus ferrooxidans or Leptospirillum ferrooxidans. Hydrometallurgy. 1999;53:57–72. [Google Scholar]
- Brierley C.L. Bacterial succession in bioheap leaching. Hydrometallurgy. 2001;59:249–255. [Google Scholar]
- Brodie E.L., Desantis T.Z., Joyner D.C., Baek S.M., Larsen J.T., Andersen G.L. Application of a high‐density oligonucleotide microarray approach to study bacterial population dynamics during uranium reduction and reoxidation. Appl Environ Microbiol. 2006;72:6288–6298. doi: 10.1128/AEM.00246-06. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhn D.F., Thompson D.N., Naoh K.S. microbial ecology assessment of a mixed copper oxide/sulfide dump leach operation. In: Amils R., Ballester A., editors. Elsevier; 1999. pp. 799–808. [Google Scholar]
- Casamayor E.O., Schäfer H., Bañeras L., Pedrós‐Alió C., Muyzer G. Identification of and spatio‐temporal differences between microbial assemblages from two neighboring sulfurous lakes: comparison by microscopy and denaturing gradient gel electrophoresis. App Environ Microbiol. 2002;66:499–508. doi: 10.1128/aem.66.2.499-508.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demergasso C.S., Galleguillos P.A., Escudero L.V., Zepeda V.J., Castillo D., Casamayor E.O. Molecular characterization of microbial populations in a low‐grade copper ore bioleaching test heap. Hydrometallurgy. 2005;80:241–253. [Google Scholar]
- Diaby N., Dold B., Pfeifer H‐R., Holliger C., Johnson D.B., Hallberg K.B. Microbial communities in a porphyry copper tailings impoundment and their impact on the geochemical dynamics of the mine waste. Environ Microbiol. 2007;9:298–307. doi: 10.1111/j.1462-2920.2006.01138.x. [DOI] [PubMed] [Google Scholar]
- Espejo R.T., Romero J. Bacterial community in copper sulfide ores inoculated and leached with solution from a commercial‐scale copper leaching plant. Appl Environ Microbiol. 1997;63:1344–1348. doi: 10.1128/aem.63.4.1344-1348.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey A., Eichler S., Flavier S., Christen R., Hüfle M.G., Guzmán C.A. Establishment of a real‐time PCR‐based approach for accurate quantification of bacterial RNA targets in water, using Salmonella as a model organism. Appl Environ Microbiol. 2004;70:3618–3623. doi: 10.1128/AEM.70.6.3618-3623.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galleguillos P., Remonsellez F., Galleguillos F., Guiliani N., Castillo D., Demergasso C. Identification of differentially expressed genes in an industrial bioleaching heap processing low‐grade copper sulphide ore elucidated by RNA arbitrarily primed polymerase chain reaction. Hydrometallurgy. 2008;94:148–154. [Google Scholar]
- Garrido P., González‐Toril E., García‐Moyano A., Moreno‐Paz M., Amils R., Parro V. An oligonucleotide prokaryotic acidophile microarray (PAM): its validation and its use to monitor seasonal variations in extreme acidic environments with total environmental RNA. Environ Microbiol. 2008;10:836–850. doi: 10.1111/j.1462-2920.2008.01477.x. [DOI] [PubMed] [Google Scholar]
- Gentry T.J., Wickham G.S., Schadt C.W., He Z., Zhou J. Microarray applications in microbial ecology research. Microb Ecol. 2006;52:159–175. doi: 10.1007/s00248-006-9072-6. [DOI] [PubMed] [Google Scholar]
- Goebel B.M., Stackebrandt E. Cultural and phylogenetic analysis of mixed microbial populations found in natural and commercial bioleaching environments. Appl Environ Microbiol. 1994;60:1614–1621. doi: 10.1128/aem.60.5.1614-1621.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg K.B., Johnson D.B. Biodiversity of acidophilic prokaryotes. Adv Appl Microbiol. 2001;49:37–84. doi: 10.1016/s0065-2164(01)49009-5. [DOI] [PubMed] [Google Scholar]
- Hallberg K.B., Coupland K., Kimura S., Johnson D.B. Macroscopic streamer growths in acidic, metal‐rich mine waters in North Wales consist of novel and remarkably simple bacterial communities. Appl Environ Microbiol. 2006;72:2022–2030. doi: 10.1128/AEM.72.3.2022-2030.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes R.B., Franzmann P.D., Plumb J.J. Moderate thermophiles including ‘Ferroplasma cyprexacervatum’ sp. nov., dominate an industrial scale chalcocite heap bioleaching operation. Hydrometallurgy. 2006;83:229–236. [Google Scholar]
- He Z., Xiao S., Xie X., Hu Y. Microbial diversity in acid mineral bioleaching systems of Dongxiang copper mine and Yinshan lead‐zinc mine. Extremophiles. 2008;12:225–234. doi: 10.1007/s00792-007-0130-x. [DOI] [PubMed] [Google Scholar]
- Johnson D.B. Biodiversity and ecology of acidophilic microorganisms. FEMS Microbiol Ecol. 1998;27:307–317. [Google Scholar]
- Johnson D.B., Hallberg K.B. The microbiology of acidic mine waters. Res Microbiol. 2003;154:466–473. doi: 10.1016/S0923-2508(03)00114-1. [DOI] [PubMed] [Google Scholar]
- Johnson D.B., Hallberg K.B. Techniques for detecting and identifying acidophilic mineral‐oxidizing microorganisms. In: Rawlings D.E., Johnson D.B., editors. Springer; 2007. pp. 237–261. [Google Scholar]
- Johnson D.B., Joulian C., D'Hugues P., Hallberg K.B. Sulfobacillus benefaciens sp. nov., and acidophilic facultative anaerobic Firmicute isolated from mineral bioleaching operations. Extremophiles. 2008;12:789–798. doi: 10.1007/s00792-008-0184-4. [DOI] [PubMed] [Google Scholar]
- Kock D., Schippers A. Geomicrobiological investigation of two different mine waste tailings generating acid mine drainage. Hydrometallurgy. 2006;83:167–175. [Google Scholar]
- Kock D., Schippers A. Quantitative microbial community analysis of three different sulfidic mine tailing dumps generating acid mine drainage. Appl Environ Microbiol. 2008;74:5211–5219. doi: 10.1128/AEM.00649-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.‐G., Plumb J., Hendry P. Rapid specific detection and quantification of bacteria and archaea involved in mineral sulfide bioleaching using real‐time PCR. Biotech Bioeng. 2006;94:330–336. doi: 10.1002/bit.20845. [DOI] [PubMed] [Google Scholar]
- Meruane G., Salhe C., Wiertz J., Vargas T. Novel electrochemical‐enzymatic model which quantifies the effect of the solutions Eh on the kinetic of ferrous iron oxidation with Acidithiobacillus ferrooxidans. Biotech Bioeng. 2002;80:280–288. doi: 10.1002/bit.10371. [DOI] [PubMed] [Google Scholar]
- Moreno‐Paz M., Parro V. Amplification of low quantity bacterial RNA for microarray studies: time‐course analysis of Leptospirillum ferrooxidans under nitrogen‐fixing conditions. Environ Microbiol. 2006;8:1064–1073. doi: 10.1111/j.1462-2920.2006.00998.x. [DOI] [PubMed] [Google Scholar]
- Muyzer G., De Waal E.C., Uitterlinden A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction‐amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59:695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadkarni M.A., Martin F.E., Jacques N.A., Hunter N. Determination of bacterial load by real‐time PCR using a broad‐range (universal) probe and primers set. Microbiology. 2002;148:257–266. doi: 10.1099/00221287-148-1-257. [DOI] [PubMed] [Google Scholar]
- Parro V., Moreno‐Paz M., González‐Toril E. Analysis of environmental transcriptomes by DNA microarrays. Environ Microbiol. 2007;9:453–464. doi: 10.1111/j.1462-2920.2006.01162.x. [DOI] [PubMed] [Google Scholar]
- Rawlings D.E. Heavy metal mining using microbes. Annu Rev Microbiol. 2002;56:65–91. doi: 10.1146/annurev.micro.56.012302.161052. [DOI] [PubMed] [Google Scholar]
- Rawlings D.E. Relevance of cell physiology and genetic adaptability of biomining microorganisms to industrial processes. In: Rawlings D.E., Johnson D.B., editors. Elsevier‐Verlag; 2007. pp. 177–198. [Google Scholar]
- Rawlings D.E., Johnson B. The microbiology of biomining: development and optimization of mineral‐oxidizing microbial consortia. Microbiology. 2007;153:315–324. doi: 10.1099/mic.0.2006/001206-0. [DOI] [PubMed] [Google Scholar]
- Rawlings D.E., Tributsch H., Hansford G.S. Reasons why ‘Leptospirillum’‐like species rather than Thiobacillus ferrooxidans are the dominant iron‐oxidizing bacteria in many commercial processes for the biooxidation of pyrite and related ores. Microbiology. 1999;145:5–13. doi: 10.1099/13500872-145-1-5. [DOI] [PubMed] [Google Scholar]
- Remonsellez F., Galleguillos F., Janse van Rensburg S., Rautenbach G.F., Galleguillos P., Castillo D., Demergasso C. Monitoring the microbial community inhabiting a low‐grade copper sulphide ore by Quantitative Real‐Time PCR analysis of 16S rRNA genes. Adv Mat Res. 2007;20(21):539–542. [Google Scholar]
- Rohwerder T., Gehrke T., Kinzler K., Sand W. Bioleaching review part A: progress in bioleaching: fundamentals and mechanisms of bacterial metal sulfide oxidation. Appl Microbiol Biotechnol. 2003;63:239–248. doi: 10.1007/s00253-003-1448-7. [DOI] [PubMed] [Google Scholar]
- Rowe O.F., Sánchez‐España J., Hallberg K.B., Johnson D.B. Microbial communities and geochemical dynamics in an extremely acidic, metal‐rich stream at an abandoned sulfide mine (Huelva, Spain) underpinned by two functional primary production systems. Environ Microbiol. 2007;9:1761–1771. doi: 10.1111/j.1462-2920.2007.01294.x. [DOI] [PubMed] [Google Scholar]
- Sand W. Ferric iron reduction by Thiobacillus ferrooxidans at extremely low pH‐values. Biogeochemistry. 1989;7:195–201. [Google Scholar]
- Sand W., Gehrke T., Jozsa P‐G., Schippers A. Bio)chemistry of bacterial leaching – direct vs. indirect bioleaching. Hydrometallurgy. 2001;59:159–175. [Google Scholar]
- Schippers A. Microorganisms involved in bioleaching and nucleic acid‐based molecular methods for their identification and quantification. In: Donati R.E., Sand W., editors. Springer; 2007. pp. 3–33. [Google Scholar]
- Schippers A., Neretin L.N. Quantification of microbial communities in near‐surface and deeply buried marine sediments on the Peru continental margin using real‐time PCR. Environ Microbiol. 2006;8:1251–1260. doi: 10.1111/j.1462-2920.2006.01019.x. [DOI] [PubMed] [Google Scholar]
- Schippers A., Sand W. Bacterial leaching of metal sulfides proceeds by two indirect mechanisms via thiosulfate or via polysulfides and sulfur. Appl Environ Microbiol. 1999;65:319–321. doi: 10.1128/aem.65.1.319-321.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schippers A., Hallmann R., Wentzien S., Sand W. Microbial diversity in uranium mine waste heaps. Appl Environ Microbiol. 1995;61:2930–2935. doi: 10.1128/aem.61.8.2930-2935.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schippers A., Jozsa P‐G., Sand W. Sulfur chemistry in bacterial leaching of pyrite. Appl Environ Microbiol. 1996;62:3424–3431. doi: 10.1128/aem.62.9.3424-3431.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schippers A., Neretin L.N., Kallmeyer J., Ferdelman T.G., Cragg B.A., Parkes R.J., Jorgensen B.B. Prokaryotic cells of the deep sub‐seafloor biosphere identified as living bacteria. Nature. 2005;433:861–864. doi: 10.1038/nature03302. [DOI] [PubMed] [Google Scholar]
- Takai K., Horikoshi K. Rapid detection and quantification of members of the archaeal community by quantitative PCR using fluorogenic probes. Appl Environ Microbiol. 2000;66:5066–5072. doi: 10.1128/aem.66.11.5066-5072.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valinsky L., Vedova G.D., Scupham A.J., Figueroa A., Yin B., Hartin R.J. Analysis of bacterial community composition by oligonucleotide fingerprinting of rRNA genes. Appl Environ Microbiol. 2002;68:3243–3250. doi: 10.1128/AEM.68.7.3243-3250.2002. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeman K., Auvinen H., Johnson D.B. Microbiological and geochemical dynamics in simulated‐heap leaching of a polymetallic sulfide ore. Biotech Bioeng. 2008;101:739–750. doi: 10.1002/bit.21951. [DOI] [PubMed] [Google Scholar]
- Watling H.R. The bioleaching of sulphide minerals with emphasis on copper suphides – a review. Hydrometallurgy. 2006;84:81–108. [Google Scholar]
- Wielinga B., Lucy J.K., Moore J.N., Seastone O.F., Gannon J.E. Microbiological and geochemical characterization of fluvially deposited sulfidic mine tailings. Appl Environ Microbiol. 1999;65:1548–1555. doi: 10.1128/aem.65.4.1548-1555.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.Y., Thompson D.K., Li G., Hurt R.A., Tiedje J.M., Zhou J. Development and evaluation of functional gene arrays for detection of selected genes in the environment. Appl Environ Microbiol. 2001;67:5780–5790. doi: 10.1128/AEM.67.12.5780-5790.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S., Xie X., Liu J., He Z., Hu Y. Composition and structures of archaeal communities in acid mineral bioleaching system of Dongxiang Copper Mine and Yinshan Lead‐Zinc Mine, China. Curr Microbiol. 2008;57:239–244. doi: 10.1007/s00284-008-9183-z. [DOI] [PubMed] [Google Scholar]
- Xie X., Xiao S., He Z., Liu J., Qiu G. Microbial populations in acid mineral bioleaching system of Tong Shankou Copper Mine, China. J Appl Microbiol. 2007;103:1227–1238. doi: 10.1111/j.1365-2672.2007.03382.x. [DOI] [PubMed] [Google Scholar]
- Yin H., Cao L., Qiu G., Wang D., Kellogg L., Zhou J. Development and evaluation of 50‐mer oligonucleotide arrays for detecting microbial populations in Acid Mine Drainages and bioleaching system. J Microb Methods. 2007;70:165–178. doi: 10.1016/j.mimet.2007.04.011. et al. [DOI] [PubMed] [Google Scholar]
- Zhou J. Microarrays for bacterial detection and microbial community analysis. Curr Opin Microbiol. 2003;6:288–294. doi: 10.1016/s1369-5274(03)00052-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Phylogenetic tree based on comparative analysis of 16S rRNA gene sequences representatives of At. ferrooxidans type strains. Sequences were aligned using the alignment tool of ARB program (Strunk and Ludwing, 1995). Phylogenetic tree was generated using the maximum-parsinomy algorithm in the ARB program. The bar indicates a 1% estimated sequence divergence. The sequences obtained in this study are indicated in bold.
Fig. S2. Phylogenetic tree based on comparative analysis of 16S rRNA gene sequences representatives of Leptospirillum strains. Sequences were aligned using the alignment tool of ARB program (Strunk and Ludwing, 1995). Phylogenetic tree was generated using the maximum-parsinomy algorithm in the ARB program. The bar indicates a 1% estimated sequence divergence. The sequences obtained in this study are indicated in bold.
Fig. S3. Assaying total industrial heap RNA with a Prokaryotic acidophile microarray (PAM).
A. Examples of two PAM images corresponding to hybridization with total RNA from strips S10 and S5. The yellow rectangles point out the universal probes UNI1392, Y2R and EU338 for a better orientation.
B. Names and the position of each probe which were described by Garrido et al. (2008). Different coloured rectangles indicate some relevant probes: universal (yellow), those showing higher intensity in earlier (S10, in green) or older (S5, red) stages of the industrial process.
C. Histograms showing the average relative signal intensity of two different hybridizations for the different analyzed strips, from younger to older ones (S10-S8-S5). BACT, Bacteria; Alpha, Alphaproteobacteria; HGC, probes for High GC containing bacteria; LGC, probes for Low GC containing bacteria, Firmicutes; Beta, Betaproteobacteria.
Table S1. Acidophilic microorganisms identified in heap processes.